

Abstract
Transforming growth factor-β (TGF-β), a prototype of multifunctional cytokine, is a key regulator of extracellular matrix (ECM) assembly and remodeling. Specifically, TGF-β isoforms have the ability to induce the expression of ECM proteins in mesenchymal cells, and to stimulate the production of protease inhibitors that prevent enzymatic breakdown of the ECM. Elevated TGF-β expression in affected organs, and subsequent deregulation of TGF-β functions, correlates with the abnormal connective tissue deposition observed during the onset of fibrotic diseases. During the last few years, tremendous progress has been made in the understanding of the molecular aspects of intracellular signaling downstream of the TGF-β receptors. In particular, Smad proteins, TGF-β receptor kinase substrates that translocate into the cell nucleus to act as transcription factors, have been studied extensively. The role of Smad3 in the transcriptional regulation of typeIcollagen gene expression and in the development of fibrosis, demonstrated both in vitro and in animal models with a targeted deletion of Smad3, is of critical importance because it may lead to novel therapeutic strategies against these diseases. This review focuses on the mechanisms underlying Smad modulation of fibrillar collagen expression and how it relates to fibrotic processes.
Keywords: Collagen, Connective tissue growth factor, Fibrosis, Smad, Transforming growth factor-β
INTRODUCTION
Fibrosis is a complex tissue disease whose predominant characteristics are the excessive and abnormal deposition of extracellular matrix (ECM) components[1,2], that may affect various organs, including lung, liver, kidney and skin. From a clinical point of view, fibrosis may be considered as a somewhat irreversible state of scar tissue, during which resolution of the healing process does not occur. Long-term activation of fibroblasts in the affected organs results in massive fibrous ECM deposition and excessive fibroblast/myofibroblast proliferation, thus contrasting with normal wound healing during which feedback mechanisms counterbalance the initial fibroblast activation into myofibroblasts[3].
Much attention is focused on the role of many cytokines and growth factors, a group of diverse molecules derived from blood cells such as platelets, or elaborated locally by mesenchymal and epithelial cells, that contribute to the fibrogenic process[1,4]. Among them, the profibrotic proteins transforming factor-β (TGF-β) and connective tissue growth factor (CTGF) are considered master switches for the induction of the fibrotic program. TGF-β induces fibroblasts to synthesize and contract ECM[5,6], and CTGF, induced by TGF-β, is considered as a critical downstream mediator of TGF-β effects on fibroblasts[7,8]. In this overview, we will discuss the progress made in understanding the central role of TGF-β in fibrotic diseases.
TGF-β AND RECEPTORS ACTIVATION
TGF-β activation
More than 60 TGF-β family members have been identified in multicellular organisms. Among these, there are three TGF-βs, five activins and at least eight Bone Morphogenetic Proteins (BMPs), all encoded by distinct genes (Figure 1)[9]. The three mammalian TGF-β isoforms, TGF-β1, 2, and 3 are secreted as latent precursor molecules (LTGF-β) that contain an amino-terminal hydrophobic signal peptide region, the latency associated peptide (LAP) region and the C-terminal potentially bioactive region[10]. The LTGF-β is usually complexed with latent TGF-β-binding proteins (LTBP), requiring activation into a mature form for receptor binding and subsequent activation of signal transduction pathways. The LTBP is removed extracellularly by either proteolic cleavage by various proteases such as plasmin, thrombin, plasma transglutaminase, or endoglycosylases, or by physical interactions of the LAP with other proteins, such as thrombospondin-1[11].
Figure 1.
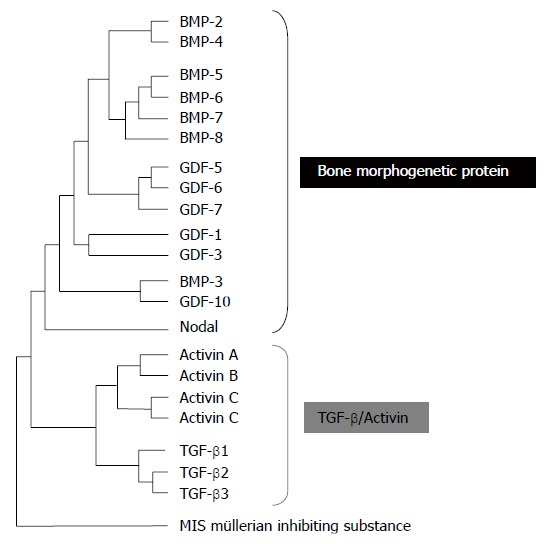
TGF-β family members.
TGF-β receptors
Signaling by TGF-β family members occurs through typeI(TβRI) and type II (TβRII) receptors (Figure 2). Five type II and seven typeIreceptors, termed Activin-receptor-like kinases (ALKs) have been identified in vertebrates[12]. TβRI and TβRII are similar transmembrane serine/threonine kinases, but typeIreceptors have a conserved Gly/Ser-rich (GS box) upstream from the kinase domain. In the absence of ligand, TβRI and TβRII are present as homodimers in the plasma membrane[13]. Ligand binding induces the assembly of typeIand type II receptors into complexes, within which TβRII phosphorylates and activates TβRI. This phosphorylation event is associated with activation of TβRI kinase and subsequent downstream signalling[12].
Figure 2.
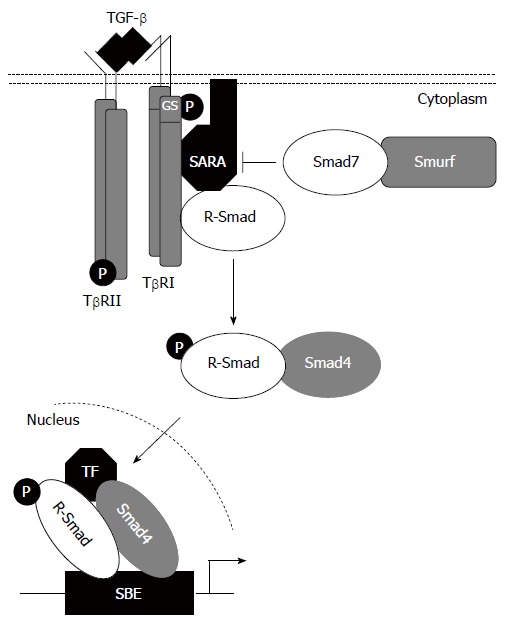
TGF-β/Smad signalling pathway.
TGF-β SIGNALLING BY SMAD PROTEINS
Smad proteins
Signaling from activated TβRI to the nucleus occurs predominantly by phosphorylation of cytoplasmic protein mediators belonging to the Smad family[9]. The receptor-associated Smads (R-Smads; Smad1, 2, 3, 5 and 8) are recruited to activated TβRI by auxiliary proteins such as Smad Anchor for Receptor Activation (SARA)[14]. They all consist of two conserved Mad-homology (MH) domains that form globular structures separated by a linker region[15]. The N-terminal MH1 domain has DNA-binding activity, whereas the C-terminal MH2 domain has protein-binding and transactivation properties. Upon phosphorylation by activated TβRI on two serine residues within a conserved-SS(M/V)S- motif at the extreme C terminus, activated R-Smads form heteromeric complexes with a Co-Smad, Smad4, and are translocated into the nucleus where they may function as transcription factors directly or in association with other DNA-binding factors[9,12,16]. Finally, the inhibitory Smads, Smad6 and Smad7, act in an opposing manner to R-Smads to antagonize signaling. They compete with R-Smads for binding to activated TβRI and thus inhibit the phosphorylation of R-Smads and/or recruit E3-ubiquitin ligases to activated TβRI, resulting in receptor degradation[16]. Additionally, they may recruit protein phosphatase-1 (PP1) to the receptor complex, resulting in the dephosphorylation, thus inactivation, of the receptors via the catalytic subunit of PP1, GADD45[9]. Once in the nucleus, Smad proteins activate transcription through physical interactions and functional cooperation of DNA-binding Smads with sequence-specific transcription factors and with the coactivators CBP and p300. The R-Smads MH1 domain can bind directly to DNA except in the case of Smad2 where a 30 amino acid insertion in this domain prevents DNA binding. The minimal Smad3/4-binding element (SBE) contains only four base pairs, 5’-AGAC-3’, but there are reports of binding to other G/C-rich sequences[9,16,17].
TGF-β REGULATION OF EXTRACELLULAR MATRIX GENE EXPRESSION
The net accumulation of collagen in tissue fibrosis is a result of an imbalance between enhanced production and deposition and impaired degradation of ECM components, mostly collagens (Figure 3). To date, about 25 types of collagens have been identified. All collagen molecules consist of three polypeptides, so-called α-chains. Some collagens are homopolymers with each of the three polypeptides being identical, while other collagens are heterotrimers with two or three distinct α-chains. TypeIcollagen, the major component of ECM is composed of two α1 (I) chains and one α2 (I) chain which are the products of two genes, COL1A1 and COL1A2. After translation, the pro-α1 (I) and pro-α2 (I) polypeptides chains enter into the endoplasmic reticulum where specific proline and lysine residues are hydroxylated to form hydroxyproline and hydroxylysine. This event allows the pro-α chains to combine with other chains by hydrogen bonds and form the triple helix procollagen structure. Procollagens are then secreted through the Golgi apparatus in the extracellular space, where the N-terminal and C-terminal propeptides are cleaved by specific proteases. The mature processed collagen molecules aggregates to form larger collagens[18]. Abnormalities in any step of typeIcollagen production may result in abnormally elevated synthesis of typeIcollagen which, in turn, causes tissue fibrosis.
Figure 3.
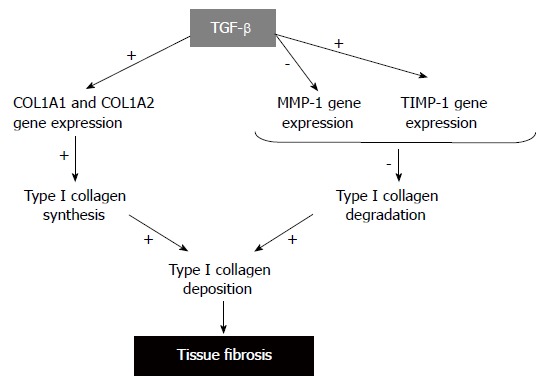
TGF-β, ECM gene expression and tissue fibrosis.
Several studies have shown that exaggerated tissue deposition of typeIcollagen during the fibrotic process is largely due to an increase in the rate of transcription of the corresponding genes[2,19,20]. To date, numerous efforts have been made to identify the signal transduction pathways involved in the transcription of typeIcollagen genes by TGF-β. Original works demonstrated that TGF-β-responsive sequences regarding the human promoter of COL1A1 are located between 174 and 84 bp from the transcription start site, which region contains a binding site for Sp1 and an element with the canonical NF-1 binding motif[21]. Regarding the human COL1A2 promoter, original works demonstrated that a 135-bp region of the promoter within 330-bp of the transcription start site could confer responsiveness to TGF-β[22,23]. The minimal TGF-β-response element was further refined to the region between nucleotides -271 and -235. The latter contains potential overlapping cis-element for Smad and AP-1, which are both implicated in COL1A2 transactivation by TGF-β[24]. Several Sp1 binding elements contribute to basal gene expression, and may represent targets for anti-fibrotic intervention[25]. Cooperation between Smad3 and Sp1 to transactivate the COL1A2 promoter have also been described, and it has been shown that Smad-p300/CBP interactions are critical for TGF-β driven COL1A2 gene transactivation[26,27]. Other transcriptional coactivators such as SRC-1 may also participate in TGF-β effects[28].
By the end of the year 2000, only approximately 12 genes were known to contain Smad-responsive regions, binding Smad complexes directly or indirectly. All Smad gene targets identified downstream TGF-β were Smad3-dependent including COL7A1[29], PAI-1[30], and COL1A2[31]. Using a combined cDNA microarray promoter transactivation approach, we have identified new Smad3/4 gene targets in cultured dermal fibroblasts: COL1A1, COL3A1, COL5A2, COL6A1, COL6A3, and TIMP-1. In addition, we identified 49 immediate-early TGF-β target genes. Their activation by TGF-β is rapid and does not require protein neo-synthesis or JNK activity. Furthermore, their activation was blocked by overexpression of the inhibitory Smad, Smad7, and did not occur in Smad3-deficient mouse fibroblasts. Thus, we demonstrated that the Smad signaling pathway is crucial for simultaneous activation of skin fibrillar collagen genes (COL1A1, COL1A2, COL3A1 and COL5A2) by TGF-β[32]. Besides playing a large part in the regulation of the expression of ECM components, Smads have been identified as capable of mediating the inhibitory activity of TGF-β on interstitial collagenase (matrix metalloproteinase-1, MMP-1) gene activation by pro-inflammatory cytokines, such as IL-1β[33], another mean by which the Smad pathway is likely to contribute to exacerbated ECM deposition.
TGF-β IN HUMAN SKIN FIBROSIS DISEASES
Keloids represent a dysregulated response to cutaneous wounding that results in an excessive deposition of collagen with a severely debilitating outcome for the affected patients. Several studies have demonstrated that TGF-β1 is expressed at greater levels in keloid fibroblasts when compared with normal dermal fibroblasts[1]. In addition, increased expression of TβRI and TBRII, and increased phosphorylation of Smad3 in keloid fibroblasts, have also been reported[34], supporting the hypothesis that TGF-β/Smad signaling plays a central role in keloid pathogenesis. Furthermore, the activation of Smad signaling, importantly that of Smad3, appears to be one facet of the complex epithelial-mesenchymal interactions in keloid pathogenesis, resulting in active keratinocyte proliferation and collagen production by fibroblasts[35].
Skin tissue fibrosis may also be a sequel of both radiotherapy or accidental exposure to gamma irradi-ation[36]. Superficial fibrosis is a sequel in humans after radiotherapy[37], and is characterized by induration of the dermis and the subcutaneous tissue. In cases of radiation accidents, high doses of radiation can be delivered to the skin and severe skin burns can be observed, resulting in the development of extensive fibronecrotic tissues[36,38]. The concept concerning the initiation of radiation damage proposes that a cascade of cytokines is initiated immediately after irradiation, during the clinically silent period, persists for long periods of times, and leads to the development of late damage[39]. The involvement of TGF-β in this early cascade has been reported in various irradiated tissues including skin, intestine, mammary gland and lung[36]. For example, in skin fibrotic samples from soldiers that suffered accidental irradiation in Lilo, Georgia, 1997, gene expression studies for collagen typeIand III, and TGF-β1 showed that these three genes are specifically overexpressed. In addition, TGF-β1 protein was overexpressed in fibronecrotic skin both in the scar epidermis and in the fibrotic dermis[36].
Systemic sclerosis (SSc) is a heterogeneous and generalized connective tissue disorder characterized by micro-vascular and larger vessel lesions, with consequent induration and thickening of the skin, fibrotic degenerative changes in muscles, joints and viscera, mainly the intestinal tract, the heart, the lungs and the kidneys. Although the mechanisms involved in the pathological increase of collagen expression in SSc have not been entirely elucidated, extensive recent efforts have been devoted to study the role of TGF-β signaling pathway by Smad proteins[40]. Immunohistochemical analysis of skin biopsies performed in non lesional areas from SSc patients and analysis of fibroblast cultures showed that Smad2 and Smad3 expression and their nuclear translocation were increased in these SSc patients[41]. More recently Dong et al[42]. reported reduction of Smad7 expression in SSc derived fibroblast cultures as compared to fibroblast cultures from unaffected areas of the same patients, suggesting that a defective Smad7 feedback inhibition could play a role in TGF-β hyper-responsiveness in SSc.
TGF-β has also been implicated as being a key mediator in a number of fibrotic diseases in organs other than skin. For example, an increased expression for TGF-β has been documented during the phase of tissue remodeling in several forms of acute or chronic lung disease[43], such as rapid progressive pulmonary fibrosis[44], idiopathic pulmonary fibrosis[45], scleroderma[46], or cystic fibrosis[47]. In the cardiovascular system, mounting evidence supports the notion that TGF-β1 stimulates the progression of cardiac fibrosis during cardiac hypertrophy and heart failure[48]. In the kidney, TGF-β is closely associated with renal interstitial fibrosis, in which normal glomerular tissue is replaced by ECM, leading to organ failure[49]. Epithelial-to-Mesenchymal transdifferentiation induced by TGF-β may contribute to tubular atrophy and generation of interstitial myofibroblasts, leading to concomitant tubulo-interstitial fibrosis[50]. In advanced liver fibrosis resulting in cirrhosis, liver failure, and portal hypertension, TGF-β fibrotic action is broadly associated with its ability to lead transdifferentiation of hepatic stellate cells into myofibroblasts[51].
Smad3, A KEY MEDIATOR OF FIBROTIC PROCESSES
The most direct evidence supporting the involvement of Smad3 in fibrosis came from the use of mice with a targeted deletion of Smad3[52]. For example, skin from Smad3-/- mice exposed to a single dose of 30 to 50 Gy of gamma-irradiation showed significantly less epidermal acanthosis and dermal influx of mast cells, macrophages, neutrophils and decreased expression of TGF-β than skin from wild type littermates suggesting that inhibition of Smad3 could decrease tissue damage and reduce fibrosis after exposure to ionizing radiations[53]. In another experimental model of fibrosis, mice deficient in Smad3 exhibited suppressed typeIprocollagen mRNA expression and reduced hydroxyproline content in the lungs compared with wild-type mice treated with bleomycin. Furthermore, loss of Smad3 greatly attenuated morphological fibrotic responses to bleomycin in the mouse lungs[54]. Likewise, transient overexpression of active TGF-β1 in lungs, using adenoviral vector-mediated gene transfer, resulted in progressive pulmonary fibrosis in wild-type mice, whereas no fibrosis was seen in the lungs of Smad3-/- animals[55]. Conversely, C57BL/6 mice with bleomycin-induced lungs receiving an intratracheal injection of a recombinant adenovirus expressing Smad7 demonstrated suppression of typeIprocollagen mRNA, reduced hydroxyproline content, and no morphological fibrotic responses in the lungs, indicated that gene transfer of Smad7 prevents bleomycin-induced lung fibrosis[56]. More recently, using mice with targeted deletion of Smad3, Roberts et al[57]. demonstrated that lack of Smad3 prevents the epithelial-to-mesenchymal transition of lens epithelial cells following injury, and attenuates the development of fibrotic sequelae. Together, these various experimental approaches demonstrate the direct implication of Smad3 activation downstream of TGF-β in the pathogenesis of pulmonary fibrosis.
CONNECTIVE TISSUE GROWTH FACTOR
Although TGF-β has long been regarded as a pivotal growth factor in the formation and maintenance of connective tissues and as a major driving influence in many progressive fibrotic diseases, attention has recently focused on the role of connective tissue growth factor (CTGF) in fibrosis. For example, Systemic sclerosis (SSc) fibroblasts demonstrate constitutive over-expression of CTGF that promotes migration, proliferation and matrix production. Specifically, in fibroblasts cultured from SSc lesions, CTGF mRNA and protein are constitutively expressed, even in the absence of exogenously added TGF-β[58]. In normal adult fibroblasts, TGF-β induces the expression of CTGF via a functional Smad3 binding site in the CTGF promoter. However, mutation of the Smad binding site does not reduce the high level of CTGF promoter activity observed in dermal fibroblasts cultured from lesional areas of scleroderma patients. Thus, the maintenance of the fibrotic phenotype in scleroderma fibroblasts, as visualized by excess CTGF expression, appears to be independent of Smad-dependent TGF-β signaling[59]. The increased level of CTGF protein and mRNA is also associated with the accumulation of fibroblasts/myofibroblasts and collagen deposition in the persistence of late intestinal radiation fibrosis[60]. Interestingly, Balb/c mice that lack CTGF induction upon stimulation with bleomycin, can be transformed into fibrosis-sensitive individuals by generation of a CTGF-rich environment using transient overexpression of CTGF by adenoviral gene transfer. In this context, silencing CTGF expression with siRNA demonstrated therapeutic potential to prevent liver fibrosis by inhibiting hepatic stellate cells activation[61]. Together these observations suggest that CTGF is an important mediator in the pathogenesis of fibrosis and can be act as an enhancer of TGF-β/Smad3 fibrotic response[62].
PERSPECTIVES FOR THERAPEUTIC INTERVENTION
Tremendous progress has been accomplished over the past several years in the understanding of the initial steps of TGF-β intracellular signalling. The identification of Smad proteins as direct links between the cell surface and the nucleus has allowed for the elucidation of critical events leading to gene activation by TGF-β. Specifically, an increasing body of evidence demonstrates that Smad3 plays a crucial role during the fibrotic process both in vitro and in vivo. These observations suggest that blocking the TGF-β/Smad3 pathways may promise opportunities for treatment of fibrotic diseases. In particular, several endogenous inhibitors of TGF-β/Smad3-mediated gene expression have been discovered. Firstly, Smad7 induction by IFN-γ, a well known anti-fibrotic cytokine, blocks TGF-β/Smad signalling pathway. In this context, halofuginone a low molecular weight plant alkaloid used as a coccidiostat for poultry, was effective in inhibiting dermal fibrosis in the tight skin mouse of scleroderma, and radiation-induced fibrosis[63-65]. Thus, halofuginone, which has demonstrated efficacy and tolerance in humans, could become an effective and novel therapy for example for liver fibrosis[66]. Secondly, activation of the MAP kinase JNK, whether by cytokines such as TNF-α or by pharmacologic molecules such as 5-fluoro-uracyl, blocks the transcriptional outcome of the TGF-β/Smad3 signaling pathway by induction of c-Jun phosphorylation which, directly interferes with Smad3-dependent transcription (Figure 4)[67-72]. Thirdly, cAMP was shown to inhibit TGF-β Smad3/4 dependent transcription via a protein kinase A-dependent mechanism[73]. However, several hurdles remain before the TGF-β/Smad3 pathway can be considered a perfect therapeutic target in situations such as fibrosis. The identification of alternate signalling pathways for TGF-β remains critically important. For example, the role of Smad2 downstream of TGF-β is rather poorly understood. Identification of Smad2 target genes will likely shed some light on alternate mechanisms by which TGF-β may affect connective tissue remodeling. Likewise, recent evidence for a role of the Rho pathway in the pathogenesis of radiation-induced enteritis suggest that inhibition of Rho pathway by pravastatin, an inhibitor of Rho isoprenylation, may also promise opportunities for new therapeutic perspectives[74].
Figure 4.
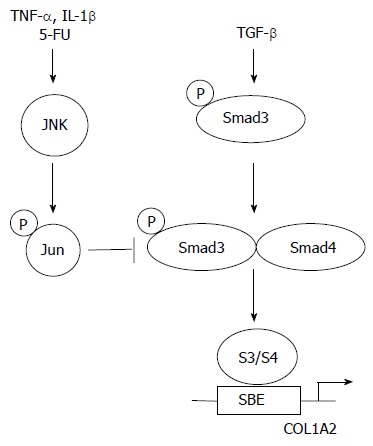
Inhibition of TGF-β-driven COL1A2 transcriptional activity by JNK.
Footnotes
Supported by Programme National de Recherche Dermatologie 2006, Institut Nationale de la Santé Et de la Recherche Médicale, Groupe Français de Recherche sur la Sclérodermie, and Association des Slérodermiques de France
S- Editor Liu Y L- Editor Alpini GD E- Editor Ma WH
References
- 1.Uitto J, Kouba D. Cytokine modulation of extracellular matrix gene expression: relevance to fibrotic skin diseases. J Dermatol Sci. 2000;24 Suppl 1:S60–S69. doi: 10.1016/s0923-1811(00)00143-2. [DOI] [PubMed] [Google Scholar]
- 2.Verrecchia F, Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 2004;16:873–880. doi: 10.1016/j.cellsig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Verrecchia F, Mauviel A. Control of connective tissue gene expression by TGF beta: role of Smad proteins in fibrosis. Curr Rheumatol Rep. 2002;4:143–149. doi: 10.1007/s11926-002-0010-4. [DOI] [PubMed] [Google Scholar]
- 4.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 5.LeRoy EC, Trojanowska MI, Smith EA. Cytokines and human fibrosis. Eur Cytokine Netw. 1990;1:215–219. [PubMed] [Google Scholar]
- 6.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8:171–179. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 8.Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol. 2004;122:1–6. doi: 10.1046/j.0022-202X.2003.22133.x. [DOI] [PubMed] [Google Scholar]
- 9.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 10.Roberts AB. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111–119. doi: 10.1159/000057358. [DOI] [PubMed] [Google Scholar]
- 11.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 12.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Gilboa L, Wells RG, Lodish HF, Henis YI. Oligomeric structure of type I and type II transforming growth factor beta receptors: homodimers form in the ER and persist at the plasma membrane. J Cell Biol. 1998;140:767–777. doi: 10.1083/jcb.140.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 17.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 18.Bornstein P, Sage H. Regulation of collagen gene expression. Prog Nucleic Acid Res Mol Biol. 1989;37:67–106. doi: 10.1016/s0079-6603(08)60695-9. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–314. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 20.Trojanowska M, LeRoy EC, Eckes B, Krieg T. Pathogenesis of fibrosis: type 1 collagen and the skin. J Mol Med (Berl) 1998;76:266–274. doi: 10.1007/s001090050216. [DOI] [PubMed] [Google Scholar]
- 21.Jimenez SA, Varga J, Olsen A, Li L, Diaz A, Herhal J, Koch J. Functional analysis of human alpha 1(I) procollagen gene promoter. Differential activity in collagen-producing and -nonproducing cells and response to transforming growth factor beta 1. J Biol Chem. 1994;269:12684–12691. [PubMed] [Google Scholar]
- 22.Inagaki Y, Truter S, Ramirez F. Transforming growth factor-beta stimulates alpha 2(I) collagen gene expression through a cis-acting element that contains an Sp1-binding site. J Biol Chem. 1994;269:14828–14834. [PubMed] [Google Scholar]
- 23.Inagaki Y, Truter S, Tanaka S, Di Liberto M, Ramirez F. Overlapping pathways mediate the opposing actions of tumor necrosis factor-alpha and transforming growth factor-beta on alpha 2(I) collagen gene transcription. J Biol Chem. 1995;270:3353–3358. doi: 10.1074/jbc.270.7.3353. [DOI] [PubMed] [Google Scholar]
- 24.Chung KY, Agarwal A, Uitto J, Mauviel A. An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem. 1996;271:3272–3278. doi: 10.1074/jbc.271.6.3272. [DOI] [PubMed] [Google Scholar]
- 25.Verrecchia F, Rossert J, Mauviel A. Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J Invest Dermatol. 2001;116:755–763. doi: 10.1046/j.1523-1747.2001.01326.x. [DOI] [PubMed] [Google Scholar]
- 26.Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J Invest Dermatol. 1999;112:49–57. doi: 10.1046/j.1523-1747.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- 27.Poncelet AC, Schnaper HW. Sp1 and Smad proteins cooperate to mediate transforming growth factor-beta 1-induced alpha 2(I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001;276:6983–6992. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]
- 28.Dennler S, Pendaries V, Tacheau C, Costas MA, Mauviel A, Verrecchia F. The steroid receptor co-activator-1 (SRC-1) potentiates TGF-beta/Smad signaling: role of p300/CBP. Oncogene. 2005;24:1936–1945. doi: 10.1038/sj.onc.1208343. [DOI] [PubMed] [Google Scholar]
- 29.Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB, Mauviel A. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc Natl Acad Sci USA. 1998;95:14769–14774. doi: 10.1073/pnas.95.25.14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen SJ, Yuan W, Lo S, Trojanowska M, Varga J. Interaction of smad3 with a proximal smad-binding element of the human alpha2(I) procollagen gene promoter required for transcriptional activation by TGF-beta. J Cell Physiol. 2000;183:381–392. doi: 10.1002/(SICI)1097-4652(200006)183:3<381::AID-JCP11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 32.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 33.Yuan W, Varga J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J Biol Chem. 2001;276:38502–38510. doi: 10.1074/jbc.M107081200. [DOI] [PubMed] [Google Scholar]
- 34.Chin GS, Liu W, Peled Z, Lee TY, Steinbrech DS, Hsu M, Longaker MT. Differential expression of transforming growth factor-beta receptors I and II and activation of Smad 3 in keloid fibroblasts. Plast Reconstr Surg. 2001;108:423–429. doi: 10.1097/00006534-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 35.Phan TT, Lim IJ, Aalami O, Lorget F, Khoo A, Tan EK, Mukhopadhyay A, Longaker MT. Smad3 signalling plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. J Pathol. 2005;207:232–242. doi: 10.1002/path.1826. [DOI] [PubMed] [Google Scholar]
- 36.Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys. 2000;47:277–290. doi: 10.1016/s0360-3016(00)00435-1. [DOI] [PubMed] [Google Scholar]
- 37.Archambeau JO, Pezner R, Wasserman T. Pathophysiology of irradiated skin and breast. Int J Radiat Oncol Biol Phys. 1995;31:1171–1185. doi: 10.1016/0360-3016(94)00423-I. [DOI] [PubMed] [Google Scholar]
- 38.Peter RU, Braun-Falco O, Birioukov A, Hacker N, Kerscher M, Peterseim U, Ruzicka T, Konz B, Plewig G. Chronic cutaneous damage after accidental exposure to ionizing radiation: the Chernobyl experience. J Am Acad Dermatol. 1994;30:719–723. doi: 10.1016/s0190-9622(08)81501-0. [DOI] [PubMed] [Google Scholar]
- 39.Rubin P, Johnston CJ, Williams JP, McDonald S, Finkelstein JN. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 1995;33:99–109. doi: 10.1016/0360-3016(95)00095-G. [DOI] [PubMed] [Google Scholar]
- 40.Verrecchia F, Mauviel A, Farge D. Transforming growth factor-beta signaling through the Smad proteins: role in systemic sclerosis. Autoimmun Rev. 2006;5:563–569. doi: 10.1016/j.autrev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Mori Y, Chen SJ, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003;48:1964–1978. doi: 10.1002/art.11157. [DOI] [PubMed] [Google Scholar]
- 42.Dong C, Zhu S, Wang T, Yoon W, Li Z, Alvarez RJ, ten Dijke P, White B, Wigley FM, Goldschmidt-Clermont PJ. Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc Natl Acad Sci USA. 2002;99:3908–3913. doi: 10.1073/pnas.062010399. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 44.Limper AH, Broekelmann TJ, Colby TV, Malizia G, McDonald JA. Analysis of local mRNA expression for extracellular matrix proteins and growth factors using in situ hybridization in fibroproliferative lung disorders. Chest. 1991;99:55S–56S. doi: 10.1378/chest.99.3_supplement.55s. [DOI] [PubMed] [Google Scholar]
- 45.Broekelmann TJ, Limper AH, Colby TV, McDonald JA. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA. 1991;88:6642–6646. doi: 10.1073/pnas.88.15.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludwicka A, Ohba T, Trojanowska M, Yamakage A, Strange C, Smith EA, Leroy EC, Sutherland S, Silver RM. Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol. 1995;22:1876–1883. [PubMed] [Google Scholar]
- 47.Wojnarowski C, Frischer T, Hofbauer E, Grabner C, Mosgoeller W, Eichler I, Ziesche R. Cytokine expression in bronchial biopsies of cystic fibrosis patients with and without acute exacerbation. Eur Respir J. 1999;14:1136–1144. doi: 10.1183/09031936.99.14511369. [DOI] [PubMed] [Google Scholar]
- 48.Euler-Taimor G, Heger J. The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res. 2006;69:15–25. doi: 10.1016/j.cardiores.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Runyan CE, Schnaper HW, Poncelet AC. Smad3 and PKCdelta mediate TGF-beta1-induced collagen I expression in human mesangial cells. Am J Physiol Renal Physiol. 2003;285:F413–F422. doi: 10.1152/ajprenal.00082.2003. [DOI] [PubMed] [Google Scholar]
- 50.Böttinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 51.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 52.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, et al. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol. 2002;160:1057–1068. doi: 10.1016/S0002-9440(10)64926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao J, Shi W, Wang YL, Chen H, Bringas P, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 55.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 56.Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 58.Denton CP, Abraham DJ. Transforming growth factor-beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol. 2001;13:505–511. doi: 10.1097/00002281-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 59.Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276:10594–10601. doi: 10.1074/jbc.M010149200. [DOI] [PubMed] [Google Scholar]
- 60.Vozenin-Brotons MC, Milliat F, Sabourin JC, de Gouville AC, François A, Lasser P, Morice P, Haie-Meder C, Lusinchi A, Antoun S, et al. Fibrogenic signals in patients with radiation enteritis are associated with increased connective tissue growth factor expression. Int J Radiat Oncol Biol Phys. 2003;56:561–572. doi: 10.1016/s0360-3016(02)04601-1. [DOI] [PubMed] [Google Scholar]
- 61.Li G, Xie Q, Shi Y, Li D, Zhang M, Jiang S, Zhou H, Lu H, Jin Y. Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J Gene Med. 2006;8:889–900. doi: 10.1002/jgm.894. [DOI] [PubMed] [Google Scholar]
- 62.Bonniaud P, Martin G, Margetts PJ, Ask K, Robertson J, Gauldie J, Kolb M. Connective tissue growth factor is crucial to inducing a profibrotic environment in "fibrosis-resistant" BALB/c mouse lungs. Am J Respir Cell Mol Biol. 2004;31:510–516. doi: 10.1165/rcmb.2004-0158OC. [DOI] [PubMed] [Google Scholar]
- 63.Pines M, Nagler A. Halofuginone: a novel antifibrotic therapy. Gen Pharmacol. 1998;30:445–450. doi: 10.1016/s0306-3623(97)00307-8. [DOI] [PubMed] [Google Scholar]
- 64.Pines M, Snyder D, Yarkoni S, Nagler A. Halofuginone to treat fibrosis in chronic graft-versus-host disease and scleroderma. Biol Blood Marrow Transplant. 2003;9:417–425. doi: 10.1016/s1083-8791(03)00151-4. [DOI] [PubMed] [Google Scholar]
- 65.Denton CP, Black CM. Targeted therapy comes of age in scleroderma. Trends Immunol. 2005;26:596–602. doi: 10.1016/j.it.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Gnainsky Y, Kushnirsky Z, Bilu G, Hagai Y, Genina O, Volpin H, Bruck R, Spira G, Nagler A, Kawada N, et al. Gene expression during chemically induced liver fibrosis: effect of halofuginone on TGF-beta signaling. Cell Tissue Res. 2007;328:153–166. doi: 10.1007/s00441-006-0330-1. [DOI] [PubMed] [Google Scholar]
- 67.Verrecchia F, Pessah M, Atfi A, Mauviel A. Tumor necrosis factor-alpha inhibits transforming growth factor-beta /Smad signaling in human dermal fibroblasts via AP-1 activation. J Biol Chem. 2000;275:30226–30231. doi: 10.1074/jbc.M005310200. [DOI] [PubMed] [Google Scholar]
- 68.Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P, Mauviel A. Induction of the AP-1 members c-Jun and JunB by TGF-beta/Smad suppresses early Smad-driven gene activation. Oncogene. 2001;20:2205–2211. doi: 10.1038/sj.onc.1204347. [DOI] [PubMed] [Google Scholar]
- 69.Verrecchia F, Tacheau C, Wagner EF, Mauviel A. A central role for the JNK pathway in mediating the antagonistic activity of pro-inflammatory cytokines against transforming growth factor-beta-driven SMAD3/4-specific gene expression. J Biol Chem. 2003;278:1585–1593. doi: 10.1074/jbc.M206927200. [DOI] [PubMed] [Google Scholar]
- 70.Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332–3340. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- 71.Verrecchia F, Wagner EF, Mauviel A. Distinct involvement of the Jun-N-terminal kinase and NF-kappaB pathways in the repression of the human COL1A2 gene by TNF-alpha. EMBO Rep. 2002;3:1069–1074. doi: 10.1093/embo-reports/kvf219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wendling J, Marchand A, Mauviel A, Verrecchia F. 5-fluorouracil blocks transforming growth factor-beta-induced alpha 2 type I collagen gene (COL1A2) expression in human fibroblasts via c-Jun NH2-terminal kinase/activator protein-1 activation. Mol Pharmacol. 2003;64:707–713. doi: 10.1124/mol.64.3.707. [DOI] [PubMed] [Google Scholar]
- 73.Schiller M, Verrecchia F, Mauviel A. Cyclic adenosine 3',5'-monophosphate-elevating agents inhibit transforming growth factor-beta-induced SMAD3/4-dependent transcription via a protein kinase A-dependent mechanism. Oncogene. 2003;22:8881–8890. doi: 10.1038/sj.onc.1206871. [DOI] [PubMed] [Google Scholar]
- 74.Haydont V, Mathé D, Bourgier C, Abdelali J, Aigueperse J, Bourhis J, Vozenin-Brotons MC. Induction of CTGF by TGF-beta1 in normal and radiation enteritis human smooth muscle cells: Smad/Rho balance and therapeutic perspectives. Radiother Oncol. 2005;76:219–225. doi: 10.1016/j.radonc.2005.06.029. [DOI] [PubMed] [Google Scholar]