

Abstract
Gangliosides are major cell-surface determinants on all vertebrate neurons. Human congenital disorders of ganglioside biosynthesis invariably result in intellectual disability and are often associated with intractable seizures. To probe the mechanisms of ganglioside functions, affinity-captured ganglioside-binding proteins from rat cerebellar granule neurons were identified by quantitative proteomic mass spectrometry. Of the six proteins that bound selectively to the major brain ganglioside GT1b (GT1b:GM1 > 4; p < 10−4), three regulate neurotransmitter receptor trafficking: Thorase (ATPase family AAA domain-containing protein 1), soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (γ-SNAP), and the transmembrane protein Nicalin. Thorase facilitates endocytosis of GluR2 subunit-containing AMPA-type glutamate receptors (AMPARs) in an ATPase-dependent manner; its deletion in mice results in learning and memory deficits (J. Zhang et al., 2011b). GluR2-containing AMPARs did not bind GT1b, but bound specifically to another ganglioside, GM1. Addition of noncleavable ATP (ATPγS) significantly disrupted ganglioside binding, whereas it enhanced AMPAR association with Thorase, NSF, and Nicalin. Mutant mice lacking GT1b expressed markedly higher brain Thorase, whereas Thorase-null mice expressed higher GT1b. Treatment of cultured hippocampal neurons with sialidase, which cleaves GT1b (and other sialoglycans), resulted in a significant reduction in the size of surface GluR2 puncta. These data support a model in which GM1-bound GluR2-containing AMPARs are functionally segregated from GT1b-bound AMPAR-trafficking complexes. Release of ganglioside binding may enhance GluR2-containing AMPAR association with its trafficking complexes, increasing endocytosis. Disrupting ganglioside biosynthesis may result in reduced synaptic expression of GluR2-contianing AMPARs resulting in intellectual deficits and seizure susceptibility in mice and humans.
Keywords: AMPA receptor, ganglioside, GluR2, N-ethylmaleimide-sensitive factor, Nicalin, Thorase
Introduction
Glutamate is the major excitatory neurotransmitter in the mammalian brain, and AMPA ionotropic glutamate receptors (AMPARs) are the predominant mediator of excitatory neurotransmission (Shepherd and Huganir, 2007; Hanley, 2014). Quantitative and qualitative regulation of AMPAR expression at postsynaptic membranes controls excitatory neurotransmission and is a key mechanism in synaptic plasticity, learning, and memory (Kessels and Malinow, 2009; Henley et al., 2011; Opazo and Choquet, 2011; Anggono and Huganir, 2012). The number and subunit composition of AMPARs at synapses is tightly regulated by activity-dependent control of the rates of receptor exocytosis, capture at the postsynaptic membrane, and endocytosis.
Gangliosides—sialic acid-bearing glycosphingolipids— are prominent cell-surface molecular determinants on all vertebrate neurons, with their lipid moieties embedded in the extracellular leaflet of the plasma membrane and their glycans extending outward (DeMarco and Woods, 2009; Sonnino and Prinetti, 2013; Schnaar et al., 2014). Gangliosides regulate cell signaling by associating laterally with membrane proteins on their own plasma membranes and mediate cell—cell recognition by interacting with complementary binding proteins on apposing cells (Hakomori, 2002; Lopez and Schnaar, 2009). Four ganglioside structures (Fig. 1)—GM1, GD1a, GD1b, and GT1b—comprise the vast majority of gangliosides in the brains of all mammals. Their biosynthesis occurs stepwise by enzymatic addition of saccharides to the growing glycan chain (Sturgill et al., 2012), and mutations in ganglioside biosynthetic enzymes result in human congenital disorders that are phenocopied in mouse genetic models (Schnaar et al., 2014). Notably, human mutations in ganglioside biosynthetic enzymes invariably result in intellectual disability and are often accompanied by seizure susceptibility (Simpson et al., 2004; Boukhris et al., 2013; Fragaki et al., 2013; Harlalka et al., 2013; Boccuto et al., 2014). The mechanisms by which changes in ganglioside biosynthesis result in learning disabilities and dysregulation of excitatory neurotransmission have not been established.
Figure 1.

Top, middle, Structures of gangliosides GM1 and GT1b. Biosynthetic genes discussed in the text are shown in red, and the double bonds that were cleaved by ozonolysis for ganglioside covalent immobilization onto magnetic beads by an arrowhead. Bottom, Stepwise biosynthesis of major brain gangliosides, with genes responsible boxed.
Using ganglioside affinity capture combined with mass spectrometric proteomics, we identified six proteins that bind selectively to the major brain ganglioside GT1b, half of which are implicated in neurotransmitter receptor trafficking. One of these, Thorase (J. Zhang et al., 2011b), is notable for ATPase-dependent regulation of the endocytosis of GluR2 AMPAR subunits, which are key to AMPAR ion channel excitability (Isaac et al., 2007; Liu and Savtchouk, 2012). Remarkably, although GluR2 did not bind GT1b, it bound specifically to another major brain ganglioside, GM1. Addition of noncleavable ATP disrupted ganglioside binding and enhanced association of GluR2 with receptor-trafficking proteins. The data support a model in which gangliosides sequester GluR2-containing AMPA receptors and GluR2 receptor-trafficking complexes in a reversible ATPase-dependent manner that may regulate GluR2-containing AMPAR endocytosis, AMPAR ion channel permeability, synaptic plasticity, learning, and memory.
Materials and Methods
Cerebellar granule cells.
Primary neurons were isolated following papain dissociation of cerebella dissected from 5-to 6-d-old Sprague Dawley rats of either sex as described previously (Mehta et al., 2007) and resuspended at 106 cells/ml in Neurobasal medium (Life Technologies) supplemented with 25 mm KCl, 2 mm glutamine, NS21 neuron survival factors (Chen et al., 2008), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were plated on poly-d-lysine-coated 35 mm or 60 mm cell culture dishes at a density of ∼250,000/cm2 and cultured for 12 d at 37°C in a 5% CO2 atmosphere with half-medium changes every 2–3 d.
Ganglioside-derivatized beads.
Gangliosides GM1 and GT1b purified from bovine brain (Matreya) were oxidized at the unique sphingosine double bond (Fig. 1) to introduce an aldehyde group that allowed stable covalent attachment to amine-derivatized beads. GT1b and GM1 were treated with freshly generated ozone in methanol essentially as described previously (Schnaar et al., 2002). Oxidation to the corresponding ganglioside aldehyde was confirmed by thin-layer chromatography and MALDI-TOF mass spectrometry (data not shown). Ganglioside aldehyde (25 nmol) was mixed with 15 mg of Dynabeads M-270 amine magnetic beads (Life Technologies) and 11 μmol (0.7 mg) of NaBH3CN in 270 μl of phosphate buffer (150 mm NaCl and 10 mm sodium phosphate, pH 7.2). After 24 h at 42°C an additional 11 μmol of freshly prepared NaBH3CN in 70 μl of phosphate buffer was added. After 48 h (total reaction time) the derivatized beads were collected magnetically, washed with phosphate buffer, and stored at 0°C until use. To confirm derivatization, aliquots of the washed beads were immunostained with specific anti-GT1b and anti-GM1 antibodies (Schnaar et al., 2002; data not shown).
Surface-protein labeling, solubilization, and enrichment.
After 12 d in culture, cerebellar granule neurons had elaborated a dense network of axons (data not shown). All protein-labeling, collection, and affinity-capture steps were performed at 0−4°C. Cultures were washed with Dulbecco's PBS then treated with 1 mm EZ-Link Sulfo-NHS-SS-Biotin, a cleavable cell-surface protein biotinylation reagent (Thermo Scientific), in PBS (3 ml per 60 mm dish). After 30 min at 4°C with gentle shaking to label cell-surface proteins, excess biotinylation reagent was quenched by addition of 50 mm Tris-HCl, pH 7.2, and the cells were collected by scraping from the culture dish into PBS. The suspension was centrifuged (500 × g, 3 min), the pellet resuspended in HEPES-saline (150 mm NaCl, and 20 mm HEPES, pH 7.2, containing a 1:1000 dilution of protease inhibitor mixture; Sigma-Aldrich P8340) and cells disrupted using a Potter–Elvehjem homogenizer (10 strokes). The resulting homogenate was centrifuged at 800 × g for 5 min to remove nuclei, and the supernatant was collected and centrifuged at 100,000 × g for 1 h to collect membranes. The membrane pellet was solubilized by the addition of solubilization buffer (HEPES-saline containing 1% Triton X-100). After gentle mixing for 1 h, the mixture was centrifuged (20,000 × g, 20 min) and the supernatant was collected and added to NeutrAvidin agarose beads (Thermo Scientific) to capture the solubilized surface-biotinylated proteins. After 60 min with gentle shaking, the beads were washed with solubilization buffer and then proteins eluted in solubilization buffer containing 10 mm DTT. The beads were removed and the eluate was diluted 1:4 with fresh solubilization buffer to reduce the DTT concentration to 2 mm.
In some experiments (as indicated) cell-surface labeling and all subsequent steps including ganglioside affinity capture were performed in solutions containing 1 mm ATPγS (EMD Millipore) and 3 mm MgCl2.
Ganglioside affinity capture.
Dynabeads covalently derivatized with GT1b or GM1 were preblocked by incubation in solubilization buffer for 1 h, washed, and then added to equivalent aliquots of the diluted eluate from NeutrAvidin beads containing surface-enriched proteins from cerebellar granule neurons. Protein–ganglioside binding was allowed to proceed for 16 h with gentle shaking. Beads with bound proteins were magnetically captured, washed with solubilization buffer and then with HEPES-saline (without detergent), and then bound proteins were eluted by heating (70°C, 10 min) in a solution of NuPAGE LDS Sample Buffer (Life Technologies) containing 50 mm DTT. To remove salts and detergents from eluted proteins for relative quantitative mass spectrometry (iTRAQ), an aliquot of the eluate was mixed with 8 volumes of 10% trichloroacetic acid (TCA) in acetone at −20°C for 2–4 h. The pellet was collected by centrifugation (14,000 × g, 10 min, 4°C), washed with −20°C acetone, recentrifuged, and the pellet briefly dried and stored at −80°C before analysis.
Relative quantitative proteomics (iTRAQ).
Samples were subjected to reduction using tris(2-carboxyethyl)phosphine and alkylation using methyl methanethiosulfonate and subsequently proteolyzed with trypsin (Promega) as described previously (Shevchenko et al., 1996). The resulting peptides eluted from GT1b and GM1 from four replicate experiments (eight samples) were individually tagged using 8plex iTRAQ reagents according to the manufacturer's protocol (AB Sciex), then the tagged peptides were combined and fractionated with strong cation exchange chromatography using a polysulfoethyl A SCX column. Peptides in the SCX column fractions were desalted using Oasis HLB reverse phase resin (Waters), dried under vacuum, resuspended in 0.1% formic acid, and loaded on a 75 μm × 2.5 cm nanobore column packed with Magic AQ C18, 5 μm diameter 100 Å pore-size beads (Microm Bioresources), then fractionated using a 2–50% acetonitrile and 0.1% formic acid gradient for 110 min at 300 nl/min. Eluting peptides were sprayed through a 10 μm emitter tip into an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific) interfaced with a 2D nanoLC system (Eksigent). Peptide sequences were identified from the Rat Refseq 40 database using Mascot v2.1 software (www.matrixscience.com) through Proteome Discoverer v1.3 (Thermo Scientific).
iTRAQ relative quantitative proteomics was performed for quadruplicate biological replicates of GT1b- and GM1-eluted proteins in a single 8plex mass spectrometry experiment. A total of 446 proteins was identified and quantified at a 1% false discovery rate threshold. For statistical consideration of relative abundance (Herbrich et al., 2013) these were considered as 446 independent multiple comparisons. The significance of the differences in relative quantitative protein binding (GT1b vs GM1) for the four biological replicates was calculated for each of the 446 proteins, and only those with differential binding that matched or exceeded the Bonferroni correction for multiple comparisons (p < 0.05/446 = 0.00011) were considered further.
Protein immunoblots.
Proteins eluted from GT1b- or GM1-derivatized beads were diluted in NuPAGE LDS buffer containing 50 mm DTT, electrophoretically resolved on 4–12% Bis-Tris NuPAGE gels, and then transferred to PVDF membranes using an iBlot system (reagents and equipment from Life Technologies). Membranes were blocked with PBS containing 5% nonfat dry milk and 0.1% Tween 20 and then subjected to immunoblot analysis using antibodies against Thorase (1:1000; Antibodies Incorporated), GluR2 (1:1000; Antibodies Incorporated), N-ethylmaleimide-sensitive fusion protein (1:2000; Abcam), glutamate receptor interacting protein (GRIP; 1:1000; BD Biosciences), or Nicalin (1:1000; Millipore). The blots were incubated overnight at 4°C, then washed and probed with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and visualized using enhanced chemiluminescence (GE Healthcare). Band intensities were quantified using ImageJ (NIH).
Comparative immunoblot analyses were performed using brain homogenates from wild-type mice, Thorase-deficient mice (Atad1-null; J. Zhang et al., 2011b), and mice with disrupted ganglioside biosynthetic genes (St3gal2-null, St3gal3-null, and St3gal2/3-double-null mice; Sturgill et al., 2012) and B4galnt1-null mice (Pan et al., 2005). Mice of either sex were anesthetized with isoflurane and their brains were rapidly removed and homogenized in ice-cold CelLytic MT tissue lysis reagent (Sigma-Aldrich) in the presence of protease inhibitors. Protein concentration was determined (BCA Protein Assay; Thermo Scientific) and equal amounts (15 μg) were resolved and transferred to PVDF as above. Immunoblot analyses were performed using the above antibodies and antibody to GAPDH (1:2000; Sigma) as loading control.
Coimmunoprecipitation and GST pull-down assays.
Coimmunoprecipitations (co-IPs) of Thorase, NSF, GluR2, and Nicalin were performed using lysates of whole mouse brain. Freshly isolated whole brain from wild-type and Thorase-deficient mice of either sex (J. Zhang et al., 2011b) were pulverized under dry ice and homogenized in Buffer A (50 mm HEPES, pH 7.5, 150 mm NaCl, 2.5 mm MgCl2, 1 mm DTT, and 5% glycerol) containing protease inhibitors with or without 2 mm ADP, ATP, or ATPγS. Triton X-100 was added to a final concentration of 1% followed by rotation for 2 h at 4°C. Extracts were centrifuged at 15,000 × g for 45 min and the supernatant was incubated for 3 h at 4°C with Protein G beads (Pierce) prebound with Thorase, NSF, GluR2, or Nicalin antibodies. The beads were washed three times with Buffer A with or without 1 mm ADP, ATP, or ATPγS (as appropriate) and bound proteins were eluted from beads using SDS-PAGE Laemmli buffer (1×; Sigma) with DTT. The eluted proteins were resolved using 10% SDS-PAGE and transferred to PVDF. Immunoblotting analyses were performed using antibodies to Thorase, NSF, GluR2, Nicalin, and GRIP1 as described above.
To determine Thorase domains that are important for GluR2, GRIP1, NSF, and Nicalin interactions, Thorase N-termini (first 100 residues) and C-termini (last 20 residues) deletion mutants were cloned into pGEX-6P-1 (Addgene) and transformed into BL21 Escherichia coli (Invitrogen). After induction with isopropyl thiogalactoside, bacterial pellets were lysed using a microfluidizer (Microfluidics) in Buffer A with protease inhibitors and centrifuged at 15,000 × g for 30 min. The supernatant was incubated with glutathione beads (Pierce) for 2 h and then washed four times with Buffer A. The glutathione beads conjugated to GST fusion Thorase proteins were incubated with freshly prepared whole-brain lysate for 2 h at 4°C in Buffer A with 2 mm ATPγS and then washed four times with Buffer A containing 1 mm ATPγS. Bound proteins were eluted from beads using Laemmli buffer (1×; Sigma) with DTT. The eluted proteins were resolved on 10% SDS-PAGE and transferred to PVDF. Immunoblotting analyses were performed using antibodies to Thorase, NSF, GluR2, Nicalin, and GRIP1 as described above.
To compare levels of GluR2, GRIP1, NSF, and Nicalin in Thorase-deficient brain, immunoblot analyses were performed using brain homogenates from wild-type mice and Thorase-deficient mice of either sex (J. Zhang et al., 2011b). Whole-brain lysates were prepared as described above. Protein concentrations were determined by BCA protein assay and 20 μg was resolved on 10% SDS-PAGE. Immunoblot analyses were performed using antibodies to Thorase, NSF, GluR2, Nicalin, GRIP1, and antibody to actin (Sigma) as control. Band intensities were quantified using ImageJ (NIH) and normalized to actin. The values obtained from ImageJ were further analyzed to determine significant differences using GraphPad Prism software.
Immunohistochemistry.
Male mice were anesthetized using Isoflurane inhalation, then perfused intracardially with PBS followed by neutralized 4% paraformaldehyde in PBS. Brains were embedded in paraffin and sectioned at 5 μm. For Thorase immunohistochemistry, antigens were retrieved by boiling the sections in 10 mm sodium citrate, pH 6.0; endogenous peroxidases were inactivated with 0.3% hydrogen peroxide; sections were blocked in 10% goat serum in PBS containing 0.1% Triton X-100; and then probed with antibodies for Thorase (1:200; Antibodies Incorporated). The sections were then incubated with biotin-conjugated secondary antibody (1:250) and avidin–biotin complex using a Vector ABC kit with diaminobenzidine substrate (Vector Laboratories). Slides were dehydrated and mounted using Kyrstalon mounting medium and analyzed using a Nikon Eclipse 90i microscope and NIS-Elements image analysis software.
Cell-surface GluR2 immunocytochemistry.
Hippocampal neurons were isolated from E18 Sprague Dawley rat pups of either sex and cultured on glass coverslips as described previously (Makuch et al., 2011). After 18 d in culture, cells were fixed in PBS containing 4% PFA and 4% sucrose for 4.5 min, which does not lead to cell permeabilization. As indicated, some cultures were treated with 20 mU/ml Vibrio cholerae sialidase (Moustafa et al., 2004) for 24 h or 48 h before fixation. For surface GluR2 detection, coverslips were incubated with an N-terminal mouse anti-GluR2 at 1:500 (15F; a kind gift from Dr. Eric Gouaux, Oregon Health Sciences University) in detergent-free GDB buffer (0.1% gelatin, 0.45 M NaCl, and 17 mm phosphate buffer, pH 7.4) for 2 h at room temperature. Coverslips were washed, then cells were permeabilized using 0.3% Triton X-100-containing GDB buffer and incubated with 1:250 rabbit anti-GluR2/3 antibody (JH4854; Hayashi et al., 2009). Coverslips were washed and incubated with fluorescent secondary antibodies (anti-mouse Alexa Fluor 546 and anti-rabbit Alexa Fluor 488; Life Technologies) for surface and total GluR2 receptors, respectively. Coverslips were washed, mounted on glass slides with Fluoromount-G (Southern Biotech), and imaged using a Zeiss LSM 510 Meta Confocal microscope.
Results
Ganglioside-binding proteins on cerebellar granule neurons
Using affinity capture, quantitative mass spectrometry (iTRAQ), and stringent statistical criteria, six surface-enriched proteins solubilized from primary cerebellar granule neurons were identified that bound selectively to the major brain ganglioside GT1b (Table 1). Half of these have been directly implicated in the control of neurotransmitter receptor trafficking. Thorase (ATPase family AAA domain-containing protein 1) is notable for regulating activity-dependent AMPAR trafficking and synaptic plasticity (Zhang et al., 2011b). Soluble NSF and its attachment proteins (SNAPs) ubiquitously function in vesicular trafficking (Söllner et al., 1993; Zhao et al., 2007). NSF binds directly and specifically to the AMPAR GluR2 subunit, regulating its insertion into membranes and stabilization at synapses (Nishimune et al., 1998; Song et al., 1998; Araki et al., 2010; Anggono and Huganir, 2012). Both NSF and Thorase are members of the AAA+ superfamily of ATPase molecular machines that regulate ATP-dependent multiprotein associations (Ogura and Wilkinson, 2001). Nicalin has not been extensively studied, but its homolog NRA-2 is implicated in regulation of neurotransmitter receptor trafficking in Caenorhabditis elegans (Haffner et al., 2004; Almedom et al., 2009). The screening data led to the preliminary hypothesis that GT1b regulates AMPAR trafficking via specific interactions with multiprotein complexes that include Thorase, NSF, and Nicalin.
Table 1.
GT1b-associated proteinsa
| Gene | Protein | Reference sequenceb | GT1b/GM1 binding ratio | p value |
|---|---|---|---|---|
| Napg | Gamma-soluble NSF attachment protein (γ-SNAP) | NP_001100854.1 | 7.3 | 1.2 × 10−8 |
| Hsd17b12 | Estradiol 17-β-dehydrogenase 12 | NP_114455.1 | 6.5 | 7.4 × 10−8 |
| Ncln | Nicalin precursor | NP_001014104.1 | 5.4 | 1.4 × 10−6 |
| Atad1 | Thorase: ATPase family AAA domain-containing protein 1 | NP_001030174.1 | 5.2 | 2.0 × 10−6 |
| Gnai2 | Guanine nucleotide-binding protein G(i) subunit α-2 | NP_112297.1 | 4.2 | 3.3 × 10−5 |
| Cct7 | T-complex protein 1 subunit η | NP_001100073.1 | 4.1 | 5.4 × 10−5 |
aCell surface-enriched detergent-solubilized proteins isolated from neonatal rat cerebellar granule neurons that bound to GT1b > GM1 as determined by iTRAQ quantitative mass spectrometry on quadruplicate biological replicates with statistical significance that exceeded the Bonferroni correction (p < 10−4).
bNCBI protein sequence identifier from the Rattus norvegicus protein database.
To validate and extend these findings, ganglioside affinity capture of cell surface-enriched proteins from cerebellar granule neurons was repeated in triplicate and the resulting captured proteins were probed by immunoblotting. Since both Thorase and NSF are AAA+ superfamily members susceptible to conformational changes upon binding of ATP (Moeller et al., 2012), ganglioside affinity capture was performed using lysates prepared in the presence or absence of the noncleavable ATP analog ATPγS, which was previously shown to stabilize Thorase-GluR2 binding (J. Zhang et al., 2011b). Both Thorase and NSF bound preferentially to GT1b via immunoblot (Fig. 2A–C), and binding was diminished or eliminated in the presence of ATPγS. Immunoblot detection of Nicalin in ganglioside eluates was too low for reliable quantification (data not shown). Unexpectedly, GluR2 was completely absent from GT1b-bead eluates but was readily detected in GM1-bead eluates (Figs. 2A,D). GluR2 binding to GM1-beads was also markedly diminished in the presence of ATPγS. Since the stable association of Thorase with GluR2 is enhanced by ATPγS (J. Zhang et al., 2011b) and association of these same proteins with GT1b and GM1, respectively, is reversed by ATPγS, we refined our hypothesis. Gangliosides and GluR2 receptors are both lipid raft components (Hou et al., 2008; Sonnino and Prinetti, 2013), and different gangliosides can segregate into different lipid rafts (Vyas et al ., 2001). We reasoned, therefore, that differential ganglioside association may physically and/or functionally segregate GluR2 from its receptor trafficking complexes, regulating GluR2 surface expression in an ATP-dependent manner (see model; Fig. 10). This hypothesis predicted that GluR2 would associate with Thorase, NSF, and Nicalin in a manner that would be enhanced by ATPγS. This was tested in brain extracts from wild-type and Thorase-deficient (Atad1-null) mice.
Figure 2.

Ganglioside-selective binding of Thorase and NSF to GT1b and GluR2 to GM1. A, Surface-enriched proteins extracted from cerebellar granule neurons were captured on GM1- or GT1b-derivatized beads, eluted under denaturing conditions, and subjected to SDS-PAGE separation and immunoblot analysis using antibodies to Thorase, NSF, or GluR2 as indicated. Preload (before ganglioside capture) control samples were analyzed using one seventh of the volume compared with GM1- and GT1b-bead eluates (indicated as GM1 and GT1b). Immunoblots are representative of triplicate determinations. B–D, Quantitative analyses of immunoblots of GM1- and GT1b-bead eluates (n = 3). For each protein, two-way ANOVA with Tukey post hoc analysis indicated that binding was ganglioside-specific, and that incubation with ATPγS significantly reduced binding to the dominant binding ganglioside (*p < 0.01; **p < 0.001).
Figure 10.

Hypothetical model. Ganglioside-regulated trafficking of GluR2-containing AMPARs. GluR2-containing AMPARs and an AMPAR trafficking complex containing Nicalin, Thorase, and NSF are proposed to be sequestered in distinct membrane rafts by association with gangliosides GM1 and GT1b, respectively. In an ATP-regulated process, the ganglioside associations are released, resulting in lateral migration, association of AMPA receptors with their trafficking complexes, and receptor endocytosis.
GluR2, Thorase, NSF, and Nicalin co-associate in an ATP-dependent manner
Wild-type (C57BL/6) mouse brains were lysed in detergent-containing buffer and the solubilized proteins were subjected to immuno-affinity capture using antibodies to Thorase, NSF, GluR2, and Nicalin (Fig. 3). Anti-Thorase IP captured NSF, GluR2, and Nicalin, as well as GRIP1. Co-IP of each protein was enhanced by ATPγS (Fig. 3A,B), whereas ATP reduced Thorase-GluR2 binding, consistent with ATP hydrolysis leading to disassembly of Thorase–AMPAR complexes (J. Zhang et al., 2011b). Likewise, Thorase, GluR2, Nicalin, and GRIP1 were coimmunoprecipitated with NSF in an ATPγS-enhanced manner (Fig. 3A,B). To test the role of Thorase in the multiprotein complex, GluR2 IP was performed using brain extracts from wild-type and Thorase-deficient mice (Fig. 3C,D). In wild-type extracts, Thorase, NSF, GRIP1, and Nicalin were coimmunoprecipitated with GluR2 in an ATPγS-dependent manner. In Thorase-deficient mice, co-IP of NSF and GRIP1 with GluR2 was sharply diminished compared with wild-type, whereas co-IP of Nicalin was less robustly affected. Consistent with these data, GluR2 was coimmunoprecipitated with Nicalin (Fig. 3C,E), and this association was diminished but still present in Thorase-deficient mice.
Figure 3.

Co-IP of Thorase, NSF, GluR2, GRIP1, and Nicalin. A, Immunoblots of Thorase and NSF IPs using wild-type whole-brain lysates in the presence of different adenine nucleotides. B, Densitometric quantification of triplicate experiments performed as in A. Asterisks denote statistical significance of pairwise comparisons between experiments performed in the presence of ATP and ATPγS. C, GluR2 IP comparing wild-type (WT) and Thorase knock-out (KO) whole-brain lysates in the presence of different adenine nucleotides. D, Densitometric quantification of triplicate experiments performed as in C in the presence of ATPγS. E, Nicalin IP comparing wild-type and Thorase knock-out whole-brain lysates in the presence of ATPγS. F, Densitometric quantification of triplicate experiments performed as in E. Asterisks in D and F denote statistical significance of pairwise comparisons between wild-type and Thorase knock-out mice. All values represent mean ± SEM (n = 3, *p < 0.05, **p < 0.001, one-way ANOVA, post-test: Tukey's multiple comparison).
NSF, Nicalin, GluR2, and GRIP1 in wild-type brain extracts were susceptible to pull down using an exogenously expressed Thorase-GST fusion protein in the presence of ATPγS (Fig. 4). Based on deletion constructs, the first 100 N-terminal amino acids of Thorase are required for optimal capture of each protein, whereas in the C-terminal 20 amino acids are less critical.
Figure 4.

GST-Thorase pull-down of GluR2, GRIP1, Nicalin, and NSF from whole mouse brain lysates. A, Whole-brain lysates were mixed with GST beads prebound to purified recombinant GST-tagged wild-type (WT), N-terminal truncated (N100), and C-terminal truncated (C20) Thorase in the presence of ATPγS. Input lanes are in the absence of lysate. The experiments were repeated three times with similar results. B, Optical densitometry quantification of pull down lanes from the blots in A with values representing the percent compared to Lysate lane, mean ± SEM (n = 3, **p < 0.001, *p < 0.05, n.s. p > 0.05, one-way ANOVA, post-test: Tukey's multiple comparison).
Since NSF, Nicalin, GRIP1, and GluR2 associate with Thorase in brain extracts, quantitative immunoblotting was used to determine whether the steady-state levels of any of these proteins were altered in Thorase-null mice (Fig. 5). Whereas NSF, Nicalin, and GRIP1 were unaltered, GluR2 was significantly increased in the Thorase mutant compared with wild-type.
Figure 5.

GluR2, GRIP1, NSF, and Nicalin in wild-type and Thorase-deficient (Atad1-null) mice. A, Western blots of total brain lysates obtained from wild-type (WT) and Thorase knock-out (KO) mice. B, Quantitative analyses of blots in A. The band intensity of each lane was normalized to the corresponding actin band intensity. The values representing the mean SEM (n = 3, *p < 0.05, n.s. p > 0.05, one-way ANOVA, post-test: Tukey's multiple comparison).
Mice with altered brain ganglioside biosynthesis express higher levels of Thorase, and Thorase-null mice express higher levels of GT1b
To test whether genetic alterations in ganglioside biosynthesis result in changes in the levels of GluR2 or GluR2-trafficking proteins, mouse genetic models with mutations in ganglioside glycosyltransferases were used (Fig. 1). Mice with a disrupted B4galnt1 gene lack all four of the major brain gangliosides; instead they express two shorter gangliosides, GM3 and GD3. Compared with wild-type mice, B4galnt1-null mice displayed a marked and significant increase in whole-brain Thorase (Fig. 6A), whereas GluR2 and Nicalin levels were unchanged. Notably, St3gal2/3 double-null mice, which have a block later in the ganglioside biosynthetic pathway, had a nearly identical ∼2-fold increase in Thorase (Fig. 6B). St3gal2/3 double-null mice lack nearly all GT1b and GD1a, but express increased GM1 and GD1b (Sturgill et al., 2012). These data imply that the lack of biosynthesis of GT1b and/or GD1a is directly or indirectly related to steady-state Thorase levels. Consistent with the immunoblot analyses, anti-Thorase immunohistochemical staining of wild-type and St3gal2/3 double-null mouse brains revealed higher levels of Thorase staining in the mutant (Fig. 7).
Figure 6.

Increased Thorase levels in ganglioside mutant mice. A, Triplicate wild-type and B4galnt1-null whole mouse brains were homogenized and equivalent aliquots subjected to SDS-PAGE separation and immunoblot analysis with antibodies to the indicated proteins. Quantification indicated that only Thorase was significantly different in the mutant mice (*p < 0.01, one-way ANOVA with Tukey post hoc analysis). B, Triplicate wild-type, St3gal2-null, St3gal3-null, and St3gal2/3 double-null whole mouse brains were homogenized and equivalent aliquots subjected to SDS-PAGE separation and immunoblot analysis with antibodies to the indicated proteins. Quantification indicated that only Thorase was significantly different, and only in the double-null mutant mice (*p < 0.02 vs wild-type, one-way ANOVA with Tukey post hoc analysis).
Figure 7.

Increased intensity of Thorase immunohistochemistry in St3gal2/3 double-null mice. Sagittal sections from 8-week-old wild-type mice (A, C) and St3gal2/3 double-null mice (B, D) were subjected to Thorase immunohistochemistry under identical conditions. Detailed images of hippocampus (A, B) and cerebellum (C, D) are shown. Scale bars: 200 μm.
The relative amount of the four major brain gangliosides is controlled within a narrow range animal-to-animal in wild-type mice, and this range is unaltered in Thorase heterozygote mice. In contrast, the relative amount of GT1b was significantly increased in Thorase-null mice, whereas of the relative amounts of GD1a and GM1 were significantly decreased (Fig. 8).
Figure 8.

Altered ganglioside levels in Thorase-deficient (Atad1-null) mice. Gangliosides were extracted from whole brains of wild-type (wt; n = 3), heterozygote (het; n = 5), and null (n = 3) mice and subjected to thin-layer chromatography separation, detected using a sialic acid-specific chemical stain and quantified by image analysis. Left, Thin layer chromatograph of representative samples of each genotype with migration positions marked based on bovine brain standards (stds). Double bands at each migration position are due to minor variations in ceramide structure. Right, Molar ratios of gangliosides based on densitometric quantification of all samples. Since stain intensity is proportional to sialic acid content, values are divided by the number of sialic acids per ganglioside for molar ratio comparisons. Two-way ANOVA with Tukey post hoc analysis indicated significant differences in ganglioside percentage composition (*p < 0.02; **p < 0.005).
Modulation of cell-surface sialoglycans alters GluR2 trafficking in cultured hippocampal neurons
Treatment of intact neurons with highly purified V. cholerae sialidase converts the major brain disialogangliosides and trisialogangliosides GD1a, GD1b, and GT1b to the monosialoganglioside GM1 (G. Zhang et al., 2011a; Mountney et al., 2013). To test whether gangliosides regulate the number of surface GluR2 receptors, well differentiated, rat embryonic hippocampal neurons in culture were treated with sialidase for 24 h or 48 h, fixed, and analyzed immunocytochemically for surface and total GluR2 receptors. Image analysis was used to quantify the size and intensity of >500 GluR2 puncta per image (on average) for 19–20 images per condition. After 24 h of sialidase treatment, GluR2 receptor clusters were significantly shifted to smaller sizes (Fig. 9B,D). The puncta were shifted slightly (but not significantly) toward lower intensity as well (Fig. 9D). The size distribution of total GluR2 puncta, detected at a separate epitope after permeabilization, was not significantly changed after 24 h of sialidase treatment, but total puncta intensities were significantly lower (Fig. 9E). These changes reverted to control levels after 48 h of continuous sialidase treatment. The reversion to control GluR2 levels was not due to loss of sialidase activity in culture; medium retrieved at the end of the 48 h treatment retained the same sialidase activity as initially added (data not shown).
Figure 9.
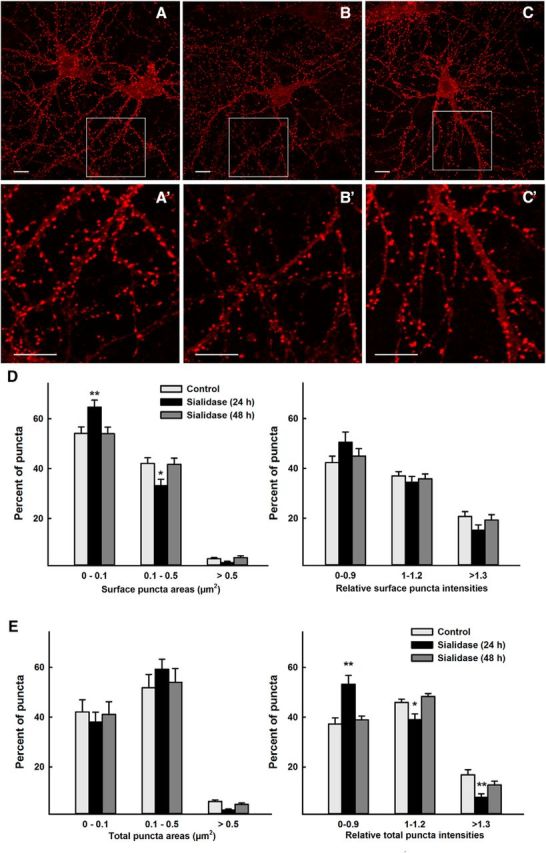
GluR2 puncta of control and sialidase-treated hippocampal neurons. Hippocampal neurons were incubated without sialidase or with 50 mU/ml sialidase for 24 h or 48 h before differential surface and total staining with anti-GluR2 antibody. Surface-stained GluR2 puncta on control (A), 24 h sialidase-treated (B), and 48 h sialidase-treated (C) hippocampal neurons are shown with enlarged areas (A′, B′, C′) from boxed regions in the corresponding image. Scale bars: 10 μm. D, Size (left) and relative intensities (right) of cell-surface GluR2 puncta. E, Size (left) and relative intensities (right) of total GluR2 puncta. Two-way ANOVA with Tukey post hoc analysis indicated that surface puncta were significantly smaller and total puncta less intense in 24 h sialidase-treated hippocampal neurons (*p < 0.05; **p < 0.01).
Consistent with a role for gangliosides in AMPAR trafficking, we conclude that altering ganglioside structures on hippocampal neurons alters trafficking of GluR2-containing AMPARs.
Discussion
Gangliosides are abundant cell-surface determinants on all mammalian nerve cells, and their structures are shared by all mammals (Schnaar et al., 2014). Genetic linkage analyses of human congenital disorders unexpectedly linked ganglioside biosynthesis to excitatory neurotransmission, learning, and memory. Congenital, refractory early onset seizures accompanied by profound psychomotor delay were traced to mutations in ST3GAL5 (Fig. 1), which codes for GM3 synthase, a sialyltransferase that acts early in the complex ganglioside biosynthetic pathway (Simpson et al., 2004; Fragaki et al., 2013; Boccuto et al., 2014). Hereditary spastic paraplegia associated with intellectual disability and occasional seizure susceptibility was traced to B4GALNT1, which codes for GM2/GD2 synthase, a subsequent step in ganglioside biosynthesis (Wilkinson et al., 2005; Boukhris et al., 2013; Harlalka et al., 2013). Our unbiased proteomic screen for ganglioside-interacting proteins unexpectedly revealed a molecular link between major brain gangliosides and AMPAR trafficking that may help explain the hyperexcitability and intellectual disability associated with human congenital disorders of ganglioside biosynthesis.
AMPARs are involved in seizure generation (Löscher and Schmidt, 2012; Rogawski, 2013) and AMPAR trafficking is a key component of learning and memory (Anggono and Huganir, 2012; Henley and Wilkinson, 2013; Hanley, 2014). AMPARs are tetrameric transmembrane proteins typically composed of a dimer of dimers constructed from four types of subunits, GluR1–GluR4 (Anggono and Huganir, 2012). The subunit composition of AMPAR tetramers defines their function, with a key being the abundant GluR2 subunit. Mature GluR2 subunits typically do not support calcium entry (Isaac et al., 2007). Since calcium-permeable AMPARs are of higher conductance (Liu and Savtchouk, 2012), selective reduction of GluR2-containing AMPARs increases excitability. For this reason, the trafficking and surface expression of GluR2 subunits, in particular, is highly regulated (Hanley, 2014). If the major brain gangliosides common to all mammals are required for optimum regulation of the trafficking of GluR2-containing AMPARs, it is reasonable to propose that the seizures and cognitive deficits associated with human congenital disorders of ganglioside biosynthesis are due to AMPAR dysregulation.
The major findings reported here are that GluR2-containing AMPARs bind selectively to ganglioside GM1, whereas AMPAR-trafficking complexes (ATCs) containing Thorase, γ-SNAP, NSF, and Nicalin bind selectively to ganglioside GT1b. Interestingly, these protein–ganglioside interactions are diminished in the presence of ATPγS (Fig. 2) whereas direct associations between GluR2-containing AMPARs and ATCs are stimulated by ATPγS (Fig. 3). These findings support a model in which GluR2-containing AMPARs and ATCs are sequestered from each other via their association with different gangliosides (GM1 and GT1b, respectively), but are in equilibrium with free-floating forms that interact leading to receptor endocytosis (Fig. 10). A prediction of this model is that altering gangliosides will result in increased GluR2-containing AMPAR endocytosis. This prediction is supported by the finding that treatment of hippocampal neurons with sialidase resulted in reduced surface expression of GluR2-containing AMPARs (Fig. 9). We interpret this result as indicating that sialidase-mediated loss of GT1b released ATCs into the free-floating pool where lateral diffusion increased interactions with AMPARs, decreasing the size of GluR2 puncta due to ATC-mediated AMPAR endocytosis at the periphery of the puncta.
Behavioral results from mouse strains blocked in brain ganglioside biosynthesis (Fig. 1) are consistent with this general model. Mice double null for St3gal5 and St8sia1, in which GM3 replaces the major brain gangliosides (GM1, GD1a, GD1b, and GT1b), are highly susceptible to early onset lethal audiogenic seizures (Kawai et al., 2001). In comparison, B4galnt1-null mice, in which gangliosides GM3 and GD3 replace the major brain gangliosides, have a milder phenotype marked by hyperactivity, late-onset audiogenic seizure susceptibility, and enhanced susceptibility to chemically induced seizures (Pan et al., 2005; Wu et al., 2005). Transgenic mice overexpressing the same enzyme (GM2/GD2 synthase, the B4galnt1 gene product) display a quantitative shift in brain gangliosides (less GT1b and GD1b, more GD1a and GM1) and impaired hippocampal long-term potentiation, a correlate to learning and memory involving changes in AMPAR trafficking (Ikarashi et al., 2011). Although there are few thorough cognitive studies on ganglioside-altered mice, B4galnt1 transgenics are learning impaired (Ikarashi et al., 2011), as are St3gal2/3 double-null mice, which lack GT1b and GD1a but overexpress GM1 and GD1b (Sturgill et al., 2012; unpublished data). The extent to which other ganglioside-altered mice share seizure susceptibility and/or learning and memory deficits is not currently known. The increase in brain Thorase upon genetic modulation of gangliosides, and the shift in brain gangliosides upon genetic loss of Thorase, are consistent with cross-regulation that may imply a functional relationship (Figs. 6–8). However, functional consequences due to these quantitative changes, and the mechanisms of cross-regulation, remain unknown.
Exogenously added gangliosides protect neurons in vitro from glutamate excitotoxicity by modulating calcium currents (de Erausquin et al., 1990; Costa et al., 1994). Whether this effect is due to changes in AMPAR trafficking (quantitative or qualitative) has not been reported. However, based on those prior studies ganglioside GM1 was administered intravenously or intramuscularly to ischemic stroke patients in over a dozen clinical trials, but failed to positively alter the overall clinical outcome (Candelise and Ciccone, 2002). A more detailed understanding of the structures and functions of gangliosides in regulating excitatory neurotransmission may provide additional insights for their potential clinical applications.
Since AMPARs are transmembrane proteins with large extracellular domains, and gangliosides are plasma membrane components with extracellularly oriented glycans, it is reasonable to propose that AMPARs directly associate laterally with GM1. A report of immunogold colocalization of GluR2 and GM1 in synaptosomes is consistent with this hypothesis (Cole et al., 2010). From a structural perspective, the AMPAR extends ∼120 Å outward from the plasma membrane, each subunit equally comprised of a membrane-proximal ligand-binding domain and a distal N-terminal domain (Sobolevsky et al., 2009). The oligosaccharide of GM1 extends ∼20 Å outward from the plasma membrane (Acquotti and Sonnino, 2000), providing ample lateral surface area for specific molecular interactions with the 60 Å ligand-binding domain of the AMPAR. Given the mild dissociation conditions we used for ganglioside capture, other adapter proteins that were not detected by mass spectrometry could also be involved. Since both gangliosides and AMPARs localize to lipid rafts (Hou et al., 2008; Sonnino and Prinetti, 2013), these small distinct lateral platforms within the plasma membrane may provide the opportunity for selective molecular clustering (Vyas et al., 2001).
Unlike AMPARs, Thorase and NSF are believed to be exclusively intracellular (Shepherd and Huganir, 2007; J. Zhang et al., 2011b). Since Nicalin (Haffner et al., 2004) is a single-pass membrane protein that associates with Thorase and NSF (Fig. 3), and binds to GT1b (Table 1), it may act as a transmembrane adapter, with the latter (intracellular) proteins remaining associated during capture. Since these three proteins also bind to GluR2 (Fig. 3), we propose that they may shuttle between GT1b-binding and GluR2-binding states (Fig. 10). In this model, the absence of GT1b, GM1, or both may result in altered association of ATCs and their target receptors, thereby shifting the quantitative and/or qualitative expression of AMPARs resulting in pathological changes in neuronal excitability and deficits in learning and memory. The discovery of a molecular link between gangliosides and AMPAR trafficking provides expanded opportunities to probe this molecular connection to human excitatory neurotransmission.
Footnotes
This work was supported by National Institutes of Health grants NS037096 (R.L.S.), AG029368 (V.L.D.), DA000266 (T.M.D. and V.L.D.), NS036715 (R.L.H.), and HHSN268201000032C (R.N.C.). T.M.D. is the Leonard and Madlyn Abramson Professor of Neurodegenerative Diseases at Johns Hopkins University. We thank Johns Hopkins colleagues Tatiana Boronina, Raghothama Chaerkady, Lauren Hitt, and Robert O'Meally (Mass Spectrometry and Proteomics Facility); Ingo Ruczinski (School of Public Health); and Christine Jelinek (Mid-Atlantic Mass Spectrometry Center) for invaluable help with proteomic analyses.
The authors declare no competing financial interests.
References
- Acquotti D, Sonnino S. Use of nuclear magnetic resonance spectroscopy in evaluation of ganglioside structure, conformation, and dynamics. Methods Enzymol. 2000;312:247–272. doi: 10.1016/S0076-6879(00)12914-3. [DOI] [PubMed] [Google Scholar]
- Almedom RB, Liewald JF, Hernando G, Schultheis C, Rayes D, Pan J, Schedletzky T, Hutter H, Bouzat C, Gottschalk A. An ER-resident membrane protein complex regulates nicotinic acetylcholine receptor subunit composition at the synapse. EMBO J. 2009;28:2636–2649. doi: 10.1038/emboj.2009.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Huganir RL. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol. 2012;22:461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Lin DT, Huganir RL. Plasma membrane insertion of the AMPA receptor GluA2 subunit is regulated by NSF binding and Q/R editing of the ion pore. Proc Natl Acad Sci U S A. 2010;107:11080–11085. doi: 10.1073/pnas.1006584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccuto L, Aoki K, Flanagan-Steet H, Chen CF, Fan X, Bartel F, Petukh M, Pittman A, Saul R, Chaubey A, Alexov E, Tiemeyer M, Steet R, Schwartz CE. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt and pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum Mol Genet. 2014;23:418–433. doi: 10.1093/hmg/ddt434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhris A, Schule R, Loureiro JL, Lourenço CM, Mundwiller E, Gonzalez MA, Charles P, Gauthier J, Rekik I, Acosta Lebrigio RF, Gaussen M, Speziani F, Ferbert A, Feki I, Caballero-Oteyza A, Dionne-Laporte A, Amri M, Noreau A, Forlani S, Cruz VT, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93:118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelise L, Ciccone A. Gangliosides for acute ischemic stroke. Stroke. 2002;33:2336. doi: 10.1161/01.STR.0000029272.13806.46. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AA, Dosemeci A, Reese TS. Co-segregation of AMPA receptors with G(M1) ganglioside in synaptosomal membrane subfractions. Biochem J. 2010;427:535–540. doi: 10.1042/BJ20091344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Armstrong DM, Guidotti A, Kharlamov A, Kiedrowski L, Manev H, Polo A, Wroblewski JT. Gangliosides in the protection against glutamate excitotoxicity. Prog Brain Res. 1994;101:357–373. doi: 10.1016/S0079-6123(08)61962-7. [DOI] [PubMed] [Google Scholar]
- de Erausquin GA, Manev H, Guidotti A, Costa E, Brooker G. Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proc Natl Acad Sci U S A. 1990;87:8017–8021. doi: 10.1073/pnas.87.20.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco ML, Woods RJ. Atomic-resolution conformational analysis of the GM3 ganglioside in a lipid bilayer and its implications for ganglioside-protein recognition at membrane surfaces. Glycobiology. 2009;19:344–355. doi: 10.1093/glycob/cwn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragaki K, Ait-El-Mkadem S, Chaussenot A, Gire C, Mengual R, Bonesso L, Bénéteau M, Ricci JE, Desquiret-Dumas V, Procaccio V, Rötig A, Paquis-Flucklinger V. Refractory epilepsy and mitochondrial dysfunction due to GM3 synthase deficiency. Eur J Hum Genet. 2013;21:528–534. doi: 10.1038/ejhg.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner C, Frauli M, Topp S, Irmler M, Hofmann K, Regula JT, Bally-Cuif L, Haass C. Nicalin and its binding partner Nomo are novel Nodal signaling antagonists. EMBO J. 2004;23:3041–3050. doi: 10.1038/sj.emboj.7600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakomori SI. The glycosynapse. Proc Natl Acad Sci U S A. 2002;99:225–232. doi: 10.1073/pnas.012540899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG. Subunit-specific trafficking mechanisms regulating the synaptic expression of Ca-permeable AMPA receptors. Semin Cell Dev Biol. 2014;27:14–22. doi: 10.1016/j.semcdb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Harlalka G, Lehman A, Chioza B, Baple EL, Maroofian R, Cross H, Sreekantan-Nair A, Priestman DA, Al-Turki S, McEntagart ME, Proukakis C, Royle L, Kozak RP, Bastaki L, Patton M, Wagner K, Coblentz R, Price J, Mezei M, Schlade-Bartusiak K, et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain. 2013;136:3618–3624. doi: 10.1093/brain/awt270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Thomas GM, Huganir RL. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron. 2009;64:213–226. doi: 10.1016/j.neuron.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci. 2013;15:11–27. doi: 10.31887/DCNS.2013.15.1/jhenley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Barker EA, Glebov OO. Routes, destinations and delays: recent advances in AMPA receptor trafficking. Trends Neurosci. 2011;34:258–268. doi: 10.1016/j.tins.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbrich SM, Cole RN, West KP, Jr, Schulze K, Yager JD, Groopman JD, Christian P, Wu L, O'Meally RN, May DH, McIntosh MW, Ruczinski I. Statistical inference from multiple iTRAQ experiments without using common reference standards. J Proteome Res. 2013;12:594–604. doi: 10.1021/pr300624g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Huang Y, Amato S, Snyder SH, Huganir RL, Man HY. Regulation of AMPA receptor localization in lipid rafts. Mol Cell Neurosci. 2008;38:213–223. doi: 10.1016/j.mcn.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikarashi K, Fujiwara H, Yamazaki Y, Goto J, Kaneko K, Kato H, Fujii S, Sasaki H, Fukumoto S, Furukawa K, Waki H, Furukawa K. Impaired hippocampal long-term potentiation and failure of learning in beta1,4-N-acetylgalactosaminyltransferase gene transgenic mice. Glycobiology. 2011;21:1373–1381. doi: 10.1093/glycob/cwr090. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kawai H, Allende ML, Wada R, Kono M, Sango K, Deng C, Miyakawa T, Crawley JN, Werth N, Bierfreund U, Sandhoff K, Proia RL. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J Biol Chem. 2001;276:6885–6888. doi: 10.1074/jbc.C000847200. [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Savtchouk I. Ca(2+) permeable AMPA receptors switch allegiances: mechanisms and consequences. J Physiol. 2012;590:13–20. doi: 10.1113/jphysiol.2011.213926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–557. doi: 10.1016/j.sbi.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. Perampanel–new promise for refractory epilepsy? Nat Rev Neurol. 2012;8:661–662. doi: 10.1038/nrneurol.2012.222. [DOI] [PubMed] [Google Scholar]
- Makuch L, Volk L, Anggono V, Johnson RC, Yu Y, Duning K, Kremerskothen J, Xia J, Takamiya K, Huganir RL. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71:1022–1029. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NR, Lopez PH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Zhao C, Fried MG, Wilson-Kubalek EM, Carragher B, Whiteheart SW. Nucleotide-dependent conformational changes in the N-Ethylmaleimide Sensitive Factor (NSF) and their potential role in SNARE complex disassembly. J Struct Biol. 2012;177:335–343. doi: 10.1016/j.jsb.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountney A, Zahner MR, Sturgill ER, Riley CJ, Aston JW, Oudega M, Schramm LP, Hurtado A, Schnaar RL. Sialidase, chondroitinase ABC, and combination therapy after spinal cord contusion injury. J Neurotrauma. 2013;30:181–190. doi: 10.1089/neu.2012.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa I, Connaris H, Taylor M, Zaitsev V, Wilson JC, Kiefel MJ, von Itzstein M, Taylor G. Sialic acid recognition by Vibrio cholerae neuraminidase. J Biol Chem. 2004;279:40819–40826. doi: 10.1074/jbc.M404965200. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM. NSF binding to GluR2 regulates synaptic transmission. Neuron. 1998;21:87–97. doi: 10.1016/S0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Pan B, Fromholt SE, Hess EJ, Crawford TO, Griffin JW, Sheikh KA, Schnaar RL. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Exp Neurol. 2005;195:208–217. doi: 10.1016/j.expneurol.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol Scand Suppl. 2013;197:9–18. doi: 10.1111/ane.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Fromholt SE, Gong Y, Vyas AA, Laroy W, Wayman DM, Heffer-Lauc M, Ito H, Ishida H, Kiso M, Griffin JW, Shiekh KA. Immunoglobulin G-class mouse monoclonal antibodies to major brain gangliosides. Anal Biochem. 2002;302:276–284. doi: 10.1006/abio.2001.5540. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease and regeneration. Physiol Rev. 2014;94:461–518. doi: 10.1152/physrev.00033.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Simpson MA, Cross H, Gurtz K, Priestman DA, Neville DC, Reinkensmeier G, Wang H, Wiznitzer M, Proukakis C, Verganelaki A, Pryde A, Patton MA, Dwek RA, Butters TD, Platt FM, Crosby AH. Infantile onset symptomatic epilepsy syndrome caused by homozygous loss of function mutations in GM3 synthase. Nat Genet. 2004;36:1225–1229. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir RL. Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron. 1998;21:393–400. doi: 10.1016/S0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- Sonnino S, Prinetti A. Membrane domains and the “lipid raft” concept. Curr Med Chem. 2013;20:4–21. [PubMed] [Google Scholar]
- Sturgill ER, Aoki K, Lopez PH, Colacurcio D, Vajn K, Lorenzini I, Majiæ S, Yang WH, Heffer M, Tiemeyer M, Marth JD, Schnaar RL. Biosynthesis of the major brain gangliosides GD1a and GT1b. Glycobiology. 2012;22:1289–1301. doi: 10.1093/glycob/cws103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas KA, Patel HV, Vyas AA, Schnaar RL. Segregation of gangliosides GM1 and GD3 on cell membranes, isolated membrane rafts, and defined supported lipid monolayers. Biol Chem. 2001;382:241–250. doi: 10.1515/BC.2001.031. [DOI] [PubMed] [Google Scholar]
- Wilkinson PA, Simpson MA, Bastaki L, Patel H, Reed JA, Kalidas K, Samilchuk E, Khan R, Warner TT, Crosby AH. A new locus for autosomal recessive complicated hereditary spastic paraplegia (SPG26) maps to chromosome 12p11.1–12q14. J Med Genet. 2005;42:80–82. doi: 10.1136/jmg.2004.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Wang J, Wang Y, Xie X, Meyenhofer MF, Ledeen RW. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci. 2005;25:11014–11022. doi: 10.1523/JNEUROSCI.3635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lehmann HC, Manoharan S, Hashmi M, Shim S, Ming GL, Schnaar RL, Lopez PH, Bogdanova N, Sheikh KA. Anti-ganglioside antibody-mediated activation of RhoA induces inhibition of neurite outgrowth. J Neurosci. 2011a;31:1664–1675. doi: 10.1523/JNEUROSCI.3829-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Chi Z, Keuss MJ, Pai YM, Kang HC, Shin JH, Bugayenko A, Wang H, Xiong Y, Pletnikov MV, Mattson MP, Dawson TM, Dawson VL. The AAA+ ATPase Thorase regulates AMPA receptor-dependent synaptic plasticity and behavior. Cell. 2011b;145:284–299. doi: 10.1016/j.cell.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Slevin JT, Whiteheart SW. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–2149. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]