

Abstract
Chronic obstructive pulmonary disease (COPD) is a complex disease with both environmental and genetic determinants, the most important of which is cigarette smoking. There is marked heterogeneity in the development of COPD among persons with similar cigarette smoking histories, which is likely partially explained by genetic variation. Genomic approaches such as genomewide association studies and gene expression studies have been used to discover genes and molecular pathways involved in COPD pathogenesis; however, these “first generation” omics studies have limitations. Integrative genomic studies are emerging which can combine genomic datasets to further examine the molecular underpinnings of COPD. Future research in COPD genetics will likely use network-based approaches to integrate multiple genomic data types in order to model the complex molecular interactions involved in COPD pathogenesis. This article reviews the genomic research to date and offers a vision for the future of integrative genomic research in COPD.
Keywords: Chronic Obstructive Pulmonary Disease, Genomewide Association Study, Genomics, Network Medicine
Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide, accounting for an estimated 3 million deaths in 2010.[1] COPD is a complex disease with both genetic and environmental determinants, the most important of which is cigarette smoking. However, only a minority of smokers develop COPD, and there is significant variability in lung function across smokers with similar cigarette exposure histories.[2] Some of this heterogeneity in the development of COPD is likely a result of genetic variation. A known genetic risk factor for COPD is severe α1-antitrypsin (AAT) deficiency, which is the result of mutations in the SERPINA1 (serpin peptidase inhibitor A1) gene.[3,4] AAT deficiency accounts for approximately 1% of COPD cases and thus is an insufficient genetic factor to account for the heterogeneity in COPD.[4]
The complex genetic component of COPD was elucidated through linkage analysis and familial aggregation studies.[5] In previous decades, a series of candidate gene association studies tested genes felt to be important in the COPD pathogenesis. As candidate gene studies by design focused on genes with known potential relationship with COPD pathogenesis, they had no ability to identify novel COPD susceptibility regions. The advent of genome wide association studies (GWAS) allowed for examinations of hundreds of thousands to millions of single nucleotide polymorphisms (SNPs) across the genome. Since 2009, GWAS have revealed replicable statistical associations for COPD. GWAS have also discovered loci related to lung function, emphysema, and other COPD phenotypes (Table 1). Despite the success of GWAS, a large portion of the heritability -- the phenotypic variability that is attributable to genetics --remains unexplained.[6,7] “Missing heritability” is not a unique feature for COPD and is a problem across all complex diseases.[8] There are many proposed explanation for the missing heritability in COPD and other diseases, and additional genomic research, including the integration of distinct sources of genomic data, is required.
Table 1. Phenotypes for COPD Genomics Studies.
| COPD diagnosis |
| Pulmonary function tests |
| Spirometry |
| Forced expiratory volume in 1 second (FEV1) |
| Ratio of FEV1 to Forced Vital Capacity |
| Decline in lung function |
| Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages [114]: Stages 1-4, from least to most severe, based on FEV1 % predicted |
| Lung Volumes |
| Diffusing capacity for carbon monoxide |
| Emphysema on chest computed tomography (CT) scans |
| Visual assessment |
| Quantitative image analysis |
| Emphysema pattern based on local histogram analysis |
| Airway disease on chest CT scans |
| Symptoms |
| Chronic bronchitis |
| Acute exacerbations |
| Physiologic impairment |
| Blood oxygen levels |
| Exercise capacity |
| Systemic effects |
| Body mass index |
| Co-morbidities |
| COPD subtypes |
| COPD-asthma overlap syndrome |
| Machine learning subtypes |
Silverman and Loscalzo describe three generations of genomics studies (Figure 1).[9] First generation studies examine the association between genetic variants and disease phenotype. This concept can be broadened to include any association studies that use a single omics technology, e.g. microarray expression profiling. Second generation studies examine associations between two different types of omics data, such as GWAS and gene expression profiling, and aim to correlate the results to disease. Third generation studies utilize multiple omics data types, combined using network methods, and address not only a disease as a whole, but also consider disease subtypes. This article will review the COPD genomics studies to date, which have largely fallen into the first generation category. We will discuss the early progress in integrative genomics studies, which represent the second generation. Future second generation and eventually third generation network medicine studies have the potential to unveil the underlying biology of COPD, which will lead to improved definition of molecular disease subtypes and potentially novel therapeutic strategies.
Figure 1.
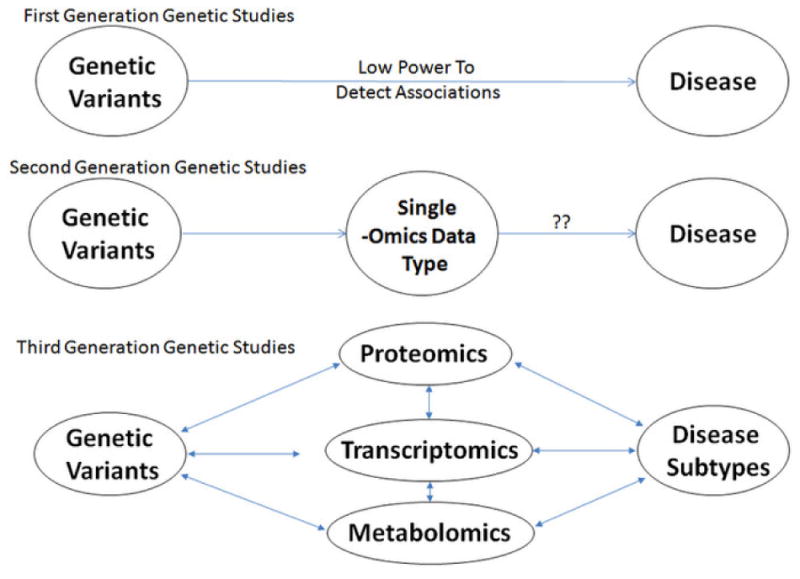
Three generations of genomic studies, described by Silverman and Loscalzo. (Discovery Medicine 2012:14:143-152). Figure reproduced with permission.
Genomewide association studies in COPD
GWAS of COPD affection status (Table 2) have found seven risk loci, which are detailed in the next sections.
Table 2.
COPD case-control genomewide association studies.
| Study | Cases | Controls | Novel Loci | Replicated Loci |
|---|---|---|---|---|
| Pillai 2009 [10] | 823 | 810 | CHRNA3/CHRNA5/IREB2, HHIP | |
| Cho 2010 [28] | 2,940 | 1,380 | FAM13A | CHRNA3/CHRNA5/IREB2, HHIP |
| Cho 2012 [31] | 3,499 | 1,922 | Chromosome 19q13 | CHRNA3/CHRNA5/IREB2, HHIP, FAM13A |
| Wilk 2012 [38] | 3,368 | 29,507 | HTR4* | CHRNA3/CHRNA5/IREB2 |
| Cho 2014 [7] | 6,633 | 5,704 | RIN3 | CHRNA3/CHRNA5/IREB2, HHIP, FAM13A |
| Cho 2014 (severe COPD) [7] | 3,125 | 1,468 | MMP12, TGFB2 | CHRNA3/CHRNA5/IREB2, HHIP, FAM13A |
Chromosome 15q25: CHRNA3/CHRNA5/IREB2
The first COPD GWAS was reported in 2009 by Pillai et al.[10] This GWAS used the GenKOLS study in Bergen, Norway[11] as the discovery population and a staged replication approach in a large family-based study, the International COPD Genetics Network (ICGN) [11,12] and a case-control population from the National Emphysema Treatment Trial (NETT) [13] and the Normative Again Study (NAS).[14] The result of the second and final phase of replication was the identification of two SNPs at the chromosome 15q25 locus reaching genome-wide significance (p value < 5 × 10-8). This initial COPD GWAS is not the only study to implicate the 15q25 locus as potentially important in COPD susceptibility.
A candidate gene study in 2009 by DeMeo et al. found associations with the iron responsive element binding protein 2 (IREB2) gene in the chromosome 15q25 locus.[15] The authors selected candidate genes based lung tissue gene expression data.[16] SNPs were genotyped in a case-control population with subsequent replication in two family studies. The authors additionally demonstrated localization of the IREB2 protein to the cilial border of human lung epithelial cells.
Chromosome 15q25 is a complex locus in its association with COPD. It encodes a family of nicotinic cholinergic receptors including nicotinic cholinergic receptors α3 and α5 (CHRNA3 and CHRNA5), which have been associated with smoking behaviors.[17,18,19,20] Siedlinski et al. used a statistical technique called mediation analysis to determine that the effect of variants in CHRNA3/5 appeared to largely be mediated by smoking.[21] In contrast, a variant at IREB2 was associated with COPD independent of smoking. This study suggests that although some of the association of the 15q25 locus with COPD affection status may be driven by nicotine dependence, there are likely associations of IREB2 with COPD independent of smoking.
Chromosome 4q31: HHIP
In addition to finding risk loci at CHRNA3/5, Pillai et al identified SNPs in hedgehog interacting protein (HHIP) that were consistently replicated, yet failed to reach a genome-wide significance level.[10] In a companion GWAS by Wilk et al., SNPs near the HHIP region were found to be associated with lung function in the general population from the Framingham Heart Study.[22] Another lung function GWAS reported a genome-wide significant association of HHIP with the ratio for forced expiratory volume in one second (FEV1) to forced vital capacity (FVC) in a meta-analysis of 20,890 subjects of European descent.[23] A separate lung function GWAS found genome-wide significant associations of the 4q31 locus with both FEV1 and FEV1/FVC ratio in two large populations (>20,000 subjects) of European ancestry.[24] The consistent statistical association of the HHIP locus with lung function traits in multiple general population GWASs increased confidence in its role in COPD susceptibility.
After confirming the association in Polish case-control study of severe COPD, Zhou et al. demonstrated reduced HHIP expression at the mRNA and protein levels in COPD lung tissues.[25] The investigators then demonstrated long-range regulation of HHIP by a genomic region ∼85 kb upstream that acted as an enhancer. This upstream HHIP enhancer region contained SNPs previously identified to be associated with COPD through GWAS. One SNP in the upstream enhancer region demonstrated increased binding of Sp3 which acts as a transcriptional repressor, consistent with the reduced HHIP expression. In a follow-up paper, the authors performed microarray expression analysis following in vitro silencing of HHIP using short hairpin RNAs. The differentially expressed genes were functionally enriched for extracellular matrix and cell proliferation pathways.[26]
Chromosome 4q22: FAM13A
In a 2010 study, Cho et al. reported association with the gene family with sequence similarity 13, member A (FAM13A) on chromosome 4q22 in a meta-analysis of COPD cases and controls from the GenKOLS and NETT-NAS studies along with subjects from the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study.[27,28] Although the top two FAM13A SNPs were not strictly genomewide significant, the association of these SNPs with COPD was replicated by genotyping in 502 cases and 504 controls from the Genetic Epidemiology of COPD (COPDGene) Study[29] as well as genotyping in 2,859 subjects in the family-based ICGN study. The FAM13A locus was also found to be associated with FEV1, an important marker of disease severity in COPD, in these studies along with the Boston Early-Onset COPD Study.[30] The FAM13A locus is one of the most consistently replicated COPD genetic associations (Table 2). However, the functional implications of FAM13A and its role in COPD pathogenesis remain to be determined.
Chromosome 19q13
Cho et al. performed a subsequent GWAS of four study populations, including NETT-NAS, GenKOLS, ECLIPSE, and the first 1000 subjects from the COPDGene study.[31] The meta-analysis resulted in a novel association at the chromosome 19q13 locus which encompasses the genes RAB4B, member RAS oncogene family (RAB4B), egl-9 family hypoxia-inducible factor 2 (EGLN2) and cytochrome P450, family 2, subfamily A, polypeptide 6 (CYP2A6), the last of which had been previously associated with smoking behavior.[18,32] It is thus reasonable to assume that part of the association with the 19q13 locus acts through cigarette smoking behavior. However, the specific susceptibility gene(s) in the locus and the functional effects of that gene or genes have yet to be clarified.
Chromosome 14q32: RIN3
The most recent COPD GWAS included approximately 10,000 subjects from the COPDGene study as well as a meta-analysis of prior GWAS results across the NETT-NAS, ECLIPSE, and GenKOLS studies.[7] In total, 6633 COPD cases were included, making it the largest COPD GWAS to date. In addition to replication of the associations at chromosome 15q25, FAM13A, and HHIP, the authors discovered a SNP at the 14q32 locus near Ras and Rab interactor 3 (RIN3) to be significantly associated with COPD. The consistently replicated results across these three COPD GWAS publications is not surprising, since they included many of the same study populations and used a consistent, objective definition of COPD based on lung function tests.
Chromosome 11q22: MMP12
In a mouse model, Mautakamki et al. demonstrated that the absence of matrix metallopeptidase 12 (MMP12, also known as macrophage elastase) protected against cigarette-smoke induced emphysema, demonstrating the importance of this gene.[33] A candidate gene study by Hunninghake et al. helped to strengthen the MMP12 relationship to COPD susceptibility. [34] A SNP in the MMP12 promoter region was associated with increased lung function and a reduced risk of the development of COPD in three populations. Ishii et al. illustrated the inverse relationship of MMP12 mRNA expression levels in alveolar macrophages with a measure of airflow obstruction, the FEV1/FVC ratio.[35]
Statistical validation of MMP12 locus association with COPD at a population level was provided in the 2014 COPD GWAS, which included a GWAS of severe COPD with cases defined FEV1<50% predicted.[7] The severe COPD GWAS meta-analysis also confirmed prior COPD associations with the 15q25, FAM13A, and HHIP loci. MMP12 is an attractive candidate in COPD pathogenesis with congruency in laboratory, candidate gene, and GWAS data suggesting variants at the MMP12 locus are protective against the development of COPD.
Chromosome 1q41: TGFB2
In the severe COPD GWAS meta-analysis, the transforming growth factor β2 (TGFB2) locus nearly reached genomewide significance.[7] When severe COPD subjects from the ICGN study were added, the meta-analysis p-value became genomewide significant. Although TGFB2 had not previously been associated with COPD, a SNP in TGFB2 was associated with the FEV1/FVC in a general population GWAS.[36]
GWAS of Pulmonary Function
Lung function GWAS have provided valuable information about the genetic architecture of the primary determinants of a diagnosis of COPD, the FEV1 and the FEV1/FVC ratio, as measured by spirometry testing.[22,23,24,36] The lung function GWAS have typically been better powered than COPD GWAS not only due to the use of quantitative rather than binary traits, but also due to the larger sample size inherit to population based studies. One disadvantage is that population based studies likely have a greater heterogeneity of smoking status than studies designed to expressly to assess COPD. Despite this, lung function GWAS have been instrumental in the replication and independent confirmation of many COPD susceptibility loci.
Hancock et al. specifically addressed the influence of smoking by performing a genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction effects in over 50,000 subjects from 19 studies.[37] They found three loci associated with FEV1 and FEV1/FVC which had not been identified in previous lung function GWAS, including delta/notch-like EGF repeat containing (DNER, chromosome 2), major histocompatibility complex, class II, DQ β1 and α2 (HLA-DQB1 and HLA-DQA2, chromosome 6), and potassium inwardly-rectifying channel, subfamily J, member 2 and sex determining region Y-box 9 (KCNJ2 and SOX9, chromosome 17).
Wilk and colleagues defined COPD subjects with reduced lung function in 15 general population samples.[38] They confirmed the association with SNPs in the chromosome 15q25 locus, with some continued evidence of association when restricted to never smokers. They also found an association with airflow obstruction in the gene 5-hydroxytryptamine (serotonin) receptor 4 (HTR4), which had been previously associated with FEV1/FVC ratio. [23,24]
COPD may be characterized by accelerated lung function decline. Hansel et al. performed a GWAS of 5-year change in FEV1 in over 4000 subjects with mild-to-moderate COPD from the Lung Health Study.[39] Interestingly, the associated loci -- ankyrin 3 (ANK3) and transmembrane protein 26 (TMEM26) on chromosome 10 and forkhead box A1 (FOXA1) on chromosome 14 – did not overlap with previous GWAS of COPD or cross-sectional lung function. The only other GWAS of lung function decline was performed in subjects with asthma.[40]
GWAS of Additional COPD Phenotypes
Outside of COPD affection status and lung function, GWAS of related phenotypes in COPD may help explain the genetic architecture of COPD heterogeneity (Table 1). For example, patients with similar degrees of airflow obstruction may have different amounts of emphysema, pathologic destruction of lung tissue.[41] GWAS focusing on emphysema may reveal unique genomic loci contributing to emphysema susceptibility. Similarly, other COPD phenotypes can be interrogated to determine genetic susceptibility to a variety of disease presentations.
One of the oldest recognized clinical COPD phenotypes is the presence of a chronic productive cough, which is referred to as chronic bronchitis. Dijkstra and colleagues performed a GWAS meta-analysis in a total of 2,704 subjects with chronic bronchitis with or without airflow obstruction (which they termed chronic mucus hypersecretion) and 7,624 controls, finding an association with an intronic SNP in the SATB homeobox 1 (SATB1) transcription factor.[42] The SNP was shown to effect SATB1 expression levels in lung tissue. In addition, SATB1 expression was increased in airway biopsies of subjects with mucus hypersecretion.
Besides chronic bronchitis, the other classic COPD phenotype is emphysema. Kong et al. performed a GWAS of both visual and quantitative emphysema assessments on chest computed tomography (CT) scans from 2,380 COPD subjects across three cohorts.[43] There was a genome-wide significant association between variants in the bicaudal D homolog 1 (BICD1) gene and visual emphysema severity. The quantitative assessment of emphysema yielded no significant associations.
A recent emphysema GWAS by Manichaikul et al. examined the quantitative chest CT emphysema measurements in multiple ethnic groups from the general population MESA study (including non-Hispanic whites, African Americans, Hispanics, and Chinese) and discovered two genome-wide significant SNPs in or near the genes small nuclear ribonucleoprotein polypeptide F (SNRPF) and palmitoyl-protein thioesterase 2 (PPT2).[44] It remains to be seen whether genetic variation related to subclinical emphysema in the general population may overlap with genes associated with emphysema in COPD.
Cachexia has long been associated with the end stages of COPD, and low body-mass index (BMI) has been validated as an independent risk factor for more severe emphysema and airflow obstruction as well as increased mortality in COPD patients.[45,46,47] Not only does low BMI have implications for COPD outcomes, but also represents a unique phenotype within the heterogeneity of COPD presentation and may have a genetic susceptibility. Wan et al. performed a GWAS of BMI in COPD subjects and discovered an association with an intronic SNP in the fat mass and obesity-associated (FTO) gene,[48] which has previously been associated with BMI in the general population.[49]
Another clinically-relevant COPD phenotype is the overlap of asthma and COPD, a phenotype for which Hardin and colleagues recently performed an epidemiology study and GWAS.[50] Approximately 12% of COPD subjects in the COPDGene study had a physician's diagnosis of asthma along spirometry-defined COPD. In the GWAS of asthma-COPD overlap subjects compared to subjects with COPD alone, no SNPs had a p-value for association meeting genome-wide significance; however, the most significant SNPs were in the G protein-coupled receptor 65 (GPR65) on chromosome 14. GPR65 plays a role in eosinophil activation and thus is a plausible candidate gene for the development of the asthma-COPD overlap syndrome.
Hypoxemia is a concerning COPD complication with variable presentation in the late stages of COPD. McDonald et al. performed a GWAS of resting oxygen saturation in COPD.[51] Within African American subjects but not in non-Hispanic white subjects, there was a genome-wide significant association of resting oxygen saturation with two loci, on chromosome 14 and 15. These loci contained genes in the hedgehog and TGF-β pathways, which have been previously implicated in COPD pathogenesis. This study of resting oxygen saturation in COPD subjects demonstrated not only the utility of GWAS in COPD phenotypes, but also highlighted the importance of genetic association studies in multiple racial populations given a variability of results between races. Ongoing and future studies will investigate additional COPD phenotypes to search for novel associations with the various clinical features and comorbidities within COPD (Table 1).
Genomewide Gene Expression Studies in COPD
Gene expression microarray studies allow genomewide assessment of transcriptional activity as a means of exploring the biologic pathways involved in COPD pathogenesis. Unlike germline SNPs, gene expression profiles demonstrate tissue-specificity. This section will review gene expression profiling studies performed in respiratory and non-respiratory tissues, including lung parenchyma, airways, and peripheral blood (Tables 3-5).
Table 3. COPD Microarray Gene Expression Studies of Lung Tissue.
| Reference | Samples (N) | Differentially expressed pathways |
|---|---|---|
| Golpon 2004 [52] | “Usual” emphysema (5) AAT deficiency (6) Normal lungs (5) |
Inflammation, Immune response, Proteolysis |
| Spira 2004 [53] | Severe emphysema (20) Mild/no emphysema (14) |
Extracellular matrix (ECM), Inflammation, Oxidative stress, Endothelium |
| Ning 2004 [54] | GOLD 2 (14)* Control smokers (12) |
Apoptosis, Inflammation, Transcription factors, ECM |
| Wang 2008 [56] | GOLD 0-3 (43)* Nonsmokers (5) |
ECM, Apoptosis, Anti-inflammatory |
| Bhattacharya 2009 [16] | Smokers (56) Cases (15)/Controls (18) |
Nucleic acid binding, DNA-dependent transcription |
| Savarimuthu Francis 2009 [62] | Mild emphysema (10) Moderate emphysema (20) |
Development, Cell differentiation, Enzyme regulation |
| Savarimuthu Francis 2011 [63] | GOLD 1 (9)* GOLD 2 (9)* |
angiogenesis, cell migration, proliferation and apoptosis, cell-matrix adhesion, leukocyte activation, cell and substrate adhesion, cell adhesion, angiogenesis, cell activation |
| Ezzie 2012 [78] | GOLD 1-4 (26)* Control smokers (9) |
transforming growth factor β signaling, Wnt, focal adhesion |
| Campbell 2012 [64] | Severe COPD (6) Donor lungs (2) 8 regions per lung |
inflammation, B-cell receptor signaling pathway, tissue repair processes, transforming growth factor β pathway, actin organization, integrin signaling |
Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages 1-4, from least to most severe, based on FEV1 %predicted [114]
Table 5. COPD Microarray Gene Expression Studies in Blood.
| References | Samples (N) | Differentially expressed pathways |
|---|---|---|
| Edmiston 2010 [68] | GOLD 1-4 (81)* Control smokers (61) |
Apoptosis, cell growth, protein and RNA transport, post-translational modification, cellular defense, inflammation |
| Poliska 2011 [70] | GOLD 2-3 (5)* Control smokers (5) |
Not described |
| Bhattacharya 2011 [71] | GOLD 2-4 (12)* Control smokers (12) |
Nucleic acid binding, transcription, nucleotide metabolism, cell signaling, inflammatory cell regulation |
| Bahr 2013 [69] | GOLD 1-4 (84)* Control smokers (42) Unclassified (10) |
Immune system, inflammatory response, sphingolioid metabolism |
Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages 1-4, from least to most severe, based on FEV1 %predicted [114]
The field started with three studies of microarray gene expression profiling in non-tumor lung tissue (Table 3). The studies by Golpon, Spira, and Ning examined small numbers of subjects, using various COPD definitions.[52,53,54] These early lung gene expression studies had minimal overlap of differentially expressed genes. This observation may have been due to the small sample sizes as well as the heterogeneity of the cell types within the tissues being studied.[55] Another consideration in the study of lung tissue gene expression is the heterogeneity of the COPD disease process (such as emphysema severity) throughout an individual's lungs.
Wang et al. profiled lung tissue from 21 smoking controls and 27 subjects with COPD and found 203 transcripts associated with the forced expiratory flow between 25 and 75% of forced expiratory volume (FEF25-75%), a measure of lung function.[56] Ingenuity Pathways Analysis illustrated that these genes were present in pathways involving transforming growth factor β1 (TGFB1) and serpin peptidase inhibitor, clade E, member 2 (SERPINE2), two prior candidate genes for COPD susceptibility.[57,58]
Zeskind et al. sought to review and integrate data from the four lung tissue gene expression studies above,[52,53,54,56] noting overlap of only 11 genes between any two studies.[59] Despite the minimal overlap of individual differentially expressed genes, Gene Ontology[60] and Gene Set Enrichment Analysis[61] showed that common biological processes potentially explain the differences in lung gene expression of COPD cases and controls.
Bhattacharya et al. performed gene expression profiling on 56 lung tissue samples.[16] They found 65 differentially expressed genes between COPD cases and controls and also reported 220 potential genomic biomarkers of lung function based on correlation of gene expression signatures with the lung function measures FEV1 and FEV1/FVC ratio. Two other small studies examined gene expression relationships with COPD severity defined by lung function testing.[62,63]
In a different approach to expression profiling in COPD, Campbell et al. examined the heterogeneity of emphysema in the lungs of COPD patients and lung donors.[64] They performed expression profiling on specimens from 8 lung regions from each of 8 subjects (64 lung tissue samples in total), noting 127 genes associated with regional emphysema differences. In the tissue regions with increasing emphysema severity, they noted increasing gene expression in inflammatory pathways including the B-cell receptor-signaling pathway, and decreased gene expression had in tissue repair pathways including the TGFβ pathway, actin organization, and integrin signaling. Based on a query of the Connectivity Map [65], they discovered a tripeptide compound GHK that could induce a gene expression pattern similar with TGFβ signaling activation. This gene expression study uniquely used gene expression signatures to search for possible molecules to reverse observed gene expression changes relevant to COPD progression in the form of increasing emphysema severity.
The lung tissue gene expression studies above have generally had small sample sizes due to the need for surgical resection to obtain tissue samples (Table 3). Sampling of the airway epithelium via bronchoscopy may be less invasive, and several groups have performed gene expression profiling in proximal bronchi and distal small airways (Table 4). In the largest such study, Steiling et al. collected bronchial brushings on current and former smokers and indetified 98 genes associated with COPD affection status.[66] The authors further demonstrated that the expression signatures of the bronchial epithelial cells were consistent with prior lung tissue and small airway gene expression studies. They proposed that a “field of injury” may exist in smokers, making bronchial epithelial an appropriate surrogate tissue for studies of lung gene expression changes in COPD. Additionally, the authors reported that a proportion of the observed COPD gene expression changes were reversed in the subset of subjects with improvement in lung function following treatment with the inhaled corticosteroid fluticasone.
Table 4. COPD Microarray Gene Expression Studies of Airway Epithelium.
| Reference | Samples (N) | Sampling | Differentially expressed pathways |
|---|---|---|---|
| Carolan 2006 [115] | Normal nonsmokers (12) Normal smokers (12) Early COPD (9) Established COPD (6) |
Small airway brushing | Only reported on 11 neuroendocrine cell genes |
| Pierrou 2007 [116] | GOLD 0-4 (38)* Healthy nonsmokers (14) Healthy smokers (18) |
Bronchial brushing | Oxidant/antioxidant, Energy metabolism, apoptosis |
| Ammous 2008 [117] | GOLD 1-2 (18)* Normal smokers (18) Normal nonsmokers (18) |
Small airway brushing | Oxidant, Signal transduction |
| Tilley 2009 [118] | GOLD 0-3 (13)* Normal smokers (15) Normal nonsmokers (20) |
Small airway brushing | Only reported on 55 Notch related genes |
| Wang 2012 [77] | GOLD 1-3 (36)* Healthy smokers (73) Healthy nonsmokers (60) |
Small airway brushing | Only reported on toll-like receptor family |
| Dunham 2013 [119] | GOLD 1-2 (6)* Healthy smokers (9) Healthy nonsmokers (3) |
Bronchial biopsy | gene expression, cellular assembly, and organization and cellular development; cell death, cancer, and reproductive disease; cellular movement, skeletal muscular development and function, and neurological disease; cardiovascular system development and function, embryonic development, and tissue development; and cell morphology, cancer, and tumor morphology |
| Steiling 2013 [66] | COPD, mostly GOLD 2 (87)* Normal smokers (151) |
Bronchial brushing | glycoproteins, proteins involved in the acute inflammatory response, EGF-like domains |
Global Initiative for Chronic Obstructive Lung Disease (GOLD) stages 1-4, from least to most severe, based on FEV1 %predicted [114]
In a study specifically designed to assess gene expression changes in response to inhaled fluticasone, van den Berge et al. obtained 221 bronchial biopsies longitudinally in 89 moderate-to-severe COPD patients before and after treatment with fluticasone with or without salmeterol (a long acting β2-agonist bronchodilator) versus placebo.[67] A total of 278 genes were reported to have changes in expression following treatment, supporting the idea that gene expression signatures in COPD patients are dynamic with therapy. Treatment with fluticasone with or without salmeterol led to down-regulation of genes related to epithelial cell signaling, oxidative stress, remodeling, and apoptosis along with up-regulation of genes related to epithelial barrier function.
Studying the longitudinal gene expression changes of respiratory tissues in COPD, particularly the changes as a result of pharmacologic treatments, is a promising way not only to define the active biologic pathways in the progression of COPD, but also to assess the genomic effects of COPD treatment. The recent studies utilizing bronchial epithelial cells as a surrogate for deeper respiratory system tissues hold promise for the ability to more easily conduct gene expression studies with a larger number of samples. In addition to larger sample sizes, the information gained from gene expression studies in COPD will also likely be enhanced by the integration of gene expression data with other genomic data.
COPD Gene Expression Studies in Peripheral Blood
Although the lung is the primary site of disease activity in COPD, lung samples are not routinely collected in epidemiologic studies or in clinical practice, in contrast to malignant diseases such as lung cancer. Peripheral blood is more easily accessible than lung or other benign tissues and can be sampled from a large number of subjects or repeatedly sampled at multiple time points. Therefore, investigators have performed expression profiling on blood leukocytes in COPD patients (Table 5). Edmiston et al. used random forest analysis on training and validation samples to identify a set of 1013 genes which predict case-control status.[68] White blood cell differential counts were entered into the analysis, but not retained in the final models. LASSO regression led to a list of 9 genes with similar predictive accuracy. Bahr et al. hypothesized that the systemic manifestations of COPD could be discovered in peripheral blood gene expression signatures and collected mononuclear cell gene expression profiles in 136 subjects with and without COPD.[69] A multiple linear regression with adjustment for covariates including smoking status and pack-years smoking was used to identify candidate genes for COPD status versus controls. Twenty-six of these candidate genes were validated and a pathway analysis demonstrated that the COPD candidate genes were found in cell signaling, transcription factor, and sphingolipid metabolism pathways. Poliska et al. found overlapping genes expressed in blood monocyte and alveolar macrophages in a limited number of COPD cases and controls.[70] Bhattacharya et al. compared peripheral blood mononuclear cell expression profiles to their previous study of lung tissue,[16] finding several genes with similar expression results across the two tissues.[71] The lung-blood expression relationships found in the latter two studies show that peripheral blood can be used for COPD genomics studies. Similar to the lung and airway expression studies, sample sizes were limited in the peripheral blood microarray studies, and COPD subtypes were not considered.
MicroRNA profiling in COPD
MicroRNAs (miRNAs) are small non-coding RNAs that play an important role in down regulating protein expression from a gene at either the transcriptional or translational level.[72] miRNAs are stable molecules which can be assayed in cells, tissues and blood. One miRNA may regulate multiple mRNAs forming a regulatory network; conversely, a single mRNA may be affected by multiple miRNAs. Therefore, miRNAs have great potential for use in integrative genomics and network analyses. To date, the high-throughput miRNA studies in COPD have been performed in a variety of specimens using different assay technologies (Table 6). The heterogeneity in study designs makes comparisons across studies difficult, similar to the early mRNA microarray studies. This field is naturally progressing toward integration of miRNA with gene expression (see below), which is likely to lead to more replicable results.
Table 6. High-throughput microRNA studies in COPD.
| Study | Subjects (N) | sample | assay | miRNAs tested | Main results |
|---|---|---|---|---|---|
| Van Pottelberge 2011 [120] | COPD (12) Never smokers (10) Current smokers (10) |
Sputum | qRT-PCR | 627 | let-7c reduced in COPD, associated with increase expression of TNFR2 |
| Leidinger 2011 [121] | COPD (24) Lung cancer (28) Healthy controls (19) |
Whole blood | microarray | 863 | 140 miRNA differentially expressed COPD vs. controls, 14 miRNA COPD vs. lung cancer |
| Akbas 2012 [122] | COPD (20) Control smokers (12) |
Serum | qRT-PCR | 72 | 5 significant miRNAs: miR-20a, miR-28-3p, miR-34c-5p, miR-100, miR-7 |
| Ezzie 2012 [78] | COPD (19) Control smokers (8) |
Lung tissue | microarray | Not shown | 70 miRNAs differentially expressed, enriched for TGF-B pathway |
| Christenson 2013 [80] | COPD (6) Controls (2) |
Lung tissue, 8 regions/subject | microarray | 467 | 63 miRNAs associated with regional emphysema |
| Chatila 2014 [123] | COPD (12) Healthy smokers (12) Healthy nonsmokers (12) |
Peripheral blood Treg cells | microarray | 743 | 6 miRNAs differentially expressed COPD vs. smokers |
Next Generation Sequencing in COPD
Although multiple GWAS and gene expression studies have been performed in COPD, the increasing affordability of massively-parallel “next generation” sequencing (NGS) will allow large scale DNA and RNA sequencing to be used on COPD patient samples. Direct sequencing information will allow for the identification of uncommon and rare genetic variants not captured by current genotyping arrays and will also allow for identification of splice variants and novel transcripts.
Wain et al. performed exome-sequencing in 100 heavy smokers with normal lung function in an effort to describe uncommon and rare genetic variants contributing to resistance to smoking. Although the study was underpowered to detect uncommon and rare variants of modest effect size, the top non-synonymous SNP was found in the coiled-coil domain containing 38 (CCDC38) gene.[73] CCDC38 has been previously associated with FEV1/FVC ratio[36] and was shown to be expressed in bronchial columnar epithelial cells.
Another application of NGS in COPD-related research was performed by Ryan et al. where purified airway basal cells were collected in a small number of subjects (10 healthy non-smokers and 7 healthy smokers) and gene expression was examined through RNA sequencing.[74] There were 676 differentially expressed genes when comparing the basal cells of smokers to non-smokers, including 13 differentially expressed genes at the chromosome 19q13 COPD locus, but no differential expression of genes at other COPD susceptibility loci. Other studies have performed RNA sequencing of small and large airway epithelium of smokers and non-smokers.[75,76,77] Future exome, genome and transcriptome sequencing projects comparing subjects with and without COPD are anticipated.
Integrating Genomic Data
Recent integrative genomics studies in COPD have fallen into two categories. Regulation of gene expression in COPD tissues has been examined through the integration of gene expression microarray data with miRNA, GWAS, or methylation datasets. GWAS data has also been used to test for genetic influences on blood biomarker levels. Both of these approaches represent the second generation studies from Silverman and Loscalzo.[9] Genomic data has been combined with machine learning subtyping analysis, in the first steps towards the third generation network medicine studies.
Regulation of Gene Expression
Understanding the utility of the integration of genomic data, Ezzie et al. profiled miRNA and mRNA expression in the lung tissue of 26 smokers with COPD and 9 without COPD.[78] Using Kyoto Encyclopedia of Genes and Genomes (KEGG) [79] pathway enrichment analysis, they reported enrichment of miRNA-mRNA pairs in Wnt and TGFβ signaling pathways.[78] Although the candidate pathways of Wnt and TGFβ signaling in COPD pathogenesis require additional research, this approach illustrated the power of integrating two genomic data types in a network context in order to generate plausible molecular pathways playing a role in a complex disease.
The heterogeneity of COPD relates not only to differences between patients, but also relates to regional emphysema severity differences within a single patient. GWAS and other omics studies have demonstrated the utility of examining phenotypes of COPD, such as emphysema. Christenson et al. studied miRNA-driven alterations in emphysema-associated gene expression networks across regional lung emphysema severity differences by integrating miRNA and gene expression profiling data in 64 lung tissue samples from 8 patients (8 samples per patient).[80] After noting 63 miRNAs were altered with regional emphysema differences, the authors created miRNA-mRNA correlation networks and discovered 1,029 significantly correlated miRNA-mRNA pairs. 51 of the 63 differentially expressed miRNAs were present in this co-expression network. The authors identified a candidate miRNA (miR-638) that may alter gene expression in oxidative stress and aging pathways in emphysematous lung tissue.
Integrative genomics have additionally been explored in COPD by identifying associations between microarray expression data and SNP variation (from GWAS datasets) to define expression Quantitative Trait Loci (eQTLs), which are SNPs that influence gene expression.[81,82] Both sputum and lung tissue samples have been used in COPD eQTL studies. Functional explanations for existing COPD susceptibility loci and novel disease associations can both be discovered in eQTL analysis.
Qiu et al. reported the first COPD eQTL study utilizing GWAS data[28] and sputum gene expression data from 131 COPD cases from the ECLIPSE study.[83] The eQTL analysis discovered two genomewide significant eQTL SNPs at the chromosome 15q25 locus with independent effects on expression of the CHRNA3/5 and IREB2 genes, suggesting two independent susceptibility signals with the locus. Seventy-four additional eQTL SNPs with nominal association with COPD were genotyped in two replication populations, identifying a novel association in psoriasis susceptibility 1 candidate 1 (PSORS1C1), near the major histocompatibility complex, class I, C (HLA-C) gene on chromosome 6.
In order to identify functional COPD variants, Lamontagne et al conducted a lung eQTL study with attention to previous COPD susceptibility loci.[84] GWAS genotyping was combined with genome-wide expression arrays from surgically resected lung tissue from subjects with and without COPD at three clinical centers. Using data from 409 subjects from one center as a discovery cohort, lung tissue eQTLs were determined and superimposed on the COPD risk loci at chromosomes 4q31 (HHIP), 4q22 (FAM13A), and 19q13. The significant eQTLs were then replicated in the two remaining site cohorts of 363 and 339 subjects. Although eQTLs at HHIP were significant in the discovery cohort, these eQTLs were only validated in one of the two other sites. At the 4q22 locus, several SNPs in linkage disequilibrium with the FAM13A COPD-susceptibility SNPs were nominally associated with FAM13A expression and were validated in one of the two replication cohorts. At the 19q13 locus, the COPD GWAS SNP was not a significant lung eQTL. The strongest eQTL association at the 19q13 locus was with the gene EGLN2. This study was successful in identifying lung eQTLs in close proximity to COPD susceptibility loci and helped to strengthen the candidacy of HHIP and FAM13A as functional genes in COPD pathogenesis while proposing that EGLN2 may be the relevant gene in the 19q13 locus. Despite having COPD subjects in this dataset, the authors did not consider COPD status in the analysis.
DNA methylation is an important epigenetic regulator of gene expression.[85] Methylation of cytosine bases in CpG sites in gene promoters can lead to silencing of gene expression, among other effects.[86] DNA methylation and other epigenetic modifications may be directly impacted by environmental exposures such as cigarette smoke, stressing the importance of this avenue of investigation in COPD.[87,88] DNA methylation arrays have been used to identify differentially methylated genes in subjects with COPD compared to smokers with normal lung function[89] and in COPD subjects treated with oral corticosteroids.[90] In an integrative study, Vucic et al. performed DNA methylation arrays and gene expression arrays on small airway epithelium from 15 COPD subjects and 23 controls, all former smokers.[91] They found 1120 differentially methylated genes, most hypermethylated, which showed enrichment for three pathways: G-protein coupled receptor signaling, aryl hydrocarbon receptor signaling, and cAMP-mediated signaling. Methylation levels of 144 of these genes were negatively correlated with gene expression, enriched for phosphatase and tensin homolog (PTEN) signaling, NF-E2-related factor 2 (Nrf2) oxidative stress response, and IL-17F in allergic inflammatory diseases.
Integration of Genomic and Biomarker Data in COPD
Multiple peripheral blood biomarkers have been studied in COPD, and several have been tied to COPD morbidity and mortality.[92,93] One general limitation of biomarkers is the temporal relationship between the biomarker level and the clinical outcome. Cross-sectional studies cannot determine whether the biomarker is the cause or the result of the outcome. Genetic variation, which by definition predates disease development, is one possible contributor to the levels of circulating biomarkers. In a search for genetic determinants of COPD biomarker levels, Kim and colleagues integrated seven blood biomarker levels with GWAS data in 1,951 COPD subjects from the ECLIPSE study.[94] The investigators discovered genome-wide significant associations with the levels of two pneumoproteins, club cell protein (CC16) and surfactant protein D (SP-D), but did not find any genome-wide significant associations for any of the five systemic inflammatory markers tested. The SNPs influencing CC16 and SP-D levels were tested for association with COPD. Several SNPs were nominally associated in a large case-control study but were not replicated in smaller COPD cohorts, which may have been due to smaller sample size or may reflect an overall heterogeneity between populations. Despite the lack of replication of the association of biomarker GWAS SNPs with COPD affection status, this biomarker GWAS represents another potential integrative tool to understand COPD pathobiology.
In contrast to most omics analyses which are hypothesis-generating, Bowler and colleagues demonstrated how integrative genomics data can be used for hypothesis testing, focusing on the cytokine Interleukin-16.[95] In COPDGene study subjects, they measured IL-16 plasma protein levels and IL-16 gene expression in peripheral blood leukocytes. eQTL analysis found a SNP associated with IL-16 mRNA levels. IL-16 was found to be a biomarker of emphysema, specifically in the upper lobes of the lungs.
Genomic Approaches to COPD Subtyping
The bulk of genomic research in COPD has focused on determining the genetic and gene expression variation contributing to COPD pathogenesis; however, genomic data can be used to address other questions in COPD. For decades, clinicians have noted two very different subgroups of patients in COPD, those with chronic bronchitis and those with emphysema.[96] COPD is increasingly being recognized as a heterogeneous disease beyond these two classic phenotypes with large variability in clinical features among patients with similar degrees of airflow obstruction.[97] Reclassification of COPD into subtypes based on the molecular mechanisms underlying pathogenesis could allow for the development of novel targeted therapeutics for COPD patients.[98]
The use of genomic data to direct (or validate) the subtyping of COPD is an area of interest. Castaldi et al. used k-means clustering to define four subgroups within the 10,192 smokers in the COPDGene study: 1) heavy smokers with relatively preserved lung function, 2) mild upper zone emphysema-predominant, 3) airway disease-predominant, and 4) severe emphysema.[99] The clinical and genetic relevance of these subgroups was evaluated by assessing the association of the clusters with COPD-related clinical measures as well as association with genetic variants from COPD susceptibility loci including HHIP, FAM13A, 15q25, and 19q13. Genetic associations were found between the mild upper zone emphysema cluster and a SNP in the HHIP locus and between the severe emphysema group and a SNP in the chromosome 15q25 region.
In the future, instead of using known genetic variants to interrogate cluster assignments, researchers can perform GWAS for the COPD subtypes, assuming large enough cluster sizes, in order to find novel genetic associations to explain the heterogeneity of COPD presentation.
Summary and Future Directions
Similar to many common complex diseases, genetics studies in COPD include a long series of poorly-replicated candidate gene studies.[100,101] With the advent of GWAS, COPD genetics now has a list of replicated disease susceptibility loci. Several of these loci harbor genes related to nicotine metabolism, which highlights the importance of cigarette smoking on the development of COPD, but does not provide new insights into disease pathogenesis. The functions of the other COPD GWAS genes have not been fully elucidated, though research on these genes is ongoing, with some early successes.[25]
Microarray gene expression studies have not had overlapping results on the gene level, but many common pathways have been uncovered using enrichment analyses, stressing the importance of pathway and network based methods. Pathway and network methods have not yet been applied to COPD GWAS, though several methods exist for this type of analysis[102,103,104] and have been applied to other diseases.[105,106]
These first-generation genomics studies have paved the way for second generation integrative studies, which have thus far focused on pairwise data integration, including GWAS data with gene expression (eQTL), GWAS with protein biomarkers (pQTL), and miRNA or DNA methylation integration with mRNA expression. Other combinations are possible, such as GWAS and methylation (mQTL), along with consideration of other omics data types like proteomics[107] and metabolomics.[108] In addition to continued pairwise integrative studies, future studies will begin to include more than two omics technologies. For example GWAS, methylation, and gene expression datasets can be combined using mediation analysis or other methods to determine relative effects of each on gene expression.[109] Studies such as ECLIPSE,[27] COPDGene (www.copdgene.org),[29] SPIROMICS (http://www.cscc.unc.edu/spir/),[110] and the Lung Genomics Research Consortium (www.lung-genomics.org) have collected or are continuing to collect the necessary samples to allow for integrative genomics. Further analyses will require continued development of statistical and network methodologies, along with new computational tools for data analysis and visualization. Public data access is essential for both methods development and applications, such as validation of network results across multiple studies.
COPD is a heterogeneous disease, which will require consideration of additional disease-related phenotypes. Molecular subtyping has been very successful in cancers,[111,112] and these lessons can be applied to benign diseases like COPD. However, in contrast to cancers, disease tissue is not usually available in patients with benign disease. Peripheral blood is more easily accessible and cost-efficient than bronchoscopy or surgical resection of lung tissue. Sample size limitations and phenotype heterogeneity can be overcome using peripheral blood for genomics studies. Additional research is required to further validate the role of blood genomics studies in COPD.
These network medicine methods have great potential to uncover novel pathways in COPD and other complex diseases.[9,113] However, methods for both replication and functional validation of genes found through networks may be more challenging. Yet these steps are necessary for the development of novel diagnostic tools and therapeutic interventions in COPD and other common diseases.
COPD is a complex disease with both environmental and genetic determinants
Genomewide association studies have discovered COPD susceptibility loci
Integration of genomic data types is offering new insights into COPD pathobiology
Network medicine approaches will be required to model complex molecular interactions
Acknowledgments
Funded by National Institutes of Health grants R01HL094635, R01NR013377, P01HL105339, T32HL007427, R01HL111759.
Footnotes
Disclosures: Dr. Hersh reports lecture fees from Novartis and consulting fees from CSL Behring. Dr. Hobbs reports no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burrows B, Knudson RJ, Cline MG, et al. Quantitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis. 1977;115:195–205. doi: 10.1164/arrd.1977.115.2.195. [DOI] [PubMed] [Google Scholar]
- 3.DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004;59:259–264. doi: 10.1136/thx.2003.006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360:2749–2757. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 5.Hersh CP, DeMeo DL, Silverman EK. Chronic Obstructive Pulmonary Disease. In: Silverman EK, Shapiro SD, Lomas DA, Weiss ST, editors. Respiratory Genetics. Hodder Arnold; New York: 2005. pp. 253–296. [Google Scholar]
- 6.Zhou JJ, Cho MH, Castaldi PJ, et al. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am J Respir Crit Care Med. 2013;188:941–947. doi: 10.1164/rccm.201302-0263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho MH, McDonald ML, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2:214–225. doi: 10.1016/S2213-2600(14)70002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman EK, Loscalzo J. Network medicine approaches to the genetics of complex diseases. Discov Med. 2012;14:143–152. [PMC free article] [PubMed] [Google Scholar]
- 10.Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu G, Warren L, Aponte J, et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. American journal of respiratory and critical care medicine. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]
- 12.Patel BD, Coxson HO, Pillai SG, et al. Airway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:500–505. doi: 10.1164/rccm.200801-059OC. [DOI] [PubMed] [Google Scholar]
- 13.Fishman A, Martinez F, Naunheim K, et al. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. The New England journal of medicine. 2003;348:2059–2073. doi: 10.1056/NEJMoa030287. [DOI] [PubMed] [Google Scholar]
- 14.Bell B, Rose CL, Damon H. The Normative Aging Study: an interdisciplinary and longitudinal study of health and aging. Aging Hum Dev. 1972;3:5–17. [Google Scholar]
- 15.DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85:493–502. doi: 10.1016/j.ajhg.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya S, Srisuma S, Demeo DL, et al. Molecular biomarkers for quantitative and discrete COPD phenotypes. Am J Respir Cell Mol Biol. 2009;40:359–367. doi: 10.1165/rcmb.2008-0114OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobacco and Genetics Consortium, Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nature genetics. 2010;42:441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thorgeirsson TE, Gudbjartsson DF, Surakka I, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nature genetics. 2010;42:448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siedlinski M, Cho MH, Bakke P, et al. Genome-wide association study of smoking behaviours in patients with COPD. Thorax. 2011;66:894–902. doi: 10.1136/thoraxjnl-2011-200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siedlinski M, Tingley D, Lipman PJ, et al. Dissecting direct and indirect genetic effects on chronic obstructive pulmonary disease (COPD) susceptibility. Hum Genet. 2013;132:431–441. doi: 10.1007/s00439-012-1262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilk JB, Chen TH, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. doi: 10.1371/journal.pgen.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. doi: 10.1038/ng.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X, Baron RM, Hardin M, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet. 2012;21:1325–1335. doi: 10.1093/hmg/ddr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou X, Qiu W, Sathirapongsasuti JF, et al. Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics. 2013;101:263–272. doi: 10.1016/j.ygeno.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE) The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2008;31:869–873. doi: 10.1183/09031936.00111707. [DOI] [PubMed] [Google Scholar]
- 28.Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. doi: 10.1038/ng.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regan EA, Hokanson JE, Murphy JR, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. doi: 10.3109/15412550903499522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silverman EK, Chapman HA, Drazen JM, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med. 1998;157:1770–1778. doi: 10.1164/ajrccm.157.6.9706014. [DOI] [PubMed] [Google Scholar]
- 31.Cho MH, Castaldi PJ, Wan ES, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21:947–957. doi: 10.1093/hmg/ddr524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saccone NL, Culverhouse RC, Schwantes-An TH, et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010;6:e1001053. doi: 10.1371/journal.pgen.1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hautamaki RD, Kobayashi DK, Senior RM, et al. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 34.Hunninghake GM, Cho MH, Tesfaigzi Y, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361:2599–2608. doi: 10.1056/NEJMoa0904006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2014;43:82–91. doi: 10.1183/09031936.00174612. [DOI] [PubMed] [Google Scholar]
- 36.Soler Artigas M, Loth DW, Wain LV, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43:1082–1090. doi: 10.1038/ng.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hancock DB, Artigas MS, Gharib SA, et al. Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function. PLoS Genet. 2012;8:e1003098. doi: 10.1371/journal.pgen.1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilk JB, Shrine NR, Loehr LR, et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. American journal of respiratory and critical care medicine. 2012;186:622–632. doi: 10.1164/rccm.201202-0366OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansel NN, Ruczinski I, Rafaels N, et al. Genome-wide study identifies two loci associated with lung function decline in mild to moderate COPD. Hum Genet. 2013;132:79–90. doi: 10.1007/s00439-012-1219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imboden M, Bouzigon E, Curjuric I, et al. Genome-wide association study of lung function decline in adults with and without asthma. The Journal of allergy and clinical immunology. 2012;129:1218–1228. doi: 10.1016/j.jaci.2012.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hersh CP, Jacobson FL, Gill R, et al. Computed tomography phenotypes in severe, early-onset chronic obstructive pulmonary disease. COPD. 2007;4:331–337. doi: 10.1080/15412550701601274. [DOI] [PubMed] [Google Scholar]
- 42.Dijkstra AE, Smolonska J, van den Berge M, et al. Susceptibility to chronic mucus hypersecretion, a genome wide association study. PLoS One. 2014;9:e91621. doi: 10.1371/journal.pone.0091621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kong X, Cho MH, Anderson W, et al. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. American journal of respiratory and critical care medicine. 2011;183:43–49. doi: 10.1164/rccm.201004-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manichaikul A, Hoffman EA, Smolonska J, et al. Genome-wide study of percent emphysema on computed tomography in the general population. The Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. American journal of respiratory and critical care medicine. 2014;189:408–418. doi: 10.1164/rccm.201306-1061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schols AM, Slangen J, Volovics L, et al. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1998;157:1791–1797. doi: 10.1164/ajrccm.157.6.9705017. [DOI] [PubMed] [Google Scholar]
- 46.Landbo C, Prescott E, Lange P, et al. Prognostic value of nutritional status in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1999;160:1856–1861. doi: 10.1164/ajrccm.160.6.9902115. [DOI] [PubMed] [Google Scholar]
- 47.Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:1005–1012. doi: 10.1056/NEJMoa021322. [DOI] [PubMed] [Google Scholar]
- 48.Wan ES, Hokanson JE, Murphy JR, et al. Clinical and radiographic predictors of GOLD-unclassified smokers in the COPDGene study. Am J Respir Crit Care Med. 2011;184:57–63. doi: 10.1164/rccm.201101-0021OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardin M, Cho M, McDonald ML, et al. The clinical and genetic features of COPD-asthma overlap syndrome. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology. 2014 doi: 10.1183/09031936.00216013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonald ML, Cho MH, Sorheim IC, et al. Common Genetic Variants Associated with Resting Oxygenation in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golpon HA, Coldren CD, Zamora MR, et al. Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol. 2004;31:595–600. doi: 10.1165/rcmb.2004-0008OC. [DOI] [PubMed] [Google Scholar]
- 53.Spira A, Beane J, Pinto-Plata V, et al. Gene expression profiling of human lung tissue from smokers with severe emphysema. Am J Respir Cell Mol Biol. 2004;31:601–610. doi: 10.1165/rcmb.2004-0273OC. [DOI] [PubMed] [Google Scholar]
- 54.Ning W, Li CJ, Kaminski N, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101:14895–14900. doi: 10.1073/pnas.0401168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen ZH, Kim HP, Ryter SW, et al. Identifying targets for COPD Treatment through gene expression profiling. Int J Chron Obstruct Pulmon Dis. 2008;3:359–370. doi: 10.2147/copd.s1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang IM, Stepaniants S, Boie Y, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med. 2008;177:402–411. doi: 10.1164/rccm.200703-390OC. [DOI] [PubMed] [Google Scholar]
- 57.Celedon JC, Lange C, Raby BA, et al. The transforming growth factor-{beta}1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD) Hum Mol Genet. 2004;13:1649–1656. doi: 10.1093/hmg/ddh171. [DOI] [PubMed] [Google Scholar]
- 58.Demeo DL, Mariani TJ, Lange C, et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am J Hum Genet. 2006;78:253–264. doi: 10.1086/499828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeskind JE, Lenburg ME, Spira A. Translating the COPD transcriptome: insights into pathogenesis and tools for clinical management. Proceedings of the American Thoracic Society. 2008;5:834–841. doi: 10.1513/pats.200807-074TH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Francis SM, Larsen JE, Pavey SJ, et al. Expression profiling identifies genes involved in emphysema severity. Respiratory research. 2009;10:81. doi: 10.1186/1465-9921-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Savarimuthu Francis SM, Larsen JE, Pavey SJ. Genes and gene ontologies common to airflow obstruction and emphysema in the lungs of patients with COPD. PLoS One. 2011;6:e17442. doi: 10.1371/journal.pone.0017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell JD, McDonough JE, Zeskind JE, et al. A gene expression signature of emphysema-related lung destruction and its reversal by the tripeptide GHK. Genome Med. 2012;4:67. doi: 10.1186/gm367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 66.Steiling K, van den Berge M, Hijazi K, et al. A dynamic bronchial airway gene expression signature of chronic obstructive pulmonary disease and lung function impairment. Am J Respir Crit Care Med. 2013;187:933–942. doi: 10.1164/rccm.201208-1449OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van den Berge M, Steiling K, Timens W, et al. Airway gene expression in COPD is dynamic with inhaled corticosteroid treatment and reflects biological pathways associated with disease activity. Thorax. 2014;69:14–23. doi: 10.1136/thoraxjnl-2012-202878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edmiston JS, Archer KJ, Scian MJ, et al. Gene expression profiling of peripheral blood leukocytes identifies potential novel biomarkers of chronic obstructive pulmonary disease in current and former smokers. Biomarkers. 2010;15:715–730. doi: 10.3109/1354750X.2010.512091. [DOI] [PubMed] [Google Scholar]
- 69.Bahr TM, Hughes GJ, Armstrong M, et al. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;49:316–323. doi: 10.1165/rcmb.2012-0230OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Poliska S, Csanky E, Szanto A, et al. Chronic obstructive pulmonary disease-specific gene expression signatures of alveolar macrophages as well as peripheral blood monocytes overlap and correlate with lung function. Respiration. 2011;81:499–510. doi: 10.1159/000324297. [DOI] [PubMed] [Google Scholar]
- 71.Bhattacharya S, Tyagi S, Srisuma S, et al. Peripheral blood gene expression profiles in COPD subjects. J Clin Bioinforma. 2011;1:12. doi: 10.1186/2043-9113-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wain LV, Sayers I, Soler Artigas M, et al. Whole Exome Re-Sequencing Implicates CCDC38 and Cilia Structure and Function in Resistance to Smoking Related Airflow Obstruction. PLoS Genet. 2014;10:e1004314. doi: 10.1371/journal.pgen.1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryan DM, Vincent TL, Salit J, et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS One. 2014;9:e88051. doi: 10.1371/journal.pone.0088051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beane J, Vick J, Schembri F, et al. Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res (Phila) 2011;4:803–817. doi: 10.1158/1940-6207.CAPR-11-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hackett NR, Butler MW, Shaykhiev R, et al. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang G, Xu Z, Wang R, et al. Genes associated with MUC5AC expression in small airway epithelium of human smokers and non-smokers. BMC Med Genomics. 2012;5:21. doi: 10.1186/1755-8794-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ezzie ME, Crawford M, Cho JH, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67:122–131. doi: 10.1136/thoraxjnl-2011-200089. [DOI] [PubMed] [Google Scholar]
- 79.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christenson SA, Brandsma CA, Campbell JD, et al. miR-638 regulates gene expression networks associated with emphysematous lung destruction. Genome Med. 2013;5:114. doi: 10.1186/gm519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheung VG, Spielman RS, Ewens KG, et al. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–1369. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stranger BE, Nica AC, Forrest MS, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiu W, Cho MH, Riley JH, et al. Genetics of sputum gene expression in chronic obstructive pulmonary disease. PLoS One. 2011;6:e24395. doi: 10.1371/journal.pone.0024395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamontagne M, Couture C, Postma DS, et al. Refining susceptibility loci of chronic obstructive pulmonary disease with lung eqtls. PLoS One. 2013;8:e70220. doi: 10.1371/journal.pone.0070220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–1350. doi: 10.1001/jama.299.11.1345. [DOI] [PubMed] [Google Scholar]
- 86.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond, Nature reviews. Genetics. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 87.Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Human Molecular Genetics. 2012;21:3073–3082. doi: 10.1093/hmg/dds135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Breitling LP, Yang R, Korn B, et al. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. American journal of human genetics. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. American Journal of Respiratory and Critical Care Medicine. 2012;185:373–381. doi: 10.1164/rccm.201108-1382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wan ES, Qiu W, Baccarelli A, et al. Systemic steroid exposure is associated with differential methylation in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2012;186:1248–1255. doi: 10.1164/rccm.201207-1280OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vucic EA, Chari R, Thu KL, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50:912–922. doi: 10.1165/rcmb.2013-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Agusti A, Sin DD. Biomarkers in COPD. Clinics in chest medicine. 2014;35:131–141. doi: 10.1016/j.ccm.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 93.Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2014;69:666–672. doi: 10.1136/thoraxjnl-2013-204778. [DOI] [PubMed] [Google Scholar]
- 94.Kim DK, Cho MH, Hersh CP, et al. Genome-wide association analysis of blood biomarkers in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 2012;186:1238–1247. doi: 10.1164/rccm.201206-1013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bowler RP, Bahr TM, Hughes G, et al. Integrative omics approach identifies interleukin-16 as a biomarker of emphysema. Omics : a journal of integrative biology. 2013;17:619–626. doi: 10.1089/omi.2013.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burrows B, Fletcher CM, Heard BE, et al. The emphysematous and bronchial types of chronic airways obstruction. A clinicopathological study of patients in London and Chicago. Lancet. 1966;1:830–835. doi: 10.1016/s0140-6736(66)90181-4. [DOI] [PubMed] [Google Scholar]
- 97.Agusti A, Calverley PM, Celli B, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. 2010;11:122. doi: 10.1186/1465-9921-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rennard SI, Vestbo J. The many “small COPDs”: COPD should be an orphan disease. Chest. 2008;134:623–627. doi: 10.1378/chest.07-3059. [DOI] [PubMed] [Google Scholar]
- 99.Castaldi PJ, Dy J, Ross J, et al. Cluster analysis in the COPDGene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax. 2014 doi: 10.1136/thoraxjnl-2013-203601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castaldi PJ, Cho MH, Cohn M, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet. 2010;19:526–534. doi: 10.1093/hmg/ddp519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hersh CP, Demeo DL, Lange C, et al. Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am J Respir Cell Mol Biol. 2005;33:71–78. doi: 10.1165/rcmb.2005-0073OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jia P, Zheng S, Long J, et al. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics. 2011;27:95–102. doi: 10.1093/bioinformatics/btq615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fehringer G, Liu G, Briollais L, et al. Comparison of pathway analysis approaches using lung cancer GWAS data sets. PLoS ONE. 2012;7:e31816. doi: 10.1371/journal.pone.0031816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holden M, Deng S, Wojnowski L, et al. GSEA-SNP: applying gene set enrichment analysis to SNP data from genome-wide association studies. Bioinformatics. 2008;24:2784–2785. doi: 10.1093/bioinformatics/btn516. [DOI] [PubMed] [Google Scholar]
- 105.Arning A, Hiersche M, Witten A, et al. A genome-wide association study identifies a gene network of ADAMTS genes in the predisposition to pediatric stroke. Blood. 2012;120:5231–5236. doi: 10.1182/blood-2012-07-442038. [DOI] [PubMed] [Google Scholar]
- 106.Han S, Yang BZ, Kranzler HR, et al. Integrating GWASs and human protein interaction networks identifies a gene subnetwork underlying alcohol dependence. American journal of human genetics. 2013;93:1027–1034. doi: 10.1016/j.ajhg.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pelaia G, Terracciano R, Vatrella A, et al. Application of Proteomics and Peptidomics to COPD. BioMed research international. 2014;2014:764581. doi: 10.1155/2014/764581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Snowden S, Dahlen SE, Wheelock CE. Application of metabolomics approaches to the study of respiratory diseases. Bioanalysis. 2012;4:2265–2290. doi: 10.4155/bio.12.218. [DOI] [PubMed] [Google Scholar]
- 109.Wagner JR, Busche S, Ge B, et al. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome biology. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Couper D, LaVange LM, Han M, et al. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS) Thorax. 2014;69:491–494. doi: 10.1136/thoraxjnl-2013-203897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haferlach T, Kohlmann A, Wieczorek L, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812–1823. doi: 10.1016/S0140-6736(11)61539-0. [DOI] [PubMed] [Google Scholar]
- 113.Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits. Nature reviews Genetics. 2014;15:34–48. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. American journal of respiratory and critical care medicine. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 115.Carolan BJ, Heguy A, Harvey BG, et al. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006;66:10729–10740. doi: 10.1158/0008-5472.CAN-06-2224. [DOI] [PubMed] [Google Scholar]
- 116.Pierrou S, Broberg P, O'Donnell RA, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:577–586. doi: 10.1164/rccm.200607-931OC. [DOI] [PubMed] [Google Scholar]
- 117.Ammous Z, Hackett NR, Butler MW, et al. Variability in Small Airway Epithelial Gene Expression Among Normal Smokers. Chest. 2008;133:1344–1353. doi: 10.1378/chest.07-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tilley AE, Harvey BG, Heguy A, et al. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179:457–466. doi: 10.1164/rccm.200705-795OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Durham AL, McLaren A, Hayes BP, et al. Regulation of Wnt4 in chronic obstructive pulmonary disease. FASEB J. 2013;27:2367–2381. doi: 10.1096/fj.12-217083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Van Pottelberge GR, Mestdagh P, Bracke KR, et al. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:898–906. doi: 10.1164/rccm.201002-0304OC. [DOI] [PubMed] [Google Scholar]
- 121.Leidinger P, Keller A, Borries A, et al. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer. 2011;74:41–47. doi: 10.1016/j.lungcan.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 122.Akbas F, Coskunpinar E, Aynaci E, et al. Analysis of serum micro-RNAs as potential biomarker in chronic obstructive pulmonary disease. Exp Lung Res. 2012;38:286–294. doi: 10.3109/01902148.2012.689088. [DOI] [PubMed] [Google Scholar]
- 123.Chatila WM, Criner GJ, Hancock WW, et al. Blunted Expression of miR-199a-5p in Treg Cells of Patients with Chronic Obstructive Pulmonary Disease Compared to Unaffected Smokers. Clinical and experimental immunology. 2014 doi: 10.1111/cei.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]