

Abstract
This review summarizes the current state of knowledge regarding the role of endothelial dysfunction in the pathogenesis of early and delayed intestinal radiation toxicity and discusses various endothelial-oriented interventions aimed at reducing the risk of radiation enteropathy. Studies published in the biomedical literature during the past four decades and cited in PubMed, as well as clinical and laboratory data from our own research program are reviewed. The risk of injury to normal tissues limits the cancer cure rates that can be achieved with radiation therapy. During treatment of abdominal and pelvic tumors, the intestine is frequently a major dose-limiting factor. Microvascular injury is a prominent feature of both early (inflammatory), as well as delayed (fibroproliferative) radiation injuries in the intestine and in many other normal tissues. Evidence from our and other laboratories suggests that endothelial dysfunction, notably a deficiency of endothelial thrombomodulin, plays a key role in the pathogenesis of these radiation responses. Deficient levels of thrombomodulin cause loss of vascular thromboresistance, excessive activation of cellular thrombin receptors by thrombin, and insufficient activation of protein C, a plasma protein with anticoagulant, anti-inflammatory, and cytoprotective properties. These changes are presumed to be critically involved in many aspects of early intestinal radiation toxicity and may sustain the fibroproliferative processes that lead to delayed intestinal dysfunction, fibrosis, and clinical complications. In conclusion, injury of vascular endothelium is important in the pathogenesis of the intestinal radiation response. Endothelial-oriented interventions are appealing strategies to prevent or treat normal tissue toxicity associated with radiation treatment of cancer.
Keywords: Endothelial cells, Thrombomodulin, Pro-teinase-activated receptors, Radiation injuries, Radiation enteropathy
INTRODUCTION
There are currently more than 10 million cancer survivors in the United States[1]. The exponential increase in the cancer survivor population has led to a stronger focus on reducing treatment-related side effects, thus prompting a more proactive approach aimed at acquiring a better understanding of the molecular and cellular basis of treatment-related side effects, and at developing interventions to ameliorate or prevent long term toxicities of cancer therapy.
Approximately 70% of all cancer patients receive radiation therapy at some point during the course of their disease and radiation therapy plays a critical role in 25% of all cancer cures[2]. Recent advances in treatment delivery, such as the development of dose-sculpting techniques, have led to an overall reduction in normal tissue exposure during radiation therapy. Nevertheless, normal tissue radiation toxicity remains the single-most important dose-limiting factor in radiation therapy and a major obstacle to uncomplicated cancer cures.
More than 200 000 patients in the United States undergo localized radiation therapy for abdominal, pelvic, and retroperitoneal malignancies each year. During treatment of such tumors, the bowel is almost invariably exposed and the risk of intestinal radiation injury (radiation enteropathy) is often the most important dose-limiting factor.
Radiation enteropathy is classified as early (acute) or delayed (chronic). Early radiation enteropathy occurs during or shortly after radiation therapy. It is a consequence of death of rapidly proliferating crypt cells, resulting in epithelial barrier breakdown and mucosal inflammation (radiation mucositis). Delayed radiation enteropathy, by convention, occurs three months or later after radiation therapy. Chronic radiation enteropathy is characterized by vascular sclerosis and progressive intestinal wall fibrosis, leading to intestinal dysfunction (e.g., dysmotility or malabsorption) and structural injury (e.g., stricture formation, fistulas, or perforation). In addition to radiation-induced cell death, radiation enteropathy is the result of a complex interplay among a plethora of pathophysiological processes, including activation of the coagulation system, inflammation, epithelial regeneration, tissue remodeling and collagen deposition. These processes are orchestrated by a large number of cell types and interacting molecular signals, including cytokines and growth factors, as well as various molecules on the endothelial cell surface[3]. Functional perturbation of these endothelial cell molecules is collectively referred to as endothelial dysfunction.
ENDOTHELIAL DYSFUNCTION IN EARLY AND DELAYED RADIATION ENTEROPATHY
Effects of ionizing radiation on the vascular endothelium
Endothelial cells form the inner lining of blood vessels and cover a total surface area of 4000-7000 m2[4]. Endothelial cells are highly dynamic and participate in a multitude of physiological functions, including maintenance of blood fluidity, control of vasomotor tone, trafficking of cells and nutrients, and growth of new blood vessels[5]. Under normal conditions, endothelial cells maintain an antithrombotic and anticoagulant balance by exerting molecular control of platelet aggregation, coagulation and fibrinolysis[6].
An increasing body of evidence shows that injury of the microvasculature plays a central role in early and delayed radiation responses in many normal tissues, including the intestine. Notably, microvascular injury may be responsible for the unique self-perpetuating nature of chronic radiation fibrosis[7-13]. A model depicting how endothelial cell dysfunction may contribute to and sustain post-radiation inflammatory and fibroproliferative responses in the intestine is shown in Figure 1.
Figure 1.
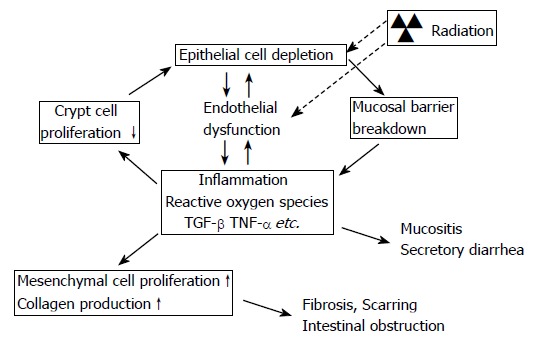
Model of interaction between epithelial and endothelial radiation injury in the intestine demonstrating how endothelial dysfunction may exacerbate the early intestinal radiation response and “drive” the cycle of chronicity of intestinal radiation fibrosis. Radiation causes epithelial crypt cell death, leading to insufficient replacement of the villus epithelium, and breakdown of the epithelial barrier that normally separates intestinal tissue from the intraluminal contents of the intestine. Simultaneously, radiation causes endothelial dysfunction, notably loss of thromboresistance and increased expression of chemokines and adhesion molecules. The combination of loss of epithelial barrier function and endothelial dysfunction enhances the post-radiation inflammatory response, inhibits restitution of the epithelium, and promotes extracellular matrix deposition.
The high radiation sensitivity of the microvasculature is to a large extent attributable to the endothelial cells[14]. Radiation induces a plethora of morphological and functional alterations in endothelial cells, including apoptosis, detachment from the basement membrane, and increased endothelial permeability, resulting in fibrin deposition in the interstitial space[15,16].
The role of endothelial apoptosis in early intestinal radiation toxicity, particularly in the so-called acute gastrointestinal radiation syndrome, has been a much debated issue for a number of years. The debate originated from reports that mice deficient in the enzyme acid sphingomyelinase are protected from radiation-induced endothelial cell apoptosis, and that these mice also exhibit decreased levels of crypt cell apoptosis and decreased lethality after total body irradiation[17]. Because endothelial cell apoptosis, but not apoptosis of the crypt epithelium, is sphingomyelin-dependent, the interpretation of this finding, together with a substantial body of additional supportive evidence, was that endothelial cell apoptosis appears to be a major contributor to early intestinal radiation toxicity and that there may be a causal relationship between endothelial cell apoptosis and crypt cell apoptosis. There has, however, been considerable controversy related to the extent and significance of endothelial apoptosis in the intestinal microvasculature after radiation exposure, and to whether or not there is a direct relationship between endothelial apoptosis and apoptosis in the crypt epithelium[18]. Despite this controversy, it may be possible to reconcile these seemingly contradictory findings. It is well known from other areas of gastrointestinal pathophysiology that genetic manipulations or pharmacologic interventions that preserve the intestinal microcirculation after an insult have a protective effect on the gut epithelium and the intestinal mucosa. Therefore, it is conceivable that radiation-induced endothelial cell apoptosis may be the bellwether, or “tip of the iceberg” that indicates a state of dysfunction of the intestinal microvasculature, and that it is the state of endothelial dysfunction that adversely affects the radiation tolerance and/or repair capacity of the crypt epithelium.
Loss of thromboresistance is a major feature of endothelial dysfunction after exposure to ionizing radiation. Radiation induces adhesion and aggregation of platelets and development of platelet-fibrin thrombi[19-22], as well as adhesion of inflammatory cells to the endothelium[23-25] with subsequent perivascular leukocyte infiltration. The molecular basis underlying the loss of endothelial thromboresistance is complex and includes increased expression of tissue factor[26,27], von Willebrand factor (vWF)[28-30], and platelet activating factor (PAF)[31]; reduction in fibrinolytic activity[32-34]; and radiation-induced reduction in the expression of prostacyclin (PGI2), the PGI2 receptor[35-37], and thrombomodulin (TM)[12,38]. Studies performed in our laboratory suggest that radiation-induced loss of TM may play a particularly important role in the pathogenesis of radiation enteropathy.
The thrombomodulin-protein C system
Endothelial TM is a transmembrane glycoprotein located on the luminal surface of endothelial cells in most normal blood vessels. TM forms a complex with thrombin, and essentially converts thrombin from a pro-coagulant to an anticoagulant by changing its substrate specificity. Thrombin, when in complex with TM, no longer cleaves fibrinogen to form fibrin and no longer activates cellular thrombin receptors, but instead activates protein C, thereby limiting further thrombin generation and counteracting thrombin’s many coagulant, inflammatory, and fibroproliferative effects (Figure 2). In addition, both TM and activated protein C (APC) have important intrinsic anti-inflammatory properties.
Figure 2.
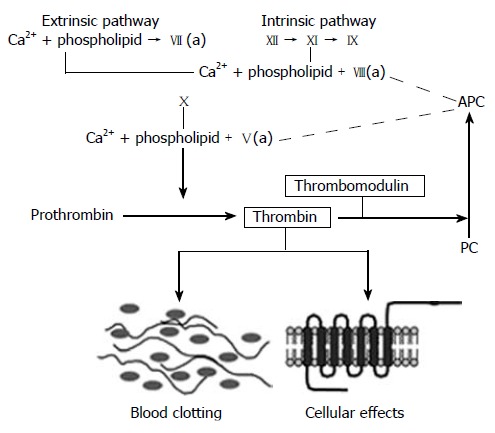
The coagulation cascade. Simplified diagram of the coagulation “cascade” with the intrinsic, extrinsic, and common pathways. Note how thrombomodulin, located on the luminal surface of endothelial cells, forms a complex with thrombin, which is converted from a pro-coagulant to an anticoagulant and how activated protein C (APC) limits thrombin generation by feed-back into the intrinsic and common coagulation pathways. See text for further details.
Recent studies have demonstrated the importance of TM in attenuation of inflammatory responses in a variety of settings, such as, endotoxin-induced tissue damage, glomerulonephritis, and atherosclerosis[39-42]. One mechanism by which TM exerts its anti-inflammatory properties involves APC. APC inhibits leukocyte chemotaxis and leukocyte adhesion, suppresses inflammatory cytokine production, reduces endothelial cell apoptosis, and maintains endothelial cell barrier function[43-48]. In addition, recent studies have shown that TM has potent intrinsic anti-inflammatory properties by virtue of its N-terminal domain binding and inhibiting high mobility group box 1 protein (HMGB1)[49].
Clinical and preclinical studies performed in our laboratory have shown that radiation causes a striking (80%-90%) and sustained reduction in endothelial TM expression in intestinal microvasculature[12,50,51]. The reduction in TM appears to be due to a combination of direct oxidative damage [52,53], and down regulation of TM at the gene expression level by radiation-induced inflammatory cytokines such as interleukin 1 (IL1), tumor necrosis factor α (TNFα) and transforming growth factor β (TGFβ)[54-57], and increased release of TM from the endothelial cell membrane into the circulation (ectodomain shedding) by granulocyte proteinases and other inflammatory mediators[58].
Thrombin and cellular thrombin receptors
In the normal situation, thrombin is rapidly removed from the microcirculation by complex formation with TM. Local deficiency of TM, such as occurs after irradiation, leads to decreased thrombin clearance and insufficient protein C activation, resulting in accumulation of thrombin. Moreover, the expression of tissue factor, a critical initiator of thrombin generation, can also be triggered by radiation, both in vitro and in vivo[26,27]. Hence, radiation enhances thrombin generation both through the intrinsic and the extrinsic pathway.
Thrombin induces gap formation between endothelial cells, resulting in increased vascular permeability[59-62]. Consequently, thrombin may pass through the endothelial cell layer into the vessel wall and extravascular tissues. Studies performed in our laboratory show that radiation causes deposition of enzymatically active thrombin on the vascular endothelium, in the vascular wall of small arteries, as well as in the extravascular connective tissue[27]. Thrombin bound to extracellular matrix remains functionally active and able to generate fibrin and interact with surrounding cells[63,64]. We have demonstrated increased deposition of fibrin in irradiated intestine that co-localizes with enzymatically active thrombin[27].
Thrombin, in addition to its central role in coagulation, activates a variety of cell types including endothelial cells, smooth muscle cells, leukocytes, and platelets, thereby enhancing many inflammatory and fibroproliferative processes. For example, thrombin has chemotactic activity for monocytes and leukocytes and stimulates the migration of these cells to sites of injury[65]. Thrombin stimulates fibroblast chemotaxis[66], fibroblast proliferation[67,68], and fibroblast procollagen production[69]. Thrombin also enhances proliferation and migration of smooth muscle cells (SMC) and promotes SMC procollagen synthesis[70-72].
The cellular effects of thrombin are mediated by activation of cell surface thrombin receptors, proteinase activated receptors (PARs), a 4-member G-protein coupled receptor subfamily. Proteinase activated receptor 1 (PAR1) is the biologically most relevant among the PARs[71-75]. Studies performed in our laboratory show that radiation upregulates PAR1 expression in endothelium, SMC, and myofibroblasts, particularly in areas of fibrosis[27]. Increased expression of PAR1 in SMC may be particularly important in the context of intestinal wall fibrosis. This is because, in the intestine, SMC rather than fibroblasts are the predominant producers of collagen. Analogous to our observations in radiation enteropathy, upregulation of PAR1 occurs in a number of other vascular disorders, including neointima formation after mechanical injury[73], as well as in response to injury-related cytokines and growth factors. Figure 3 depicts a model for how deficient levels of TM after radiation exposure, with subsequent increased thrombin formation and upregulation of PAR1, may contribute to and sustain inflammatory and fibroproliferative responses in irradiated tissues. Consistent with this model, in vivo studies performed in our laboratory have confirmed that scavenging active TGFβ1[76], inhibiting platelet aggregation[77], inhibiting thrombin function[27], mucosal immunomodulation[78], or inhibiting PAR1 (unpublished data, 2005) all ameliorate various aspects of early and/or delayed radiation enteropathy. These studies are consistent with the notion that thrombin is a key link between downregulated TM and radiation-induced vascular and intestinal fibrosis.
Figure 3.
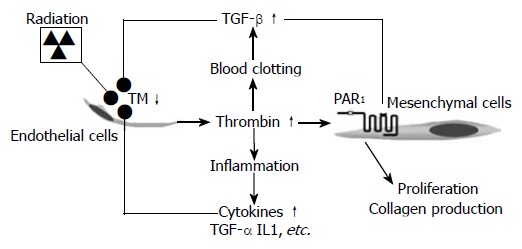
Proposed model linking radiation-induced endothelial dysfunction to chronic inflammation and progressive intestinal fibrosis via chronic PAR1 activation. Radiation causes TM deficiency in endothelial cells, leading to insufficient “scavenging” of locally formed thrombin. Thrombin exerts pro-coagulant, pro-inflammatory, mitogenic, and pro-fibrogenic effects on mesenchymal cells (smooth muscle cells, fibroblasts, and myofibroblasts), as well as other cell types in the irradiated tissue. Feed-back by cytokines and other inflammatory mediators sustains the endothelial TM deficiency and thus contributes to the chronicity of radiation injury.
Platelets
Thrombin, in addition to the properties described above, also has major effects on blood platelets. Platelets are the first cellular elements at the site of endothelial injury, where they initiate the hemostatic and inflammatory responses and contribute to the local cytokine milieu[79]. In vivo and in vitro studies have demonstrated that radiation enhances platelet adhesion[80] and platelet aggregation in the microvascular network[19,20]. The anti-platelet agent, acetylsalicylic acid (ASA, aspirin) may ameliorate certain aspects of intestinal and renal radiation toxicity[81-83].
Platelet adhesion, aggregation, and secretion are regulated by several mediators that are recognized by platelet surface receptors. Thrombin is a powerful platelet agonist and PARs mediate most of the actions of thrombin on platelet function[84]. Hence, PAR1 activating peptide (PAR1-AP) triggers complete platelet aggregation similar to the aggregation induced by thrombin. Adenosine diphosphate (ADP) is stored in platelet granules and is released in response to primary agonists, including thrombin. Thus, part of the response of platelets to thrombin is via autocrine and paracrine effects by secreted ADP[85]. In fact, some studies suggest that PAR1-AP-induced aggregation may be entirely dependent on release of ADP[86]. ADP potentiates multiple platelet responses including the initiation of platelet aggregation (by receptor P2Y1) and the subsequent full aggregation and stabilization of platelet aggregates (by receptor P2Y12)[87,88]. Recent studies from our laboratory and others show that inhibition of ADP-induced platelet aggregation by clopidogrel or ticlopidine ameliorates early and delayed intestinal radiation toxicity[77,89].
Activated platelets directly elicit an inflammatory response by the production of free radicals and by the release of potent inflammatory mediators, such as, TGFβ, PAF, thromboxane, platelet derived growth factor, and IL1, which all contribute to chemoattraction and activation of inflammatory cells[79,90]. The ubiquitous proinflammatory, immunosuppressive, and fibrogenic growth factor, TGFβ, has been implicated in radiation injury such as skin, liver, heart, kidney, lung and intestine[76,91-93]. Platelets contain TGFβ in about 100-fold higher amounts than other types of cells or tissues. We have observed that TGFβ is expressed at significantly higher than normal levels after irradiation[94-96]. Moreover, our studies in radiation enteropathy were the first to demonstrate a mechanistic role for TGFβ in radiation-induced tissue toxicity[76].
ENDOTHELIAL-ORIENTED APPROACHES TO MODULATE RADIATION ENTEROPATHY
As described in the previous sections of this review, radiation induces a plethora of changes in the microvascular endothelium. Some of these changes are transient, but may contribute to aspects of early radiation enteropathy. Other changes are sustained and may play direct roles in the pathogenesis of intestinal radiation fibrosis and in the mechanisms of chronicity and progression of injury. The postradiation shift in the thrombohemorrhagic balance toward procoagulation and the accompanying cellular effects that are the consequences of this shift represent particularly promising targets for intervention (Table 1).
Table 1.
Potential pharmacological strategies for modulating post-radiation endothelial dysfunction to ameliorate development of radiation enteropathy and some of their respective limitations
| Intervention | Major limitation |
| Platelet aggregation inhibitors | Narrow therapeutic window (bleeding) |
| Direct thrombin inhibitors | Narrow therapeutic window (bleeding) |
| Thrombin receptor blockers | Blocks only cellular thrombin effects |
| Recombinant thrombomodulin | Does not restore endothelial thrombomodulin |
| Activated protein C | Only partly blocks the effects of preformed thrombin |
| Statins | Non-specificity |
| Pentoxifylline | Non-specificity |
| Vitamin E | Non-specificity and variable efficacy |
Many of the conventional inhibitors of blood clotting have been tested in the attempt to ameliorate normal tissue radiation toxicity. The inconsistent results of these interventions are likely a result of the use of non-specific drugs with multiple actions, use of compounds with dose-limiting side-effects (primarily bleeding), and/or a too narrow focus on coagulation without appropriate consideration of the cellular effects of thrombin and the anti-inflammatory properties of the TM-protein C pathway. For example, while heparin is a highly effective anticoagulant, at therapeutic concentrations heparin reduces the affinity of thrombin for TM and the rate of protein C activation[97], and heparin administered at the time of irradiation actually exacerbates radiation-induced intestinal tissue injury[98]. The direct thrombin inhibitor, hirudin, ameliorates radiation enteropathy, but is less effective than an inhibitor of ADP-induced platelet aggregation, clopidogrel[27]. A possible explanation of these findings may be that thrombin inhibition also reduces thrombin-induced protein C activation and thereby the anti-inflammatory actions of APC. In contrast, inhibition of ADP-induced platelet aggregation targets processes downstream of thrombin and does not influence APC in the same manner. These observations are consistent with results from other studies showing that direct thrombin inhibition enhances leukocyte-endothelial cell interaction in endotoxin-induced sepsis[99] and, despite a favorable effect on collagen accumulation, does not affect inflammatory cell recruitment in bleomycin-induced lung injury[100].
Particularly attractive and presumably safe approaches to modulate radiation-induced endothelial dysfunction are to administer exogenous recombinant TM and APC, to restore endothelial cell TM, and/or to block the downstream effector of thrombin, PAR1.
Recombinant human soluble TM (rhsTM) is composed of the active, extracellular domain of TM. rhsTM activates protein C[101], reduces thrombin generation[102], and prevents thrombosis in vivo[103-105]. The efficacy of rhsTM has been demonstrated in other situations associated with deficiency of endothelial TM, such as disseminated intravascular coagulation, experimental sepsis, and multiple system organ failure[42,105,106]. rhsTM also inhibits smooth muscle proliferation and vascular neointimal hyperplasia[107,108]. Although rhsTM has not yet been tested in the context of radiation toxicity, it is conceivable that rhsTM may be beneficial in normal tissue radiation toxicity. The objective would be to provide TM by the exogenous route for a limited period of time and thus allow TM to regenerate on the endothelial surface.
Synthetic TM mimics are compounds that change thrombin’s substrate specificity in a fashion similar to TM and thus cause thrombin to activate protein C[109]. This is a new class of antithrombotic agents that exploits the powerful natural protein C anticoagulant pathway. This approach may be particularly appealing in the context of radiation enteropathy, because localized radiation does not cause protein C deficiency, but rather induces a decrease in local protein C activation due to lack of functional TM. However, while the TM mimics may have a superior therapeutic profile compared to direct thrombin inhibitors, TM mimics suitable for use in vivo are not yet available.
Replacement therapy with recombinant APC (rAPC) is another strategy that might allow endothelial function to recover and thus interrupt the vicious cycle that leads to radiation-induced organ dysfunction. APC possesses a number of properties that are different from those of conventional anticoagulants, including potent anti-inflammatory and cytoprotective activities[110-113]. Studies by others have shown that rAPC prevents the lethal effects of E. coli-associated sepsis in animal models and improves the outcome of patients with severe sepsis[114], and that short-term rAPC administration ameliorates lung fibrosis in bleomycin-induced lung injury[115]. Administration of rAPC during the early postradiation phase warrants investigation as an approach to mitigate radiation enteropathy development.
A particularly interesting approach to upregulate and/or restore endothelial TM is treatment with inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, for example, the lipid-lowering statins. In 2003, we and a Japanese group demonstrated independently that statins, in addition to inhibiting the biosynthesis of cholesterol, strongly upregulate TM gene expression, protein levels, and function[116,117] and counteract the effects of TNFα on endothelial TM[116]. It was subsequently shown that statins attenuate radiation pneumonitis[118] and early radiation-induced intestinal toxicity[119]. Whether the radioprotective properties of statins are indeed attributable to their effect on TM expression or to other non-lipid-related statin effects, and whether the beneficial effect of statins in experimental models of normal tissue radiation toxicity can be translated to the clinical situation remains to be shown.
Pentoxifylline as monotherapy or in combination with tocopherol (vitamin E) is another approach that may ameliorate normal tissue radiation toxicity in some tissues by decreasing endothelial dysfunction and restoring endothelial TM. Pentoxifylline is a methylxanthine derivative with potent hemorrheologic properties. It improves blood fluidity by multiple effects such as increasing the deformability of red blood cells and leukocytes, preventing the aggregation of platelets, and decreasing plasma viscosity. It was originally developed for treatment of regional microcirculation disorders such as intermittent claudication and cerebrovascular disease. However, recent studies have shown that pentoxifylline possesses anti-inflammatory and immunomodulatory properties[120-122] and can be used as an adjuvant in the treatment of a diverse group of diseases, including sepsis and severe acute respiratory distress syndrome. Pentoxifylline increases endothelial TM expression and prevents hypoxic- and TNFα-induced reduction in TM expression[123,124]. Pentoxifylline also inhibits TF expression and counteracts activation of the coagulation cascade by endotoxin[125]. Clinical studies suggest that pentoxifylline may reverse radiation-induced chronic skin and subcutaneous tissue fibrosis[126]. Beneficial effects have also been observed in radiation-induced ulcer healing, as well as in radiation-induced toxicity in lung, intestine, uterine, breast, and jaw muscles[127-131]. Nevertheless, a number of negative animal studies[132,133] and several inconclusive clinical reports highlight the need for further studies to define the benefits, indications, and mechanisms of action of pentoxifylline in radiation fibrosis.
Inhibition of PAR1 may prove to be a particularly effective strategy to reduce radiation-induced normal tissue toxicity. Because PAR1 antagonists are specific for the cellular actions of thrombin, it does not interfere with formation of the thrombin-TM complex and therefore does not reduce activation of protein C. Furthermore, since PAR1 inhibitors do not interfere with fibrin generation, they will likely be associated with fewer bleeding complications than other anticoagulants. Several peptide and non-peptide (small molecule) PAR1 antagonists are under development[134,135]. Some act on the extracellular portion of the receptors[134], whereas others act as intracellular inhibitors of signal transduction from receptors to G proteins[135]. Studies of PAR1 inhibition as an approach to reduce normal tissue radiation toxicity are currently underway in our laboratory.
CONCLUSIONS
Normal tissue toxicity, including intestinal radiation toxicity, is the main dose-limiting factor during radiation therapy of cancer. Radiation enteropathy adversely impacts the therapeutic efficacy of radiation therapy, as well as the quality of life of long term cancer survivors. Clinical and preclinical evidence strongly suggests that endothelial dysfunction plays a critical role in the pathogenesis of early and delayed radiation enteropathy. Various endothelial-oriented pharmacological interventions are currently under development for the purpose of preventing or treating radiation enteropathy. Strategies aimed at restoring or preserving endothelial TM or blocking the thrombin receptor, PAR1, hold particular promise, especially if interventions can be targeted to specific tissues or cellular compartments.
Footnotes
Supported by National Institutes of Health, Grant CA83719 and US Department of Veterans Affairs
S- Editor Liu Y L- Editor Alpini GD E- Editor Wang HF
References
- 1.National Cancer Policy Board CoCS, Hewitt M, Greenfield S, Stovall E. From Cancer Patient to Cancer Survivor: Lost in Transition. Hewitt M, Greenfield S, Stovall E, editors. Washington DC: The National Academies Press; 2006. [Google Scholar]
- 2.DeVita VT, Hellman S, Rosenberg SA. Cancer: Principles and Practice of Oncology. Philadelphia: Lippincott Williams & Wilkins; 2005. [Google Scholar]
- 3.Denham JW, Hauer-Jensen M. The radiotherapeutic injury--a complex 'wound'. Radiother Oncol. 2002;63:129–145. doi: 10.1016/s0167-8140(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 4.Wolinsky H. A proposal linking clearance of circulating lipoproteins to tissue metabolic activity as a basis for understanding atherogenesis. Circ Res. 1980;47:301–311. doi: 10.1161/01.res.47.3.301. [DOI] [PubMed] [Google Scholar]
- 5.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- 6.Pearson JD. Endothelial cell function and thrombosis. Baillieres Best Pract Res Clin Haematol. 1999;12:329–341. doi: 10.1053/beha.1999.0028. [DOI] [PubMed] [Google Scholar]
- 7.Baker DG, Krochak RJ. The response of the microvascular system to radiation: a review. Cancer Invest. 1989;7:287–294. doi: 10.3109/07357908909039849. [DOI] [PubMed] [Google Scholar]
- 8.Hopewell JW, Calvo W, Jaenke R, Reinhold HS, Robbins ME, Whitehouse EM. Microvasculature and radiation damage. Recent Results Cancer Res. 1993;130:1–16. doi: 10.1007/978-3-642-84892-6_1. [DOI] [PubMed] [Google Scholar]
- 9.Jaenke RS, Robbins ME, Bywaters T, Whitehouse E, Rezvani M, Hopewell JW. Capillary endothelium. Target site of renal radiation injury. Lab Invest. 1993;68:396–405. [PubMed] [Google Scholar]
- 10.Lyubimova N, Hopewell JW. Experimental evidence to support the hypothesis that damage to vascular endothelium plays the primary role in the development of late radiation-induced CNS injury. Br J Radiol. 2004;77:488–492. doi: 10.1259/bjr/15169876. [DOI] [PubMed] [Google Scholar]
- 11.Rezvani M, Hopewell JW, Robbins ME. Initiation of non-neoplastic late effects: the role of endothelium and connective tissue. Stem Cells. 1995;13 Suppl 1:248–256. doi: 10.1002/stem.5530130730. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Zheng H, Ou X, Fink LM, Hauer-Jensen M. Deficiency of microvascular thrombomodulin and up-regulation of protease-activated receptor-1 in irradiated rat intestine: possible link between endothelial dysfunction and chronic radiation fibrosis. Am J Pathol. 2002;160:2063–2072. doi: 10.1016/S0002-9440(10)61156-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fajardo LF. The pathology of ionizing radiation as defined by morphologic patterns. Acta Oncol. 2005;44:13–22. doi: 10.1080/02841860510007440. [DOI] [PubMed] [Google Scholar]
- 14.Fajardo LF. The complexity of endothelial cells. A review. Am J Clin Pathol. 1989;92:241–250. doi: 10.1093/ajcp/92.2.241. [DOI] [PubMed] [Google Scholar]
- 15.Heckmann M, Douwes K, Peter R, Degitz K. Vascular activation of adhesion molecule mRNA and cell surface expression by ionizing radiation. Exp Cell Res. 1998;238:148–154. doi: 10.1006/excr.1997.3826. [DOI] [PubMed] [Google Scholar]
- 16.Langley RE, Bump EA, Quartuccio SG, Medeiros D, Braunhut SJ. Radiation-induced apoptosis in microvascular endothelial cells. Br J Cancer. 1997;75:666–672. doi: 10.1038/bjc.1997.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–297. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 18.Schuller BW, Binns PJ, Riley KJ, Ma L, Hawthorne MF, Coderre JA. Selective irradiation of the vascular endothelium has no effect on the survival of murine intestinal crypt stem cells. Proc Natl Acad Sci USA. 2006;103:3787–3792. doi: 10.1073/pnas.0600133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang HF, Li XD, Chen YM, Yuan LB, Foye WO. Radiation-protective and platelet aggregation inhibitory effects of five traditional Chinese drugs and acetylsalicylic acid following high-dose gamma-irradiation. J Ethnopharmacol. 1991;34:215–219. doi: 10.1016/0378-8741(91)90040-k. [DOI] [PubMed] [Google Scholar]
- 20.Amoaku WM, Mahon GJ, Gardiner TA, Frew L, Archer DB. Late ultrastructural changes in the retina of the rat following low-dose X-irradiation. Graefes Arch Clin Exp Ophthalmol. 1992;230:569–574. doi: 10.1007/BF00181780. [DOI] [PubMed] [Google Scholar]
- 21.Schneider MD. Functional aspects of blood platelets in irradiated burros. Am J Vet Res. 1977;38:209–216. [PubMed] [Google Scholar]
- 22.Bicher HI, D'Agostino L, Doss LL, Kaufman N, Amigone J. Prevention of ionizing radiation-induced liver microcirculation changes by the use of flow improvers. Adv Exp Med Biol. 1977;94:383–389. doi: 10.1007/978-1-4684-8890-6_50. [DOI] [PubMed] [Google Scholar]
- 23.Dunn MM, Drab EA, Rubin DB. Effects of irradiation on endothelial cell-polymorphonuclear leukocyte interactions. J Appl Physiol (1985) 1986;60:1932–1937. doi: 10.1152/jappl.1986.60.6.1932. [DOI] [PubMed] [Google Scholar]
- 24.Chan CC, Nathaniel DJ, Yusko PJ, Hall RA, Ford-Hutchinson AW. Inhibition of prostanoid-mediated platelet aggregation in vivo and in vitro by 3-hydroxymethyl-dibenzo(b,f)thiepin 5,5-dioxide (L-640,035) J Pharmacol Exp Ther. 1984;229:276–282. [PubMed] [Google Scholar]
- 25.Hallahan D, Clark ET, Kuchibhotla J, Gewertz BL, Collins T. E-selectin gene induction by ionizing radiation is independent of cytokine induction. Biochem Biophys Res Commun. 1995;217:784–795. doi: 10.1006/bbrc.1995.2841. [DOI] [PubMed] [Google Scholar]
- 26.Verheij M, Dewit LG, van Mourik JA. The effect of ionizing radiation on endothelial tissue factor activity and its cellular localization. Thromb Haemost. 1995;73:894–895. [PubMed] [Google Scholar]
- 27.Wang J, Zheng H, Ou X, Albertson CM, Fink LM, Herbert JM, Hauer-Jensen M. Hirudin ameliorates intestinal radiation toxicity in the rat: support for thrombin inhibition as strategy to minimize side-effects after radiation therapy and as countermeasure against radiation exposure. J Thromb Haemost. 2004;2:2027–2035. doi: 10.1111/j.1538-7836.2004.00960.x. [DOI] [PubMed] [Google Scholar]
- 28.van Kleef E, Verheij M, te Poele H, Oussoren Y, Dewit L, Stewart F. in vitro and in vivo expression of endothelial von Willebrand factor and leukocyte accumulation after fractionated irradiation. Radiat Res. 2000;154:375–381. doi: 10.1667/0033-7587(2000)154[0375:ivaive]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 29.Boerma M, Kruse JJ, van Loenen M, Klein HR, Bart CI, Zurcher C, Wondergem J. Increased deposition of von Willebrand factor in the rat heart after local ionizing irradiation. Strahlenther Onkol. 2004;180:109–116. doi: 10.1007/s00066-004-1138-0. [DOI] [PubMed] [Google Scholar]
- 30.Stewart FA, Te Poele JA, Van der Wal AF, Oussoren YG, Van Kleef EM, Kuin A, Verheij M, Dewit LG. Radiation nephropathy--the link between functional damage and vascular mediated inflammatory and thrombotic changes. Acta Oncol. 2001;40:952–957. doi: 10.1080/02841860152708233. [DOI] [PubMed] [Google Scholar]
- 31.McManus LM, Ostrom KK, Lear C, Luce EB, Gander DL, Pinckard RN, Redding SW. Radiation-induced increased platelet-activating factor activity in mixed saliva. Lab Invest. 1993;68:118–124. [PubMed] [Google Scholar]
- 32.Henderson BW, Bicher HI, Johnson RJ. Loss of vascular fibrinolytic activity following irradiation of the liver--an aspect of late radiation damage. Radiat Res. 1983;95:646–652. [PubMed] [Google Scholar]
- 33.Svanberg L, Astedt B, Kullander S. On radiation-decreased fibrinolytic activity of vessel walls. Acta Obstet Gynecol Scand. 1976;55:49–51. doi: 10.3109/00016347609156783. [DOI] [PubMed] [Google Scholar]
- 34.Ts'ao CH, Ward WF, Port CD. Radiation injury in rat lung. III. Plasminogen activator and fibrinolytic inhibitor activities. Radiat Res. 1983;96:301–308. [PubMed] [Google Scholar]
- 35.Hosoi Y, Yamamoto M, Ono T, Sakamoto K. Prostacyclin production in cultured endothelial cells is highly sensitive to low doses of ionizing radiation. Int J Radiat Biol. 1993;63:631–638. doi: 10.1080/09553009314450821. [DOI] [PubMed] [Google Scholar]
- 36.Leigh PJ, Cramp WA, MacDermot J. Identification of the prostacyclin receptor by radiation inactivation. J Biol Chem. 1984;259:12431–12436. [PubMed] [Google Scholar]
- 37.Eldor A, Vlodavsky I, Riklis E, Fuks Z. Recovery of prostacyclin capacity of irradiated endothelial cells and the protective effect of vitamin C. Prostaglandins. 1987;34:241–255. doi: 10.1016/0090-6980(87)90247-4. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Q, Zhao Y, Li P, Bai X, Ruan C. Thrombomodulin as a marker of radiation-induced endothelial cell injury. Radiat Res. 1992;131:285–289. [PubMed] [Google Scholar]
- 39.Uchiba M, Okajima K, Murakami K, Johno M, Okabe H, Takatsuki K. Recombinant thrombomodulin prevents endotoxin-induced lung injury in rats by inhibiting leukocyte activation. Am J Physiol. 1996;271:L470–L475. doi: 10.1152/ajplung.1996.271.3.L470. [DOI] [PubMed] [Google Scholar]
- 40.Kaido T, Yoshikawa A, Seto S, Yamaoka S, Furuyama H, Arii S, Takahashi Y, Imamura M. Pretreatment with soluble thrombomodulin prevents intrasinusoidal coagulation and liver dysfunction following extensive hepatectomy in cirrhotic rats. Thromb Haemost. 1999;82:1302–1306. [PubMed] [Google Scholar]
- 41.Waugh JM, Li-Hawkins J, Yuksel E, Kuo MD, Cifra PN, Hilfiker PR, Geske R, Chawla M, Thomas J, Shenaq SM, et al. Thrombomodulin overexpression to limit neointima formation. Circulation. 2000;102:332–337. doi: 10.1161/01.cir.102.3.332. [DOI] [PubMed] [Google Scholar]
- 42.Hasegawa N, Kandra TG, Husari AW, Veiss S, Hart WT, Hedgpeth J, Wydro R, Raffin TA. The effects of recombinant human thrombomodulin on endotoxin-induced multiple-system organ failure in rats. Am J Respir Crit Care Med. 1996;153:1831–1837. doi: 10.1164/ajrccm.153.6.8665042. [DOI] [PubMed] [Google Scholar]
- 43.Hoffmann JN, Vollmar B, Laschke MW, Fertmann JM, Jauch KW, Menger MD. Microcirculatory alterations in ischemia-reperfusion injury and sepsis: effects of activated protein C and thrombin inhibition. Crit Care. 2005;9 Suppl 4:S33–S37. doi: 10.1186/cc3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW. Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-gamma, or phorbol ester. J Immunol. 1994;153:3664–3672. [PubMed] [Google Scholar]
- 45.Yuksel M, Okajima K, Uchiba M, Horiuchi S, Okabe H. Activated protein C inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappa B and activator protein-1 in human monocytes. Thromb Haemost. 2002;88:267–273. [PubMed] [Google Scholar]
- 46.Schmidt-Supprian M, Murphy C, While B, Lawler M, Kapurniotu A, Voelter W, Smith O, Bernhagen J. Activated protein C inhibits tumor necrosis factor and macrophage migration inhibitory factor production in monocytes. Eur Cytokine Netw. 2000;11:407–413. [PubMed] [Google Scholar]
- 47.White B, Schmidt M, Murphy C, Livingstone W, O'Toole D, Lawler M, O'Neill L, Kelleher D, Schwarz HP, Smith OP. Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor kappaB (NF-kappaB) and tumour necrosis factor alpha (TNF-alpha) production in the THP-1 monocytic cell line. Br J Haematol. 2000;110:130–134. doi: 10.1046/j.1365-2141.2000.02128.x. [DOI] [PubMed] [Google Scholar]
- 48.Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32 Suppl 1:49–60. doi: 10.1055/s-2006-939554. [DOI] [PubMed] [Google Scholar]
- 49.Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, Uchimura T, Ida N, Yamazaki Y, Yamada S, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richter KK, Fink LM, Hughes BM, Sung CC, Hauer-Jensen M. Is the loss of endothelial thrombomodulin involved in the mechanism of chronicity in late radiation enteropathy? Radiother Oncol. 1997;44:65–71. doi: 10.1016/s0167-8140(97)00063-7. [DOI] [PubMed] [Google Scholar]
- 51.Richter KK, Fink LM, Hughes BM, Shmaysani HM, Sung CC, Hauer-Jensen M. Differential effect of radiation on endothelial cell function in rectal cancer and normal rectum. Am J Surg. 1998;176:642–647. doi: 10.1016/s0002-9610(98)00280-3. [DOI] [PubMed] [Google Scholar]
- 52.Glaser CB, Morser J, Clarke JH, Blasko E, McLean K, Kuhn I, Chang RJ, Lin JH, Vilander L, Andrews WH, et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest. 1992;90:2565–2573. doi: 10.1172/JCI116151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ross CC, MacLeod SL, Plaxco JR, Stites WEl, Froude JW, Fink LM, Hauer-Jensen M. Direct inactivation of endothelial thrombomodulin by ionizing radiation (Abstr.) Radia Res Society. 2006;53:111. doi: 10.1667/RR1148.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohji T, Urano H, Shirahata A, Yamagishi M, Higashi K, Gotoh S, Karasaki Y. Transforming growth factor beta 1 and beta 2 induce down-modulation of thrombomodulin in human umbilical vein endothelial cells. Thromb Haemost. 1995;73:812–818. [PubMed] [Google Scholar]
- 55.Conway EM, Rosenberg RD. Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol Cell Biol. 1988;8:5588–5592. doi: 10.1128/mcb.8.12.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A. 1986;83:3460–3464. doi: 10.1073/pnas.83.10.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lentz SR, Tsiang M, Sadler JE. Regulation of thrombomodulin by tumor necrosis factor-alpha: comparison of transcriptional and posttranscriptional mechanisms. Blood. 1991;77:542–550. [PubMed] [Google Scholar]
- 58.Boehme MW, Deng Y, Raeth U, Bierhaus A, Ziegler R, Stremmel W, Nawroth PP. Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: in vivo and in vitro studies. Immunology. 1996;87:134–140. [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol. 1986;128:96–104. doi: 10.1002/jcp.1041280115. [DOI] [PubMed] [Google Scholar]
- 60.Laposata M, Dovnarsky DK, Shin HS. Thrombin-induced gap formation in confluent endothelial cell monolayers in vitro. Blood. 1983;62:549–556. [PubMed] [Google Scholar]
- 61.Bogatcheva NV, Garcia JG, Verin AD. Role of tyrosine kinase signaling in endothelial cell barrier regulation. Vascul Pharmacol. 2002;39:201–212. doi: 10.1016/s1537-1891(03)00009-0. [DOI] [PubMed] [Google Scholar]
- 62.Johnson A, Tahamont MV, Kaplan JE, Malik AB. Lung fluid balance after pulmonary embolization: effects of thrombin vs. fibrin aggregates. J Appl Physiol Respir Environ Exerc Physiol. 1982;52:1565–1570. doi: 10.1152/jappl.1982.52.6.1565. [DOI] [PubMed] [Google Scholar]
- 63.Bar-Shavit R, Eldor A, Vlodavsky I. Binding of thrombin to subendothelial extracellular matrix. Protection and expression of functional properties. J Clin Invest. 1989;84:1096–1104. doi: 10.1172/JCI114272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bar-Shavit R, Benezra M, Eldor A, Hy-Am E, Fenton JW, Wilner GD, Vlodavsky I. Thrombin immobilized to extracellular matrix is a potent mitogen for vascular smooth muscle cells: nonenzymatic mode of action. Cell Regul. 1990;1:453–463. doi: 10.1091/mbc.1.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bar-Shavit R, Benezra M, Sabbah V, Bode W, Vlodavsky I. Thrombin as a multifunctional protein: induction of cell adhesion and proliferation. Am J Respir Cell Mol Biol. 1992;6:123–130. doi: 10.1165/ajrcmb/6.2.123. [DOI] [PubMed] [Google Scholar]
- 66.Dawes KE, Gray AJ, Laurent GJ. Thrombin stimulates fibroblast chemotaxis and replication. Eur J Cell Biol. 1993;61:126–130. [PubMed] [Google Scholar]
- 67.Huang L, Ogushi F, Tani K, Ogawa H, Kawano T, Endo T, Izumi K, Sono N, Ueno J, Nishitani H, et al. Thrombin promotes fibroblast proliferation during the early stages of experimental radiation pneumonitis. Radiat Res. 2001;156:45–52. doi: 10.1667/0033-7587(2001)156[0045:tpfpdt]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 68.Tani K, Yasuoka S, Ogushi F, Asada K, Fujisawa K, Ozaki T, Sano N, Ogura T. Thrombin enhances lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 1991;5:34–40. doi: 10.1165/ajrcmb/5.1.34. [DOI] [PubMed] [Google Scholar]
- 69.Chambers RC, Dabbagh K, McAnulty RJ, Gray AJ, Blanc-Brude OP, Laurent GJ. Thrombin stimulates fibroblast procollagen production via proteolytic activation of protease-activated receptor 1. Biochem J. 1998;333(Pt1):121–127. doi: 10.1042/bj3330121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stouffer GA, Runge MS. The role of secondary growth factor production in thrombin-induced proliferation of vascular smooth muscle cells. Semin Thromb Hemost. 1998;24:145–150. doi: 10.1055/s-2007-995833. [DOI] [PubMed] [Google Scholar]
- 71.Rauch BH, Millette E, Kenagy RD, Daum G, Fischer JW, Clowes AW. Syndecan-4 is required for thrombin-induced migration and proliferation in human vascular smooth muscle cells. J Biol Chem. 2005;280:17507–17511. doi: 10.1074/jbc.M410848200. [DOI] [PubMed] [Google Scholar]
- 72.Dabbagh K, Laurent GJ, McAnulty RJ, Chambers RC. Thrombin stimulates smooth muscle cell procollagen synthesis and mRNA levels via a PAR-1 mediated mechanism. Thromb Haemost. 1998;79:405–409. [PubMed] [Google Scholar]
- 73.Wilcox JN. Thrombotic mechanisms in atherosclerosis. Coron Artery Dis. 1994;5:223–229. doi: 10.1097/00019501-199403000-00007. [DOI] [PubMed] [Google Scholar]
- 74.Kanthou C, Parry G, Wijelath E, Kakkar VV, Demoliou-Mason C. Thrombin-induced proliferation and expression of platelet-derived growth factor-A chain gene in human vascular smooth muscle cells. FEBS Lett. 1992;314:143–148. doi: 10.1016/0014-5793(92)80961-f. [DOI] [PubMed] [Google Scholar]
- 75.Walker TR, Cadwallader KA, MacKinnon A, Chilvers ER. Thrombin induces DNA synthesis and phosphoinositide hydrolysis in airway smooth muscle by activation of distinct receptors. Biochem Pharmacol. 2005;70:959–967. doi: 10.1016/j.bcp.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Zheng H, Wang J, Koteliansky VE, Gotwals PJ, Hauer-Jensen M. Recombinant soluble transforming growth factor beta type II receptor ameliorates radiation enteropathy in mice. Gastroenterology. 2000;119:1286–1296. doi: 10.1053/gast.2000.19282. [DOI] [PubMed] [Google Scholar]
- 77.Wang J, Albertson CM, Zheng H, Fink LM, Herbert JM, Hauer-Jensen M. Short-term inhibition of ADP-induced platelet aggregation by clopidogrel ameliorates radiation-induced toxicity in rat small intestine. Thromb Haemost. 2002;87:122–128. [PubMed] [Google Scholar]
- 78.Boerma M, Wang J, Richter KK, Hauer-Jensen M. Orazipone, a locally acting immunomodulator, ameliorates intestinal radiation injury: a preclinical study in a novel rat model. Int J Radiat Oncol Biol Phys. 2006;66:552–559. doi: 10.1016/j.ijrobp.2006.05.067. [DOI] [PubMed] [Google Scholar]
- 79.Collins CE, Rampton DS. Platelet dysfunction: a new dimension in inflammatory bowel disease. Gut. 1995;36:5–8. doi: 10.1136/gut.36.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Verheij M, Dewit LG, Boomgaard MN, Brinkman HJ, van Mourik JA. Ionizing radiation enhances platelet adhesion to the extracellular matrix of human endothelial cells by an increase in the release of von Willebrand factor. Radiat Res. 1994;137:202–207. [PubMed] [Google Scholar]
- 81.Mennie AT, Dalley VM, Dinneen LC, Collier HO. Treatment of radiation-induced gastrointestinal distress with acetylsalicylate. Lancet. 1975;2:942–943. doi: 10.1016/s0140-6736(75)90358-x. [DOI] [PubMed] [Google Scholar]
- 82.Ludgate CM. Preliminary report: acetylsalicylic acid therapy in the treatment of complications following abdominal radiation. J Can Assoc Radiol. 1985;36:138–140. [PubMed] [Google Scholar]
- 83.Verheij M, Stewart FA, Oussoren Y, Weening JJ, Dewit L. Amelioration of radiation nephropathy by acetylsalicylic acid. Int J Radiat Biol. 1995;67:587–596. doi: 10.1080/09553009514550701. [DOI] [PubMed] [Google Scholar]
- 84.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 85.Nylander S, Mattsson C, Ramström S, Lindahl TL. Synergistic action between inhibition of P2Y12/P2Y1 and P2Y12/thrombin in ADP- and thrombin-induced human platelet activation. Br J Pharmacol. 2004;142:1325–1331. doi: 10.1038/sj.bjp.0705885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chung AW, Jurasz P, Hollenberg MD, Radomski MW. Mechanisms of action of proteinase-activated receptor agonists on human platelets. Br J Pharmacol. 2002;135:1123–1132. doi: 10.1038/sj.bjp.0704559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gachet C. Platelet activation by ADP: the role of ADP antagonists. Ann Med. 2000;32 Suppl 1:15–20. [PubMed] [Google Scholar]
- 88.Woulfe D, Yang J, Brass L. ADP and platelets: the end of the beginning. J Clin Invest. 2001;107:1503–1505. doi: 10.1172/JCI13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Akyurek S, Atahan L, Cengiz M, Sokmensuer C, Haberal I, Yildiz F, Onal C. Effect of ticlopidine in the prevention of radiation enteropathy. Br J Radiol. 2006;79:409–414. doi: 10.1259/bjr/16265085. [DOI] [PubMed] [Google Scholar]
- 90.Weksler BB. Platelets. In: Gallin JI, Goldstein IM, Snyderman R, editors. Inflammation: Basic Principles and Clinical Correlates. 3rd ed. PMCid. New York: Raven Press; 1999. p. PMC1220509. [Google Scholar]
- 91.Barcellos-Hoff MH. Radiation-induced transforming growth factor beta and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res. 1993;53:3880–3886. [PubMed] [Google Scholar]
- 92.Martin M, Lefaix JL, Pinton P, Crechet F, Daburon F. Temporal modulation of TGF-beta 1 and beta-actin gene expression in pig skin and muscular fibrosis after ionizing radiation. Radiat Res. 1993;134:63–70. [PubMed] [Google Scholar]
- 93.Finkelstein JN, Johnston CJ, Baggs R, Rubin P. Early alterations in extracellular matrix and transforming growth factor beta gene expression in mouse lung indicative of late radiation fibrosis. Int J Radiat Oncol Biol Phys. 1994;28:621–631. doi: 10.1016/0360-3016(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 94.Langberg CW, Hauer-Jensen M, Sung CC, Kane CJ. Expression of fibrogenic cytokines in rat small intestine after fractionated irradiation. Radiother Oncol. 1994;32:29–36. doi: 10.1016/0167-8140(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 95.Richter KK, Langberg CW, Sung CC, Hauer-Jensen M. Association of transforming growth factor beta (TGF-beta) immunoreactivity with specific histopathologic lesions in subacute and chronic experimental radiation enteropathy. Radiother Oncol. 1996;39:243–251. doi: 10.1016/0167-8140(95)01735-6. [DOI] [PubMed] [Google Scholar]
- 96.Richter KK, Langberg CW, Sung CC, Hauer-Jensen M. Increased transforming growth factor beta (TGF-beta) immunoreactivity is independently associated with chronic injury in both consequential and primary radiation enteropathy. Int J Radiat Oncol Biol Phys. 1997;39:187–195. doi: 10.1016/s0360-3016(97)00290-3. [DOI] [PubMed] [Google Scholar]
- 97.De Cristofaro R, De Candia E, Landolfi R. Effect of high- and low-molecular-weight heparins on thrombin-thrombomodulin interaction and protein C activation. Circulation. 1998;98:1297–1301. doi: 10.1161/01.cir.98.13.1297. [DOI] [PubMed] [Google Scholar]
- 98.Wang J, Zheng H, Qiu X, Kulkarni A, Fink LM, Hauer-Jensen M. Modulation of the intestinal response to ionizing radiation by anticoagulant and non-anticoagulant heparins. Thromb Haemost. 2005;94:1054–1059. doi: 10.1160/TH05-05-0330. [DOI] [PubMed] [Google Scholar]
- 99.Hoffmann JN, Vollmar B, Inthorn D, Schildberg FW, Menger MD. The thrombin antagonist hirudin fails to inhibit endotoxin-induced leukocyte/endothelial cell interaction and microvascular perfusion failure. Shock. 2000;14:528–534. doi: 10.1097/00024382-200014050-00006. [DOI] [PubMed] [Google Scholar]
- 100.Howell DC, Goldsack NR, Marshall RP, McAnulty RJ, Starke R, Purdy G, Laurent GJ, Chambers RC. Direct thrombin inhibition reduces lung collagen, accumulation, and connective tissue growth factor mRNA levels in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2001;159:1383–1395. doi: 10.1016/S0002-9440(10)62525-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mohri M, Sugimoto E, Sata M, Asano T. The inhibitory effect of recombinant human soluble thrombomodulin on initiation and extension of coagulation--a comparison with other anticoagulants. Thromb Haemost. 1999;82:1687–1693. [PubMed] [Google Scholar]
- 102.Ohishi R, Watanabe N, Aritomi M, Gomi K, Kiyota T, Yamamoto S, Ishida T, Maruyama I. Evidence that the protein C activation pathway amplifies the inhibition of thrombin generation by recombinant human thrombomodulin in plasma. Thromb Haemost. 1993;70:423–426. [PubMed] [Google Scholar]
- 103.Gomi K, Zushi M, Honda G, Kawahara S, Matsuzaki O, Kanabayashi T, Yamamoto S, Maruyama I, Suzuki K. Antithrombotic effect of recombinant human thrombomodulin on thrombin-induced thromboembolism in mice. Blood. 1990;75:1396–1399. [PubMed] [Google Scholar]
- 104.Solis MM, Vitti M, Cook J, Young D, Glaser C, Light D, Morser J, Wydro R, Yu S, Fink L. Recombinant soluble human thrombomodulin: a randomized, blinded assessment of prevention of venous thrombosis and effects on hemostatic parameters in a rat model. Thromb Res. 1994;73:385–394. doi: 10.1016/0049-3848(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 105.Mohri M, Gonda Y, Oka M, Aoki Y, Gomi K, Kiyota T, Sugihara T, Yamamoto S, Ishida T, Maruyama I. The antithrombotic effects of recombinant human soluble thrombomodulin (rhsTM) on tissue factor-induced disseminated intravascular coagulation in crab-eating monkeys (Macaca fascicularis) Blood Coagul Fibrinolysis. 1997;8:274–283. doi: 10.1097/00001721-199707000-00003. [DOI] [PubMed] [Google Scholar]
- 106.Uchiba M, Okajima K, Murakami K, Nawa K, Okabe H, Takatsuki K. Recombinant human soluble thrombomodulin reduces endotoxin-induced pulmonary vascular injury via protein C activation in rats. Thromb Haemost. 1995;74:1265–1270. [PubMed] [Google Scholar]
- 107.Li J, Garnette CS, Cahn M, Claytor RB, Rohrer MJ, Dobson JG, Gerlitz B, Cutler BS. Recombinant thrombomodulin inhibits arterial smooth muscle cell proliferation induced by thrombin. J Vasc Surg. 2000;32:804–813. doi: 10.1067/mva.2000.107992. [DOI] [PubMed] [Google Scholar]
- 108.Li JM, Singh MJ, Itani M, Vasiliu C, Hendricks G, Baker SP, Hale JE, Rohrer MJ, Cutler BS, Nelson PR. Recombinant human thrombomodulin inhibits arterial neointimal hyperplasia after balloon injury. J Vasc Surg. 2004;39:1074–1083. doi: 10.1016/j.jvs.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 109.Berg DT, Wiley MR, Grinnell BW. Enhanced protein C activation and inhibition of fibrinogen cleavage by a thrombin modulator. Science. 1996;273:1389–1391. doi: 10.1126/science.273.5280.1389. [DOI] [PubMed] [Google Scholar]
- 110.Van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24:1374–1383. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 111.Mosnier LO, Griffin JH. Protein C anticoagulant activity in relation to anti-inflammatory and anti-apoptotic activities. Front Biosci. 2006;11:2381–2399. doi: 10.2741/1977. [DOI] [PubMed] [Google Scholar]
- 112.Espana F, Medina P, Navarro S, Zorio E, Estellés A, Aznar J. The multifunctional protein C system. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:119–131. doi: 10.2174/1568016053544336. [DOI] [PubMed] [Google Scholar]
- 113.Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 114.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 115.Yasui H, Gabazza EC, Tamaki S, Kobayashi T, Hataji O, Yuda H, Shimizu S, Suzuki K, Adachi Y, Taguchi O. Intratracheal administration of activated protein C inhibits bleomycin-induced lung fibrosis in the mouse. Am J Respir Crit Care Med. 2001;163:1660–1668. doi: 10.1164/ajrccm.163.7.9911068. [DOI] [PubMed] [Google Scholar]
- 116.Shi J, Wang J, Zheng H, Ling W, Joseph J, Li D, Mehta JL, Ponnappan U, Lin P, Fink LM, et al. Statins increase thrombomodulin expression and function in human endothelial cells by a nitric oxide-dependent mechanism and counteract tumor necrosis factor alpha-induced thrombomodulin downregulation. Blood Coagul Fibrinolysis. 2003;14:575–585. doi: 10.1097/00001721-200309000-00010. [DOI] [PubMed] [Google Scholar]
- 117.Masamura K, Oida K, Kanehara H, Suzuki J, Horie S, Ishii H, Miyamori I. Pitavastatin-induced thrombomodulin expression by endothelial cells acts via inhibition of small G proteins of the Rho family. Arterioscler Thromb Vasc Biol. 2003;23:512–517. doi: 10.1161/01.ATV.0000060461.64771.F0. [DOI] [PubMed] [Google Scholar]
- 118.Williams JP, Hernady E, Johnston CJ, Reed CM, Fenton B, Okunieff P, Finkelstein JN. Effect of administration of lovastatin on the development of late pulmonary effects after whole-lung irradiation in a murine model. Radiat Res. 2004;161:560–567. doi: 10.1667/rr3168. [DOI] [PubMed] [Google Scholar]
- 119.Wang J, Qiu X, Zheng H, Joseph J, Ponnappan U, Mehta JL, Fink LM, Hauer-Jensen M. Effect of statins on endothelial thrombomodulin in vitro and the intestinal radiation response in vivo (Abstr.) Radia Res Society. 2004;51:37. [Google Scholar]
- 120.Bruynzeel I, Stoof TJ, Willemze R. Pentoxifylline and skin inflammation. Clin Exp Dermatol. 1998;23:168–172. doi: 10.1046/j.1365-2230.1998.00316.x. [DOI] [PubMed] [Google Scholar]
- 121.Reynolds H. Pentoxifylline--more evidence that it improves host defenses during sepsis. Crit Care Med. 1999;27:681–683. doi: 10.1097/00003246-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 122.Samlaska CP, Winfield EA. Pentoxifylline. J Am Acad Dermatol. 1994;30:603–621. doi: 10.1016/s0190-9622(94)70069-9. [DOI] [PubMed] [Google Scholar]
- 123.Seigneur M, Dufourcq P, Belloc F, Lenoble M, Renard M, Boisseau MR. Influence of pentoxifylline on membrane thrombomodulin levels in endothelial cells submitted to hypoxic conditions. J Cardiovasc Pharmacol. 1995;25 Suppl 2:S85–S87. doi: 10.1097/00005344-199500252-00018. [DOI] [PubMed] [Google Scholar]
- 124.Ohdama S, Takano S, Ohashi K, Miyake S, Aoki N. Pentoxifylline prevents tumor necrosis factor-induced suppression of endothelial cell surface thrombomodulin. Thromb Res. 1991;62:745–755. doi: 10.1016/0049-3848(91)90378-a. [DOI] [PubMed] [Google Scholar]
- 125.de Prost D. Pentoxifylline: a potential treatment for thrombosis associated with abnormal tissue factor expression by monocytes and endothelial cells. J Cardiovasc Pharmacol. 1995;25 Suppl 2:S114–S118. [PubMed] [Google Scholar]
- 126.Delanian S, Balla-Mekias S, Lefaix JL. Striking regression of chronic radiotherapy damage in a clinical trial of combined pentoxifylline and tocopherol. J Clin Oncol. 1999;17:3283–3290. doi: 10.1200/JCO.1999.17.10.3283. [DOI] [PubMed] [Google Scholar]
- 127.Dion MW, Hussey DH, Doornbos JF, Vigliotti AP, Wen BC, Anderson B. Preliminary results of a pilot study of pentoxifylline in the treatment of late radiation soft tissue necrosis. Int J Radiat Oncol Biol Phys. 1990;19:401–407. doi: 10.1016/0360-3016(90)90549-y. [DOI] [PubMed] [Google Scholar]
- 128.Hille A, Christiansen H, Pradier O, Hermann RM, Siekmeyer B, Weiss E, Hilgers R, Hess CF, Schmidberger H. Effect of pentoxifylline and tocopherol on radiation proctitis/enteritis. Strahlenther Onkol. 2005;181:606–614. doi: 10.1007/s00066-005-1390-y. [DOI] [PubMed] [Google Scholar]
- 129.Letur-Könirsch H, Guis F, Delanian S. Uterine restoration by radiation sequelae regression with combined pentoxifylline-tocopherol: a phase II study. Fertil Steril. 2002;77:1219–1226. doi: 10.1016/s0015-0282(02)03120-5. [DOI] [PubMed] [Google Scholar]
- 130.Steeves RA, Robins HI. Pentoxifylline treatment of radiation mastitis. Int J Radiat Oncol Biol Phys. 1998;42:1177. [PubMed] [Google Scholar]
- 131.Chua DT, Lo C, Yuen J, Foo YC. A pilot study of pentoxifylline in the treatment of radiation-induced trismus. Am J Clin Oncol. 2001;24:366–369. doi: 10.1097/00000421-200108000-00010. [DOI] [PubMed] [Google Scholar]
- 132.Ward WF, Kim YT, Molteni A, Ts'ao C, Hinz JM. Pentoxifylline does not spare acute radiation reactions in rat lung and skin. Radiat Res. 1992;129:107–111. [PubMed] [Google Scholar]
- 133.Tamou S, Trott KR. Modification of late radiation damage in the rectum of rats by deproteinized calf blood serum (ActoHorm) and pentoxifylline (PTX) Strahlenther Onkol. 1994;170:415–420. [PubMed] [Google Scholar]
- 134.Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci USA. 1999;96:12257–12262. doi: 10.1073/pnas.96.22.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760. [DOI] [PubMed] [Google Scholar]