

Abstract
Accumulating evidence support the notion that acute myeloid leukemia (AML) is organized in a hierarchical system, originating from a special proportion of leukemia stem cells (LSC). Similar to their normal counterpart, hematopoietic stem cells (HSC), LSC possess self-renewal capacity and are responsible for the continued growth and proliferation of the bulk of leukemia cells in the blood and bone marrow. It is believed that LSC are also the root cause for the treatment failure and relapse of AML because LSC are often resistant to chemotherapy. In the past decade, we have made significant advancement in identification and understanding the molecular biology of LSC, but it remains a daunting task to specifically targeting LSC, while sparing normal HSC. In this review, we will first provide a historical overview of the discovery of LSC, followed by a summary of identification and separation of LSC by either cell surface markers or functional assays. Next, the review will focus on the current, various strategies for eradicating LSC. Finally, we will highlight future directions and challenges ahead of our ultimate goal for the cure of AML by targeting LSC.
Keywords: Acute myeloid leukemia, Leukemia stem cell, Immunotherapy, Cancer stem cell, Cell therapy
Core tip: Acute Myeloid Leukemia (AML) remains an incurable disease in most of cases. Leukemia stem cells (LSC) are a subpopulation of leukemic cells responsible for the continued proliferation and propagation of bulk leukemic cells. Growing evidence support the notion that LSCs are the root source of disease relapse and treatment resistance. Here we review the literature on historical overview of the discovery of LSC, identification and separation of LSC and strategies of targeting LSC as a potential cure for AML.
INTRODUCTION
Acute myeloid leukemia (AML) remains a hefty challenge for hematologists and oncologists. There are approximate 18800 new cases diagnosed with AML each year in United States alone, but estimated death cases is as high as 10000, ranking AML as the 6th highest cancer-related death in male population (Cancer Facts and Figures 2014, American Cancer Society). AML is a group of morphologically, genetically and epigentically heterogeneous disorders characterized by the accumulation of differentiation-arrested abnormal hematopoietic progenitor cells in the bone marrow and blood. The complexity of AML is further complicated by the existence of a spectrum of functionally diverse leukemic and preleukemic clones. Recent strides in massively parallel sequencing technology and powerful bioinformatic tools enable us to gain a deep and panoramic insight of AML genome and epigenome at unprecedented level. Elegant studies tracking clonal evolution from diagnosis to relapse revealed the greater clonal heterogeneity in AML than we previously estimated[1-3]. Some clones either founding clone (major clone) or subclones (minor clone) at diagnosis, can survive chemotherapy. These survival clones may gain a small number of cooperating mutations, eventually leading to a relapse[1-3]. For example, a subclone within the founding clones containing somatic mutations in some well-characterized pivot genes such as DNMT3A, FLT3, NPM1, etc., can develop into dominant clone after acquiring additional mutations in ETV6 and MYO18B. The mutations in these pivot genes are recurrent in AML[1].
From the identification of chromosomal translocation in the 1970s, leukemia has been a prime and pioneering paradigm for the breakthrough discoveries in cancer genetics and the development of novel therapeutics[4]. For example, the demonstration of the presence of leukemia stem cells (LSC) has preceded the discovery of the first cancer stem cells (CSC) in solid tumor (breast cancer) by almost 10 years[5]. LSC, or leukemia initiating cells (LIC), are a subpopulation of cells that acquire self-renewal function and sustain the disease. AML LSC is the not only the first identified CSC, but also the best characterized CSC. It has become increasingly apparent that AML LSCs are generally insensitive to the conventional chemotherapy. They reside in the bone marrow microenvironment and are poised to propagate, leading to the treatment failure and relapse. This suggests that the LSC subpopulation is the culprit for the poor outcome of AML patients and selectively targeting LSC will be a important strategy towardscuring AML.
IDENTIFICATION OF LSC-CELL SURFACE MARKERS IN COMBINATION WITH FUNCTION ASSAYS
CD34+CD38-: the beginning of LSC hunting
Pioneer studies from John Dick’s group in 1990s firmly established the AML LSC model, that AML is a hierarchical disease which is initiated and sustained by a rare subset of LSC. Only the subset of immature CD34+CD38- leukemia cells is capable of not only initiating leukemia in sublethally irradiated immunodeficiency mice, but also transplantable in second and third generation mice. In contrast, the fraction of more mature CD34+CD38+ leukemia blasts failed to imitate disease under the same condition. The estimated frequency of LSC in the CD34+CD38- cells is one in one million, thus LSC represent a very rare of unique population of leukemia cells sharing the similar cell surface marker as normal immature hematopoietic cells. Importantly, several clinically observatory studies demonstrated that high frequency of CD34+CD38- cells, but not total CD34+ cells, amongst blast cells at diagnosis correlates with poor survival in both adult and pediatric AML patients[6,7]. More recently, gene expression profiles generated from this rare subset of CD34+CD38- cells support their clinical impact that high expression of LSC signature predicts worse outcome[8-11].
However, recently findings derived from newly generated NOD/ShiLtSz-scid/IL2Rγnull (NSG) and NOD/ShiJic-scid/IL2Rγnull (NOG) mice, the most immunodeficient strains, cast new light on the origin of LSC. These two strains of mice don’t express the IL-2 receptor common gamma chain, which allow more efficient engraftments of human hematopoietic cells than SCID or NOD/SCID mice in previous studies. Using these more immuosupressive mice as hosts, CD34+CD38+ cells from some primary AML can induce transplantable disease, indicating CD34+CD38+ cells have LSC activity too[12,13]. Works from Bonnet’s laboratory unveiled the possibly confounding factor that the anti-CD38 antibody used for separation of primary AML cells has significant inhibitory effect on engraftment of leukemia cells[13]. Taken together, these studies suggest LSC might co-exist in CD34+CD38- and CD34+CD38+ subpopulation.
Cell surface markers differentially expressed between LSC and normal HSC
Because LSC and HSC sharing similar CD34+CD38- surface immunophenotype, the search of cell surface markers unique to LSC (ideal circumstances) or at least differentially expressed has attracted intensive enthusiasm in hematology and oncology field. Such makers will provide excellent therapeutic windows for specifically targeting LSC, while sparing normal HSC. Such therapies are expected to be much tolerable for AML patients.
CD90
CD90, also known as Thy-1, is a small glycosylphosphatidylinositol (GPI)-anchored protein (25-37 kDa) regulating multiple signaling cascades which control cellular survival, proliferation, adhesion and response to cytokines[14]. One of the early studies reported that the majority of AML blasts did not express CD90 and CD34+CD90- cells were capable of maintaining the disease in vitro and in vivo as demonstrated by production of leukemic clonogenic cells (CFU) and engraftments in nonobese diabetic severe combined immune deficient (NOD/SCID) mice, respectively[15]. However, independent study to validate CD90 as a possible LSC marker is scarce in the literature. In contrast, CD90 expression was detected at high frequency of a group of high-risk AML, such as secondary AML (40%) and elderly > 60 years AML (24%) patients[16]. Univariate analysis revealed that CD90 expression was an independent prognostic factor for a shorter survival[16]. This finding appears to contradict to the proposal of CD34+CD90- fraction is the source of LSCs because it is generally believed that abundant level of LSC markers is associated with poor survival. Interestingly, CD90 has been identified as marker of cancer stem cell (CSC) of hepatocellular carcinoma[17], esophageal cancer[18] and high-grade gliomas[19].
CD96
CD96 (also known as TACTILE), a type I membrane protein, belongs to the immunoglobulin superfamily. CD96 plays a role in the antigen presentation of immune response the adhesive interactions of activated T and NK cells. CD96 is expressed on the majority of CD34+CD38- AML cells and vice versa[20]. In contrast, CD96 is weakly expressed in cells in the normal HSC-enriched population [Lin(-)CD34(+)CD38(-)CD90(+)]. Significant level of engraftment is only achieved in mice implanted with CD96+ AML cells, but not CD96- AML cells[20]. From a therapeutic point view, this LSC marker offers a few new avenues for treatment of AML disease. Firstly, CD96 specific monoclonal antibody can be used to selectively eradicate AML-LSCs before autologous stem cell transplantation[21]. Secondly, Fc-engineered mini-antibodies directed against CD96 shows enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) activity of affinity and the highest cytolytic potential[22].
CD123
CD123 is also known as interleukin 3 receptor, alpha (IL-3Rα). IL3R is a heterodimeric cytokine receptor comprised of the alpha unit and beta unit, which is activated by the ligand binding and necessary of IL-3 activity[23]. IL-3 is one of the prominent cytokines that controls proliferation, growth and differentiation of hematopoietic cells[24]. Compared to all other cell surface antigens as potential LSC markers, the studies on CD123 have been investigated into much more details and targeting CD123 is now in clinical trials[23].
Jordan and colleagues[25] first reported that CD123 was aberrantly expressed on CD34+CD38- cells from AML patients, but not detectable on CD34+CD38- cells from healthy controls. Moreover, purified CD34+CD123+ cells from AML patients were capable of establishing and propagating leukemia disease in NOD/SCID mice[25]. This result functionally validated CD123 as a LSC marker. A following-up study from the same group further revealed that NFκB activity was constitutively activated in the CD123+ LSCs, but not CD123+ normal HSC, providing a molecular difference between these two cell entities[26]. Higher level of spontaneous signal transducer and activator of transcription 5 (STAT5) activity is another factor contributing to the proliferative advantage and resistance to apoptosis of AML blasts with elevated CD123[27]. It is well documented that enhanced STAT pathway activity confers drug resistance in AML[28], possibly through two distinct mechanisms: upregulation of anti-apoptotic survivin (BIRC5), Bcl-xL (BCL2L1) genes and ATP-binding cassette (ABC) family genes, which encode multidrug-resistance (MDR) transport proteins.
The utility of CD123 as a LSC marker has been convincingly confirmed by many other studies[29,30]. A flow cytometric analysis of CD123 expression of diagnostic blasts from 111 de novo AML patients younger than 65 years old shows the presence of more than 1% population of CD34(+)CD38(low/-)CD123(+) cells adversely affected the disease-free-survival and over-all survival[30]. Notably, not only the percentage of CD123+ cells, but also the expression level of CD123+ predicts clinical outcome. Patients whose AML blasts have higher CD123 expression have a lower complete remission (CR) rate and shorter survival duration than those showing normal CD123 expression level[27]. In AML arsing from Fanconi anemia (FA) background, only CD123+ cells achieve significant level of engraftment and cause leukemia in a "humanized" FA xenotransplant model[29].
Other studies have depicted what other molecules are co-expressed with CD123 in AML-LSCs. High CD123 AML cells often exhibit elevated level of receptor tyrosine kinases (RTKs) such as FLT3 (Fms-Related Tyrosine Kinase 3), c-Kit[31], N-cadherin and Tie2 (Tunica Interna Endothelial Cell Kinase)[32]. Both FLT3 and c-Kit are important RTKs for the survival of hematopoietic stem/progenitor cells. N-Cadherin and Tie2 play a pivotal role in regulation of interaction between LSCs and their niche in the bone marrow microenvironment. These findings reinforce the role of CD123 as a LSC marker because these co-expression molecules provide CD123+ cells survival advantages and sanctuary in their niche environments.
Antibody therapy specifically targeting CD123 has been advanced to clinical development over a short 5-year period since the first report of in vivo preclinical study[33]. Anti-CD123 monoclonal antibody 7G3 has been shown to completely inhibit bone marrow engraftment by ex vivo treatment and partially impede bone marrow engraftment in a pre-established disease model in mice. CSL360, a recombinant chimeric IgG1 mAb derived from 7G3, was evaluated in phase I clinical trial against AML. The preliminary results showed that anti-CD123 mAb therapy with CSL360 is safe and tolerable and biological effects have been observed (ClinicalTrials.gov Identifier: NCT00401739). A humanized, affinity-matured version of anti-CD123 antibody, CSL362, was developed through engineering the Fc-domain for increased affinity for human CD16 (FcγRIIIa) on (natural killer) NK cells. CSL362 exhibits greater ADCC against both bulks of AML blasts and CD34+CD38-CD123+ LSCs[34]. Currently, CSL362 is under phase I clinical trials in patients with CD123+ AML in complete remission (CR) or CR with incomplete platelet recovery at high risk for early relapse (Clinical Trials.gov identifier: NCT01632852). Novel molecules targeting both CD123 and CD33 have been shown to have stronger anti-AML effect than mono-targeting agents in vitro[35]. It will be interesting to test these dual-targeting or triple-targeting molecules in animal studies or even in human clinical trials against LSC.
Adoptive T cell therapy is an alternatively attractive approach for the treatment of cancer utilizing chimeric antigen receptors (CARs)[36]. The third generation of CARs consist of an extracellular antigen-binding domain and three or more intracellular signaling domains[36]. CD123 chimeric antigen receptor (CAR) redirected T cells/cytokine-induced killer (CIK) cells show robust activity against CD123+ cell lines, primary AML cells and mouse xenograft models transplanted with patient AML cells[37-39]. One important advantage of this approach lies on the observation that relapsed or refractory AML cells which often are chemotherapy-resistant are still vulnerable to CD123 CAR T cell therapy[37]. However, depletion of normal human myelopoiesis caused by CD123 CAR T cells as a potential side effect should be taken account when planning a clinical trial[38].
Taken together, novel immunotherapy approaches such as improved variants of anti-CD123 monoclonal Ab and CD123 CAR T cell therapy hold great promising for AML treatment.
CD47
CD47 (also known as Integrin-associated protein, IAP) is one of the unique member of the Ig superfamily, consisting of a V-type Ig-like extracellular domain at its N-terminus, five hydrophobic membrane-spanning segments and a variably spliced (3-36 amino acids) cytoplasmic tail at its C-terminus[40]. CD47 is a receptor for the C-terminal cell binding domain of thrombospondin-1 (TSP-1) and a ligand for the extracellular region of signal-regulatory protein alpha (SIRPα)[41]. CD47 is ubiquitously expressed on human cells and involved in many fundamental cellular processes including immune and angiogenic responses[40].
Majeti and co-workers first discovered higher expression of CD47 on AML LSC compared to their normal counterparts, HSC and multipotent progenitor cells (MPP), by flow cytometer and microarray gene expression analysis[42,43]. The association between increased CD47 expression with worse outcome has been validated in 3 independent, large clinical cohorts with total 664 AML patients. Moreover, increased CD47 expression remains a prognostic factor for poor event-free survival and over-all survival in multivariable analysis considering age, FLT3-ITD status[42]. SIRPα serves as inhibitory receptor expressed on phagocytic cells such as macrophages and dendritic cells. It was previously reported CD47 expressed on red blood cells (RBC) as a marker of self and interaction of CD47 and SIRPα on phagocytic cells delivered a “do not eat me” message, limiting clearance of circulating RBC by the means of phagocytosis[44]. Similarly, upregulation of CD47 on AML LSCs prevents themselves from the attack of phagocytic cells through the interaction of CD47 with its inhibitory ligand SIRPα. This conclusion is supported by several lines of evidence. Firstly, human AML cell line with low endogenous CD47 level fails to engraft in immunodeficient mice, while ectopic expression of mouse CD47 in this cell line improves engraftment[45]. In an inducible and controlled expression of CD47 in vitro and in vivo models, it has shown that the level of CD47 expression negatively correlates the percentage of phagocytosis by the macrophages[45]. Secondly, transgenic mice expressing SIRPα variants with differential ability to bind human CD47 demonstrates that the engraftment of AML LSCs depends on the interaction of CD47 with SIRPα and AML LSCs are eliminated by macrophage-mediated phagocytosis in the absence of SIRPα signaling. In addition, pharmacological disruption of CD47-SIRPα binding by SIRPα-Fc fusion protein augments phagocytosis of AML cells by both mouse and human macrophages and damages engraftment of CD34+CD38- AML LSCs in mice[46]. Thirdly, AML patients with high SIRPα mRNA expression on AML blasts have poor survival and inhibition of SIRPα signaling lead to reduced cell proliferation and enhanced apoptosis of AML cells[47]. Based on the aforementioned evidence generated from in vitro experiments, in vivo mouse model and clinical data, we believe elevation of CD47 expression in AML LSCs appears to enable them to evade host immune surveillance.
A few anti-CD47 monoclonal antibodies have been tested in vitro and animal models. Two antibodies that block CD47/SIRPα interaction induce phagocytosis of AML cells in vitro and in vivo and eradicate LSCs in xenograft mouse and isogenic mouse leukemia models, while an anti-CD47 antibody that does not disrupt CD47 binding to SIRPα fails to promote phagocytosis of AML cells[42]. The other promising strategy to target this interaction is to use soluble SIRPα-Fc fusion proteins to neutralize CD47[46]. Treatment of SIRPα-Fc fusion proteins leads to activate macrophages mediated phagocytosis, resulting in potent anti-AML effect and clearance of LSCs[46].
Taken together, these evidences indicate that delivering a “do not eat” signal to phagocytic cells is a prime consequence of CD47/SIRPα interaction, which suppresses phagocytosis. Disruption of this interaction would successfully initiate innate immune response to eliminate LSCs through macrophage phagocytosis.
CD44
CD44 belongs to a family of transmembrane glycoproteins that act primarily as a receptor for hyaluronan acid (HA), but it also binds to other receptors including osteopontin, collagens, matrix metalloproteinases (MMPs), etc.[48]. Hyaluronan is one of the major components of the extracellular matrix[49]. The major function of CD44 is to regulate cell-cell adhesion and cell-matrix interaction through binding to HA and other receptors[49]. Specifically, the roles of CD44 in haematopoiesis include cell migration, proliferation, differentiation, survival and bone marrow homing of hematopoietic stem/progenitor cells[50].
It has been long recognized that CD44 is expressed in normal and leukemic CD34+ early hematopoietic cells and empowers them to seek intramedullary or extramedullary sanctuary[51]. It has been postulated that such protective ability resulted from CD44 interaction with various cellular receptors and matrix components allows small numbers of leukemic cells to survive from the attack of cytotoxic chemotherapy[52].
Detection of CD44 and coexpression of CD123 (abovementioned) on CD34+CD38- AML cells indicates the CD44 is a potential candidate of LSC marker[53]. Jin et al[54] first comprehensively characterized CD44 as a critical regulator of AML LSCs in a few mouse models. Treatment with H90, a monoclonal antibody targeting CD44, significantly prolonged survival of NOD/SCID mice transplanted with CD34+CD38- AML LSCs and reduced the number of LSCs in mouse bone marrow as compared to control IgG treatment. Furthermore, in a secondary transplantation experiment, leukemic cells obtained from H90 treated mice (primary mice) failed to engraft into the secondary receipt mice. However, in the parallel experiment, leukemic cells harvested from primary mice treated with control IgG initiated robust engraftment in the secondary receipt mice[54]. The power of eliminating LSCs by anti-CD44 monoclonal antibody treatment could be explained by three different mechanisms by which targeting CD44 induces leukemic cell differentiation[54-58], inhibits cell cycle progression and cell proliferation[58,59] and impedes LSCs homing to bone marrow niches[54]. Collectively, these data conclusively demonstrate that CD44 is functional important for LSCs.
CD32 OR CD25
CD32 is a member of a family of immunoglobulin Fc receptors, expressed on macrophages, neutrophils and nature killer cells[60]. CD32 binds to the Fc region of immunoglobulins gamma (Igγ) and executes phagocytosis and clearing of immune complexes[60]. CD25 is also known as interleukin 2 (IL2) receptor alpha (IL2RA)[61]. IL-2 cytokine regulates cell proliferation, differentiation, survival and apoptosis[62].
CD32 and CD25 were discovered to be overexpressed on quiescent and chemotherapy-resistant human AML LSCs by microarray study of LSCs vs normal HSCs. Normal CD34+CD38-CD133+ HSCs are negative for CD32 or CD25 expression[63]. In xenotransplantation experiments with sorted human AML cells injected into immunodeficient mice, CD32+CD34+CD38- or CD25+CD34+CD38- cells were capable of engraftment and inducing AML. On the contrary, no engraftment was detected in mice inoculated with CD32-CD34+CD38- or CD25-CD34+CD38- cells[63]. The CD32+CD34+CD38- or CD25+CD34+CD38- cells not only survived after treating the mice with cytosine arabinoside (Ara-C), but also initiated in vivo AML when injected into the secondary receipt mice in a serial transplantation model[63].
CLL-1
C-type lectin-like molecule-1 (CLL-1) is a member of type II transmembrane receptor family containing C-type lectin/C-type lectin-like domain (CTL/CTLD). CLL-1 was initially identified as a novel surface marker of AML cells through phage display technology combined with flow cytometry[64]. Further studies revealed that CLL-1 was expressed on CD34+CD38- cells in 87% of AML patients, but was not expressed in normal HSCs[65]. Successful engraftment was observed in all 3 NOD/SCID mice transplanted with CD34+CLL-1+ AML cells[65]. The same group also reported that side population (SP) cells isolated from AML samples which were highly enriched for LSCs also expressed CLL-1[66].
A series of monoclonal antibodies against CLL-1 was developed and two lead antibodies were chosen based on their high affinity and potent cytotoxic activity[67]. These antibodies induced dose-dependent complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity against AML cell lines, primary AML patient cells and xenograft mice implanted with HL-60 AML cells. However, the possibility of targeting LSCs was not assessed in this study[67]. It would be of interesting to further evaluate the impact of these anti-CLL-1 antibodies on LSCs in animal experiments. Nanomicelles decorated with CLL1-targeting peptides can specifically binds to CD34+CLL-1+ primary AML cells and delivered chemodrug daunorubicin directly to target cells[68]. Importantly, these nanomicelles did not bind to normal CD34+ cells, so it was not expected to harm normal hematopoiesis[68]. The challenge of in vivo delivery of nanomicelles remains a concern.
TIM-3
T cell immunoglobulin-3 (TIM-3) belongs to the mucin domain-containing molecule (Tim) superfamily and is a member of the T cell Ig[69]. TIM-3 is expressed on CD4+ Th1, CD8+ T cytotoxic 1 (Tc1) cells, monocytes/macrophages, dendritic cells and mast cells[70]. TIM-3 plays an important role in T cell response and regulation of innate immunity[69,70].
TIM-3 was found to be expressed on CD34+CD38- fraction of AML cells except FAB M3 subtype (acute promyelocytic leukemia, APL) but absent on normal CD34+CD38- HSCs through comparative analysis of transcriptome of these two populations[71]. TIM-3 expression was significantly higher in a distinct subtype of AML with core binding factor (CBF) translocation or CEBPα mutation[72]. This association was a bit puzzling because AML patients with CBF and CEBPα abnormalities often have favourable prognosis[73]. Reconstitution of AML in immunodeficient mice was established only when TIM-3+ AML cells were transplanted, but not TIM-3- AML cells. Treatment of mice injected with human primary AML cells with an anti-TIM-3 monoclonal antibody, ATIK2a, effectively blocked reconstitution of AML. Importantly, human CD45+ AML cells harvested from the primary recipient mice treated with ATIK2a lose the ability to initiate AML retransplanted into secondary recipient mice[71]. Normal HSCs were not damaged by ATIK2a treatment because normal HSCs appear to residue in TIM-3- population[71,72]. These data suggest that TIM-3 could serve as a useful marker to distinguish LSCs from HSCs and monoclonal antibody against TIM-3 holds promise to eradicate LSCs.
Aldehyde dehydrogenase
Aldehyde dehydrogenase (ALDH) gene superfamily consists of 19 functional genes and three pseudogenes. ALDH oxidise a wide range of endogenous and exogenous aldehyde substrates, thus detoxifying large portion of adverse aldehydes to the cells. ALDH is highly expressed in primitive stem cells from several tissue origins, including bone marrow and intestine[74]. HSCs have high level of ALDH activity[75] and can be distinguished using a fluorescent aldehyde, dansyl aminoacetaldehyde (DAAA) in conjunction with FACS analysis[74,76].
Since LSCs share some functional similarity with HSCs, researchers soon started to investigate the role of ALDH in AML LSCs. In total, 3 distinct patterns of ALDH activity were documented. In the first pattern, the subpopulation of AML cells with high ALDH activity was rare, which was similar to the pattern seen in normal core blood. In the second pattern, the frequency of cells with ALDH activity was more frequent and their side scatter profiles were higher than normal stem/ progenitor cells. No fraction of cells with high ALDH activity was present in the third pattern[77]. Xenograft transplantation experiments demonstrated that ALDH+ cells were enriched for LSCs and engrafted better than ALDH- cells[77,78]. From a clinical point of view, higher ALDH activity is associated with dismal prognosis, drug resistance and relapse[78-80].
SMALL MOLECULE INHIBITORS TARGETING LSCS
Parthenolide and analogs
Dimethylamino-parthenolide (DMAPT), modified analog of parthenolide (PTL) which is a major active component of herbal medicine Feverfew, possesses improved pharmacologic properties and is orally bioavailable[81,82]. DMAPT and PTL preferentially kill AML leukemia stem/progenitor cells through mechanisms involved in inhibition of NFκB pathway, induction of tumor suppressor p53 and reactive oxygen species (ROS) production[81,82]. DMAPT shows potent in vivo biological activity in spontaneous canine acute leukemia and mouse xenotransplantation models[82]. DMAPT is a novel compound that is specifically target LSCs and now is being evaluated in a phase 1-2 “first in man” in clinical trial in AML in Cardiff University, United Kingdom.
Epigenetic inhibitors
AR-42 (OSU-HDAC42), a novel histone deacetylase inhibitor (HDACi), inhibits NFκB activity and HSP90 interaction with its various client proteins, leading to robust and selective apoptosis of AML LSCs[83]. Currently, AR-42 is being tested in advanced or relapsed multiple myeloma (MM), chronic lymphocytic leukemia (CLL), or lymphoma in clinical trials (ClinicalTrials.gov Identifier: NCT01129193).
BRD4 (Bromodomain-containing protein 4) was identified as a promising anti-AML target in a whole-genome RNAi screening[84,85]. BRD4 is a chromatin “reader” that recognizes and binds acetylated histones. JQ1 is a novel small molecule inhibitor that competes with BRD4 to bind acetyl-lysine recognition motifs[86]. JQ1 can induce apoptosis in CD34+CD38- and CD34-CD38+ stem- and progenitor cells from both de novo AML and refractory AML patients[87].
3-Deazaneplanocin A (DZNep), is a newly discovered S-adenosyl-methionine-dependent methyltransferase inhibitor[88]. DZNep inhibits EZH2, disrupts polycomb-repressive complex 2 (PRC2), and preferentially induces apoptosis in cancer cells[88]. We and another group showed that DZNep promoted cell death in CD34+CD38- AML cells, but not normal CD34+ progenitor cells[89,90].
Apoptosis pathway modulators
ABT-737, a BCL-2 homology domain 3 mimetic inhibitor, have been shown to target Lin-/Sca-1(+)/c-Kit(+) primitive cells, and progenitor population in a myelodysplastic syndrome (MDS)-AML transgenic mouse model[91].
Using reversed-phase protein array, Carter BZ and colleagues[92] found that CD34+CD38- AML stem/progenitor cells expressed increased caspase 8 and increased ratio of cIAP (Baculoviral IAP Repeat Containing 2, BIRC2) to SMAC (second mitochondrial-derived activator of caspases) compared to bulk AML cells. Birinapant is a novel bivalent SMAC mimetic with high affinity for IAP proteins. Treatment with birinapant induced apoptosis of AML stem/progenitor cells involving in activation of DR (death receptor)/caspase-8 complex. In human AML xenograft mouse model, diseased mice treated with birinapant or in combination with 5-azacytadine (5-Aza), decitabine (DAC), survived significantly longer than mice administrated with vehicle control[92].
Kinase inhibitors
Rapamycin is the first generation of mTOR (mammalian target of rapamycin), a downstream target of phosphatidylinositol 3-kinase (PI3K)-Akt pathway, inhibitor[93]. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) negatively regulates PI3K-AKT-mTOR activity. Tissue specific deletion of PTEN in hematopoietic cells led the mice to develop AML and acute lymphoid leukemia (ALL) and all mice succumbed to disease rapidly in one month[94]. One out of 81 Flk-2-Sca-1+Lin-c-Kit+CD48- (enriched for LSCs) from PTEN null AML mice was able to initiate AML in serial transplantation experiments[94]. A search of “ClinicalTrials.gov” database on 10 July 2014 identified a total of 40 clinical trials that test Rapamycin or its analogs, Temsirolimus (CCI-779) or Everolimus (RAD001), in AML either by alone or in combination with chemotherapy or kinase inhibitors or transplantation. However, it appears that the evaluation of the effect of mTOR inhibitors against LSCs is not included in these trials.
Dasatinib is a multiple kinase inhibitors targeting Abl, Src family and c-Kit. Dos Santos et al[95] reported that combination of dasatinib and daunorubicin enhanced the eradication of AML LSCs in mouse xenotransplantation model through increasing p53 activity[95].
Hematopoietic Cell Kinase (HCK) belongs to the Src family of tyrosine kinases. HCK is mostly expressed in hematopoietic cells, particularly phagocytes. HCK was reported by Saito Y, et al. to overexpress on quiescent, chemotherapy-resistant LSCs compared to normal HSCs[63]. The same group performed integrated, multiple platform analysis to uncover RK-20449, a pyrrolo-pyrimidine derivative as a potent inhibitor of these LSCs in vitro and in vivo[96].
CONCLUSION
The advance in high-throughput and whole genome techniques in conjunction with the development of more immunocompromised mouse strains helps deepen and broaden our understanding of LSCs, the enigmatic fraction of leukemic cells which is the origin of the disease. From the single pattern of CD34+CD38- as phenotypic hallmark for LSCs, a long-list of additional cell surface antigens such as CD123, CD47, CD44, CLL-1, CD96, CD90, CD32, CD25, and TIM-3, has been identified to separate LSCs from normal HSCs (Figure 1). From the notion that LSC is extremely rare, it is now clear that the frequency of LSC among AML patients is highly heterogenous, ranging from very low to frequent. From the concept that LSCs only reside in CD34+CD38- subpopulation, emerging study reveals that CD34+CD38+ fraction also harbours LSCs. From the idea that one patient only has one population of LSCs, we now understand that some patients may have more than one populations of LSCs.
Figure 1.
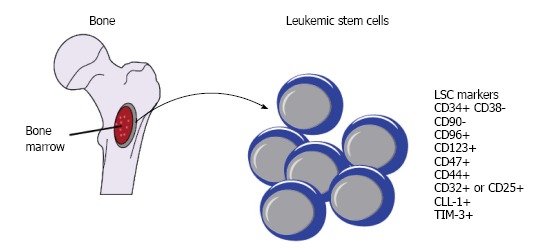
Diagram of leukemia stem cells, bone marrow microenvironment and phenotypic markers of leukemia stem cell. LSC: Leukemic stem cell.
Along the advance in our understanding of LSC, a growing list of strategies for targeting LSC has been proposed and some of these agents as summarized above have advanced into clinical trials. Currently, monoclonal antibodies targeting CD123 or their related immunoconjugate therapy or CD123 CAR T cell therapy appear to be the front runner leading the way to eliminate LSC and eventually cure AML. The second gold mine for the discovery of drug targets is how LSCs employ “epigenetic machinery” to program or reprogram themselves because epigenetic changes are reversible and epigenetic enzymes are often targetable. The first generation of some of these small molecule inhibitors such as DZNep, JQ1, already showed potent effect in killing LSCs. We shall witness the second generation of these compounds or novel small molecule inhibitors with favourable pharmacological profiles and safety profiles entering clinical trials in the next few years.
However, the real impact on clinical management of AML is far less promising than the remarkable response observed in ex vivo cell culture models or xenotransplanted mouse experiments as reported in numerous “sophisticated” studies. In our opinion, although many surface antigens have been identified to be aberrantly expressed on LSCs, it is probably impossible for any single monoclonal antibody targeting one of these surface antigens to eradicate LSCs, given such heterogeneity and dynamics of LSC properties in AML patients. Synergistic therapies in combination with immunotherapy, cell therapy and epigenetic drugs may provide a better opportunity to achieve our ultimate goal of targeting LSCs and curing AML (Figure 2). By using CD123 target as an example, it is hoped that combination of CD123 CAR T cells which bind to CD123 on the surface of LSC or mono- or dual- targeting antibody with small molecule inhibitors targeting epigenetic machinery, such as Brd4 inhibitor or HMTi or HDACi, will be effective for the treatment of AML.
Figure 2.
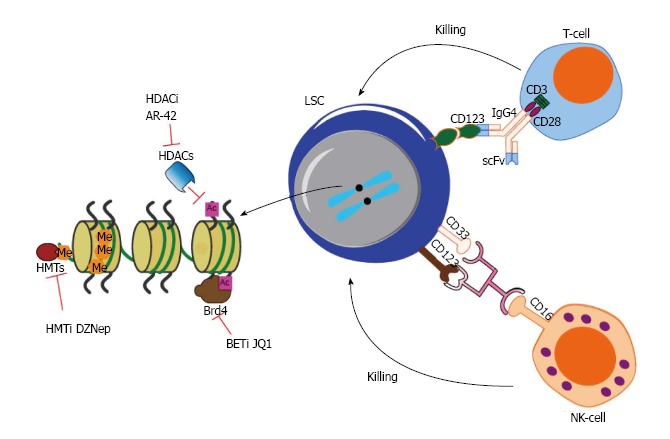
This illustration shows combination therapies aiming to achieve maximal and synergistic anti-leukemia stem cells effect. HDACi: Histone deacetylase inhibitor; HMTi: Histone methyltransferase inhibitor; BETi: Bromodomain and Extra-Terminal inhibitor; Brd4: Bromodomain-containing protein 4; Ac: acetylation; Me: methylation; NK-cell: Natural killer-cell.
ACKNOWLEDGMENTS
The authors thank Mr Ching Ying Qing for his excellent illustration of Figures 1 and 2. Due to space limit, some of important works in this field were not cited and we sincerely apologize to those authors whose important studies were not summarized.
Footnotes
Supported by National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative, NMRC Clinician-Scientist IRG Grant CNIG11nov38 and NMRC Clinician Scientist Investigator award
P- Reviewer: Chen SS, Fukuda S, Krimerk DB S- Editor: Tian YL L- Editor: A E- Editor: Lu YJ
References
- 1.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, Larson DE, McLellan MD, Dooling D, Abbott R, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandts CH, Berdel WE, Serve H. Oncogenic signaling in acute myeloid leukemia. Curr Drug Targets. 2007;8:237–246. doi: 10.2174/138945007779940197. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 6.van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, van der Pol MA, Waisfisz Q, Ossenkoppele GJ, Schuurhuis GJ. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–6527. doi: 10.1158/1078-0432.CCR-05-0468. [DOI] [PubMed] [Google Scholar]
- 7.Witte KE, Ahlers J, Schäfer I, André M, Kerst G, Scheel-Walter HG, Schwarze CP, Pfeiffer M, Lang P, Handgretinger R, et al. High proportion of leukemic stem cells at diagnosis is correlated with unfavorable prognosis in childhood acute myeloid leukemia. Pediatr Hematol Oncol. 2011;28:91–99. doi: 10.3109/08880018.2010.528171. [DOI] [PubMed] [Google Scholar]
- 8.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 9.Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, Nagler A, Tavor S, Ein-Dor L, Lapidot T, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia. 2006;20:2147–2154. doi: 10.1038/sj.leu.2404401. [DOI] [PubMed] [Google Scholar]
- 10.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304:2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krivtsov AV, Wang Y, Feng Z, Armstrong SA. Gene expression profiling of leukemia stem cells. Methods Mol Biol. 2009;538:231–246. doi: 10.1007/978-1-59745-418-6_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, Swider CR, Strzelecki AC, Cavelier C, Récher C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121:384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 14.Rege TA, Hagood JS. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim Biophys Acta. 2006;1763:991–999. doi: 10.1016/j.bbamcr.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89:3104–3112. [PubMed] [Google Scholar]
- 16.Buccisano F, Rossi FM, Venditti A, Del Poeta G, Cox MC, Abbruzzese E, Rupolo M, Berretta M, Degan M, Russo S, et al. CD90/Thy-1 is preferentially expressed on blast cells of high risk acute myeloid leukaemias. Br J Haematol. 2004;125:203–212. doi: 10.1111/j.1365-2141.2004.04883.x. [DOI] [PubMed] [Google Scholar]
- 17.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Tang KH, Dai YD, Tong M, Chan YP, Kwan PS, Fu L, Qin YR, Tsao SW, Lung HL, Lung ML, et al. A CD90(+) tumor-initiating cell population with an aggressive signature and metastatic capacity in esophageal cancer. Cancer Res. 2013;73:2322–2332. doi: 10.1158/0008-5472.CAN-12-2991. [DOI] [PubMed] [Google Scholar]
- 19.He J, Liu Y, Zhu T, Zhu J, Dimeco F, Vescovi AL, Heth JA, Muraszko KM, Fan X, Lubman DM. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol Cell Proteomics. 2012;11:M111.010744. doi: 10.1074/mcp.M111.010744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, Weissman IL. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci USA. 2007;104:11008–11013. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staudinger M, Humpe A, Gramatzki M. Strategies for purging CD96(+) stem cells in vitro and in vivo: New avenues for autologous stem cell transplantation in acute myeloid leukemia. Oncoimmunology. 2013;2:e24500. doi: 10.4161/onci.24500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohseni Nodehi S, Repp R, Kellner C, Bräutigam J, Staudinger M, Schub N, Peipp M, Gramatzki M, Humpe A. Enhanced ADCC activity of affinity maturated and Fc-engineered mini-antibodies directed against the AML stem cell antigen CD96. PLoS One. 2012;7:e42426. doi: 10.1371/journal.pone.0042426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2:4. doi: 10.1186/2050-7771-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas D, Vadas M, Lopez A. Regulation of haematopoiesis by growth factors - emerging insights and therapies. Expert Opin Biol Ther. 2004;4:869–879. doi: 10.1517/14712598.4.6.869. [DOI] [PubMed] [Google Scholar]
- 25.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 26.Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, Luger SM, Jordan CT. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 27.Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, Latagliata R, Mariani G, Rossini A, Battistini A, et al. Elevated expression of IL-3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100:2980–2988. doi: 10.1182/blood-2002-03-0852. [DOI] [PubMed] [Google Scholar]
- 28.Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, Poon LF, Xie Z, Palaniyandi S, Yu H, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:4052–4062. doi: 10.1182/blood-2008-05-156422. [DOI] [PubMed] [Google Scholar]
- 29.Du W, Li XE, Sipple J, Pang Q. Overexpression of IL-3Rα on CD34+CD38- stem cells defines leukemia-initiating cells in Fanconi anemia AML. Blood. 2011;117:4243–4252. doi: 10.1182/blood-2010-09-309179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vergez F, Green AS, Tamburini J, Sarry JE, Gaillard B, Cornillet-Lefebvre P, Pannetier M, Neyret A, Chapuis N, Ifrah N, et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96:1792–1798. doi: 10.3324/haematol.2011.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riccioni R, Rossini A, Calabrò L, Diverio D, Pasquini L, Lococo F, Peschle C, Testa U. Immunophenotypic features of acute myeloid leukemias overexpressing the interleukin 3 receptor alpha chain. Leuk Lymphoma. 2004;45:1511–1517. doi: 10.1080/104281090310001646031. [DOI] [PubMed] [Google Scholar]
- 32.Zhi L, Wang M, Rao Q, Yu F, Mi Y, Wang J. Enrichment of N-Cadherin and Tie2-bearing CD34+/CD38-/CD123+ leukemic stem cells by chemotherapy-resistance. Cancer Lett. 2010;296:65–73. doi: 10.1016/j.canlet.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 33.Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, Thomas D, Barry EF, Boyd A, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, Braley H, Panousis C, Roberts AW, He SZ, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014 doi: 10.1038/leu.2014.128. [DOI] [PubMed] [Google Scholar]
- 35.Kügler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, Schubert I, Singer H, Oduncu F, Stockmeyer B, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. 2010;150:574–586. doi: 10.1111/j.1365-2141.2010.08300.x. [DOI] [PubMed] [Google Scholar]
- 36.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, Hoffman L, Aguilar B, Chang WC, Bretzlaff W, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 40.Sick E, Jeanne A, Schneider C, Dedieu S, Takeda K, Martiny L. CD47 update: a multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br J Pharmacol. 2012;167:1415–1430. doi: 10.1111/j.1476-5381.2012.02099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barclay AN. Signal regulatory protein alpha (SIRPalpha)/CD47 interaction and function. Curr Opin Immunol. 2009;21:47–52. doi: 10.1016/j.coi.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, van Rooijen N, Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Majeti R, Becker MW, Tian Q, Lee TL, Yan X, Liu R, Chiang JH, Hood L, Clarke MF, Weissman IL. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci USA. 2009;106:3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 45.Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, Ho JM, Poeppl AG, van Rooijen N, Minden MD, Danska JS, et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209:1883–1899. doi: 10.1084/jem.20120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irandoust M, Alvarez Zarate J, Hubeek I, van Beek EM, Schornagel K, Broekhuizen AJ, Akyuz M, van de Loosdrecht AA, Delwel R, Valk PJ, et al. Engagement of SIRPα inhibits growth and induces programmed cell death in acute myeloid leukemia cells. PLoS One. 2013;8:e52143. doi: 10.1371/journal.pone.0052143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 49.Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, Ghatak S. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011;278:1429–1443. doi: 10.1111/j.1742-4658.2011.08071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hertweck MK, Erdfelder F, Kreuzer KA. CD44 in hematological neoplasias. Ann Hematol. 2011;90:493–508. doi: 10.1007/s00277-011-1161-z. [DOI] [PubMed] [Google Scholar]
- 51.Liesveld JL, Dipersio JF, Abboud CN. Integrins and adhesive receptors in normal and leukemic CD34+ progenitor cells: potential regulatory checkpoints for cellular traffic. Leuk Lymphoma. 1994;14:19–28. doi: 10.3109/10428199409049647. [DOI] [PubMed] [Google Scholar]
- 52.Bradstock KF, Gottlieb DJ. Interaction of acute leukemia cells with the bone marrow microenvironment: implications for control of minimal residual disease. Leuk Lymphoma. 1995;18:1–16. doi: 10.3109/10428199509064917. [DOI] [PubMed] [Google Scholar]
- 53.Florian S, Sonneck K, Hauswirth AW, Krauth MT, Schernthaner GH, Sperr WR, Valent P. Detection of molecular targets on the surface of CD34+/CD38-- stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47:207–222. doi: 10.1080/10428190500272507. [DOI] [PubMed] [Google Scholar]
- 54.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 55.Charrad RS, Gadhoum Z, Qi J, Glachant A, Allouche M, Jasmin C, Chomienne C, Smadja-Joffe F. Effects of anti-CD44 monoclonal antibodies on differentiation and apoptosis of human myeloid leukemia cell lines. Blood. 2002;99:290–299. doi: 10.1182/blood.v99.1.290. [DOI] [PubMed] [Google Scholar]
- 56.Song G, Liao X, Zhou L, Wu L, Feng Y, Han ZC. HI44a, an anti-CD44 monoclonal antibody, induces differentiation and apoptosis of human acute myeloid leukemia cells. Leuk Res. 2004;28:1089–1096. doi: 10.1016/j.leukres.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Gadhoum Z, Delaunay J, Maquarre E, Durand L, Lancereaux V, Qi J, Robert-Lezenes J, Chomienne C, Smadja-Joffe F. The effect of anti-CD44 monoclonal antibodies on differentiation and proliferation of human acute myeloid leukemia cells. Leuk Lymphoma. 2004;45:1501–1510. doi: 10.1080/1042819042000206687. [DOI] [PubMed] [Google Scholar]
- 58.Zada AA, Singh SM, Reddy VA, Elsässer A, Meisel A, Haferlach T, Tenen DG, Hiddemann W, Behre G. Downregulation of c-Jun expression and cell cycle regulatory molecules in acute myeloid leukemia cells upon CD44 ligation. Oncogene. 2003;22:2296–2308. doi: 10.1038/sj.onc.1206393. [DOI] [PubMed] [Google Scholar]
- 59.Gadhoum Z, Leibovitch MP, Qi J, Dumenil D, Durand L, Leibovitch S, Smadja-Joffe F. CD44: a new means to inhibit acute myeloid leukemia cell proliferation via p27Kip1. Blood. 2004;103:1059–1068. doi: 10.1182/blood-2003-04-1218. [DOI] [PubMed] [Google Scholar]
- 60.Morel PA, Ernst LK, Metes D. Functional CD32 molecules on human NK cells. Leuk Lymphoma. 1999;35:47–56. doi: 10.3109/10428199909145704. [DOI] [PubMed] [Google Scholar]
- 61.Driesen J, Popov A, Schultze JL. CD25 as an immune regulatory molecule expressed on myeloid dendritic cells. Immunobiology. 2008;213:849–858. doi: 10.1016/j.imbio.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 62.Mahmud SA, Manlove LS, Farrar MA. Interleukin-2 and STAT5 in regulatory T cell development and function. JAKSTAT. 2013;2:e23154. doi: 10.4161/jkst.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra9. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, Jongeneelen MA, Visser TJ, Bijl N, Geuijen CA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004;64:8443–8450. doi: 10.1158/0008-5472.CAN-04-1659. [DOI] [PubMed] [Google Scholar]
- 65.van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, Zweegman S, Ossenkoppele GJ, Jan Schuurhuis G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–2666. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 66.Moshaver B, van Rhenen A, Kelder A, van der Pol M, Terwijn M, Bachas C, Westra AH, Ossenkoppele GJ, Zweegman S, Schuurhuis GJ. Identification of a small subpopulation of candidate leukemia-initiating cells in the side population of patients with acute myeloid leukemia. Stem Cells. 2008;26:3059–3067. doi: 10.1634/stemcells.2007-0861. [DOI] [PubMed] [Google Scholar]
- 67.Zhao X, Singh S, Pardoux C, Zhao J, Hsi ED, Abo A, Korver W. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica. 2010;95:71–78. doi: 10.3324/haematol.2009.009811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H, Luo J, Li Y, Henderson PT, Wang Y, Wachsmann-Hogiu S, Zhao W, Lam KS, Pan CX. Characterization of high-affinity peptides and their feasibility for use in nanotherapeutics targeting leukemia stem cells. Nanomedicine. 2012;8:1116–1124. doi: 10.1016/j.nano.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu C, Anderson AC, Kuchroo VK. TIM-3 and its regulatory role in immune responses. Curr Top Microbiol Immunol. 2011;350:1–15. doi: 10.1007/82_2010_84. [DOI] [PubMed] [Google Scholar]
- 70.Han G, Chen G, Shen B, Li Y. Tim-3: an activation marker and activation limiter of innate immune cells. Front Immunol. 2013;4:449. doi: 10.3389/fimmu.2013.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki Y, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–717. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 72.Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, Majeti R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA. 2011;108:5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88:318–327. doi: 10.1002/ajh.23404. [DOI] [PubMed] [Google Scholar]
- 74.Jones RJ, Barber JP, Vala MS, Collector MI, Kaufmann SH, Ludeman SM, Colvin OM, Hilton J. Assessment of aldehyde dehydrogenase in viable cells. Blood. 1995;85:2742–2746. [PubMed] [Google Scholar]
- 75.Kastan MB, Schlaffer E, Russo JE, Colvin OM, Civin CI, Hilton J. Direct demonstration of elevated aldehyde dehydrogenase in human hematopoietic progenitor cells. Blood. 1990;75:1947–1950. [PubMed] [Google Scholar]
- 76.Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA. 1999;96:9118–9123. doi: 10.1073/pnas.96.16.9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pearce DJ, Taussig D, Simpson C, Allen K, Rohatiner AZ, Lister TA, Bonnet D. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005;23:752–760. doi: 10.1634/stemcells.2004-0292. [DOI] [PubMed] [Google Scholar]
- 78.Cheung AM, Wan TS, Leung JC, Chan LY, Huang H, Kwong YL, Liang R, Leung AY. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia. 2007;21:1423–1430. doi: 10.1038/sj.leu.2404721. [DOI] [PubMed] [Google Scholar]
- 79.Ran D, Schubert M, Pietsch L, Taubert I, Wuchter P, Eckstein V, Bruckner T, Zoeller M, Ho AD. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Exp Hematol. 2009;37:1423–1434. doi: 10.1016/j.exphem.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 80.Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, Galkin S, Collector MI, Perkins B, Levis MJ, et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood. 2012;119:3571–3577. doi: 10.1182/blood-2011-06-364182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, Becker MW, Bennett JM, Sullivan E, Lachowicz JL, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guzman ML, Yang N, Sharma KK, Balys M, Corbett CA, Jordan CT, Becker MW, Steidl U, Abdel-Wahab O, Levine RL, et al. Selective Activity of the Histone Deacetylase Inhibitor AR-42 against Leukemia Stem Cells: A Novel Potential Strategy in Acute Myelogenous Leukemia. Mol Cancer Ther. 2014;13:1979–1990. doi: 10.1158/1535-7163.MCT-13-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blobel GA, Kalota A, Sanchez PV, Carroll M. Short hairpin RNA screen reveals bromodomain proteins as novel targets in acute myeloid leukemia. Cancer Cell. 2011;20:287–288. doi: 10.1016/j.ccr.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Müllauer L, Vakoc CR, Sperr WR, Horny HP, Bradner JE, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget. 2012;3:1588–1599. doi: 10.18632/oncotarget.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- 91.Beurlet S, Omidvar N, Gorombei P, Krief P, Le Pogam C, Setterblad N, de la Grange P, Leboeuf C, Janin A, Noguera ME, et al. BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells. Blood. 2013;122:2864–2876. doi: 10.1182/blood-2012-07-445635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carter BZ, Mak PY, Mak DH, Shi Y, Qiu Y, Bogenberger JM, Mu H, Tibes R, Yao H, Coombes KR, et al. Synergistic targeting of AML stem/progenitor cells with IAP antagonist birinapant and demethylating agents. J Natl Cancer Inst. 2014;106:djt440. doi: 10.1093/jnci/djt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs. 2009;18:1333–1349. doi: 10.1517/14728220903136775. [DOI] [PubMed] [Google Scholar]
- 94.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 95.Dos Santos C, McDonald T, Ho YW, Liu H, Lin A, Forman SJ, Kuo YH, Bhatia R. The Src and c-Kit kinase inhibitor dasatinib enhances p53-mediated targeting of human acute myeloid leukemia stem cells by chemotherapeutic agents. Blood. 2013;122:1900–1913. doi: 10.1182/blood-2012-11-466425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saito Y, Yuki H, Kuratani M, Hashizume Y, Takagi S, Honma T, Tanaka A, Shirouzu M, Mikuni J, Handa N, et al. A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci Transl Med. 2013;5:181ra52. doi: 10.1126/scitranslmed.3004387. [DOI] [PubMed] [Google Scholar]