

Abstract
This review is part two of three, which will present an update on the classification of gastrointestinal submucosal tumors. Part one treats of the diagnosis and part three of the therapeutic methods regarding gastrointestinal submucosal tumors. In the past there has been some confusion as to the classification of gastrointestinal submucosal tumors. Changes in classifications have emerged due to recent advances in mainly immunohistochemistry and electron microscopy. The aim of this paper is to update the reader on the current classification. Literature searches were performed to find information related to classification of gastrointestinal submucosal tumors. Based on these searches the twelve most frequent submucosal tumor types were chosen for description of their classification. The factors that indicate whether tumors are benign or malignant are mainly size and number of mitotic counts. Gastrointestinal stromal tumors are defined mainly by their CD117 positivity. In the future, there should be no more confusion between gastrointestinal stromal tumors and other types of submucosal tumors.
Keywords: Submucosal tumor, Immunohistochemistry, Smooth muscle derived submucosal tumors, Submucosal tumors of neurogenic origin, Gastrointestinal stromal tumor, Malignant, Benign
INTRODUCTION
Submucosal tumors (SMTs) are mesenchymal tumors and as such, they may have very diverse origins. SMTs were originally divided into being of muscular or neural derivation. However, in the past decade it has become more obvious that the SMT group, gastrointestinal stromal tumors (GISTs), cannot be placed in any of these groups. This conclusion has been drawn based on the electron microscopic and immunohistochemical features, since GISTs in about 95% of cases stain positively for the protein CD117[1-3]. This protein is not expressed by any of the other SMTs, except for heterotopic pancreatic tissue[3], which however does not pose a differential diagnosis since it is easily differentiated from GISTs by light microscopy. However, as metastases from various sites may also present as SMTs, there is almost no limitation to the origin of SMTs.
Differing between benign and malignant SMTs may course few problems, whereas it creates a special obstacle to distinguish between a benign SMT and the potentially malignant GIST. GISTs may appear benign both in mitotic counts and lack of cellular atypia, but still behave malignantly[4]. Therefore it is of great value that immunohistochemistry has been introduced, since staining for few proteins provides the information for this classification.
The implementation of immunohistochemistry in the definition of GISTs and other recent chances regarding the classification of SMTs, have created a need for a review in this field. The aim of this paper is to update the reader on particularly immunohistochemistry, morphology, and other characteristics of the 12 most frequently encou-ntered SMTs for classification purposes, grouped as benign or malignant.
BENIGN SUBMUCOSAL TUMORS
SMTs smaller than 3 cm are generally considered benign tumors. The number of mitotic counts allowed for benign SMTs varies among the different SMTs.
Leiomyomas
On cut section leiomyomas have a pale, firm, rubbery or whorled appearance[6]. Microscopically, they constitute of a bland spindle cell population arranged in fascicles and whorls. Mitoses are lacking or few in number and necrosis is normally absent[7]. Concerning immunohistochemistry, leiomyomas are globally positive for desmin and smooth muscle actin, but negative for CD34 and CD117 proteins[8].
Malignant change is very rare in leiomyomas. Thus, a leiomyoma does not represent a presage of a leiomyosarcoma in most cases[10-12].
Schwannomas
On cut section, Schwannomas are grey in color[13]. Microscopically, spindle cells are seen with vague nuclear palisading. There are often sprinkled lymphocytes and a nodular lymphoid cuff[8,9,13]. Immunohistochemically, Schwannomas are positive for S100-protein and vimentin[3,9,13].
Schwannomas are always benign and have never been reported to develop malignancy[8,9]. Therefore, it is important to differentiate them from GISTs, which they resemble both grossly and clinically. Immunohistochemistry provides the sufficient distinction[9,13].
Granular cell tumors
This benign neoplasm is of neural origin (Schwann cell) and often involves peripheral nerves in mucosa or connective tissue[14,15].
Microscopically, granular cell tumors typically infiltrate between adjacent tissues and the overlying mucosa may show pseudocarcinomatous hyperplasia[15,16]. Furthermore, they most commonly appear as sheets of uniform histiocyte-like cells with an abundant, eosinophilic, periodic acid-Schiff-reaction-positive cytoplasm containing lysosomal granules and small vesicular nuclei[7]. Immunohistochemically, granular cell tumors are S100 protein- and neuronspecific enolase-positive, lending support to their neural derivation[1,7,9,16].
Malignant change is very rare and based strictly on the presence of metastases[7].
The firm consistency of granular cell tumors makes it difficult to achieve a biopsy[14]. If the biopsy is too superficial, granular cell tumors may be confused with squamous cell carcinoma, since the overlying mucosa may show pseudocarcinomatous hyperplasia[16].
Heterotopic pancreatic tissue
The cut surface of heterotopic pancreatic tissue is typically tan[17]. If the covering mucosa is intact, the heterotopia appears smooth-walled and well circumscribed[18]. Microscopically and immunohistochemically, heterotopic pancreas may contain all features of a normal panc-reas[1,18,19].
Though rare, malignancy in heterotopic pancreas must be considered[1,19-21].
Differential diagnoses. If mucus retention is present, heterotopic pancreatic tissue can be hard to differ from duplication of the stomach and mucinous carcinoma[19]. If acini and ducts are missing, it may be misinterpreted as an adenomyoma[17].
Lipomas
The cut surface in a lipoma is homogenously yellow, lobulated and has the appearance of adipose tissue[17,22]. Microscopically, lipomas are composed of mature adipose tissue surrounded by a fibrotic capsule[1,9]. They arise mostly from submucosal fat, but infrequently from subserosal fat[22,23]. Fat cells are S100 positive, and CD34 positive spindle cells may be seen, but immunohistochemistry plays little role in the diagnosis of lipomas[24].
Neither solitary lipomas nor lipomatosis has a malignant potential. Liposarcomas are exceptionally rare and will therefore not be mentioned further in this review[1].
Neurofibromatosis
Neurofibromas are classified into three groups as either being localized, diffuse or plexiform, the latter being pathognomonic for neurofibromatosis type 1 (von Recklinghausen Disease). Diffuse neurofibromas are rare in the GI tract[25,26]. They normally involve the myenteric nerve plexus[25].
Macroscopically, localized neurofibromas are fusiform or diffuse tumors with a gray or tan cut surface. Plexiform neurofibromas have a ropelike appearance, when they involve non-branching nerves, but are described as “a bag of worms”, when they involve highly branching nerves[25].
Localized and plexiform neurofibromas have the same microscopic appearance, but the latter is organized into multiple fascicular units. The tumors consist of spindle cells loosely arranged (Schwann cells and fibroblasts) with varying amounts of intervening collagen. Frequently, accumulation of mucopolysaccharides results in a gelatinous or myxoid tumor[25,27]. Immunostaining for S-100 may reveal residual myelinated nerve fibers[25].
Malignant progression may be seen especially in patients with plexiform neurofibromas forming malignant peripheral nerve sheath tumors[28].
Vascular tumors
Hemangiomas: Hemangiomas are classified into three major types: capillary, cavernous or mixed. The former is the commonest and results in small tumors, contrary to the cavernous, which may involve long segments and all wall layers of the ileum[1]. Hemangiomas represent either true neoplasms or hamartomas[29]. Microscopically, sheets of spindle cells are seen, interspersed by clusters of erythrocytes[9]. Immunohistochemically, hemangiomas are positive for CD31, CD34 and factor VIII[3,9].
Lymphangiomas: Histologically, the presence of lymphocytes in lymphangiomas aids the differentiation from hemangiomas[1]. Immunohistochemically, lymphangiomas are typically factor VIII and D2-40 positive, where D2-40 is more specific and aids the differentiation from hemangiomas[3].
MALIGNANT SUBMUCOSAL TUMORS
SMTs larger than 3-5 cm, with mitotic counts greater than 2 per 10 high power fields or that involve more layers are generally considered high-risk tumors for malignancy. GISTs have another classification, as described below[2,9].
Leiomyosarcoma
Leiomyosarcomas are predominantly exophytic and macros-copically visible (Figures 1 and 2)[30,31]. Microscopically, necrosis, cellular and nuclear pleomorphism, mitotic figures and atypical mitoses are typically seen (Figure 3)[17,30,32]. There may be areas of fibrosis, hyalinization or necrosis[17]. Leiomyosarcomas are positive for desmin and smooth muscle actin, but negative for CD34 and CD117[9].
Figure 1.
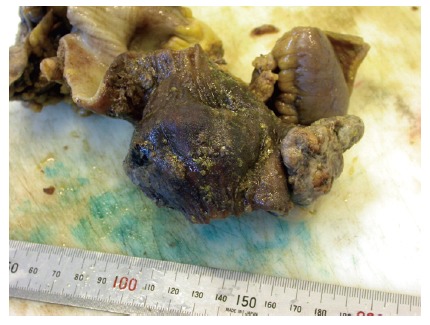
Exophytic leiomyosarcoma of the ileum, measuring 6 cm x 5 cm x 3 cm. A fibrin coated mucosa with stigmata of hemorrhage and discolorations of the adjacent mucosa can be seen (Courtesy by S Duun).
Figure 2.
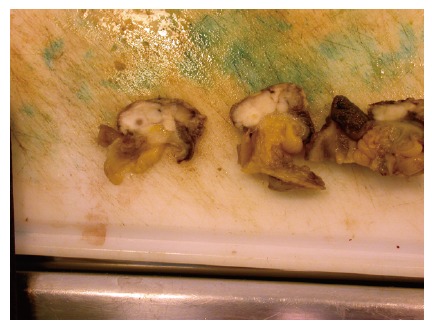
Cut surface of the leiomyosarcoma presented in Figure 1, showing a possible necrosis and a white, fish-flesh-like color, as typical for sarcomas. The surface did not bulge on incision (Courtesy by S Duun).
Figure 3.
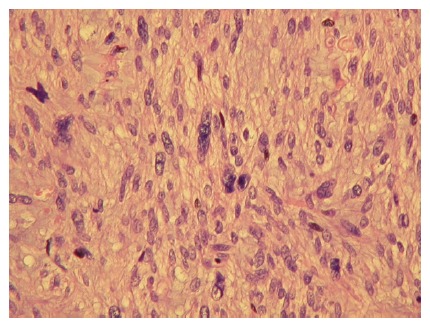
The leiomyosarcoma presented in Figures 1 and 2. It is of low malignancy, but shows nuclear atypia, pleomorphism and mitoses. (HE, x 100) (Courtesy by S Duun).
Differentiation between leiomyosarcomas and leiomyomas is difficult, but leiomyosarcomas may possess typically malignant features as disorganized microscopic appearance, a high mitotic index and the presence of metastasis[31].
Gastrointestinal Kaposi’s sarcoma
Microscopically, Kaposi’s sarcoma exhibit erythrocytes trapped in clefts in pleomorphic spindle cells, and may therefore be classified as a vascular tumor[1,9]. Kaposi's sarcomas are positive for vimentin and smooth muscle actin and typically also for CD31 and CD34. A little more than 50% are positive for factor VIII[3]. Furthermore, human herpes virus 8 can be demonstrated by polymerase chain reaction[9,33].
The most important differential diagnosis is bacillary angiomatosis[34]. Additionally, flat Kaposi’s sarcomas may be confused with a cytomegalovirus (CMV) lesion[7].
Metastases in the gastrointestinal tract
Microscopic and immunohistochemical similarity between the primary tumor and another tumor suggests metastasis[35,36]. Immunohistochemistry may therefore be essential in determining the origin of the metastasis[36]. Differential diagnoses to metastases are mainly primary tumors[35,37].
Gastrointestinal stromal tumors
Immunohistochemistry was the reason for the introduction of the GIST appellation in 1983[38]. Still, many SMTs were misclassified as GISTs and vise versa until recently[5,39].
Macroscopically, low-risk GISTs are typically circumscribed but not encapsulated. The cut surface is without whorls and has a characteristic grey color. High-risk GISTs are sarcomatous on the cut surface, white, fish flesh like, and may show signs of hemorrhage, calcification, ulceration, necrosis, cystic areas and myxoid degeneration. However, these features may also be seen in larger low-risk GISTs. Neither endophytic features nor ulcerations necessarily equals malignancy[1,4,40].
Microscopically, GISTs typically have spindle cell morphology, but epithelioid morphology may be seen[1,8,41]. Immunohistochemically, CD117 protein is a rather specific marker for GISTs with 95% positivity for the protein (Figure 4)[1-3]. The World Health Organization suggests that this may be the single, best defining feature of GISTs. The 5% of GISTs negative for CD117 are due to artifacts, sampling errors, clonal evolution (perhaps in imatinib treatment) and only 2% actually lack CD117[3,5]. The latter seem to have mutations in platelet derived growth factor receptor alpha (a CD117-related tyrosine kinase receptor) instead[42]. About 70% of GISTs are positive for CD34[3]. Furthermore, nearly all GISTs will show diffuse and strong staining for vimentin[3]. A recent investigation has shown no significant correlation between survival, histological tumor type (epithelioid or spindle cell) and CD34 immunoreactivity (positive versus negative)[43]. Electron microscopic features are a mixture of autonomic nerve and smooth muscle cells[9,44].
Figure 4.
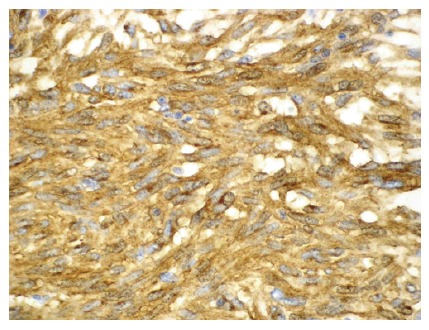
Histological findings of a GIST showing a positive CD117 immunohisto-chemical reaction. (× 200) (Courtesy by B Vainer).
About 20%-30% of all GISTs display malignant behavior[45,46]. All GISTs are potentially malignant and thus cannot be classified as benign versus malignant. Instead, they are regarded as being of very low risk (tumor < 2 cm and < 5 mitoses/50 high power fields), low risk, intermediate risk or high risk (tumor > 5 cm and > 5 mitoses/50 high power fields or tumor >10 cm regardless of mitotic activity) for recurrence and metastasis or overtly malignant (proven metastases at initial diagnosis)[1,43,47]. A recent study has found a perhaps more clinically useful classification focusing on three factors: tumor size (smaller or larger than 5 cm), hemorrhage/necrosis (absence or presence) and Ki-67 LI (proliferation marker; more or less than 3%), which shows significant difference between benign and malignant defined this way[45].
As typical for sarcomas, GISTs generally do not metastasize to the regional lymph nodes[48], but instead spread hematogenously to the liver or metastasize to the peritoneum[41,49,50]. These are also the commonest sites of recurrence[47,51]. A few GISTs seem to lack mitotic activity, but still metastasize[4,5]. Due to this unpredictable behavior, all GISTs must be treated as potentially malignant.
Microscopic differential diagnoses are leiomyoma, leiomyosarcoma, Schwannoma (if the nuclei have a palisade conformation), neurofibroma and more[48]. Differentiation is made with immunohistochemistry or electron microscopy.
CONCLUSION
The combination of size, histological, immunohisto-chemical and, if possible, ultrastructural criteria is the most precise way of classifying SMTs and defining benign or malignant properties. Concerning the possibility of malignancy, this should always be considered when SMTs are larger than 3 cm or with mitotic counts greater than 2 per 10 high power fields. However, GISTs should always be considered potentially malignant.
Smooth muscle derived SMTs (e.g. leiomyomas and leiomyosarcomas) stain strongly and diffusely for desmin and smooth muscle actin and are negative for CD34 and CD117 as opposed to GISTs, which are mostly positive for the CD34 and CD117 biomarkers, with the latter being an almost specific marker for GISTs. Accordingly mesenchymal tumors are now less likely to be misclassified.
As a third group, SMTs of neurogenic origin (e.g. Schwannomas, granular cell tumors and neurofibromas) typically show positivity for S100 and negativity for desmin, actin and CD117. Vascular tumors (e.g. hemangiomas, lymphangiomas and Kaposi’s sarcomas) are typically factor VIII positive. Immunohistochemistry plays little role in the diagnosing of lipomas and heterotopic pancreatic tissue, as their microscopic appearance is easily recognized.
ACKNOWLEDGMENTS
We thank Ben Vainer for photographs of a gastrointestinal stromal tumor and Susanne Duun for photographs of a leiomyosarcoma.
Footnotes
S- Editor Zhu LH L- Editor Rampone B E- Editor Lu W
References
- 1.Day D, Jass J, Price AB, Shepherd NA, Sloan JM, Talbot IC, Warren BF, Williams GT. Morson & Dawson's Gastrointestinal Pathology. Massachusetts: Blackwell Science Ltd; 2003. pp. 205–209, 383-388, 615. [Google Scholar]
- 2.Giuly JA, Picand R, Giuly D, Monges B, Nguyen-Cat R. Von Recklinghausen disease and gastrointestinal stromal tumors. Am J Surg. 2003;185:86–87. doi: 10.1016/s0002-9610(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 3.Frisman D. June 2002. Available from: www.immunoquery.com.
- 4.Miettinen M, El-Rifai W, H L Sobin L, Lasota J. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. Hum Pathol. 2002;33:478–483. doi: 10.1053/hupa.2002.124123. [DOI] [PubMed] [Google Scholar]
- 5.Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti H, Rubin BP, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459–465. doi: 10.1053/hupa.2002.123545. [DOI] [PubMed] [Google Scholar]
- 6.Gill SS, Heuman DM, Mihas AA. Small intestinal neoplasms. J Clin Gastroenterol. 2001;33:267–282. doi: 10.1097/00004836-200110000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Odze RD, Antonioli DA, Wallace MB, Thomas Jr CR, Keohan ML, Hibshoosh H, Antman KH. Gastrointestinal Cancers – A comparison to Sleisenger and Fordtran's Gastrointestinal and Liver Disease. Spain: Elsevier Science Limited; 2003. pp. 265–266, 671, 724. [Google Scholar]
- 8.Miettinen M, Lasota J. Gastrointestinal stromal tumors--definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001;438:1–12. doi: 10.1007/s004280000338. [DOI] [PubMed] [Google Scholar]
- 9.Miettinen M, Blay JY, Sobin LH, Wotherspoon A, Chott A, Gascoyne RD, M�ller-Hermelink HK, Kindblom LG. World Health Organization classification of tumors -- Pathology and genetics of tumours of the digestive system. Lyon: IARC Press; 2000. pp. 29, 58, 65, 142–143. [Google Scholar]
- 10.Davis GB, Blanchard DK, Hatch GF, Wertheimer-Hatch L, Hatch KF, Foster RS, Skandalakis JE. Tumors of the stomach. World J Surg. 2000;24:412–420. doi: 10.1007/s002689910066. [DOI] [PubMed] [Google Scholar]
- 11.Lee YT. Leiomyosarcoma of the gastro-intestinal tract: general pattern of metastasis and recurrence. Cancer Treat Rev. 1983;10:91–101. doi: 10.1016/0305-7372(83)90007-5. [DOI] [PubMed] [Google Scholar]
- 12.Hatch GF, Wertheimer-Hatch L, Hatch KF, Davis GB, Blanchard DK, Foster RS, Skandalakis JE. Tumors of the esophagus. World J Surg. 2000;24:401–411. doi: 10.1007/s002689910065. [DOI] [PubMed] [Google Scholar]
- 13.Inagawa S, Hori M, Shimazaki J, Matsumoto S, Ishii H, Itabashi M, Adachi S, Kawamoto T, Fukao K. Solitary schwannoma of the colon: report of two cases. Surg Today. 2001;31:833–838. doi: 10.1007/s005950170060. [DOI] [PubMed] [Google Scholar]
- 14.Palazzo L, Landi B, Cellier C, Roseau G, Chaussade S, Couturier D, Barbier J. Endosonographic features of esophageal granular cell tumors. Endoscopy. 1997;29:850–853. doi: 10.1055/s-2007-1004320. [DOI] [PubMed] [Google Scholar]
- 15.Maureen Barlow Pugh, editor . Stedman's Medical Dictionary. Maryland: Lippincott Williams & Wilkins; 2000. p. 815, 1894. [Google Scholar]
- 16.Nakachi A, Miyazato H, Oshiro T, Shimoji H, Shiraishi M, Muto Y. Granular cell tumor of the rectum: a case report and review of the literature. J Gastroenterol. 2000;35:631–634. doi: 10.1007/s005350070064. [DOI] [PubMed] [Google Scholar]
- 17.Wiech T, Walch A, Werner M. Histopathological classification of nonneoplastic and neoplastic gastrointestinal submucosal lesions. Endoscopy. 2005;37:630–634. doi: 10.1055/s-2005-870127. [DOI] [PubMed] [Google Scholar]
- 18.Nickels J, Laasonen EM. Pancreatic heterotopia. Scand J Gastroenterol. 1970;5:639–640. [PubMed] [Google Scholar]
- 19.Ikematsu Y, Nishiwaki Y, Kida H, Iwaoka Y, Nagashima S, Ozawa T, Hasegawa S, Okawada T, Waki S. Gastric outlet obstruction caused by a heterotopic pancreas in a pregnant woman: report of a case. Surg Today. 2003;33:952–955. doi: 10.1007/s00595-003-2614-3. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Wasserman PG. Acinar cell carcinoma arising in the stomach: a case report with literature review. Hum Pathol. 2004;35:263–265. doi: 10.1016/j.humpath.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita Y, Maekawa T, Sakai T, Shirakusa T. Transgastrostomal endoscopic surgery for early gastric carcinoma and submucosal tumor. Surg Endosc. 1999;13:361–364. doi: 10.1007/s004649900990. [DOI] [PubMed] [Google Scholar]
- 22.Fernandez MJ, Davis RP, Nora PF. Gastrointestinal lipomas. Arch Surg. 1983;118:1081–1083. doi: 10.1001/archsurg.1983.01390090065015. [DOI] [PubMed] [Google Scholar]
- 23.Agha FP, Dent TL, Fiddian-Green RG, Braunstein AH, Nostrant TT. Bleeding lipomas of the upper gastrointestinal tract. A diagnostic challenge. Am Surg. 1985;51:279–285. [PubMed] [Google Scholar]
- 24.Fletcher C, Unni K, Mertens F. WHO Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. pp. 19–23. [Google Scholar]
- 25.Levy AD, Patel N, Dow N, Abbott RM, Miettinen M, Sobin LH. From the archives of the AFIP: abdominal neoplasms in patients with neurofibromatosis type 1: radiologic-pathologic correlation. Radiographics. 2005;25:455–480. doi: 10.1148/rg.252045176. [DOI] [PubMed] [Google Scholar]
- 26.Leslie A, Virjee JP, Moorghen M. Plexiform neurofibroma of the small bowel infiltrated with metastatic adenocarcinoma. Br J Radiol. 1999;72:604–606. doi: 10.1259/bjr.72.858.10560344. [DOI] [PubMed] [Google Scholar]
- 27.Stevens A, Lowe J, Young B. Wheater's Basic Histopathology -- a colour atlas and text. Edinburgh, London, New York, Philadelphia, St. Louis, Sydney, Toronto: Churchill Livingstone; 2002. p. 282 PMCid: PMC1746283. [Google Scholar]
- 28.Hirsch NP, Murphy A, Radcliffe JJ. Neurofibromatosis: clinical presentations and anaesthetic implications. Br J Anaesth. 2001;86:555–564. doi: 10.1093/bja/86.4.555. [DOI] [PubMed] [Google Scholar]
- 29.Rubin E. Essential Pathology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 271–273, 709. [Google Scholar]
- 30.Gourtsoyiannis N, Grammatikakis J, Prassopoulos P. Role of conventional radiology in the diagnosis and staging of gastrointestinal tract neoplasms. Semin Surg Oncol. 2001;20:91–108. doi: 10.1002/ssu.1023. [DOI] [PubMed] [Google Scholar]
- 31.Gourtsoyiannis N, Makó E. Imaging of primary small intestinal tumours by enteroclysis and CT with pathological correlation. Eur Radiol. 1997;7:625–642. doi: 10.1007/BF02742916. [DOI] [PubMed] [Google Scholar]
- 32.Fockens P. Current endosonographic possibilities in the upper gastrointestinal tract. Baillieres Clin Gastroenterol. 1994;8:603–619. doi: 10.1016/0950-3528(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 33.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 34.Dezube BJ. Acquired immunodeficiency syndrome-related Kaposi's sarcoma: clinical features, staging, and treatment. Semin Oncol. 2000;27:424–430. [PubMed] [Google Scholar]
- 35.Choi SH, Sheehan FR, Pickren JW. Cancer. Vol. 17. Philadelphia: Lippincott Williams & Wilkins; 1964. Metastatic involvement of the stomach by breast cancer; pp. 791–797. [DOI] [PubMed] [Google Scholar]
- 36.Gupta RK, Naran S, Lallu S, Fauck R. Cytodiagnosis of neoplasms of the central nervous system in cerebrospinal fluid samples with an application of selective immunostains in differentiation. Cytopathology. 2004;15:38–43. doi: 10.1046/j.1365-2303.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 37.Hsu CC, Chen JJ, Changchien CS. Endoscopic features of metastatic tumors in the upper gastrointestinal tract. Endoscopy. 1996;28:249–253. doi: 10.1055/s-2007-1005437. [DOI] [PubMed] [Google Scholar]
- 38.Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7:507–519. doi: 10.1097/00000478-198309000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Joensuu H, Fletcher C, Dimitrijevic S, Silberman S, Roberts P, Demetri G. Management of malignant gastrointestinal stromal tumours. Lancet Oncol. 2002;3:655–664. doi: 10.1016/s1470-2045(02)00899-9. [DOI] [PubMed] [Google Scholar]
- 40.El-Zohairy M, Khalil el-SA, Fakhr I, El-Shahawy M, Gouda I. Gastrointestinal stromal tumor (GIST)'s surgical treatment, NCI experience. J Egypt Natl Canc Inst. 2005;17:56–66. [PubMed] [Google Scholar]
- 41.Blay JY, Bonvalot S, Casali P, Choi H, Debiec-Richter M, Dei Tos AP, Emile JF, Gronchi A, Hogendoorn PC, Joensuu H, et al. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST Consensus Conference of 20-21 March 2004, under the auspices of ESMO. Ann Oncol. 2005;16:566–578. doi: 10.1093/annonc/mdi127. [DOI] [PubMed] [Google Scholar]
- 42.Miettinen M, Hirota S, Nishida T, Kitamura Y, Shirao K, Yamao K, Koseki M, Okamura T, Ohtsu A, Sugiyama T. Gastrointestinal stromal tumor (GIST): from pathology to molecular target therapy. Tokyo: Japan Scientific Societies Press; 2004. p. 6, 35, 155, 156. [Google Scholar]
- 43.Nilsson B, Bümming P, Meis-Kindblom JM, Odén A, Dortok A, Gustavsson B, Sablinska K, Kindblom LG. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era--a population-based study in western Sweden. Cancer. 2005;103:821–829. doi: 10.1002/cncr.20862. [DOI] [PubMed] [Google Scholar]
- 44.Knoop M, St Friedrichs K, Dierschke J. Surgical management of gastrointestinal stromal tumors of the stomach. Langenbecks Arch Surg. 2000;385:194–198. doi: 10.1007/s004230050264. [DOI] [PubMed] [Google Scholar]
- 45.Yokoi K, Tanaka N, Shoji K, Ishikawa N, Seya T, Horiba K, Kanazawa Y, Yamashita K, Ohaki Y, Tajiri T. A study of histopathological assessment criteria for assessing malignancy of gastrointestinal stromal tumor, from a clinical standpoint. J Gastroenterol. 2005;40:467–473. doi: 10.1007/s00535-005-1571-6. [DOI] [PubMed] [Google Scholar]
- 46.Nowain A, Bhakta H, Pais S, Kanel G, Verma S. Gastrointestinal stromal tumors: clinical profile, pathogenesis, treatment strategies and prognosis. J Gastroenterol Hepatol. 2005;20:818–824. doi: 10.1111/j.1440-1746.2005.03720.x. [DOI] [PubMed] [Google Scholar]
- 47.Lau S, Tam KF, Kam CK, Lui CY, Siu CW, Lam HS, Mak KL. Imaging of gastrointestinal stromal tumour (GIST) Clin Radiol. 2004;59:487–498. doi: 10.1016/j.crad.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 48.Rubin B, Demetri G. Gastrointestinal Oncology -- principles and practice. Philadelphia: ippincott Williams & Wilkins; 2002. pp. 922–929. [Google Scholar]
- 49.Miettinen M, Sarlomo-Rikala M, Lasota J. Gastrointestinal stromal tumours. Ann Chir Gynaecol. 1998;87:278–281. [PubMed] [Google Scholar]
- 50.Reddy MP, Reddy P, Lilien DL. F-18 FDG PET imaging in gastrointestinal stromal tumor. Clin Nucl Med. 2003;28:677–679. doi: 10.1097/01.RLU.0000079395.25949.c1. [DOI] [PubMed] [Google Scholar]
- 51.Bucher P, Taylor S, Villiger P, Morel P, Brundler MA. Are there any prognostic factors for small intestinal stromal tumors? Am J Surg. 2004;187:761–766. doi: 10.1016/j.amjsurg.2003.09.010. [DOI] [PubMed] [Google Scholar]