

Abstract
We have recently developed aged cortical neuron cultures from autopsied human brains with Alzheimer's disease (AD). During the culturing process, we found that glutamatergic cortical neurons from the AD brain lacked a response to glial cell line-derived neurotrophic factor (GDNF), including no axonal regrowth, and were starting to undergo apoptosis. Here we showed that, in cortical neurons from age- and gender-matched cognitively normal control (NC) subjects (NC neurons), GDNF enhanced the expression of GDNF family receptor subtype α1 (GFRα1), but not the other three subtypes (GFRα2, GFRα3, and GFRα4), whereas GDNF failed to induce GFRα1 expression in cortical neurons from the AD brain (AD neurons). The exogenous introduction of GFRα1, but not of its binding partner α1-neural cell adhesion molecule, or RET into AD neurons restored the effect of GDNF on neuronal survival. Moreover, between NC and AD neurons, the AMPA receptor blocker CNQX and the NMDA receptor blocker AP-5 had opposite effects on the GFRα1 expression induced by GDNF. In NC neurons, the presence of glutamate receptors was necessary for GDNF-linked GFRα1 expression, while in AD neurons the absence of glutamate receptors was required for GFRα1 expression by GDNF stimulation. These results suggest that, in AD neurons, specific impairments of GFRα1, which may be linked to glutamatergic neurotransmission, shed light on developing potential therapeutic strategies for AD by upregulation of GFRα1 expression.
Keywords: neurodegeneration, neuron
Introduction
The accumulation of β-amyloid (Aβ) peptide is a primary characteristic in Alzheimer's disease (AD) pathogenesis. According to the amyloid hypothesis, alleviating Aβ-induced damage is one of the therapeutic strategies (Hardy and Selkoe, 2002); however, approaches based on the amyloid hypothesis have been challenged by the disappointing results of recent clinical studies (Hardy, 2009). Although numerous hypothesis-based (including the amyloid hypothesis) disease-modifying drug candidates are still under development for clinical trials (Alzheimer's Association et al., 2011; Imbimbo et al., 2011), drug development efforts should be diversified to fully address the multifactoriality of the disease (Seabrook et al., 2007; Huang and Mucke, 2012). Given the past failure of monotherapies for AD treatments, it might be necessary to use a combination of different disease-modifying approaches [i.e., neurotrophic factor (NTF) therapy] to halt the different disease-causing mechanisms simultaneously (Weissmiller and Wu, 2012), regardless of whether neuronal damage is primarily caused by Aβ or other mechanisms. Although this type of treatment is actually under development for clinical trials, it has not yet provided satisfactory outcomes to fulfill its potential as a neurorestorative therapy (Weissmiller and Wu, 2012; Allen et al., 2013).
The potential use of NTFs, such as neurotrophins, including nerve growth factor and brain-derived neurotrophic factor, for the treatment of neurodegenerative diseases including AD, has long been suggested (Siegel and Chauhan, 2000). Glial cell line-derived neurotrophic factor (GDNF) is recognized as a potent NTF in the nervous system (Walton, 1999). GDNF family ligands, such as GDNF, signal through receptors consisting of RET (Treanor et al., 1996) and one of four ligand-binding proteins [GDNF family receptor subtype α1 (GFRα1), GFRα2, GFRα3, and GFRα4; Rosenthal, 1999], in which GFRα1 preferentially binds to GDNF (Sariola and Saarma, 2003). GDNF uses the α1-neural cell adhesion molecule (α1-NCAM)-dependent signaling pathway instead of the RET-dependent pathway, particularly in hippocampal and cortical neurons (Paratcha et al., 2003), which are vulnerable to AD pathology. Clinical use of GDNF for the treatment of Parkinson's disease is currently under trial (Ramaswamy et al., 2009), and GDNF has a high potential for therapeutic applications in neurologic disorders (Arenas, 1996); however, no study so far has addressed the role of GDNF in the treatment of AD.
Here we isolated and cultured cortical neurons from autopsied elderly human brains (Konishi et al., 2002) and found a significant decrease in GFRα1 levels in AD neurons. The application of GFRα1 ligands, GDNF and artemin (Baloh et al., 1998), could not promote neurite growth or the cell survival of AD neurons, unlike the positive role of GDNF and artemin in normal control (NC) neurons. The introduction of GFRα1 into AD neurons under treatment with GDNF and artemin could improve cell survival.
Materials and Methods
Human subjects.
All subjects or their legally authorized representatives signed an informed consent form for autopsy and research. The standard research protocol was approved by the Institutional Review Board. NC and AD brain tissues were obtained from the Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2008). The subjects and patients consisted of six NC subjects (82.7 ± 5.9 years old, 3 males and 3 females) and six patients with AD (84.3 ± 6.4 years old, disease duration of 9.7 ± 2.7 years, 3 males and 3 females). The subject and patient profiles are shown in Table 1.
Table 1.
Clinical and neuropathological characteristics of the NC subjects and AD patients
| Case no. | Age (years)* | Gender | PMI (h)* | ApoE allele | Clinical diagnosis | Disease duration (years) | MMSE score | Neuropathological summary | Brain weight (g)** | CERAD plaque score | Braak staging | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | AD-1 | 81 | M | 3 | 3/4 | AD | 13 | 6 | AD | 1090 | Frequent | V |
| AD-2 | 76 | M | 2.3 | 3/3 | AD | 11 | 0 | AD, cerebral white matter rarefaction | 1045 | Frequent | VI | |
| AD-3 | 79 | M | 2 | 3/3 | AD | 7 | 0 | AD, cerebral white matter rarefaction | 1110 | Frequent | V | |
| AD-4 | 90 | F | 3 | 3/3 | AD, osteoarthritis, depression | 11 | 5 | AD, cerebral white matter rarefaction, hippocampal sclerosis | 905 | Frequent | V | |
| AD-5 | 89 | F | 3 | 3/4 | AD | 10 | 0 | AD, cerebral white matter rarefaction in the frontal cortex | 1010 | Frequent | V | |
| AD-6 | 91 | F | 3 | 3/3 | AD | 6 | 0 | AD, argyrophilic grains in the temporal cortex, acute infarctions in the right inferior temporal and occipital cortex | 995 | Frequent | V | |
| NC | NC-1 | 85 | M | 3.2 | 3/3 | Control | 0 | 30 | Control, old lacunar and microscopic infarcts in the left postcentral cortex | 1280 | None | II |
| NC-2 | 86 | F | 2.5 | 3/3 | Control, chronic lung fibrosis, rheumatoid arthritis | 0 | 27 | Control, recent small infarctions in the left frontal, left temporal cortex, old cortical microinfarction in the left precentral cortex, argyrophilic grains in mesial temporal cortex | 1145 | None | III | |
| NC-3 | 88 | F | 3 | 3/4 | Control | 0 | 30 | Normal brain showing minimal age-related changes | 1030 | None | II | |
| NC-4 | 73 | M | 2 | 3/4 | Control | 0 | 28 | Control, brain showing only normal aging changes | 1410 | None | II | |
| NC-5 | 86 | F | 2 | 2/3 | Control, liver cancer, right leg thrombosis | 0 | 29 | Control, brain showing only normal aging changes | 1150 | None | II | |
| NC-6 | 78 | M | 1.7 | 3/3 | Control | 0 | 28 | Control, cerebral white matter rarefaction and gliosis in the right temporal cortex | 1460 | None | I |
M, Male; F, female; PMI, postmortem interval; MMSE, Mini-Mental State Examination; ApoE, apolipoprotein E.
*Not significantly different;
**p < 0.05 between AD and NC groups.
Clinical diagnosis and pathological confirmation.
The criteria of AD patients are defined by the National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease (1997) “high likelihood” and pathological Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuritic plaque density (Mirra et al., 1991) as well as Braak staging (Braak and Braak, 1991). The details are presented in Table 1. NC subjects were selected based on the absence of a clinical history of dementia and on the results of neuropathological examination.
Cultures of cortical neurons from rapidly autopsied brains.
Neurons from the frontal cortex of the NC and AD brains (NC and AD neurons, respectively) were isolated and cultured as described previously (Konishi et al., 2002). Briefly, ∼20 g of brain tissue was taken from the frontal cortex at 0.5–2.5 h postmortem, digested with papain (Worthington), and processed to increase the purity of the neuronal population. The neurons (1 × 106/ml) were incubated with tetanus toxin C (TTC) fragments (Boehringer-Ingelheim) followed by an anti-TTC fragment mouse monoclonal antibody (Boehringer-Ingelheim). Microbeads coated with anti-mouse polyclonal antibodies (Miltenyi Biotec) were added for magnetic cell sorting (Miltenyi Biotec). These beads of 50 nm diameter do not affect cell function or viability and do not need to be removed after sorting, according to the manufacturer's instructions. Approximately 1 × 106 neurons per gram of brain tissue weight were obtained with no significant differences in yield between NC and AD neurons. The neurons were cultured in Neurobasal A with B27 (Invitrogen) in the presence or absence of recombinant GDNF, artemin, neurturin, or persephin (R&D Systems) for further studies.
Immunocytochemistry.
The isolated and cultured cortical neurons were immunostained with antibodies against neurons and neurotransmitters as described previously (Konishi et al., 2002). For neuronal identification, antibodies against neurofilament protein (SMI33; Sternberger), microtubule-associated protein-2 (MAP2; Millipore) and neuronal class III β-tubulin (TUJ1; Covance) were used. Antibodies against glial fibrillary acidic protein (GFAP; DAKO), human leukocyte antigen-DR (LN-3; ICN), von Willebrand factor (vWF; DAKO), and fibronectin (Sigma) were used for non-neuronal identification. Moreover, antibodies that detect neural multipotent progenitors and neural stem cells, anti-NG2 (Millipore) and anti-Musashi (a gift from Dr. H. Okano, Keio University, Tokyo, Japan; Sakakibara and Okano, 1997), respectively, were also used. To detect neurotransmitter-synthesizing enzymes, antibodies against phosphate-activated glutaminase (PAG; a gift from Dr. T. Kaneko, Kyoto University, Kyoto, Japan; Kaneko et al., 1987), glutamate decarboxylase (GAD; Millipore), and choline acetyltransferase (ChAT; Millipore) were used. To detect glutamate receptors, antibodies against the NMDA glutamate receptor subtype 1 (GluRN1; Pharmingen, BD Biosciences) and the AMPA-type glutamate receptor types 2 and 4 (GluRA2/4; Pharmingen, BD Biosciences) were used. Secondary antibodies conjugated to Alexa Fluor 488 (Invitrogen) were used for visualization. Sudan Black B (1%) in 70% ethanol was used to quench autofluorescence, which is present in large amounts in aged neurons (Schnell et al., 1999).
Cell viability tests and calcium imaging.
Three different assays of cell viability were conducted for the cultured neurons using acetoxymethy (AM) ester of calcein (calcein AM) plus ethidium homodimer (EthD-1; LIVE/DEAD Viability/Cytotoxicity test; Invitrogen), SYTO 10 plus DEAD Red (LIVE/DEAD Reduced Biohazard Viability/Cytotoxicity test; Invitrogen), and tetrazolium salts such as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Invitrogen). For the calcium imaging test, fluo-3 AM (Invitrogen) was used. Methods were briefly described in our previous report (Konishi et al., 2002).
Quantification of neurite extension.
For the quantification of neurite extension, as described previously (Chang et al., 1987; Lozano et al., 1995; Savoca et al., 1995), the cortical neurons were plated at a density of 3 × 104 cells per well in six-well plates coated with polyethyleneimine (Sigma), cultured in the presence or absence of GDNF at 30 ng/ml, and evaluated as the ratio of total neurite length on day 2 and 7 to that on day 0. Among different parameters of neurite outgrowth, we measured the length of neurites directly from the base of the soma to the neurite apex, including the length of the cell soma, using the ImageJ program. It is reported that the total neurite length or longest neurite length is a more sensitive measure than neurons with neurite measurement for quantitative assessment of neurite outgrowth in dissociated neuronal cultures (Mitchell et al., 2007). Neurites were counted only if they had not collided with other test neurites and did not come from a clump of cells (Chang et al., 1987).
Determination of mRNA levels of each GFR subtype by RT-PCR analyses.
Total RNA was extracted from the cultured cortical neurons using a reagent for total RNA isolation (Trizol; Invitrogen) and reverse transcribed with MMLV-RT (Invitrogen). The first-strand cDNA was amplified by PCR using AmpliTaq Gold DNA polymerase (Applied Biosystems) and the following specific forward and reverse primers: GFRα1 forward: 5′-GCACAGCTACGGAATGCTCTTCTG-3′; GFRα1 reverse: 5′-GTAGTTGGGGGTCATGACTGTGCCAA-3′; GFRα2 forward: 5′-GAATCCAACTGCAGCTCT-3′; GFRα2 reverse: 5′-AAGCAAGCCTGAAGATGTCC-3′; GFRα3 forward: 5′-GGAACTTGTGCAACAGAGCA-3′; GFRα3 reverse: 5′-ACAGCAAAGGTAGGGTGTGG-3′; GFRα4 forward: 5′-TGCCCTTTGTAGGTTTGGAC-3′; GFRα4 reverse: 5′-TTCTGGGATTCTGGATGGTC-3′; β-actin forward: 5′-TGGTGGGCATGGGTCAGAAGGATTC-3′; and β-actin reverse: 5′-CATGGCTGGGGTGTTGAAGGTCTCA-3′. PCR was performed as follows: 12 min at 94°C, followed by 15 and 37 cycles of amplification, respectively, for β-actin and GFRα1–4, with each cycle consisting of 45 s at 94°C, 45 s at the primer-specific annealing temperature, and 1 min at 72°C. A final elongation step at 72°C for 10 min completed the reaction. The annealing temperatures were as follows: 60°C for GFRα1 and GFRα2; 58°C for GFRα3 and GFRα4; and 56°C for β-actin.
Western blotting.
Western blotting was performed as described previously (He et al., 2007). The cultured cortical neurons were lysed in Tris-NaCl buffer containing 0.5% Triton X-100, 1% β-octylglucoside plus protease and phosphatase inhibitors. The total cell lysate was electrophoresed and electroblotted onto nitrocellulose membranes. For the detection of proteins, the membranes were incubated with specific antibodies against GFRα1–4 (R&D Systems) before the addition of corresponding secondary antibodies conjugated to horseradish peroxidase (Bio-Rad). The enhanced chemiluminescence method (ECL Advance; GE Healthcare) was applied for detection with quantification of the detected bands performed by optical densitometry with FluorChem 8900 (Alpha Innotech). We confirmed the ability of each antibody to detect the correct molecule of the expected size using recombinant protein (data not shown).
Cell transfection.
The Adenovirus expression vector kit (Takara Bio) was used for the transduction of target genes into the neurons (Miyake et al., 1996) according to the manufacturer's instructions. Briefly, cDNA for GFRα1, α1-NCAM, or RET was inserted into the cassette cosmid containing the entire adenovirus serotype 5 genome, except for genes E1 and E3, and CAG promoter [cytomegalovirus enhancer, chicken β-actin promoter, and rabbit β-globin poly (A) signal]. The cosmid has BspT140 I and PacI restriction sites outside both terminal ends of the virus genome. The recombinant adenoviruses expressing GFRα1, α1-NCAM, or Ret were generated by transfection into HEK293 cells with PacI-digested recombinant cosmid, using FuGENE6 transfection reagent (Roche), and subsequently propagated in HEK293 cells. The quaternary virus seeds were purified by sequential centrifugation in cesium chloride as described previously (Kanegae et al., 1994). The neurons were seeded onto Laminine (BD Bioscience)-coated 12-well plates in Neurobasal A with B27 at a density of 6 × 103 cells per well and infected after 24 h with purified virus solution for 2 h. Successful transfection was confirmed on day 2 by immunofluorescence using an antibody against GFRα1 (R&D Systems), α1-NCAM, or RET (Millipore), and also by checking the expression of β-galactosidase.
To check the β-galactosidase expression, the cortical neurons were infected with the recombinant adenovirus expressing LacZ (provided in this kit) and stained with X-gal. At a multiplicity of infection (MOI) of ≥10, ∼15–20% of neurons were positive for β-galactosidase on day 2. There were no significant differences in infection efficiencies between NC and AD neurons. Infection efficiencies seemed to be more dependent on patient age and postmortem intervals than on disease status. Western blot analysis on day 2 showed that the expression levels of GFRα1 protein in AD neurons infected with recombinant adenoviruses expressing GFRα1 at an MOI of 50 recovered to the levels corresponding to ∼30% of the levels in uninfected NC neurons, and that the transfection of GFRα1 at an MOI of 50 made the expression levels of GFRα1 protein in NC neurons 1.2–1.3 times higher.
Inhibition of GFRα1 with the antisense oligonucleotide.
The following locked nucleic acid-containing oligonucleotides (Wahlestedt et al., 2000) were used for antisense experiments: 5′-GGCACCATGTTCCTA-3′ for GFRα1 sense; 5′-TAGGAACATGGTGCC-3′ for GFRα1 antisense; 5′-TGGTGCGTAGCCATG-3′ for α1-NCAM sense; and 5′-CATGGCTACGCACCA-3′ for α1-NCAM antisense (Wahlestedt et al., 2000; Blaheta et al., 2004). These oligonucleotides were commercially synthesized, purified by HPLC, and transduced into the neurons using an oligofectamine reagent (Invitrogen). Successful transfection was confirmed on days 2 and 5 with Western blot analyses using an antibody against GFRα1 or α1-NCAM.
Evaluation of neuronal survival.
Following the transduction of adenoviruses or oligonucleotides, the cortical neurons were cultured in Neurobasal A with B27 in the presence or absence of recombinant GDNF, artemin, neurturin, or persephin for 0, 2, and 5 d; and were subjected to RT-PCR, Western blot, and immunocytochemical analyses. For the cell survival assays, the neurons were cultured for 0, 2, and 7 d. The numbers of surviving neurons were identified by positive staining for NeuN (Millipore) or SMI-33 (Covance) and counted in a blinded manner from triplicate wells of six independent cultures. Neuronal survival rates are shown as the ratio of the percentage of the number of labeled neurons on days 2 and 7 to those on day 0.
Data analysis.
Researchers were always blinded during the experiments, including neuron cultures, and when analyzing raw data from each case. The clinical and pathological diagnoses were unknown until final data analysis. The cases shown in Table 1 are 12 cases of our 30 trials of isolation and in vitro maintenance of neurons from freshly autopsied human brain tissues. For the final data analysis, from these 30 cases we randomly selected six NC subjects and six AD patients who fulfill the clinical and neuropathological criteria for NC and AD. Results are expressed as the mean ± 1 SD. All analyses were performed using appropriate software (Excel Statistics 2010; Social Survey Research Information). Differences among three or more groups were evaluated by the Steel–Dwass test following the Kruskal–Wallis test. The level of significance was considered to be p ≤ 0.05.
Results
Neuronal and neurotransmitter characterizations
Our neurons from the frontal cortex of rapidly autopsied brains (six NC and six AD) were characterized with immunocytochemistry for neuron-specific markers, as previously reported (Konishi et al., 2002). Quantitative evaluations of these cells revealed that 76 ± 5%, 70 ± 4%, and 71 ± 4%, respectively, were positive for SMI33, MAP2, and TUJ1. In addition, such neuron-rich cultures were found to contain 9.4 ± 7.3%, 8.9 ± 2.5%, 9.5 ± 7.8%, and 6.8 ± 1.0%, respectively, of astrocytes, microglia, endothelial cells, and fibroblasts by using specific antibodies against each cell type of marker, including GFAP, LN-3, vWF, and fibronectin. The percentage of cells immunoreactive to NG2 was 3.7 ± 3.6%. The neuron-rich cultures lacked immunoreactivity for the Musashi. These types of cells showed no changes in percentage throughout the culture periods, and there were no significant differences in the percentage of cells labeled with any antibody mentioned above between NC and AD neurons, suggesting that our primary cells were not differentiated from immature progenitors or stem cells in cultures. Furthermore, to determine neurotransmitter phenotypes, these neurons were evaluated for synthesizing enzymes of glutamate, GABA, and acetylcholine (images not shown), showing that PAG-, GAD-, and ChAT-positive neurons comprised 61 ± 10%, 49 ± 11%, and 8.1 ± 1.6%, which are presumed to be glutamatergic, GABAergic, and cholinergic neurons, respectively. These proportions of PAG-, GAD-, and ChAT-positive neurons in our cultures are almost consistent with the estimated 75%, 35%, and ∼10% prevalence of glutamatergic, GABAergic, and cholinergic neurons, respectively, in the intact human or primate adult neocortex (Hendry et al., 1987; Akiyama et al., 1990; Kasashima et al., 1999). The percentage of glutamatergic, GABAergic, and cholinergic neurons isolated from the autopsied AD brains was not different from that isolated from the autopsied NC brains, suggesting that there is no phenotypic preference for cell loss in AD neuron culture processes, compared with NC neuron culture processes, as judged from data on neuronal and neurotransmitter markers.
Biochemical evaluation of neuronal activity
Three different assays of cell viability were performed for our neurons (six NC subjects and six AD patients) after 7 d in vitro in the presence of GDNF. The LIVE/DEAD Viability/Cytotoxicity test uses 30 min incubation of cells with calcein AM and EthD-1. Green fluorescence indicates living cells, while red fluorescence indicates dead cells. In the presence of esterase activity, calcein AM is metabolized to calcein, producing green fluorescence, according to the manufacturer's instructions. NC neurons consisted of 75 ± 12% living cells and 18 ± 6% dead cells, whereas the AD neurons consisted of 35 ± 9% living cells and 61 ± 11% dead cells. The LIVE/DEAD Reduced Biohazard Viability/Cytotoxicity test uses 15 min incubation of cells with SYTO10 and DEAD Red. With the staining method recommended by the manufacturer's instructions, living cells appear fluorescent green and dead cells fluoresce red. NC neurons consisted of 65 ± 10% living cells and 28 ± 5% dead cells, whereas AD neurons consisted of 39 ± 12% living cells and 66 ± 13% dead cells. Tetrazolium salts are water-soluble colorless compounds that form highly colored water-insoluble formazans after reduction by the respective specific components of the electron transport chain in mitochondria of only living cells (Abe and Saito, 1998). In 65 ± 11% of NC neurons and 37 ± 10% of AD neurons, purple formazan granules were recognized when 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was used as tetrazolium salts, indicating that they are living cells.
In the calcium imaging test, the neurons after 7 d in vitro (six NC subjects and six AD patients) were loaded with fluo-3 AM and exposed to 50–100 mmol/L KCl to induce calcium influx. We found that 58 ± 12% NC neurons and 24 ± 9% AD neurons exhibited increments in calcium influx. In addition, cells loaded with fluo-3 AM were exposed to glutamate at 100–500 μmol/L, showing that 55 ± 9% of NC neurons and 20 ± 7% of AD neurons exhibited increases in calcium influx.
GFRα1 expression is downregulated in AD neurons
In the presence of GDNF, there was no obvious axonal growth of AD neurons, while NC neurons were able to grow neurites (Fig. 1A); therefore, neurite extension was quantitatively evaluated under a phase-contrast microscope (Fig. 1B). These observations led us to consider further challenging experiments to determine whether NC and AD neurons could express GDNF receptors differently, since GDNF functions through its receptor subtypes, GFRα1–4 (Sariola and Saarma, 2003).
Figure 1.
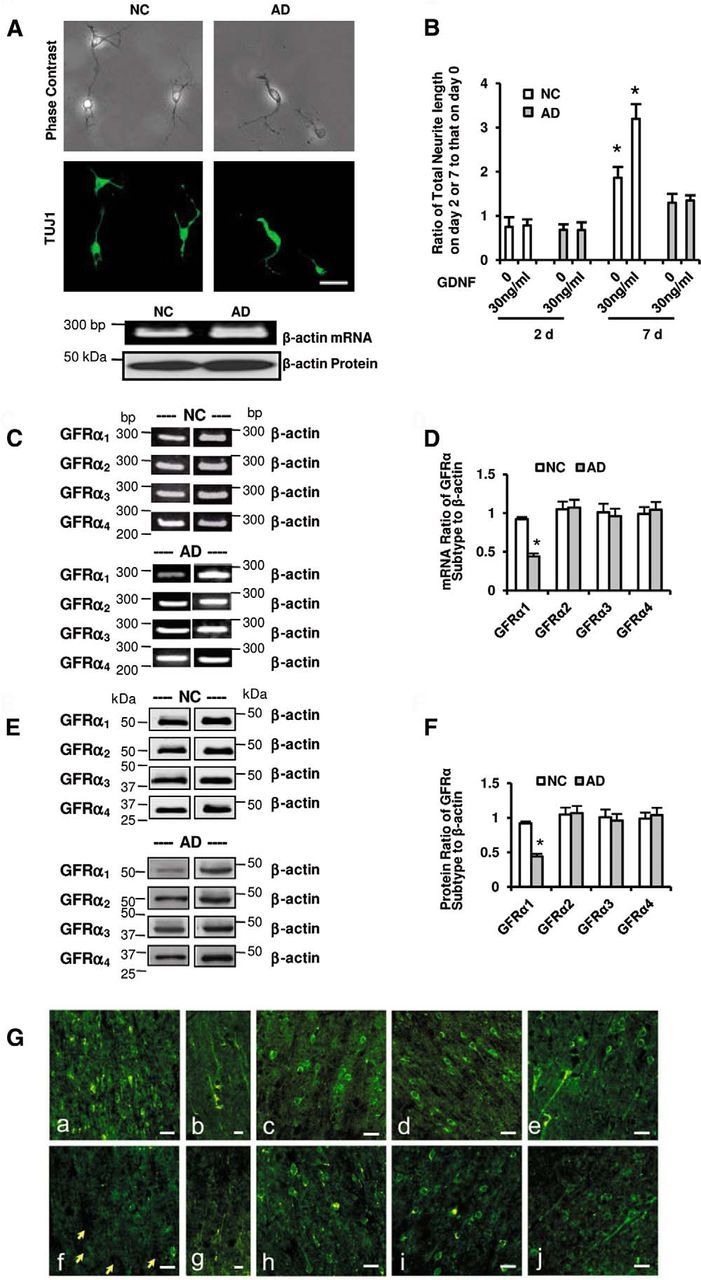
Expression of GFRα in NC and AD neurons. A, Cortical neurons isolated from NC and AD brains (NC and AD neurons) were cultured for 7 d in the presence of 30 ng/ml GDNF. Representative neurons were verified by phase contrast microscopy and TUJ1 immunostaining. Scale bar, 100 μm. The β-actin expression was not different between NC and AD neurons at both mRNA and protein levels. B, Neurite extension of NC and AD neurons was quantitatively evaluated as the ratio of total neurite length in the presence or absence of GDNF on days 2 and 7 to that on day 0. We measured the length of neurites directly from the cell base to the neurite apex, including the length of the cell soma. Data were obtained in a blinded manner from triplicate wells of six independent cultures (mean ± 1 SD; n = 6; *p < 0.05 by the Steel–Dwass test). C–F, Representative images of GFRα mRNA (C, D) and protein (E, F) expressions in NC and AD neurons are shown, and their semiquantitative RT-PCR mRNA (D) and protein (F) levels were analyzed by RT-PCR and Western blotting, respectively. The levels of GFRα mRNA and protein were normalized to the corresponding β-actin. Data were obtained in triplicate from six independent cultures (mean ± 1 SD; n = 6; *p < 0.05 by the Steel-Dwass test). Ga–j, Representative images of the immunohistochemistry of GFRα1 (a, b, f, g), GFRα2 (c, h), GFRα3 (d, i), and GFRα4 (e, j) in the temporal cortex from the NC (a–e) and AD (f–j) brain. Tissue sections were incubated with specific antibodies against GFRα1–4 (R&D Systems) before the addition of corresponding secondary biotinylated antibodies (Vector Laboratories), followed by visualization with streptavidin-Alexa Fluor 488 (Invitrogen). Yellow arrows depict unstained neurons. Scale bars: b, g, 50 μm; a, c–f, h–j, 100 μm.
With semiquantitative RT-PCR analysis, all four GFRα subtypes, GFRα1–4, were demonstrated to be expressed equally in NC neurons (Fig. 1C,D). Surprisingly, GFRα1 expression, but not GFRα2, GFRα3, or GFRα4 expression, was significantly reduced in AD neurons compared with NC neurons (Fig. 1C,D). We looked at each subtype individually using Western blotting to see whether the protein levels corresponded to their mRNA levels (Fig. 1E,F). In NC neurons, GFRα1–4 were equally expressed at moderate levels, whereas the expression of GFRα1, but not the other three subtypes, was significantly reduced in AD neurons (Fig. 1E,F). This selective reduction of GFRα1 in AD neurons in vitro was confirmed in noncultured postmortem brain tissues from five NC subjects (84.0 ± 9.6 years old, three males and two females) and five AD patients (79.4 ± 6.9 years old, three males and two females; Fig. 1G), which were different from the six NC subjects and six AD patients used in the present in vitro culture study.
GDNF enhances the expression of GFRα1 in NC but not in AD neurons
To examine whether GDNF regulates the expression of its own receptor subtype in neurons, GDNF was added daily to cultured neurons. The neurons were then harvested on days 2 and 5 to conduct Western blot analysis to determine the protein levels of GFRα1–4. In NC neurons, the expression of GFRα1 was selectively enhanced in a dose-dependent manner by GDNF treatment for 5 d, but no significant change in the expression of the other three GFRα subtypes was observed (Fig. 2A). In AD neurons, however, there were no significant changes in the mRNA or protein expression of any of the four GFRα subtypes with GDNF treatment (Fig. 2B,C; data for GFRα2, GFRα3, or GFRα4 are not shown). The time course of the regulation of GFRα1 expression by GDNF in NC and AD neurons (n = 6 for each) was examined, as follows: increases in GFRα1 mRNA and protein levels in NC neurons with GDNF treatment for 5 and 7 d were significant compared with those in AD neurons, although there was no significant change between NC and AD neurons in the expression of GFRα1 with 2 d treatment (data not shown).
Figure 2.
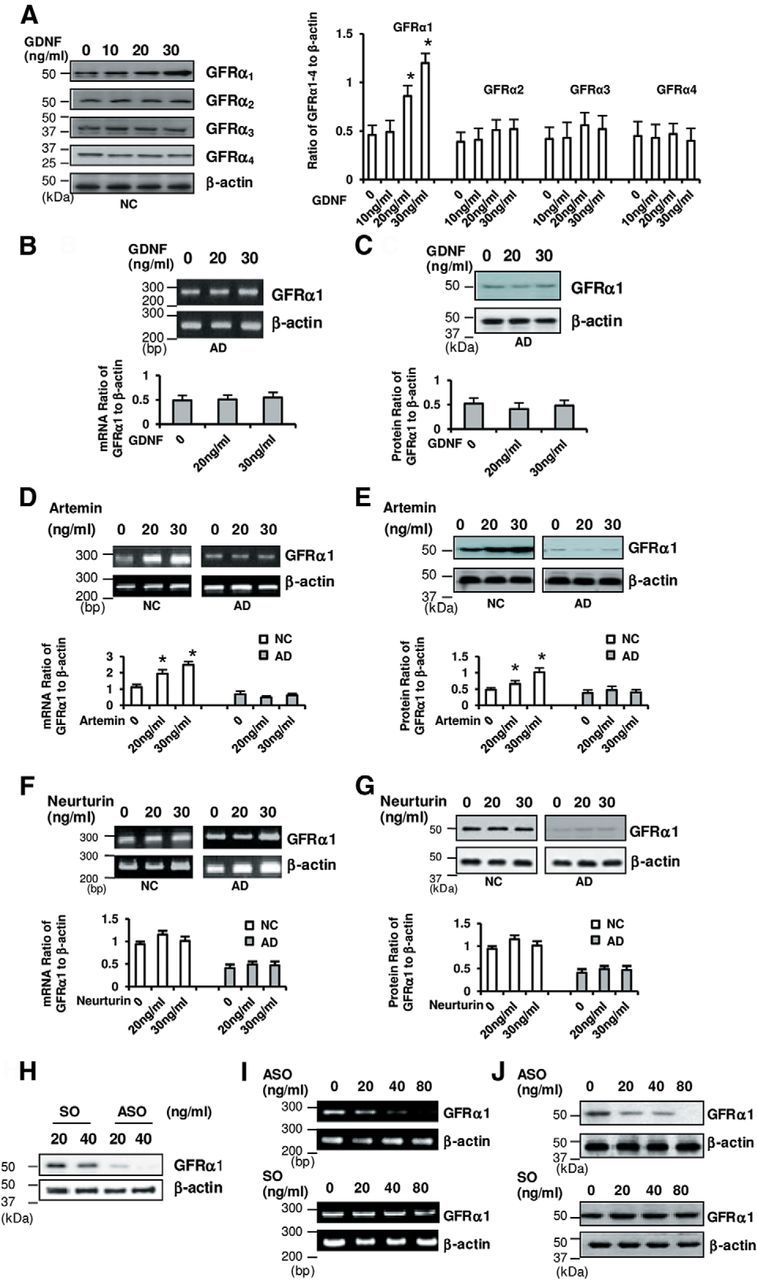
GDNF enhances the expression of GFRα1 in NC but not AD neurons. A, Representative images of GFRα expression in NC neurons are shown, and semiquantitative analyses of the expression levels are presented. GFRα1 expressions, but not GFRα2, GFRα3, or GFRα4 expressions, were upregulated in response to exogenous GDNF in a dose-dependent manner in NC neurons. B, C, Representative images of GFRα1 mRNA (B) and protein (C) expressions in AD neurons are shown. The levels of GFRα1 mRNA (B) or protein (C) expressions were not significantly elevated in the presence of GDNF in AD neurons. D–G, Representative images of GFRα1 mRNA (D, F) and protein (E, G) expressions in NC and AD neurons are shown. The levels of GFRα1 mRNA (D) and protein (E) were upregulated in NC but not in AD neurons in the presence of artemin. In contrast, there was no significant elevation of mRNA (F) or protein (G) levels in either NC or AD neurons in the presence of neurturin. All of the results of GFRα mRNA and protein levels were normalized to the corresponding β-actin. A–G, Data were obtained in triplicate from six independent cultures (mean ± 1 SD; n = 6; *p < 0.05 by the Steel–Dwass test). H, Photoimages show, in the presence of GDNF for 5 d, downregulation of GFRα1 protein expression in NC neurons with the introduction of GFRα1 ASOs but not with the introduction of GFRα1 SOs. I, J, Similar to the finding in the presence of GDNF, in the presence of artemin for 5 d GFRα1 mRNA (I) and protein (J) expression were downregulated in NC neurons with the introduction of GFRα1 ASOs, but not with introduction of GFRα1 SOs.
A new GDNF member, artemin (Baloh et al., 1998), shares GFRα1 with GDNF (Sariola and Saarma, 2003). To examine whether artemin regulates the expression of GFRα1 similarly to GDNF, it was added daily to NC and AD neurons for 2 and 5 d. All experiments were performed exactly as those for GDNF, except that artemin was used instead of GDNF. GFRα1 mRNA (Fig. 2D) and protein (Fig. 2E) expressions were significantly enhanced in NC neurons by artemin treatment, although GFRα2, GFRα3, or GFRα4 expressions were not. In contrast, no change was seen in AD neurons treated with artemin (Fig. 2D,E).
Neurturin is known to activate GFRα1 and GFRα2 receptors (Rosenthal, 1999). To examine whether neurturin affects GFRα1 expression in both NC and AD neurons, it was added daily to cultured neurons for 2 and 5 d. RT-PCR and Western blot analyses were used for the detection of GFRα1 mRNA and protein, respectively. No significant changes were seen in GFRα1 mRNA or protein expression after incubation with neurturin in NC or AD neurons (Fig. 2F,G). In addition, persephin did not upregulate GFRα1 expression in NC or AD neurons (n = 6 for each; data not shown); therefore, GFRα1 expression is independent of neurturin or persephin in cortical neurons; that is, neurturin or persephin does not upregulate GFRα1 expression in NC or AD neurons.
To further verify the effect of GDNF on its specific cell-surface receptor, GFRα1, we used antisense oligonucleotides (ASOs) against GFRα1 in the presence of GDNF for 5 d. GFRα1 protein expression was knocked down in NC neurons. In the presence of GDNF, the introduction of GFRα1 ASOs inhibited GFRα1 protein expression in NC neurons in a dose-dependent manner (Fig. 2H). The introduction of sense oligonucleotides (SOs) for GFRα1 did not inhibit GFRα1 expression in NC neurons (Fig. 2H). Similarly, to verify the specific regulatory effects of artemin on its specific cell-surface receptor, GFRα1, NC neurons were transfected with the ASO against GFRα1 for 5 d. In the presence of artemin, GFRα1 ASOs inhibited GFRα1 mRNA (Fig. 2I) and protein (Fig. 2J) expression in NC neurons in a dose-dependent manner, while the GFRα1 SOs did not have any effect on GFRα1 expression in NC neurons (Fig. 2I,J). The introduction of either ASOs against GFRα2, GFRα3 or GFRα4, or SOs for them had no effect on GFRα1 expression in NC neurons under treatment with GDNF or artemin. Also, levels of mRNA and protein of β-actin showed no alteration even if an ASO against GFRα1 was added. These results indicated that these oligonucleotides were not toxic to neurons.
Survival and neurite outgrowth improve in AD neurons transfected with GFRα1
The present data suggest that artemin may act like GDNF to influence neuronal survival through GFRα1 expression. NC and AD neurons were cultured for 7 d, and the ability of artemin to support neuronal survival was compared with that of GDNF, neurturin, and persephin. The surviving neurons were identified by immunocytochemistry for NeuN (Fig. 3A). The survival rates of NC and AD neurons infected with GFRα1 under treatment with artemin were 66 ± 8% and 60 ± 7%, respectively, and those of NC and AD neurons infected with GFRα1 under treatment with GDNF were 72 ± 7% and 65 ± 5%, respectively (Fig. 3B). Thus, treatment with artemin or GDNF resulted in much higher survival rates of NC and AD neurons infected with GFRα1 than treatment with neurturin or persephin. With artemin or GDNF, the survival rates of NC neurons not infected with GFRα1 (infected with empty vector) increased to some extent (43 ± 5% and 44 ± 6%, respectively), but those of AD neurons not infected with GFRα1 did not (Fig. 3B); that is, GDNF and artemin support the survival of AD neurons only when the cells are infected with GFRα1. Interestingly, in NC neurons, GDNF and artemin supported survival, even without the introduction of GFRα1; this was possibly due to the original expression of GFRα1 in NC neurons. Neither neurturin nor persephin supported the survival of NC or AD neurons to the same extent as GDNF and artemin, even when GFRα1 was overexpressed (Fig. 3B). The length of neurite outgrowth in AD neurons was significantly shorter than that of NC neurons; that is, in the presence of GDNF, the neurites of NC neurons were significantly extended; however, the result was not observed in AD neurons (Fig. 1A,B). Here we showed that the survival of AD neurons was enhanced with GFRα1 infection. Whether neurite outgrowth could also be significantly promoted by GFRα1 infection is still unclear; therefore, we introduced GFRα1 or empty vector into isolated AD neurons. Neurite extension was evaluated as the average neurite length. For this, AD neurons were cultured for 7 d and the average neurite length was measured in a blinded manner from triplicate wells of six independent cultures. We found that, under treatment with GDNF, the average neurite length of AD neurons infected with GFRα1 was significantly increased by twofold (58 ± 9 μm) compared with that of AD neurons infected with empty vector (20 ± 6 μm). Similarly, under treatment with artemin, the average neurite length of AD neurons infected with GFRα1 was increased to 48 ± 8 μm compared with empty vector-infected AD neurons (Fig. 3C); however, enhanced neurite outgrowth was not observed in AD neurons infected with GFRα1 in the presence of neurturin or persephin (Fig. 3C).
Figure 3.
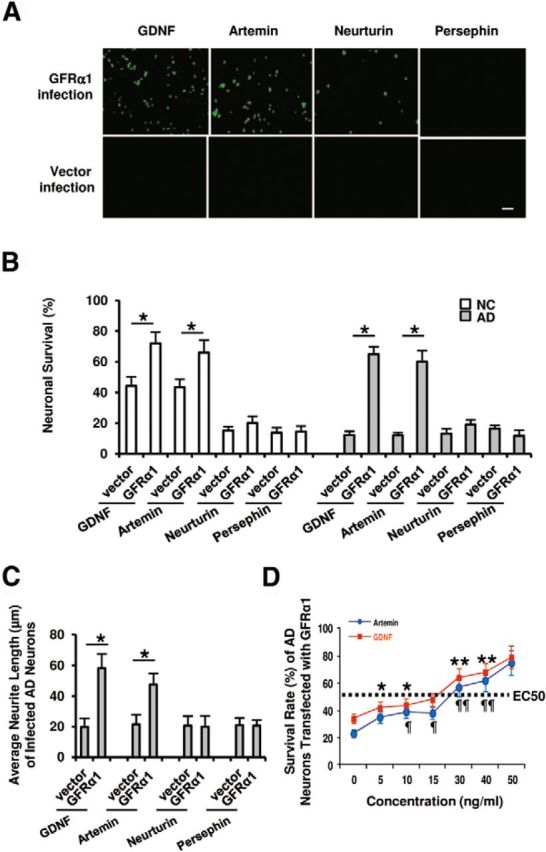
GDNF and artemin promote cortical neuronal survival. A, AD neurons were cultured in the presence of GDNF, artemin, neurturin, or persephin for 7 d. During cultures, AD neurons were infected with GFRα1. The survival of neurons was verified by the antibody against NeuN. With GFRα1 infection, the number of surviving neurons was increased in the presence of GDNF or artemin, but not with treatment with neurturin or persephin. Scale bar, 200 μm. B, NC and AD neurons were infected with GFRα1 or empty vector in the presence of GDNF, artemin, neurturin, or persephin for 7 d. GDNF and artemin significantly supported cell survival in NC and AD neurons infected with GFRα1, compared with those infected with the corresponding empty vectors. *p < 0.05 by the Steel–Dwass test. Note that GDNF and artemin supported the survival of AD neurons only when the cells were infected with GFRα1. In contrast, neurturin or persephin did not play a role. C, AD neurons were infected with GFRα1 or empty vector for 7 d. The neurite outgrowth of AD neurons infected with GFRα1 was significantly promoted in the presence of GDNF or artemin compared with those infected with the corresponding empty vector (*p < 0.05 by the Steel–Dwass test); however, neurite extension was not significantly increased in AD neurons in the presence of neurturin or persephin even when GFRα1 was infected. The neurites from each neuron in designated fields (minimum of 150 cells of two to four wells in four-well dishes) were counted using photoimages taken with a phase-contrast microscope. The experiments were independently repeated six times. D, Dose response of the survival-promoting effect of GDNF and artemin on AD neurons infected with GFRα1. The effect of GDNF and artemin was dose dependent. p < 0.05, in comparison with data at two different concentrations of GDNF or artemin, except for comparison between the data with GDNF at 5 and 10 (*) and 30 and 40 (**), and artemin at 10 and 15 (¶) and 30 and 40 (¶¶) ng/ml (Steel–Dwass test). B–D, The data were obtained in a blinded manner from triplicate wells of six independent cultures (n = 6; mean ± 1 SD).
Dose–response analysis of the survival-promoting effect of artemin and GDNF on AD neurons infected with GFRα1 revealed an EC50 of 15–29 ng/ml (Fig. 3D).
α1-NCAM contributes to the regulation of NC but not AD neurons by GDNF
To examine whether GDNF, via its interaction with α1-NCAM, an alternative signaling receptor for GDNF (Paratcha et al., 2003; Iwase et al., 2005), could enhance neuronal survival, NC and AD neurons were grown in the presence or absence of GDNF for 7 d, accompanying the infection of α1-NCAM (Fig. 4A,B). The surviving neurons were identified by immunocytochemistry for SMI-33 (Fig. 4A). α1-NCAM infection induced an ∼1.5-fold increase in the survival rate of NC neurons in the presence of GDNF for 7 d (Fig. 4B), similar to the finding that GFRα1 infection resulted in an ∼1.5-fold increase in NC neurons (Fig. 3B). In contrast, the survival rates of AD neurons were barely influenced by α1-NCAM infection, even under treatment with GDNF (Fig. 4B).
Figure 4.
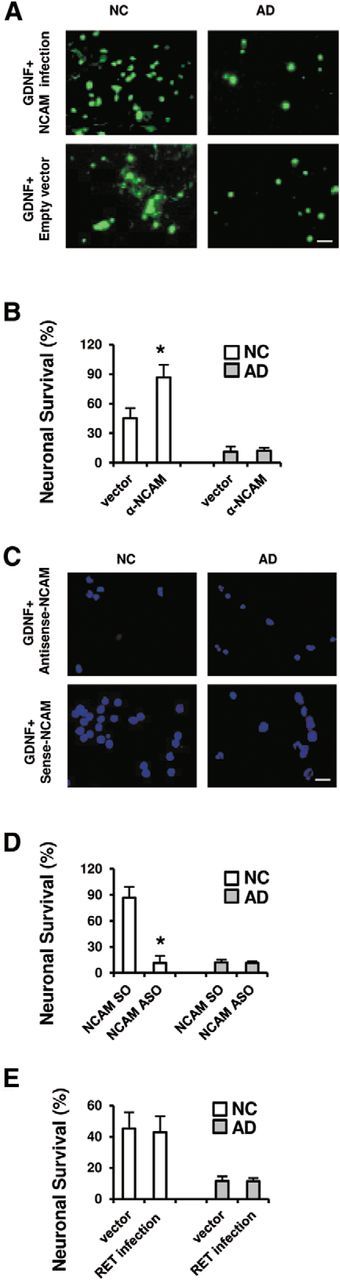
α1-NCAM contributes to the regulation of NC neurons, but not AD neurons, by GDNF. A, Neurofilament protein (SMI-33) immunofluorescence images show that, with α1-NCAM infection, the survival of NC neurons was increased, but that of AD neurons was not. Scale bar, 100 μm. B, In the presence of GDNF, NC and AD neurons were grown and infected with either α1-NCAM or empty vector. Neurons positive for SMI-33 were calculated on day 7. The number of surviving neurons in the presence of GDNF was significantly increased in NC neurons infected with α1-NCAM (*p < 0.05 by the Steel–Dwass test), but not in AD neurons infected with α1-NCAM. C, Immunofluorescence images of SMI-33 show the effect of ASOs and SOs of α1-NCAM. Scale bar, 100 μm. D, NC and AD neurons were transfected with α1-NCAM ASOs or SOs, combined with the presence of GDNF for 7 d. α1-NCAM ASOs significantly decreased the survival of NC neurons. Due to the low survival rate of AD neurons, transfection with α1-NCAM ASOs had little effect on survival. E, In the presence of GDNF, NC and AD neurons were grown and infected with either RET or empty vector. No significant differences were seen between infections of empty vector and RET in NC neurons as well as in AD neurons. B, D, E, Data were obtained in a blinded manner from triplicate wells of six independent cultures (n = 6; mean ± 1 SD).
Next, NC and AD neurons were transfected with α1-NCAM ASOs or SOs in the presence of GDNF for 7 d. Immunofluorescence images for SMI-33 showed the effect of α1-NCAM ASOs and SOs (Fig. 4C). α1-NCAM ASOs significantly decreased the survival of NC neurons (Fig. 4D). The introduction of α1-NCAM ASOs had little effect on the low survival rate of AD neurons (Fig. 4D). In addition, no significant changes were observed in the survival of either NC or AD neurons in the presence of GDNF after RET infection (Fig. 4E). Even when RET was introduced into AD neurons in the presence of GDNF (Fig. 4E), the survival rate of AD neurons was comparable to that with α1-NCAM infection (Fig. 4B). Thus, the introduction of GFRα1 but not α1-NCAM or RET into AD neurons can restore the ability of GDNF to rescue AD neurons.
Blockage of glutamate receptors regulates levels of GFRα1 expression in cortical neurons
It is generally accepted that glutamate receptors are important for the expression and function of NTFs and their receptors (Levine et al., 1998; Nicole et al., 2001). To understand whether glutamate receptors are involved in GFRα1 regulation by GDNF, neurons were incubated with 20 ng/ml GDNF in the presence of either the AMPA receptor blocker CNQX (10 μm) or the NMDA receptor blocker AP-5 (50 μm; Sigma-Aldrich) for 7 d. In NC neurons, CNQX and AP-5 were found to completely abolish GDNF-enhanced GFRα1 protein expression (Fig. 5, left column). In AD neurons, on the other hand, CNQX and AP-5 significantly enhanced GFRα1 protein expression in the presence of GDNF (Fig. 5, right column). Therefore, it appears that glutamate receptors are involved in the regulation of GFRα1 by GDNF. In NC neurons, the presence of glutamate receptors is necessary for GDNF-linked GFRα1 expression, while in AD neurons the absence of glutamate receptors is absolutely required for GFRα1 expression by GDNF stimulation. Without treatment with glutamate receptor antagonists in AD neurons, GFRα1 expression was neither induced nor enhanced by GDNF administration alone (Fig. 5, right column). This finding also suggests that, to prevent or rescue neuronal death/damage in AD brains, the treatment of glutamate receptor antagonists alone may not be sufficient. The combination of NTFs such as GDNF with glutamate receptor antagonists may be necessary to rescue neuronal damage in AD neurodegeneration. The administration of GDNF alone is also unlikely to show some beneficial effects on damaged AD neurons. For healthy elderly NC neurons, on the other hand, the combination of GDNF with glutamate receptor antagonists may show opposite adverse or deteriorating effects, although treatment with glutamate receptor antagonists alone may be somewhat effective.
Figure 5.

Blockage of glutamate receptors regulates levels of GFRα1 expression in cortical neurons. Our neurons (six NC subjects and six AD patients) were incubated with GDNF (20 ng/ml) in the absence or presence of either the AMPA receptor blocker CNQX (10 μm) or the NMDA receptor blocker AP-5 (50 μm) for 7 d. CNQX and AP-5 completely abolished the GDNF-enhanced GFRα1 protein expression in NC neurons (left panels). On the other hand, in AD neurons, CNQX and AP-5 greatly enhanced GFRα1 protein expression under GDNF treatment (right panels), whereas, without CNQX or AP-5, GFRα1 expression was neither induced nor enhanced by GDNF administration alone.
Discussion
GDNF is a potent NTF for a variety of neuronal populations (Walton, 1999). Our report is the first to show a key deficit in GDNF–GFRα1 signaling in human AD neurons. Here we demonstrated a significant decrease in the GFRα1 levels in AD neurons in vitro. This was confirmed in noncultured postmortem brain tissues from AD patients; that is, the selective reduction of GFRα1 expression was also detectable in cortical neurons in AD brain tissues, but not in NC brain tissues. This indicates that the decreased expression of GFRα1 is a manifestation of AD pathology, but is not a consequence of in vitro culturing processes. The introduction of GFRα1, but not α1-NCAM or RET, into AD neurons restored the enhancement of cell survival by GDNF. Although α1-NCAM is required as an essential component of GFRα1-linked signal cascades for neuronal survival (Paratcha et al., 2003), it is likely that a deficit of GFRα1 is one of the key factors for cell death in AD neurons. Thus, for effective GDNF-mediated AD therapy, it is essential to induce or enhance GFRα1 expression. We will further address the selectivity of GFRα1, and why GFRα2, GFRα3, and GFRα4 are not changed, using GFRα subtype knock-out mice.
The time course of GFRα1 expression in NC and AD neurons can exclude the possibility of decreased turnover of GFRα1 over time in NC neuron cultures and/or enhanced turnover of GFRα1 over time in AD neuron cultures. We found that, between NC and AD neurons, there were no significant changes in the expression of GFRα1 with 2 d treatment with GDNF, but that, with 5 and 7 d GDNF treatment, the mRNA and protein levels of GFRα1 subtype in AD neurons were significantly lower than those in NC neurons; therefore, enhanced turnover of GFRα1 in AD neurons cannot be considered. If this is true, mRNA levels of GFRα1 in AD neurons should be higher than those in NC neurons, which was not the case. In addition, since enhanced turnover generally became depressed over time in cultures, the differences between mRNA and protein levels of NC and AD neurons should have become smaller in this case; however, in fact, the differences became larger over time in our neuron cultures. Treatment with GDNF increased the mRNA and protein levels of GFRα1 in NC neurons, while those in AD neurons remained stable, suggesting that mRNA expression and protein production of GFRα1 were enhanced in NC neurons, but were not increased in the turnover of AD neurons.
Surprisingly, our primary neurons were partly alive, but they were, of course, sick; in particular, AD neurons were less healthy and less responsive. AD neurons were dying or degenerating. Under in vitro isolation, treating with papain and complicated purification procedures from brain samples, AD neurons are thought to be more vulnerable than NC neurons. Basically, they were taken from autopsied aged and sick brains of AD patients. As one of the mechanisms underlying cell vulnerability, we found and demonstrated selective reduction of GFRα1 in neurons, probably induced by AD-related pathology, such as enhanced glutamate neurotoxicity.
Why were the levels of GFRα1 expression not increased, even when GDNF was added to AD neurons? Our present study suggests that glutamatergic neurotransmission may participate in the reduced expression levels of GFRα1 in AD neurons. In our neurons, ∼70–75% were positive for GluRN1 and GluRA2/4. This was confirmed by immunocytochemistry in the present study. When the AMPA receptor blocker CNQX or the NMDA receptor blocker AP-5 was added to uninfected AD neurons in advance, the levels of GFRα1 expression were increased by GDNF treatment. By contrast, GDNF treatment did not enhance the levels of GFRα1 expression in uninfected NC neurons when CNQX or AP5 was added in advance. These results suggest that, in AD neurons, there is an excess of glutamatergic neurotransmission, which may chronically reduce GFRα1 expression. There is clinical and laboratory evidence of excessive glutamate receptor activity in the AD brain, which is possibly involved in Aβ-induced neuronal death (Mattson et al., 1992; Lipton, 2005). This receptor activity possibly remains excessive in AD neurons in vitro, regardless of whether Aβ or glutamate is increased in culture medium.
Once excess glutamatergic neurotransmission has been settled down by CNQX or AP-5 in vitro, GDNF is expected to increase GFRα1 expression, even in AD neurons. Our neurons were seeded at the same density (5 × 105/ml) in NC and AD neuron cultures at the beginning. As described above, our neuron cultures contained ∼9% astrocytes identified by anti-GFAP, with no differences between NC and AD neuron cultures. Our neurons were cultured in serum-free Neurobasal A with B27, and subsequently the conditioned medium of neuron cultures treated with GDNF or KCl was processed to measure glutamate and aspartate release, revealing no differences in the release between NC and AD neuron cultures (unpublished data; n = 6 for each), which might be due to the in vitro dissociated neuron-rich cultures. In the reality of in vivo conditions of AD, an excess amount of extracellular glutamate may be contributed not only by neurons, but also by astrocytes, microglial cells, and furthermore by cerebral vascular cells (i.e., endothelial cells). Here we indeed observed that the response to GDNF treatment with blockage of glutamatergic neurotransmission was different between NC and AD neurons in vitro. This is possibly due to treatment with glutamate receptor blockers, which may block intracellular signals for glutamatergic neurotransmission that have already shifted to being toxic or detrimental to AD neurons, while, in NC neurons, CNQX and AP-5 may inhibit intracellular signals that have already shifted to being beneficial or trophic. This different change in signal transduction may lead to differences in the GDNF-induced GFRα1 expression. The different responses to GDNF show that it is irrelevant whether glutamate is increased in the medium of AD neuron cultures.
We found here that GDNF enhanced the survival of NC neurons infected with α1-NCAM, an alternative signaling receptor for GDNF, but not with RET. Generally, GDNF uses RET-independent pathways, particularly in cortical neurons (Paratcha et al., 2003); however, recently, RET was reported to be involved in axon guidance signals (Bonanomi et al., 2012). It is unlikely that, in our cortical neurons, RET is involved in GDNF-enhanced survival.
Artemin, one of the four structurally related members of GDNF family ligands (Rosenthal, 1999), is known to support the differentiation and survival of neuronal populations (Baloh et al., 1998). Studies have suggested artemin to be a NTF for midbrain dopaminergic neurons (Zihlmann et al., 2005), whereas the specific function of artemin in other brain regions is not yet known. We demonstrated herein the trophic effects of artemin on aged human cortical neurons. Biochemical experiments have shown that artemin preferentially binds to GFRα3 in general: this binding specificity has largely been studied for the receptor complex consisting of RET and one of four GFRα subtypes (Rosenthal, 1999; Sariola and Saarma, 2003; Wang et al., 2006). In the case of α1-NCAM as an alternative GFRα partner, it remains to be determined whether artemin signals through GFRα1 or GFRα3 to elicit biological effects on aged cortical neurons. Herein we demonstrated that, similar to the effects of GDNF (Schmutzler et al., 2011), artemin increased the expression levels of GFRα1, not GFRα2, GFRα3, or GFRα4; this effect was dose and time dependent, and 20–30 ng/ml artemin was enough to elicit an effect on GFRα1, indicating its specific effect (Schmutzler et al., 2011). Artemin, as well as GDNF, promoted cell survival and neurite extension of AD neurons when they were infected with GFRα1, but not with the corresponding empty vectors. These results suggest that artemin elicits its trophic effects on cortical neurons through receptor complex, including GFRα1.
Footnotes
This work was supported by National Institutes of Health Grants RO1AG025888, RO1032441, and RO1025888S; by Arizona Biomedical Research Commission ABRC002; by the Alzheimer's Association Zenith Award II Initiated Research Grants IIRG-07-59510 and IIRG-09-61521 and New Investigator Research Grant NIRG-08-91471; by American Health Assistance Foundation Grant G2006-118; and a grant from the Research Resource Network (RRN) of the National organization in Japan.
B.L. works at GlaxoSmithKline (GSK) Pharmaceutical. The remaining authors declare no competing financial interests.
References
- Abe K, Saito H. Amyloid β protein inhibits cellular MTT reduction not by suppression of mitochondrial succinate dehydrogenase but by acceleration of MTT formazan exocytosis in cultured rat cortical astrocytes. Neurosci Res. 1998;31:295–305. doi: 10.1016/S0168-0102(98)00055-8. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Kaneko T, Mizuno N, McGeer PL. Distribution of phosphate-activated glutaminase in the human cerebral cortex. J Comp Neurol. 1990;297:239–252. doi: 10.1002/cne.902970207. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Watson JJ, Shoemark DK, Barua NU, Patel NK. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol Ther. 2013;138:155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association. Thies W, Bleiler L. Alzheimer's Association Report. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011;7:208–244. doi: 10.1016/j.jalz.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Arenas E. GDNF, a multispecific neurotrophic factor with potential therapeutic applications in neurodegenerative disorders. Mol Psychiatry. 1996;1:179–182. [PubMed] [Google Scholar]
- Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Jr, Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/S0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaheta RA, Beecken WD, Engl T, Jonas D, Oppermann E, Hundemer M, Doerr HW, Scholz M, Cinatl J. Human cytomegalovirus infection of tumor cells downregulates NCAM (CD56): a novel mechanism for virus-induced tumor invasiveness. Neoplasia. 2004;6:323–331. doi: 10.1593/neo.03418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, Chivatakarn O, Bai G, Abdesselem H, Lettieri K, Marquardt T, Pierchala BA, Pfaff SL. Ret is a multifunctional coreceptor that integrates diffusible- and contact-axon guidance signals. Cell. 2012;148:568–582. doi: 10.1016/j.cell.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Chang S, Rathjen FG, Raper JA. Extension of neurites on axons is impaired by antibodies against specific neural cell surface glycoproteins. J Cell Biol. 1987;104:355–362. doi: 10.1083/jcb.104.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry SH, Schwark HD, Jones EG, Yan J. Numbers and proportions of GABA-immunoreactive neurons in different areas of monkey cerebral cortex. J Neurosci. 1987;7:1503–1519. doi: 10.1523/JNEUROSCI.07-05-01503.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, Panza F, Frisardi V, Solfrizzi V, D'Onofrio G, Logroscino G, Seripa D, Pilotto A. Therapeutic intervention for Alzheimer's disease with γ-secretase inhibitors: still a viable option? Expert Opin Investig Drugs. 2011;20:325–341. doi: 10.1517/13543784.2011.550572. [DOI] [PubMed] [Google Scholar]
- Iwase T, Jung CG, Bae H, Zhang M, Soliven B. Glial cell line-derived neurotrophic factor-induced signaling in Schwann cells. J Neurochem. 2005;94:1488–1499. doi: 10.1111/j.1471-4159.2005.03290.x. [DOI] [PubMed] [Google Scholar]
- Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Urade Y, Watanabe Y, Mizuno N. Production, characterization, and immunohistochemical application of monoclonal antibodies to glutaminase purified from rat brain. J Neurosci. 1987;7:302–309. doi: 10.1523/JNEUROSCI.07-01-00302.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasashima S, Kawashima A, Muroishi Y, Futakuchi H, Nakanishi I, Oda Y. Neurons with choline acetyltransferase immunoreactivity and mRNA are present in the human cerebral cortex. Histochem Cell Biol. 1999;111:197–207. doi: 10.1007/s004180050349. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lindholm K, Yang LB, Li R, Shen Y. Isolation of living neurons from human elderly brains using the immunomagnetic sorting DNA-linker system. Am J Pathol. 2002;161:1567–1576. doi: 10.1016/S0002-9440(10)64435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res. 2005;2:155–165. doi: 10.2174/1567205053585846. [DOI] [PubMed] [Google Scholar]
- Lozano AM, Schmidt M, Roach A. A convenient in vitro assay for the inhibition of neurite outgrowth by adult mammalian CNS myelin using immortalized neuronal cells. J Neurosci Methods. 1995;63:23–28. doi: 10.1016/0165-0270(95)00081-X. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to exciyoyoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/WNL.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Hanson JC, Quets-Nguyen AT, Bergeron M, Smith RC. A quantitative method for analysis of in vitro neurite outgrowth. J Neurosci Methods. 2007;164:350–362. doi: 10.1016/j.jneumeth.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci U S A. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Nicole O, Ali C, Docagne F, Plawinski L, MacKenzie ET, Vivien D, Buisson A. Neuroprotection mediated by glial cell line-derived neurotrophic factor: involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J Neurosci. 2001;21:3024–3033. doi: 10.1523/JNEUROSCI.21-09-03024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibáñez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/S0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Soderstrom KE, Kordower JH. Trophic factors therapy in Parkinson's disease. Prog Brain Res. 2009;175:201–216. doi: 10.1016/S0079-6123(09)17514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal A. The GDNF protein family: gene ablation studies reveal what they really do and how. Neuron. 1999;22:201–203. doi: 10.1016/S0896-6273(00)81077-6. [DOI] [PubMed] [Google Scholar]
- Sakakibara S, Okano H. Expression of neural RNA-binding proteins in the postnatal CNS: implications of their roles in neuronal and glial cell development. J Neurosci. 1997;17:8300–8312. doi: 10.1523/JNEUROSCI.17-21-08300.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariola H, Saarma M. Novel functions and signalling pathways for GDNF. J Cell Sci. 2003;116:3855–3862. doi: 10.1242/jcs.00786. [DOI] [PubMed] [Google Scholar]
- Savoca R, Ziegler U, Sonderegger P. Effects of L-serine on neurons in vitro. J Neurosci Methods. 1995;61:159–167. doi: 10.1016/0165-0270(95)00038-V. [DOI] [PubMed] [Google Scholar]
- Schmutzler BS, Roy S, Pittman SK, Meadows RM, Hingtgen CM. Ret-dependent and Ret-independent mechanisms of Gfl-induced sensitization. Mol Pain. 2011;7:22. doi: 10.1186/1744-8069-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Ray WJ, Shearman M, Hutton M. Beyond amyloid. The next generation of Alzheimer's disease therapeutics. Mol Interv. 2007;7:261–270. doi: 10.1124/mi.7.5.8. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Chauhan NB. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Brain Res Rev. 2000;33:199–227. doi: 10.1016/S0165-0173(00)00030-8. [DOI] [PubMed] [Google Scholar]
- Treanor JJ, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hökfelt T, Broberger C, Porreca F, Lai J, Ren K, Ossipov M, Koshkin A, Jakobsen N, Skouv J, Oerum H, Jacobsen MH, Wengel J. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci U S A. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KM. GDNF: a novel factor with therapeutic potential for neurodegenerative disorders. Mol Neurobiol. 1999;19:43–59. doi: 10.1007/BF02741377. [DOI] [PubMed] [Google Scholar]
- Wang X, Baloh RH, Milbrandt J, Garcia KC. Structure of artemin complexed with its receptor GFRα3: convergent recognition og glial cell line-derived neurotrophic factors. Structure. 2006;14:1083–1092. doi: 10.1016/j.str.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Weissmiller AM, Wu C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl Neurodegener. 2012;1:14. doi: 10.1186/2047-9158-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zihlmann KB, Ducray AD, Schaller B, Huber AW, Krebs SH, Andres RH, Seiler RW, Meyer M, Widmer HR. The GDNF family members neurturin, artemin and persephin promote the morphological differentiation of cultured ventral mesencephalic dopaminergic neurons. Brain Res Bull. 2005;68:42–53. doi: 10.1016/j.brainresbull.2004.10.012. [DOI] [PubMed] [Google Scholar]