

Abstract
Bioactive peptides are packaged in large dense-core secretory vesicles, which mediate regulated secretion by exocytosis. In a variety of tissues, the regulated release of neurotransmitters and hormones is dependent on calcium levels and controlled by vesicle-associated synaptotagmin (SYT) proteins. Drosophila express seven SYT isoforms, of which two (SYT-α and SYT-β) were previously found to be enriched in neuroendocrine cells. Here we show that SYT-α and SYT-β tissue expression patterns are similar, though not identical. Furthermore, both display significant overlap with the bHLH transcription factor DIMM, a known neuroendocrine (NE) regulator. RNAi-mediated knockdown indicates that both SYT-α and SYT-β functions are essential in identified NE cells as these manipulations phenocopy loss-of-function states for the indicated peptide hormones. In Drosophila cell culture, both SYT-α and neuropeptide cargo form DIMM-dependent fluorescent puncta that are coassociated by super-resolution microscopy. DIMM is required to maintain SYT-α and SYT-β protein levels in DIMM-expressing cells in vivo. In neurons normally lacking all three proteins (DIMM−/SYT-α−/SYT-β−), DIMM misexpression conferred accumulation of endogenous SYT-α and SYT-β proteins. Furthermore transgenic SYT-α does not appreciably accumulate in nonpeptidergic neurons in vivo but does so if DIMM is comisexpressed. Among Drosophila syt genes, only syt-α and syt-β RNA levels are upregulated by DIMM overexpression. Together, these data suggest that SYT-α and SYT-β are important for NE cell physiology, that one or both are integral membrane components of the large dense-core vesicles, and that they are closely regulated by DIMM at a post-transcriptional level.
Keywords: DIMM, Drosophila, LDCV, neuropeptide, super-resolution, synaptotagmin
Introduction
Neurotransmitters and neurohormones are stored in specialized vesicles that undergo regulated release by exocytosis. Vesicle exocytosis is triggered by increases in calcium levels sensed by various vesicular synaptotagmin (SYT) isoforms (Südhof, 2008). SYT proteins typically contain two calcium-binding domains (C2A and C2B) and a single transmembrane domain. Calcium binding permits SYT interactions with SNARE complexes, leading to vesicle anchoring at plasma membrane phospholipids and its eventual exocytosis (Chapman, 2002). There are several SYT isoforms, each with different calcium affinities and distinct expression patterns (Sugita et al., 2002).
In neuroendocrine (NE) cells, bioactive peptides are processed and packaged in large dense-core vesicles (LDCVs). The regulated exocytosis of LDCV is also controlled by calcium levels, although generally much lower calcium levels are needed than for exocytosis of small synaptic vesicles (Mansvelder and Kits, 2000). However, because they are not generally located near calcium channels, LDCV secretion often requires more prolonged stimulation periods to permit calcium levels to rise sufficiently high. Several SYT isoforms have been associated with LDCVs in diverse tissues (e.g., Fukuda et al., 2004; Gustavsson et al., 2008; Schonn et al., 2008). In the Drosophila genome, there are seven annotated SYT isoforms (Adolfsen et al., 2004); and of these, two (syt-α, CG5559 and syt-β, CG3020) appear enriched in NE cells. Whether syt-α and syt-β has NE-specific physiological roles remains to be determined.
In Drosophila, the bHLH protein DIMMED regulates the innate genetic machinery that distinguishes professional secretory cells. DIMM expression is limited to NE neurons, but it is not correlated with any specific secretory peptide (Hewes et al., 2003; Park et al., 2008a). It is correlated with periods of differentiation and function. Genetic analyses suggest that DIMM helps organize the regulated secretory pathway in NE cells (Allan et al., 2005; Gauthier and Hewes, 2006; Park et al., 2008b; Hamanaka et al., 2010). Ectopic expression of DIMM within conventional Drosophila neurons confers display of numerous LDCVs (which can store and process transgenic neuropeptides) as well as the downregulation of SSVs and presynaptic active zones (Hamanaka et al., 2010). Thus, DIMM promotes the peptidergic regulated secretory pathway at the expense of trafficking pathways that produce conventional neurotransmitters. In mammals, Mist1 organizes corresponding features of the secretory pathway in serous exocrine cells (Mills and Taghert, 2012).
We previously identified several of DIMM's direct transcriptional targets (Park et al., 2008b, 2011) and more recently performed genome-wide profiling of all its binding sites (Hadžić et al., manuscript submitted). Some targets encode critical LDCV proteins that play well-defined roles in neuropeptide biosynthesis, including Phm (CG3324) and Cytochrome C b561 (CG1275) (Park et al., 2011). To broaden the understanding of LDCV biology and its regulation by DIMM, here we consider additional peptidergic cell-specific functions and focus on potential calcium-sensing SYT isoforms. We show that SYT-α and SYT-β are associated with overlapping subsets of peptidergic NEs, that SYT-α is an integral membrane protein of the LDCV, and that SYT-α and SYT-β expressions are controlled by DIMM post-transcriptionally.
Materials and Methods
Fly stocks.
The following fly lines were used in this study: dimm mutant alleles (Rev4 and Rev8) and UAS-dimm-myc (Hewes et al., 2003); we created two transgenic fly lines: UAS-syt-α-mCherry and UAS-syt-α-myc using P-element germline transformation (Genetic Services). We used elav-gal4, ap-gal4, c929-gal4 (Hewes et al., 2003), CCAP-gal4 (Park et al., 2003), and ETH-Geneswitch GAL4 to drive the UAS transgenes. Behavioral test were performed by crossing the transgenic dsRNAi lines, obtained from the Vienna Drosophila RNAi centers (dimm:KK100204, syt-α: KK106350, GD1754; syt-β: KK11946; syt-4: GD2842; syt1: KK108653, JF1234; eth: GD5964), with UAS-dicer2; ETH-geneswitch GAL4. To activate geneswitch gal4, flies were fed food containing 50 μm RU486 (mifepristone, M8046, Sigma) and raised at 29°C. We measured viability by computing the number of experimental (GAL4 > RNAi) versus control (GAL4-only) siblings. In all experiments, we studied flies of either sex.
Quantitative real-time PCR.
RNA preparation followed by protocols previously described (Lin et al., 2002). For these experiments, we crossed UAS-dimm-myc flies (II) with the pan-neuronal driver, elav-gal4 to misexpress DIMM protein. The crossed flies were reared at 18°C to reduce the lethality, and the embryos of either sex (24–36 h AEL at 18°C) were collected and total RNA is prepared. For the mutant, Rev4/Rev8 larvae were collected at room temperature. Total RNA was isolated with Trizol reagent (Sigma), digested with RNase-free DNase I, and purified with RNAeasy columns (QIAGEN). Reverse transcription reactions were performed following the manufacturer's protocols (NEB). We measured transcript quantities using SYBR-green incorporation on an ABI 7000 machine and made genotypic comparisons with the double δCt method (Pfaffl, 2001). Levels of RP49 RNA were used for normalization. The primers used are listed (CG5559 (syt-α): 5′-AAACGCTAT CGCCTGAAC TGG-3′ and 5′-TTGTCCACA TTGTAGCCC ACG-3′; CG3020 (syt-β): 5′-GCATTTGCG CGTTAAGTA CGA-3′ and 5′-TGCAACATC AGGCGGACA T-3′; CG10047 (syt-4): 5′-GCGGCTAAG CCAAATCCA TT-3′ and 5′-GGCTCTGTT TCTTGTTCC GGA-3′; CG3139 (syt-1): 5′-AACATGAAG TAAGGCGGC AACA-3′ and 5′-TGTCTGACG GTTCGCGTA CAT-3′; CG2381 (syt-7): 5′-GCTATCACC CGTCGAACT CAAT-3′ and 5′-GCCAAACTG TAGCCACAC CTTT-3′; CG10617 (syt-12): 5′-TCGGACTAT CCAATCACA AGCA-3′ and 5′-CGCTTGAAA CTACCACTG GTGC-3′; CG9778 (syt-14): 5′-AGTGCAGCT CGAACGATT CCA-3′ and 5′-TCCCGTTCC TTGTCCAGC TTT-3′; Rp49: 5′-GTTCGATCC GTAACCGAT GT-3′ and 5′-TCGGATCGA TATGCTAAG CTG-3′).
Antibodies, immunocytochemistry, and fluorescence imaging.
For primary antibodies, we used affinity-purified guinea pig anti-DIMM (1:200) (Allan et al., 2005); rabbit anti-SYTα (1:500) and rabbit anti-SYTβ (1:1000) (Adolfsen et al., 2004) (anti-SYT antibodies were gifts from Dr. Troy Littleton, MIT); rabbit anti-dGM130 (1:1000; Nakamura et al., 1995), rabbit anti-Lava Lamp (lva; 1:500; Sisson et al., 2000) and rabbit anti-Sec16 (1:500; Ivan et al., 2008) (all three were gifts from Dr. Aaron DiAntonio, Washington University); mouse monoclonal anti-myc (1:1000, Sigma); mouse monoclonal anti-GFP 3E6 (1:800, Invitrogen); and rabbit anti-GFP (1:500; rabbit polyclonal, #AB3080 Millipore Bioscience Research Reagents). For secondary antibodies, we used Cy3-conjugated or Alex-488-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Confocal images were acquired on an Olympus FV500 laser scanning confocal microscope at the Bakewell Neuroimaging facility, Washington University and manipulated by Adobe Photoshop software to adjust contrast and/or levels. To permit direct comparisons between genotypes, microscope settings were held constant within single experiments. The imageJ program was used to measure the mean gray value within the selected area.
Luciferase assay.
sytα genomic fragments were subcloned into the sv40-pGL3 luciferase vectors (Promega). For luciferase assays, we followed the experiment previously described (Park et al., 2008b). hEK293 cells were transiently transfected with lipofectamine 2000 (Invitrogen). The luciferase activity was measured from lysates with a Victor-Wallac2 plate reader (PerkinElmer) and normalized with thymidine kinase-Renilla luciferase (TK-Rluc). Each transfection was performed at least three times independently. As a positive control, we used Phm-sv-luc construct (Park et al., 2008b).
Drosophila cell cultures and transfection.
BG3-c2 cells were maintained at room temperature in Schneider's medium (#S0146, Sigma) + 10% FCS (Serum Source International) + 10 μg/ml insulin + 2 mm l-glutamine + 50 U penicillin/streptomycin. For transient transfections, we used FuGene HD reagent (Roche) and followed the manufacturer's instructions. PDF-EGFP was constructed by fusing pdf (as a BamHI/SpeI PCR-generated fragment), to EGFP (as a KpnI/NotI fragment derived from pEGFP; Invitrogen); PDF-mEOS2 (SacI/NotI for mEOS2), syta-6xmyc was made by fusing Sytα (as a BamHI/SacII PCR-generated fragment) to a SacII/XbaI fragment that contained a 6xmyc sequence derived from P{UAS-6xmyc}); sytα-mCherry was made by fusing Sytα (the same BamHI/SacII fragment as above) to a SacII/XbaI fragment containing mCherry (from pcDNA–mCherry (Invitrogen). cDNA constructs were subcloned sequentially into p{PAC} vector containing a strong actin5C promoter. In a 12-well plate, we split the cells with 5.0 × 105 number and transfected with the mixture of 2 μg DNA + 10 μl FuGene HD reagent. After 24 h incubation, we performed conventional immunostaining.
Super-resolution microscopy optical setup and imaging.
STORM/PALM imaging was performed on a custom-built microscopy rig. Briefly, the optical rig was constructed around a Nikon Ti-Eclipse inverted microscope fitted with the Nikon perfect focus system for focus stabilization, a motorized stage (Marzhauser Wetzlar) and a 100× 1.4 NA objective (Olympus UPLSAPO). Illumination lasers, 642 nm (Vortran laser), 561 nm, 488 nm (Coherent, Sapphire), and 405 nm (Coherent, Cube) were shuttered individually using an acoustic-optical tunable filter (Crystal Technologies). Laser beams were combined, expanded, collimated, and focused at the back focal plane of the 100× objective. Total internal reflection fluorescence illumination was achieved with an objective-type total internal reflection fluorescence geometry using a custom-built translation stage. For super-resolution SIM, we used a Zeiss LSM 780, ELYRA PS.1 system, a 63×/1.4 Plan-Apochromat objective, and a PCO egde sCMOS camera (50 ms exposure times). Data were analyzed with commercial (Zen) software, using the SIM module.
BG3-c2 cells cultured on No 1.0 coverglass (ThermoFisher) were transiently transfected with plasmids encoding DIMM, SYT-α-MYC, and PDF-mEOS2. At 40 h after transfection, cells were fixed with 4% PFA in PBS, washed with excess PBS, blocked, and permeabilized using 3% BSA + 0.1% Triton X-100 in PBS. Cells were immunolabeled using a 1:10 dilution of the anti-Myc 9E10 monoclonal antibody supernatant (Developmental Studies Hybridoma Bank) and a donkey anti-mouse IgG secondary antibody (Jackson ImmunoResearch Laboratories) custom conjugated with the fluorophores Alexa-647 (Invitrogen) and Cy2 (GE Healthcare). Coverslips with labeled cells were inverted onto a slide with imaging buffer containing glucose, mercaptoethylamine, and an oxygen scavenging system as described previously (Bates et al., 2005). Sparse single molecule images were obtained at 60 Hz frequency by alternating pairs of imaging and activation lasers in the following sequence: 561 nm (250 W/cm2) + weak 405 nm for the mEOS2 channel, followed by 642 nm (560 W/cm2) + weak 488 nm for Alexa-647. The amount of 405 or 488 nm activation lasers was adjusted to ensure sparse single molecule events in each camera frame and was typically ∼4 W/cm2 (488 nm) and 0.085 W/cm2 (405 nm) measured at the sample. Fluorescence from ∼8000 frames per channel was collected by the same objective, separated using a quad band dichroic zt/zet-405,488,561,642rpc (Chroma), and filtered with a dual band zet561/640m emission filter (Chroma). Image stacks were captured on an EMCCD camera (iXon+ DU897, Andor) and analyzed using custom software. A647, and mEOS2 raw image stacks were fitted with an elliptical Gaussian function to determine the centroid positions of fluorescent pixel intensity peaks. These STORM localizations were rendered as images or analyzed quantitatively. Images of 100 nm TetraSpec fluorescent beads (Invitrogen) obtained using the above optics show a high degree of correspondence between the two channels as shown by the difference in the X and Y bead positions (mean ΔX = 3.35 nm and ΔY = 1.79 nm).
Quantification of SYT-α-MYC and PDF-mEOS2 localizations was performed by first calculating the average dark-off time for the two channels from the raw images followed by applying a blinking correction factor independently to the two channels as described previously (Annibale et al., 2011). Clusters of PDF-mEOS2 localizations representing putative LDCVs were digitally isolated by cropping a 500 nm × 500 nm ROI, around each cluster. To align selected clusters, we used the k-means algorithm to independently identify centroid positions of PDF-mEOS2 and SYT-α-MYC localizations for each cluster. Each cluster was shifted such that the PDF-mEOS2 centroid positions were aligned at origin point. Further, for each cluster, we identified the vector from the origin point to the centroid of SYT-α-MYC localizations and aligned all vectors along the positive z-axis by rotating each cluster. Finally, localizations from all aligned clusters were summed and projected onto the z-axis to generate a normalized histogram. Quantification of PDF-mEOS2 and SYT-α-MYC localizations was performed in MATLAB (MathWorks) followed by plotting the histogram in Origin software.
Results
SYT-α- and SYT-β-expressing cells are mostly DIMM-positive
We used immunocytochemistry to determine the degree to which SYT-α and SYT-β are coextensive with DIMM. In the third instar larval CNS, the patterns of the two SYT antibody stains were congruent in many places. Furthermore, most DIMM-positive cells expressed either or both of these specific SYT proteins (Fig. 1). We measured the incidence and coincidence of DIMM, SYT-α, and SYT-β within a limited set of neuromeres (segments T2-A3) (Table 1) and observed the following. First, there were approximately equivalent numbers of SYT-α+ and SYT-β+ neurons. Second, these neuronal subsets were partially different because the percentage of each that was DIMM+ was different: SYT-β+ neurons were more likely to also be DIMM+ than SYT-α+ neurons. We emphasize that SYT-α and SYT-β antibody stains were not restricted to DIMM cells. For example, Kenyon Cells of the Mushroom Body (which contain the neuropeptide short NPF) (Johard et al., 2008) are DIMM−, but they are both SYT-α+ and SYT-β+ (Adolfsen et al., 2004). Outside the larval CNS, we observed coexpression of DIMM with SYT-α and SYT-β in peripheral NE centers, such as the corpora cardiaca of the Ring Gland (data not shown). Also, Adolfsen et al. (2004) reported SYT-α is selectively present in the embryonic LBD neuron and that SYT-β is selectively expressed in the embryonic Inka cells, both of which are DIMM+ peripheral peptidergic cells (Hewes et al., 2003; Park et al., 2011). Together, these results indicate extensive coexpression of either, or both, SYT-α and SYT-β with DIMM. These observations prompted us to investigate the physiological roles of these specific SYT isoforms in NE cells.
Figure 1.
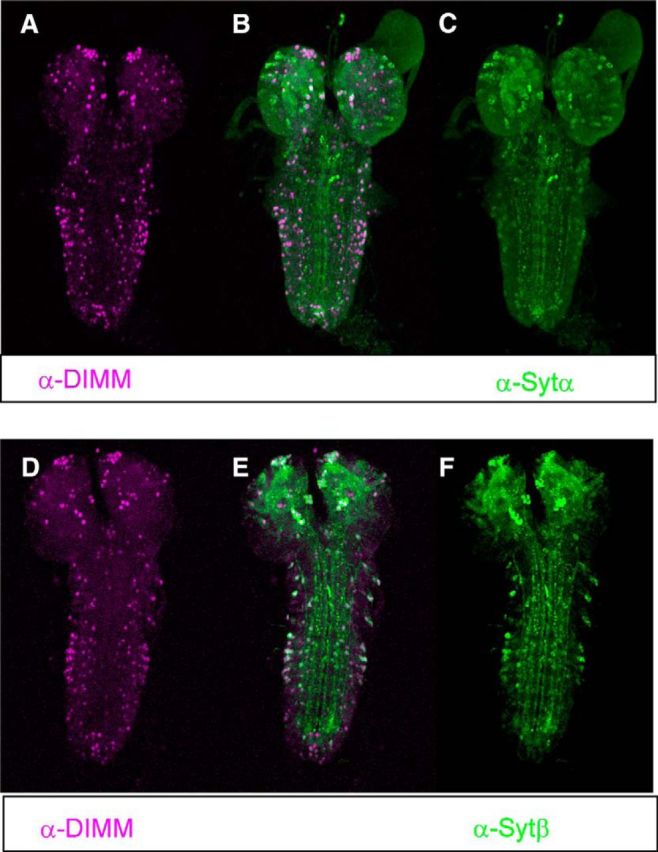
DIMM colocalizes with high-level expression of SYT-α and SYT-β. A–C, Double immunostaining for DIMM (magenta) and SYT-α (green). D–F, Double-immunostaining for DIMM and SYT-β in the larval ventral nerve cord.
Table 1.
The number of the cells staining positive for DIMM and/or SYT-α, and DIMM and/or SYT-β, within a five-neuromere region (T2–A3) of the larval CNSa
| SYT-α/DIMM (n = 3) | SYT-α+ | DIMM+/SYT-α+ |
| 55.67 ± 6.77 | 27.67 ± 3.71 (49.70%) | |
| SYT-β/DIMM (n = 4) | SYT-β+ | DIMM+/SYT-β+ |
| 41.00 ± 2.58 | 37.00 ± 2.48 (90.24%) |
aData are mean ± SEM. Student's t test indicates that the difference between the percentages is significantly different (p < 0.016).
The SYT-α and SYT-β proteins are critical for peptidergic cell control of innate behaviors
Many SYT proteins act as essential calcium sensors to control release of neurotransmitters and peptide hormones (Pang and Südhof, 2010). In insects, the pace of growth and metamorphosis is set by the process of molting, during which the cuticle is replaced by a new and (typically) larger one. The shedding of the old cuticle and hardening of the new one are therefore critical developmental and behavioral events (Fabre, 1917). This complex coordination is effected by a set of neuroendocrine peptides and peptide hormones that dictate the timing and sequence of innate behaviors (for review, see Ewer and Reynolds, 2002). One critical triggering peptide is called the Ecdysis Triggering Hormone (ETH): it is produced by the set of DIMM-positive, peripheral endocrine cells called Inka's (Zitnan et al., 1996; O'Brien and Taghert, 1998; Hewes et al., 2003). We therefore measured the importance of SYT-α and SYT-β in DIMM cell physiology by their transgenic RNAi knockdown in Inka cells.
Drosophila has 14 Inka cells that release the peptides ETH and P-ETH to trigger ecdysial behaviors (Zitnan et al., 1996). Both the ablation of the cells and the deletion of the eth gene lead to early lethality due to failure to complete ecdysis. In the few survivors, loss of the eth gene leads to a failure to display normal coordination of larval ecdysial behaviors (Park et al., 2002). ETH secretion from Inka cells is dependent on increases in intracellular calcium levels (Kingan et al., 2001). Inka cells are DIMM+ and in the embryo are also SYT-β+ (Adolfsen et al., 2004; Park et al., 2008a). We used an Inka cell-specific eth-geneswitch-GAL4 to test the effects of RNAi for various syt isoforms. Adult viability for flies of the GAL4 > RNAi genotype was compared with that of RNAi-only siblings. The ratio in the control cross was 1.19 ± 0.07. However, expression of either eth RNAi or dimm RNAi produced severe lethality, with relative ratios of 0.07 ± 0.05 and 0.10 ± 0.10, respectively. In the same conditions, syt-α and syt-β RNAi constructs also showed significant loss of viability (syt-α, 0.01 ± 0.01; syt-β, 0.01 ± 0.01). In contrast, syt-4 knockdown in Inka cells showed no significant difference in viability with control siblings (0.87 ± 0.22). Interestingly, syt1 knockdown also produced significantly reduced viability (0.39 ± 0.11) (Fig. 2A). Importantly, RNAi knockdown did not lead to loss of Inka cells, as these could still be observed with anti-DIMM or anti-PHM (O'Brien and Taghert, 1998) antibody staining (data not shown).
Figure 2.
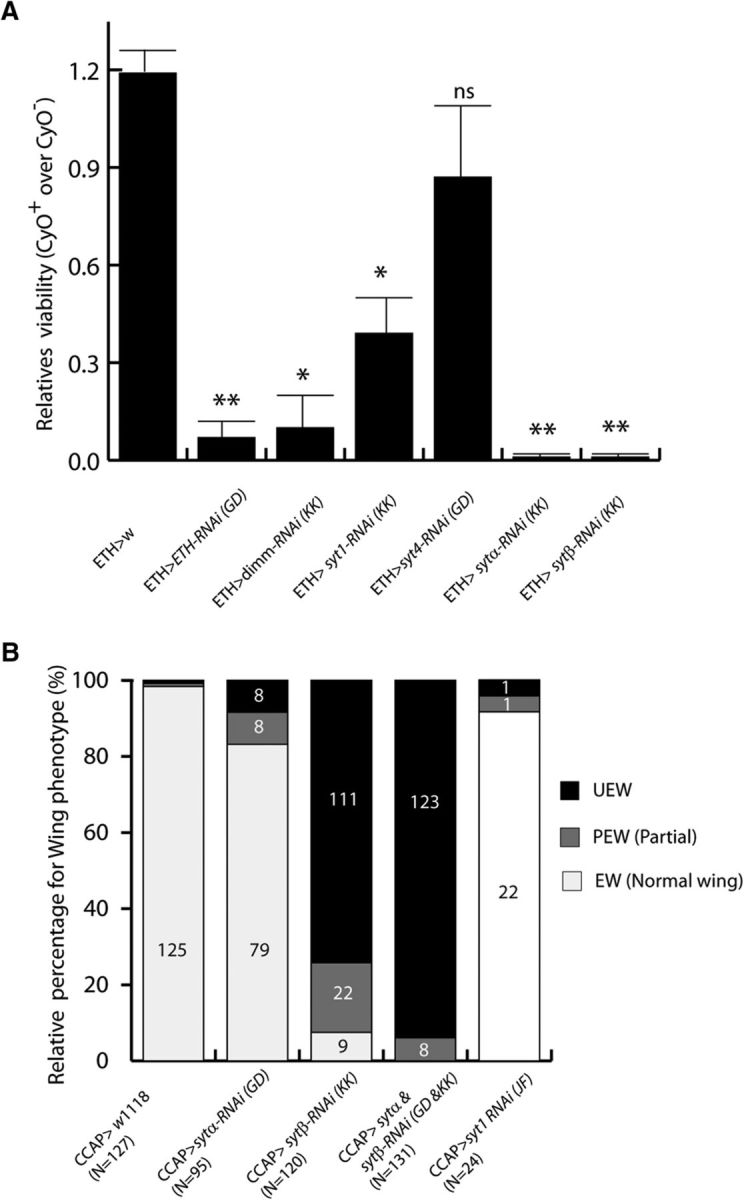
sytα and sytβ expression is necessary for neuropeptide-related innate behaviors. A, Knocking down either sytα or sytβ levels reduces developmental viability. Each graph represents the relative ratio of sibling progeny: experimental (ETH-GS-Gal4/RNAi) versus control (RNAi/CyO). Results are summed from two independent crosses. Student's t test was used for statistical analysis. **p < 0.01. *p < 0.05. ns, Not significant. B, Knocking down sytα and sytβ levels produces a cooperative wing expansion defect. Each graph represents the percentage of flies displaying wing expansion during eclosion in adult female flies: CCAP > w1118 (n = 127); CCAP > sytα-RNAi (n = 95); CCAP > sytβ-RNAi (n = 120); CCAP > sytα and sytβ-RNAi (n = 131); CCAP > syt1-RNAi (n = 24). EW, Fully expanded wing; PEW, partial expanded wing; UEW, unexpanded wing.
Ablation or altered function of crustacean cardioactive peptide (CCAP)-expressing neurons impairs adult eclosion or subsequent success in wing expansion (Park et al., 2003). Subsets of CCAP-expressing neurons express DIMM and have been specifically implicated in wing expansion and postecdysial cuticular tanning (Peabody et al., 2008). We used the same transgenic RNAi for syt-α and syt-β and the selective CCAP-GAL4 driver to knock down each gene alone and/or together. Control flies showed normal levels of wing expansion (n = 127 flies). More than 92.5% (111 of 120) of syt-β RNAi flies failed to properly expand their wings. However, only 17% (16 of 95) of sytα RNAi flies displayed this same unexpanded wing phenotype. When syt-α and syt-β RNAi constructs were knocked down together (CCAP-GA4 > UAS-syt-α RNAi, UAS-syt-β RNAi), all 131 flies failed to expand their wings (Fig. 2B). Finally, we observed that knocking down syt-1 in CCAP neurons had little effect on wing expansion rates (8.4% of 24 flies). These results indicate that both SYT-α and SYT-β have fundamental roles in NE cell physiology, although these roles may differ according to cell type. To investigate the functional role of SYT-α specifically, here we focused on its subcellular localization. As a putative LDCV component, we compared its position with LDCV cargo following in vitro coexpression with DIMM.
SYT-α localizes with LDCV neuropeptide cargo in DIMM-expressing cultured cells
Many SYT proteins are associated with secretory vesicles and help to regulate activity-dependent exocytosis. If SYT-α or SYT-β is indeed associated with NE cells, their subcellular distributions should be correlated with markers of bioactive neuropeptides within LDCVs. We previously demonstrated that ectopic DIMM expression within non-NE neurons (photoreceptors) promotes generation of functional LDCVs in vivo: ones that permit accumulation and complete processing of a transgenic peptide precursor (Hamanaka et al., 2010). To examine this issue at the light microscope level, we studied a Drosophila neuronal cell line (BG3-c2 cell, derived from the larval CNS), which normally express very low levels of dimm, all syt isoforms, the neuropeptide Pdf, as well as most other neuropeptides (Schaaf et al., 2009). As expected, we found that accumulation of a PDF-GFP fusion within these cells is markedly elevated by dimm cotransfection (Fig. 3A vs E), as is the accumulation of a SYT-α-mCherry fusion (Fig. 3B vs F). Without dimm cotransfection, some BG3 cells produce low levels of PDF-GFP and SYT-α-mCherry expression: these fluorescent puncta can extend throughout the cell but are few in number and small on average. In contrast, dimm cotransfection produced many more, larger puncta that in total produced significantly more fluorescent intensities (Fig. 3H). We also confirmed that the fluorescent puncta are DIMM-dependent by using dimm dsRNA (data not shown). Importantly, the subcellular patterns of PDF-GFP and SYT-α-mCherry puncta were highly congruent (Fig. 3I–K). Additionally, we asked whether the fluorescent puncta reflect localization of neuropeptide precursor or intermediates with ER of Golgi, due to the fragmentation of these organelles in Drosophila (Kondylis and Rabouille, 2009). The cis-Golgi marker, dGM130, the peripheral Golgi marker, Golgin-like protein, Lava Lamp (LVA), and the endoplasmic reticulum exit site marker, SEC16 all displayed little overlap with the peptide fluorescent punta (Fig. 3L,M).
Figure 3.

SYT-α puncta colocalize with neuropeptide-GFP puncta in cultured neurons. The cotransfection of dimm, PDF-GFP, and SYT-α-mCherry in the Drosophila neuronal BG3–c2 cell line. There is enhanced accumulation and colocalization of neuropeptide and SYT-α protein, dependent on DIMM expression. A–C, Representative levels of PDF-GFP (green) and SYT-α-mCherry (magenta) fusions without DIMM. D–G, The same but now with dimm cotransfection. H, Quantitative measurement of fluorescence intensity within the selected area indicates significant increases with DIMM cotransfection. **p < 0.01. I–K, Higher magnification of processes from a dimm-transfected cell showing correspondence of the two fluorescent signals indicating a subcellular association between puncta containing neuropeptide cargo and SYT-α (arrow). L–N, Representative labelings of PDF-GFP (green) and the intracellular markers (magenta), including two Golgi markers (dGM130 and lava lamp) (L, M) and the ER exit site marker (Sec16, N).
We further investigated the ultrastructural localization of Syt-α as a putative LDCV membrane protein along with the neuropeptide cargo PDF by using super-resolution fluorescence microscopy. These imaging approaches include methods that fall into two broad categories: those such as stimulated emission depletion (STED) (Klar and Hell, 1999) and structured illumination microscopy (SIM) (Gustafsson, 2005) achieve subdiffraction limit resolution imaging by engineering the pattern of excitation light, as well as sparse single molecule localization based methods of (fluorescence) photoactivable localization microscopy (F-PALM) (Betzig et al., 2006; Hess et al., 2006) and stochastic optical reconstruction microscopy (STORM) (Rust et al., 2006). Here we used STORM/PALM as well as SIM to resolve the spatial arrangement of Syt-α and the neuropeptide cargo, PDF. We transiently cotransfected BG3 cells with DIMM along with Syt-α fused with a MYC tag (Syt-α-MYC) and the PDF neuropeptide precursor fused to the photoconvertible fluorescent protein mEOS2 (McKinney et al., 2009). Conventional fluorescence images showed a colocalization pattern in BG3 cells, similar to that observed in Figure 3, indicating that the choice of fusion protein did not affect the localization pattern (data not shown). STORM imaging of Syt-α-MYC along with PDF-mEOS2 showed that the peptide cargo PDF-mEOS2 and Syt-α-MYC were distributed throughout the distal processes of BG3 cells and were tightly correlated with each other (Fig. 4A–D). Interestingly, in a majority of clusters, the Syt-α-MYC localizations were either partially overlapping or polarized with respect to the PDF-mEOS2 localizations. However, the distribution of the two molecules is directional but not unidirectional for each punctum. We quantified the relative distribution of localizations by digitally isolating PDF-mEOS2-Syt-α-MYC clusters followed by rotating the clusters in 3D and aligning them by their centroid positions. A histogram constructed by aligning, overlaying multiple clusters, and projecting their localizations from 50 clusters derived from six independent BG3 cells reveals a considerable overlap with a slight shift between the two localization patterns. Furthermore, we used SIM, an independent super-resolution method, to confirm the coassociation between PDF and Syt-α. In these experiments, we coexpressed the fluorescent proteins Syt-α-mCherry and PDF-EGFP in BG3-c2 cells (along with DIMM). SIM images show colocalized puncta corresponding to PDF and Syt-α fusion proteins throughout BG3 cell processes and a polarized arrangement of Syt-α around individual PDF puncta (Fig. 4E,F). The observations made with SIM were produced without the use of antibodies to detect either the SYT-α or the cargo. They nevertheless independently suggest a polarized aspect to the coassociation of these two proteins. Together, these data strongly indicate that Syt-α is coassociated with LDCV cargo in putative LDCVs, when these are coexpressed as transgenes along with DIMM within the neuronal BG3-c2 cell line.
Figure 4.
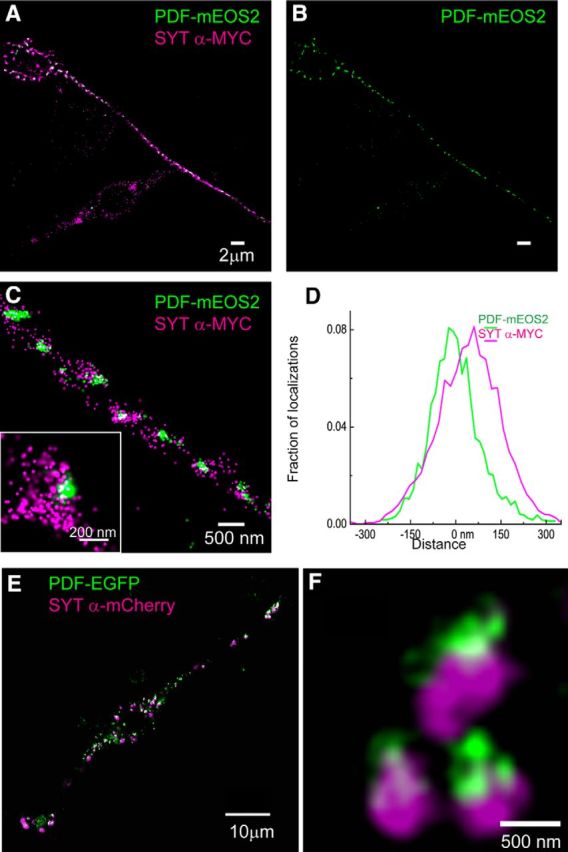
Coassociation of SYT-α and neuropeptide cargo with LDCV-like vesicles in cultured Drosophila neurons. STORM/PALM image of a Drosophila neuronal BG3–c2 cell transiently transfected with plasmids encoding DIMM (data not shown), PDF-mEOS2, and SYT-α-MYC (A–C). Punctate clusters of PDF-mEOS2 are distributed throughout the neuronal process (B), where they appear highly coassociated with SYT-a-MYC localizations. Zoomed-in images indicate a structure with LDCV-like dimensions, wherein SYT-α-MYC localizations decorate PDF-mEOS2 clusters (C). Frequently, SYT-α-MYC localizations appear polarized adjacent to those of PDF-mEOS2 (C, inset). A projection histogram of these localizations obtained from ∼50 aligned clusters. D, Quantitative analysis represents this polarized localization pattern. SIM images of a BG3–c2 cell coexpressing DIMM, PDF-eGFP, and SYT-α-mCherry indicate a similar polarized association of SYT-α-MYC adjacent to cargo (E, F).
Loss of DIMM reduces steady-state SYT-α and SYT-β protein levels
Based on the foregoing observations, we next asked whether DIMM affects the in vivo expression of SYT-α and SYT-β. dimm mutants display mild-to-dramatic reductions in the levels of neuropeptides and neuropeptide processing enzymes (Hewes et al., 2003; Allan et al., 2005). The losses are found only in NE cells that are normally DIMM-expressing; there are many peptidergic neurons in the Drosophila CNS that do not contain appreciable DIMM, and these do not show such phenotypes (Hewes et al., 2003; Park et al., 2008a; Luo et al., 2013). Severe dimm mutants die primarily at embryonic and early larval stages (Hewes et al., 2003). We therefore focused attention on young larvae. In the CNS of control second instar larvae, we detected both SYT-α and SYT-β proteins, although the number of antibody-positive cells was considerably reduced from that found in later larval stages of the same control genotype (Fig. 5A,C). In severe dimm hypomorphs (Rev4/Rev8), these SYT-α and SYT-β signals were clearly reduced (Fig. 5B,D). However, the (DIMM−)-Kenyon Cells displayed normal SYT-α and SYT-β immunostaining in the dimm mutant background (data not shown). These observations suggest that SYT-α and SYT-β exhibit cell-autonomous DIMM regulation (compare Hewes et al., 2003). In summary, we demonstrated a regulatory interaction between dimm and the two NE-specific SYTs: the loss of DIMM caused a reduction in expression of both SYT-α and SYT-β within cells that normally express DIMM.
Figure 5.

SYT-α and SYT-β protein levels are reduced in dimm mutants. SYT-α-like immunoreactivity (A, B) and SYT-β-like immunoreactivity (C, D) are severely diminished in the larval CNS of dimm mutants. A, C, Wild-type. B, D, Mutant (Rev4/Rev8). Green represents SYT-α and SYT-β. Magenta represents DIMM. N ≥ 12.
Misexpression of DIMM elevates SYT-α and SYT-β protein accumulations
Next, we turned to a gain-of-function model: we asked whether misexpressed DIMM increases SYT-α or SYT-β levels, in vivo as we showed it could do in vitro (in BG3 cells; Fig. 3). We used the ap-GAL4 driver and focused on the four cell Tv cluster of third instar larvae, of which two are normally DIMM+ peptidergic neurons, whereas the other two neurons are DIMM− (Benveniste et al., 1998; Park et al., 2004; Allan et al., 2005). Within that four cell cluster, the two peptidergic neurons are called Tv (dFMRFa+) and Tvb (NPLP1+) (Baumgardt et al., 2009). The DIMM− neurons are called Tva and Tvc. These four cells are also called Ap1, Ap2, Ap3, and Ap4 (Baumgardt et al., 2009). Consistent with their peptidergic NE character, both Tv and Tvb are SYT-α+ and SYT-β+; Tv-cell staining (distinguished from Tvb by weaker ap-gal4 activity; compare Park et al., 2004) was typically stronger than Tvb staining for both SYT isoforms. Notably, Tva and Tvc were not positive for either SYT antibody (Fig. 6A,C). However, when DIMM was misexpressed by the ap-gal4 driver (ap>dimm) throughout all four Tv cluster neurons, all four neurons now displayed clear SYT-α and SYT-β immunosignals (Fig. 6B,D). These results indicate that DIMM is sufficient to elevate levels of endogenous SYT-α and SYT-β proteins. We reasoned that, because DIMM is a potent transcription factor (Park et al., 2008b), DIMM could directly transactivate syt-α and/or syt-β, and so we next tested that hypothesis.
Figure 6.
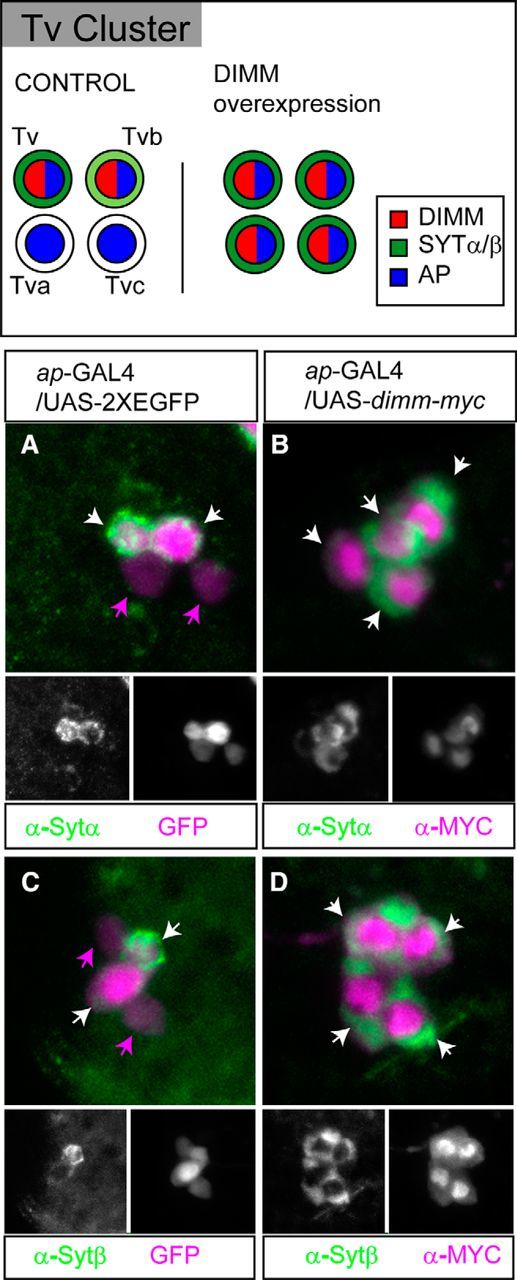
Misexpression of DIMM produces ectopic SYT expression. Top, Schematized drawing of the Tv cluster in the larval CNS. Bottom, Ectopic expression of DIMM within Tv cells produces ectopic SYT expression. ap-gal4 is expressed throughout the four cell Tv cluster, but only two Tv cells are SYT-α-positive (A) and SYT-β-positive (C). Overexpression of DIMM in all four Tv neurons generates ectopic expression of SYT-α (B) and SYT-β (D). A, C, ap-gal4/UAS-2XEGFP. B, D, ap-GAL4/UAS-dimm-myc. Arrow indicates SYT-α/β-positive cells.
DIMM does not directly activate the syt-α or syt-β genes
In a previous report, we used in vitro and in vivo analyses to demonstrate that DIMM transactivates Phm and other gene targets directly, via multiple E boxes in their first introns (Park et al., 2008b, 2011). To test the hypothesis that syt-α and syt-β genes are likewise direct DIMM targets, we first identified 10 putative DIMM binding sites (E box sequences CATATG or CAGCTG) (compare Park et al., 2008b) near and within the gene regions (Fig. 7, top). Second, we tested eight syt-α::luciferase fusion constructs following transfection of mammalian hEK cells, with or without dimm cotransfection. We chose potential regulatory fragments that spanned the entire upstream and first intronic regions; however, we found no evidence that DIMM could transactivate syt-α genomic fragments in this system (Fig. 7). Independently, we found by in vivo CHIP-chIP analysis that DIMM is not resident at either the syt-α or syt-β genomic locus in adult heads (Hadžić et al., manuscript submitted). These observations suggest that the elevation of endogenous SYT-α and SYT-β proteins in vivo by ectopic DIMM could reflect a post-transcriptional mechanism.
Figure 7.

DIMM-dependent transactivation of putative syta regulatory fragments. In vitro transactivation assay presents a systematic analysis of eight fragments derived from the syta genomic locus fused to luciferase. PHM is a fragment of the first intron of the Phm gene, which is an established DIMM target. Top, Map of the genomic fragments within the syta locus. Small black boxes represent positions of E-boxes (putative DIMM binding sites) that have the dinucleotide GC or TA in the middle two positions.
DIMM generates a permissive environment that permits accumulation of SYT-α and SYT-β proteins
The data just presented suggest that DIMM does not directly target the genes encoding the two NE-specific SYT isoforms. Yet, our data also suggest a clear indication of DIMM's control over their protein levels (Figs. 2, 3, 5, and 6). Indeed, there is clear precedent for DIMM control of key peptidergic LDCV cargo (neuropeptide protein levels) by a post-transcriptional mechanism (Hewes et al., 2003, Hamanaka et al., 2010). Hence, to evaluate whether DIMM likewise affects SYT-α protein accumulation post-transcriptionally, we generated a UAS-syt-α-mCherry fusion transgenic line. First, we asked whether the accumulation of transgenic SYT-α protein is dependent on the expression of DIMM in non-DIMM (nonpeptidergic) neurons. In the larval ventral nerve cord, the ap-positive dAP cells normally express NPLP1 peptide and are also DIMM+; ap+ vAP cells are normally nonpeptidergic and DIMM− (Park et al., 2004) (Fig. 8A,B). When ap-GAL4 drove only UAS-syt-α-mCherry, we detected mCherry fluorescence in the peptidergic dAP neuron, but not in the nonpeptidergic vAP neuron (Fig. 8C). However, comisexpression of dimm and syt-α-mCherry transgenes (ap-GAL4 > UAS-dimm, UAS-syt-α-mCherry) produced increased accumulation of SYT-α-mCherry throughout many regions of the CNS, specifically including the vAP cells (Fig. 8E).
Figure 8.
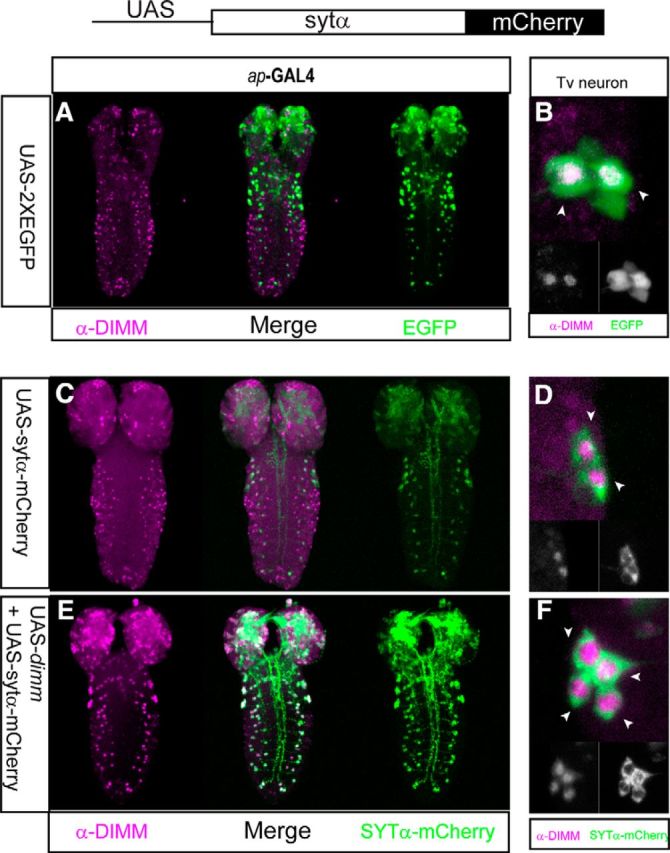
SYT-α protein accumulates by a post-transcriptional mechanism. A, B, The expression pattern of the ap-gal4 driver detected by a GFP reporter. A, Some GFP-positive cells express DIMM in the larval CNS. B, In the Tv cluster, only two cells are DIMM-positive. C, D, Misexpression of SYT-α-mCherry driven by ap-gal4. C, SYT-α protein is only detected in a limited number of cells, probably the two peptidergic ones. D, In the four cell-Tv cluster, misexpressed SYT-α is only detected in two cells (arrowheads), which are also DIMM-positive. E, F, Comisexpression of SYT-α-mCherry and DIMM is driven by ap-gal4. E, SYT-α protein was detectable throughout all levels of the CNS (compare the green channels in C vs E), matching the pattern of DIMM. F, Within the Tv cluster, all four cells now accumulate SYT-α protein.
Likewise, when the transgenic syt-α-mCherry transgene was driven by ap-gal4 driver throughout the Tv cluster, SYT-α-mCherry protein was only detected in two of the four neurons, similar to endogenous SYT-α (Fig. 8B; see also Fig. 6, drawing). Presumably, these two neurons are the peptidergic Tv and Tvb neurons, both of which are normally both DIMM+ and SYT-α+ (Fig. 8D). Importantly, comisexpression of DIMM along with SYT-α-mCherry (ap-GAL4 > UAS-dimm, UAS-syt-α-mCherry) led to accumulation of SYT-α-mCherry within all four cells of the cluster (Fig. 8F). We also asked whether SYT-α could mediate DIMM's ability to promote neuropeptide (cargo) accumulation (compare Hamanaka et al., 2010). Indeed, overexpressing SYT-α in neurons that do not normally express DIMM did not permit accumulation of ectopic neuropeptide (data not shown). Next, we repeated this experiment but now in a severe dimm mutant background (Rev8/R6). In this dimm mutant background, transgenic SYT-α proteins were barely detected in Tv neurons (neurons identified by GFP expression; Fig. 9A–D). However, the comisexpression of DIMM and SYT-α in the same dimm mutant background (ap-GAL4 > UAS-dimm, UAS-syt-α-mCherry) increased the intensity of SYT-α-mCherry fluorescence dramatically (Fig. 9E–G). Combined, these results strongly suggest that DIMM can significantly contribute to accumulation of SYT-α transgenic protein in vivo by a post-transcriptional mechanism.
Figure 9.
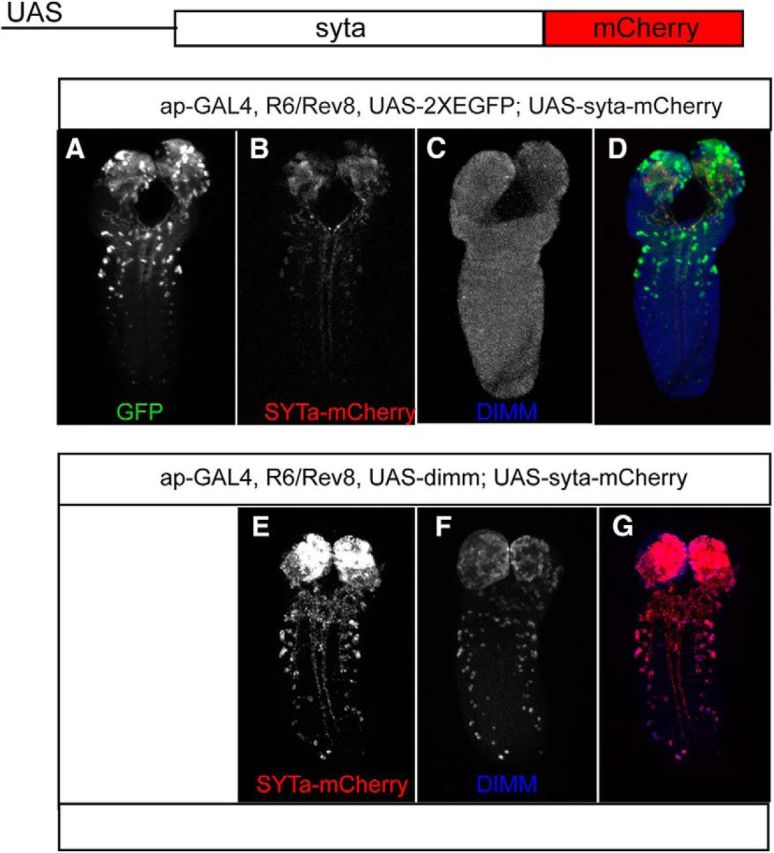
Post-translational mechanisms in a dimm LOF state. A–D, In a severe dimm mutant background (R6/rev8), UAS-syt-a-mCherry expression driven by ap-GAL4 displays minimal accumulation. E–G, However, DIMM co-misexpression (ap > UAS-syt-a-mCherry, UAS-dimm) rescues SYT-α accumulation.
Of seven syt genes, only syt-α and syt-β are responsive to DIMM
To broadly define potential regulation of SYT isoforms by DIMM, we performed qRT-PCR to ask which, if any, of the seven reported syt RNAs (Adolfsen et al., 2004) are responsive to dimm manipulation in vivo. We first misexpressed DIMM using the pan-neuronal elav-GAL4 driver at 18°C and collected mRNA from 24 to 32 h embryos. We compared results with those obtained from comparably treated samples of the GAL4-only, single transgenic stock, designated as control. DIMM overexpression significantly increased mRNA levels of syt-α and syt-β, 3.5-fold and 1.7-fold, respectively (Fig. 10, closed bar), but the five other syt RNAs were not responsive to this treatment. Next, we analyzed the same RNA set in a dimm loss-of-function condition: we prepared mRNA of second instar larvae that were trans-heterozygous for deletions (Rev8/Rev4) and that produce a severe dimm phenotype (compare Hewes et al., 2003). Surprisingly, there was no change in levels of any syt RNAs (Fig. 10, open bar). In summary, two of seven SYT isoforms RNAs, syt-α and syt-β, which are normally enriched in NE cell types, are responsive to a gain of function for DIMM, but not to its loss of function.
Figure 10.

DIMM levels selectively affect sytα and sytβ mRNAs. Levels of seven Drosophila syt mRNAs measured by quantitative real-time PCR in dimm-overexpressing embryos (elavG > dimm) versus in control embryos (elavG > +, closed bar) (n = 4) (ANOVA, p < 0.0001) and in dimm mutant larvae (dimmRev8/Rev4) versus control larvae (dimmRev4/+, open bar) (n = 2) (ANOVA, p < 0.270). The rp49 gene was used for normalization. The one-way ANOVA followed by Tukey's multiple-comparison test was used for statistical analysis. *p < 0.01.
Discussion
The LDCV is the critical subcellular organelle for peptidergic neurotransmission: to understand peptide cell biology in detail, it is critical to provide an in-depth understanding of how LDCVs are made, trafficked, and released. In pursuit of this goal, we have studied the two “peptidergic-specific” Drosophila synaptotagmin isoforms: syt-α (CG5559) and syt-β (CG3020). Our results provide a foundation for future genetic and cell biological studies of LDCV synthesis and regulation.
NE-specific SYT proteins are LDCV components and have a role in NE physiology
The subcellular distribution of SYT-α in DIMM-transformed Drosophila neurons in vitro displayed extensive overlap with puncta of a fluorescently labeled neuropeptide. The major protein components of LDCV have been identified in a variety of systems. In this study, we used super-resolution microscopy to reveal that SYT-α is strongly associated with LDCV peptide cargo in Drosophila neuronal cell culture and is detected unevenly at LDCVs with two different super-resolution microsopy. That result is novel in that it identifies SYT-α as an integral membrane protein of the LDCV and supports the hypothesis that it may serve as a calcium sensor for LDCV exocytosis. Different SYT isoforms have been associated with diverse LDCVs in a variety of different cell types, both functionally (Fukuda et al., 2004; Gustavsson et al., 2008; Schonn et al., 2008) and by expression (e.g., Zhang et al., 2011). Future studies on the functional roles of different SYT isoforms in LDCV will benefit greatly from the genetic repertoire available for Drosophila studies. These experiments are further significant because they indicate that the nanoscale arrangement of numerous LDCV proteins and their structure–function relationships could be dissected in subsequent studies using super-resolution light microscopy.
We tested the effects of knocking down syt-α and syt-β RNA levels specifically in two small peptidergic NE populations: the peripheral ETH-secreting Inka cells and the CCAP-secreting cells of the CNS. Peptides released from either population normally trigger and/or shape characteristic innate patterns of behavior, ecdysis and wing-spreading, respectively. The loss of either peptide signal, and also the loss of the peptide-secreting cell population, produces severe disruptions in the regulated behaviors (Park et al., 2002; Park et al., 2003). syt-α and syt-β RNAi produced strong phenotypes in Inka cells, which mimicked the genetic loss of the Inka bioactive peptide (ETH/PETH) and the effects of Inka cell ablation (Park et al., 2002). Because Inka cells were present and appeared healthy and because we used a conditional RNAi transgene design, we presume the Inka cell phenotypes were due to altered function of mature Inka cells. Hence, we conclude that SYT-α and SYT-β play critical roles in the physiology of Inka cells, presumably by contributing to the episodic release of the ETH and PETH peptides to drive ecdysial behaviors (Fig. 2).
Notably, the RNAi construct directed to Inka cells for syt-α was as effective in creating lethality as was the one for syt-β. Adolfsen et al. (2004) reported that embryonic Inka cells contain syt-β but not syt-α. To explain our results, we speculate that the α isoform may appear later in postembryonic stages of Inka cell function and hence be vulnerable to RNAi-mediated attack during larval stages. Alternatively, the syt-α RNAi construct may have produced a nonspecific effect on syt-β. We consider the possibility of nonspecific RNAi effects to be remote because we did not observe them in parallel experiments in the case of CCAP neurons. RNAi for syt-β, but not for syt-α, show the clear phenocopy of the CCAP loss of function syndrome, affecting a fixed wing spreading routine (Park et al., 2003). On this basis, we argue that the different syt RNAi constructs displayed overall specificity of effects. Together, the genetic results support the hypothesis that the SYT-α and SYT-β isoforms play critical roles for neuropeptide release in DIMM+ NE cells, most likely acting as calcium sensors.
SYT-α and SYT-β proteins are highly enriched in DIMM+ NE cells and regulated by DIMM
Although expression of SYT-α and SYT-β proteins was not exclusively associated with DIMM, their strongest expression sites are well-known peripheral and central DIMM+ NE cells. These observations strongly support the prediction by Adolfsen et al. (2004) that SYT-α and SYT-β would be closely aligned with peptidergic cell physiology. Based on this conclusion, the regulation of SYT-α and SYT-β expression levels and their precise subcellular localizations become significant issues. dimm loss of function alleles produced marked declines in SYT-α and SYT-β expression. Additionally, DIMM contributed to endogenous and transgenic SYT protein stability. DIMM therefore appears both necessary and sufficient to control SYT-α and -β expression. Overexpressing dimm upregulated syt-α and syt-β RNAs selectively among seven syt genes tested (Fig. 6); paradoxically, however, syt-α and syt-β RNA levels did not decline in a loss-of-dimm background. We propose that dimm contributes to syt RNA stability, but that it is not normally an important activator of syt-α or syt-β gene expression (other regulators must control their transcription).
These general effects mirror previous findings on how DIMM controls levels of secretory peptides, such as PDF. In that case, DIMM indirectly dictated peptide accumulation in DIMM neurons, without directly activating neuropeptide gene expression (Hewes et al., 2003; Hamanaka et al., 2010). In the present example, we found that accumulation of ectopic SYT-α and SYT-β transgenic proteins is largely dependent on whether or not DIMM is also misexpressed. Likewise, average Drosophila neurons cannot accumulate neuropeptides ectopically, suggesting that only peptidergic neurons have an enhanced ability to accumulate and/or release neuropeptides compared with neurons that primarily release classical neurotransmitters (Helfrich-Förster et al., 2000; Rao et al., 2001). At the molecular level, DIMM cannot transactivate syt-α promoter fragments in an in vitro assay, in which regulation of true DIMM direct targets is readily measured (Park et al., 2008a, 2011). Also, in vivo ChIP-chip genomic surveys indicate that DIMM protein is not normally resident at either the syt-α or syt-β loci (Hadžić et al., manuscript submitted). Thus, both cellular and molecular analyses suggest that DIMM critically regulates syt-α or syt-β levels post-transcriptionally, not transcriptionally.
Which mammalian SYT isoforms are orthologous to SYT-α and SYT-β?
There are at least 17 synaptotagmin isoforms in mammals: as yet, none appears to be a clear molecular ortholog of SYT-α and SYT-β, although SYT-IX and SYT-XVII are the most closely related. In mammals, several SYT isoforms (especially synaptotagmins 1, 4, 7, 9, and 10) are implicated in regulating exocytosis of peptide-containing LDCVs (Fukuda et al., 2004; Gustavsson et al., 2008; Schonn et al., 2008). The detailed roles of SYT isoforms in regulating the trafficking and exocytosis of peptidergic LDCVs appear complex. Zhang et al. (2011) reported that different SYT isoforms are associated with different size DCVs. SYT-1 regulates an early phase of calcium-dependent exocytosis from chromaffin cells vesicles (Wegrzyn et al., 2007), whereas SYT-7 regulates a second, slower one (Schonn et al., 2008). Likewise, SYT-4, SYT-7, SYT-9, SYT-10, and SYT-17 have all been implicated by expression and/or by functional studies to be associated with release of bioactive peptides (Lee et al., 2001; Iezzi et al., 2005; Gustavsson et al., 2008; Cao et al., 2011; Zhang et al., 2011). Further analysis of these issues in a genetic model system, such as Drosophila, will help clarify some of these essential contributions.
DIMM regulates the molecular composition of LDCV directly and indirectly
Our results suggest a model whereby DIMM organizes the regulated secretory pathway in NE cells by both direct and indirect mechanisms. Direct mechanisms refer to DIMM's transcriptional activation of a defined set of target genes. Using several independent methods, we have already identified six direct targets (Park et al., 2011), and these include neuropeptide biosynthetic enzymes, putative transporters, and chromatin remodeling factors. There are many additional direct DIMM targets that are identified by genome-wide methods, such as ChiP-CHIP (Hadzic et al., manuscript submitted). A significant outcome of these studies when summed is that DIMM does not target genes that encode neuropeptides or peptide hormones.
Indirect DIMM mechanisms could influence protein sorting by directing certain specific seed proteins (e.g., PHM and/ or Cytb-561) to exceed threshold concentrations. Seed proteins could permit accumulation and stabilization of “indirect targets” (e.g., SYT-α and SYT-β) that are regulated by transcription factors other than DIMM. Importantly, the possibility that interactions between proteins in the LDCV membrane and those in the LDCV lumen may contribute to protein sorting has already been proposed (Arvan and Castle, 1998). Thus, direct DIMM targets may represent rate-limiting components that can affect the protein composition of LDCV. Regardless of the precise molecular mechanism, we have shown that this general explanation holds for SYT-α and SYT-β, and we therefore propose that a significant fraction of LDCV-associated components may be likewise dependent indirectly on DIMM actions. Cell-specific neuronal gene expression is most often ascribed to direct gene control by specific transcription factors (e.g., Hendricks et al., 1999; Liu et al., 2003; Flames and Hobert, 2009; Kratsios et al., 2011). We now extend that general hypothesis to include what we term “indirect transcription factor effects” as major contributors to cellular differentiation. Thus, patterns of cell-specific protein expression reflect in part underlying indirect support by critical transcription factors.
Footnotes
This work was supported by a P30 Neuroscience Core Grant NS057105 to Washington University and the National Institutes of Health Grant R21MH-099798 to A.D. and Grant R01NS-21749 to P.H.T. The funding institutes had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Weihua Li and Jennifer Trigg for technical support; Tarik Hadzic for sharing unpublished data; the Bloomington Stock Center (Bloomington, IN), Vienna Drosophila RNAi Center (Vienna, Austria), and Dale Dorsett (St. Louis University) for the BG3-c2 cell line; Yoonseong Park and Mike Adams (University of California at Riverside, Riverside, CA) for Eth-geneswitch-GAL4; Loren Looger (Howard Hughes Medical Institute, Janelia Farms, Ashburn, VA) for the mEOS2 plasmid; Troy Littleton, Aaron DiAntonio, and Sungsu Kim for generously providing antibodies; Mike Nonet and Jim Skeath for reading an earlier draft of the manuscript; and members of our laboratories for helpful commentary.
The authors declare no competing financial interests.
References
- Adolfsen B, Saraswati S, Yoshihara M, Littleton JT. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J Cell Biol. 2004;166:249–260. doi: 10.1083/jcb.200312054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan DW, Park D, St Pierre SE, Taghert PH, Thor S. Regulators acting in combinatorial codes also act independently in single differentiating neurons. Neuron. 2005;45:689–700. doi: 10.1016/j.neuron.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PLoS One. 2011;6:e22678. doi: 10.1371/journal.pone.0022678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvan P, Castle D. Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem J. 1998;332:593–610. doi: 10.1042/bj3320593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Blosser TR, Zhuang X. Short-range spectroscopic ruler based on a single-molecule optical switch. Phys Rev Lett. 2005;94:108101. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgardt M, Karlsson D, Terriente J, Díaz-Benjumea FJ, Thor S. Neuronal subtype specification within a lineage by opposing temporal feed-forward loops. Cell. 2009;139:969–982. doi: 10.1016/j.cell.2009.10.032. [DOI] [PubMed] [Google Scholar]
- Benveniste RJ, Thor S, Thomas JB, Taghert PH. Cell type-specific regulation of the Drosophila FMRF-NH2 neuropeptide gene by Apterous, a LIM homeodomain transcription factor. Development. 1998;125:4757–4765. doi: 10.1242/dev.125.23.4757. [DOI] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewer J, Reynolds S. Neuropeptide control of molting in insects. In: Pfaff DW, Arnold AP, Fahrbach SE, Etgen AM, Rubin RT, editors. Hormones, brain and behavior. San Diego: Academic; 2002. pp. 1–92. [Google Scholar]
- Fabre JH. The insect world of Jean Henri Fabre. Boston: Beacon; 1917. [Google Scholar]
- Flames N, Hobert O. Gene regulatory logic of dopamine neuron differentiation. Nature. 2009;458:885–889. doi: 10.1038/nature07929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Satoh M, Saegusa C, Yamamoto A. Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+ -dependent exocytosis in PC12 cells. J Biol Chem. 2004;279:52677–52684. doi: 10.1074/jbc.M409241200. [DOI] [PubMed] [Google Scholar]
- Gauthier SA, Hewes RS. Transcriptional regulation of neuropeptide and peptide hormone expression by the Drosophila dimmed and cryptocephal genes. J Exp Biol. 2006;209:1803–1815. doi: 10.1242/jeb.02202. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, Li C, Radda GK, Südhof TC, Han W. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci U S A. 2008;105:3992–3997. doi: 10.1073/pnas.0711700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka Y, Park D, Yin P, Annangudi SP, Edwards TN, Sweedler J, Meinertzhagen IA, Taghert PH. Transcriptional orchestration of the regulated secretory pathway in neurons by the bHLH protein DIMM. Curr Biol. 2010;20:9–18. doi: 10.1016/j.cub.2009.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich-Förster C, Täuber M, Park JH, Mühlig-Versen M, Schneuwly S, Hofbauer A. Ectopic expression of the neuropeptide pigment-dispersing factor alters behavioral rhythms in Drosophila melanogaster. J Neurosci. 2000;20:3339–3353. doi: 10.1523/JNEUROSCI.20-09-03339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks T, Francis N, Fyodorov D, Deneris ES. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewes RS, Park D, Gauthier SA, Schaefer AM, Taghert PH. The bHLH protein Dimmed controls neuroendocrine cell differentiation in Drosophila. Development. 2003;130:1771–1781. doi: 10.1242/dev.00404. [DOI] [PubMed] [Google Scholar]
- Iezzi M, Eliasson L, Fukuda M, Wollheim CB. Adenovirus-mediated silencing of synaptotagmin 9 inhibits Ca2+-dependent insulin secretion in islets. FEBS Lett. 2005;579:5241–5246. doi: 10.1016/j.febslet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- Ivan V, de Voer G, Xanthakis D, Spoorendonk KM, Kondylis V, Rabouille C. Drosophila Sec16 mediates the biogenesis of tER sites upstream of Sar1 through an arginine-rich motif. Mol Biol Cell. 2008;19:4352–4365. doi: 10.1091/mbc.E08-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johard HA, Enell LE, Gustafsson E, Trifilieff P, Veenstra JA, Nässel DR. Intrinsic neurons of Drosophila mushroom bodies express short neuropeptide F: relations to extrinsic neurons expressing different neurotransmitters. J Comp Neurol. 2008;507:1479–1496. doi: 10.1002/cne.21636. [DOI] [PubMed] [Google Scholar]
- Kingan TG, Cardullo RA, Adams ME. Signal transduction in eclosion hormone-induced secretion of ecdysis-triggering hormone. J Biol Chem. 2001;276:25136–25142. doi: 10.1074/jbc.M102421200. [DOI] [PubMed] [Google Scholar]
- Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Optics Lett. 1999;24:954–956. doi: 10.1364/OL.24.000954. [DOI] [PubMed] [Google Scholar]
- Kondylis V, Rabouille C. The Golgi apparatus: lessons from Drosophila. FEBS Lett. 2009;583:3827–3838. doi: 10.1016/j.febslet.2009.09.048. [DOI] [PubMed] [Google Scholar]
- Kratsios P, Stolfi A, Levine M, Hobert O. Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene. Nat Neurosci. 2011;15:205–214. doi: 10.1038/nn.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Choi SH, Shin SL, Chin H, Kwon OJ. Distribution of B/K protein in rat brain. Cell Tissue Res. 2001;303:47–56. doi: 10.1007/s004410000221. [DOI] [PubMed] [Google Scholar]
- Lin Y, Han M, Shimada B, Wang L, Gibler TM, Amarakone A, Awad TA, Stormo GD, Van Gelder RN, Taghert PH. Influence of the period-dependent circadian clock on diurnal, circadian, and aperiodic gene expression in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2002;99:9562–9567. doi: 10.1073/pnas.132269699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Goshu E, Wells A, Fan CM. Identification of the downstream targets of SIM1 and ARNT2, a pair of transcription factors essential for neuroendocrine cell differentiation. J Biol Chem. 2003;278:44857–44867. doi: 10.1074/jbc.M304489200. [DOI] [PubMed] [Google Scholar]
- Luo J, Liu Y, Nässel DR. Insulin/IGF-regulated size scaling of neuroendocrine cells expressing the bHLH transcription factor Dimmed in Drosophila. PLoS Genet. 2013;9:e1004052. doi: 10.1371/journal.pgen.1004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, Kits KS. Calcium channels and the release of large dense-core vesicles from neuroendocrine cells: spatial organization and functional coupling. Prog Neurobiol. 2000;62:427–441. doi: 10.1016/S0301-0082(00)00003-4. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Taghert PH. Scaling factors: transcription factors regulating subcellular domains. Bioessays. 2012;34:10–16. doi: 10.1002/bies.201100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien MA, Taghert PH. A peritracheal neuropeptide system in insects: release of myomodulin-like peptides at ecdysis. J Exp Biol. 1998;201:193–209. doi: 10.1242/jeb.201.2.193. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Taghert PH. Peptidergic neurosecretory cells in insects: organization and control by the bHLH protein DIMMED. Gen Comp Endocrinol. 2009;162:2–7. doi: 10.1016/j.ygcen.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Park Y, Filippov V, Gill SS, Adams ME. Deletion of the ecdysis-triggering hormone gene leads to lethal ecdysis deficiency. Development. 2002;129:493–503. doi: 10.1242/dev.129.2.493. [DOI] [PubMed] [Google Scholar]
- Park JH, Schroeder AJ, Helfrich-Förster C, Jackson FR, Ewer J. Targeted ablation of CCAP neuropeptide-containing neurons of Drosophila causes specific defects in execution and circadian timing of ecdysis behavior. Development. 2003;130:2645–2656. doi: 10.1242/dev.00503. [DOI] [PubMed] [Google Scholar]
- Park D, Han M, Kim YC, Han KA, Taghert PH. Ap-let neurons: a peptidergic circuit potentially controlling ecdysial behavior in Drosophila. Dev Biol. 2004;269:95–108. doi: 10.1016/j.ydbio.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Park D, Veenstra JA, Park JH, Taghert PH. Mapping peptidergic cells in Drosophila: where DIMM fits in. PLoS One. 2008a;3:e1896. doi: 10.1371/journal.pone.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Shafer OT, Shepherd SP, Suh H, Trigg JS, Taghert PH. The Drosophila bHLH protein Dimmed directly activates Phm, a gene encoding a neuropeptide amidating enzyme. Mol Cell Biol. 2008b;28:410–421. doi: 10.1128/MCB.01104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Hadžić T, Yin P, Rusch J, Abruzzi K, Rosbash M, Skeath JB, Panda S, Sweedler JV, Taghert PH. Molecular organization of Drosophila neuroendocrine cells by Dimmed. Curr Biol. 2011;21:1515–1524. doi: 10.1016/j.cub.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peabody NC, Diao F, Luan H, Wang H, Dewey EM, Honegger HW, White BH. Bursicon functions within the Drosophila CNS to modulate wing expansion behavior, hormone secretion, and cell death. J Neurosci. 2008;28:14379–14391. doi: 10.1523/JNEUROSCI.2842-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Lang C, Levitan ES, Deitcher DL. Visualization of neuropeptide expression, transport, and exocytosis in Drosophila melanogaster. J Neurobiol. 2001;49:159–172. doi: 10.1002/neu.1072. [DOI] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CA, Misulovin Z, Sahota G, Siddiqui AM, Schwartz YB, Kahn TG, Pirrotta V, Gause M, Dorsett D. Regulation of the Drosophila Enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS One. 2009;4:e6202. doi: 10.1371/journal.pone.0006202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JC, Field C, Ventura R, Royou A, Sullivan W. Lava lamp, a novel peripheral golgi protein, is required for Drosophila melanogaster cellularization. J Cell Biol. 2000;151:905–918. doi: 10.1083/jcb.151.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Neurotransmitter release. Handb Exp Pharmacol. 2008;184:1–21. doi: 10.1007/978-3-540-74805-2_1. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Südhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J, Lee J, Neveu JM, Lane WS, Hook V. Proteomics of neuroendocrine secretory vesicles reveal distinct functional systems for biosynthesis and exocytosis of peptide hormones and neurotransmitters. J Proteome Res. 2007;6:1652–1665. doi: 10.1021/pr060503p. [DOI] [PubMed] [Google Scholar]
- Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, Guariglia S, Meng Q, Cai D. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. 2011;69:523–535. doi: 10.1016/j.neuron.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wu Y, Wang Z, Dunning FM, Rehfuss J, Ramanan D, Chapman ER, Jackson MB. Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell. 2011;22:2324–2336. doi: 10.1091/mbc.E11-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitnan D, Kingan TG, Hermesman JL, Adams ME. Identification of ecdysis-triggering hormone from an epitracheal endocrine system. Science. 1996;271:88–91. doi: 10.1126/science.271.5245.88. [DOI] [PubMed] [Google Scholar]