

Abstract
Similar to peripheral immune/inflammatory cells, neuroglial cells appear to rely on calcineurin (CN) signaling pathways to regulate cytokine production and cellular activation. Several studies suggest that harmful immune/inflammatory responses may be the most impactful consequence of aberrant CN activity in glial cells. However, newly identified roles for CN in glutamate uptake, gap junction regulation, Ca2+ dyshomeostasis, and amyloid production suggest that CN’s influence in glia may extend well beyond neuroinflammation. The following review will discuss the various actions of CN in glial cells, with particular emphasis on astrocytes, and consider the implications for neurologic dysfunction arising with aging, injury, and/or neurodegenerative disease.
Keywords: Alzheimer’s disease, Amyloid, Astrocytes, Ca2+ regulation, Calcineurin, Gap junction, Glutamate, Neurodegeneration, Neuroinflammation
Background
Calcineurin (CN) is a Ca2+/calmodulin (Ca2+/CaM)-dependent protein phosphatase expressed in most mammalian tissues, but found at especially high levels in brain. The CN holoenzyme is a heterodimeric protein containing a 60 kDa catalytic subunit (CN A) and a 19 kDa regulatory subunit (CN B). Multiple isoforms of both subunits have been identified and are expressed differentially throughout brain and most other tissues (for review see [1-3]). CN A contains the catalytic core as well as binding sites for the CN B subunit and Ca2+/CaM. There is also a critical autoinhibitory domain located near the C terminus of CN A that suppresses catalytic activity when Ca2+ levels are low. The CN B subunit contains four Ca2+-binding EF-hand motifs and is generally physically bound to CN A at resting Ca2+ levels. Allosteric interactions between CN B, Ca2+/CaM, and the autoinhibitory domain allow CN to respond to rapid Ca2+ fluctuations with relatively high fidelity [4,5]. Within the cell, CN can be found throughout the cytosol and the nucleus, and is also commonly associated with membrane receptors, ion channels, and pumps via physical interactions with a variety of anchoring proteins [1,3].
Immunohistochemical and in situ hybridization studies on healthy adult rat brain performed in the mid-1980s to early 1990s reported high levels of CN in neurons of the striatum, hippocampus, amygdala, and neocortex [6-9], with little to no expression observed in glial cells [6,7]. Many functional studies performed around this time found that CN plays an integral role in coupling glutamate receptor activation to the regulation of cytoskeletal proteins and dendritic spine morphology [10,11]. Since these initial studies, CN has been shown to interact with numerous neuronal substrates and to modulate diverse cellular functions including receptor and ion channel trafficking, ion channel function, apoptosis, and gene regulation, to name a few [2]. In the last 10 years, there have been a steadily increasing number of studies identifying neuronal CN as a primary suspect in synapse loss, dendritic atrophy, synaptic dysfunction, and neuronal vulnerability [12,13]. Nevertheless, despite the apparently selective association of CN with neurons and neuronal signaling cascades, a handful of reports in the mid- to late 1990s found that CN can also appear in primary glial cells and glial cells of intact brain tissue, notably following inflammatory insult [14-17]. The clear connection between glial cells and neuro-immune/inflammatory signaling, in addition to the well-defined role of CN in cytokine production in peripheral immune cells, suggested a strong linkage between glial CN and the neuroinflammation inherent to most acute and chronic neurodegenerative diseases. Recent and ongoing work from our group and others has not only largely confirmed CN as a major modulator of immune/inflammatory processes in glial cells, but has also identified other possible functions for glial CN signaling that may have a major impact on neurologic function. This article reviews the functional implications associated with glial CN expression.
Review
CN and glial cells
Though primarily localized to neurons in healthy nervous tissue, CN may also be strongly expressed in glia during aging, injury, and/or disease. Greater numbers of CN-positive astrocytes have been reported in the hippocampus of aged, β-amyloid (Aβ)-bearing transgenic amyloid precursor protein/presenilin 1 (APP/PS1) mice, particularly in the immediate vicinity of extracellular Aβ deposits [18]. A similar relationship between CN-positive astrocytes and Aβ pathology was shown in postmortem brain tissue obtained from human subjects diagnosed with Alzheimer’s disease (AD) [19-21]. Though closely associated with Aβ deposits, the expression of CN in astrocytes does not necessarily depend on the presence of Aβ. Indeed, aged wild-type mice showed a far greater number of CN-positive astrocytes compared to younger wild-type mice, while expression of CN in astrocytes of APP/PS1 mice could be detected as early as three months of age, long before the appearance of extensive plaque pathology in this model [18]. The presence of numerous CN-positive astrocytes in human hippocampus at very early stages of cognitive decline [19] suggests that the upregulation of CN in astrocytes is an antecedent to dementia found at later disease stages. In addition to aging and AD, CN expression in astrocytes has also been reported for animal models of acute injury. In an early study by Hashimoto et al. [16], an increase in astrocytic CN labeling was observed in gerbil hippocampus following bilateral carotid artery occlusion, even though whole tissue levels of CN were reduced (as measured by Western blot). Shifting expression patterns for CN during aging, injury, and disease are notable because of the timing, i.e., CN can appear in astrocytes before widespread pathology is observed, and also because of the selectivity, i.e., increased CN expression only appears to occur in a subset of astrocytes. These findings suggest that changes in CN serve unique and perhaps critical roles in the initiation and progression of neurodegeneration and cognitive decline.
Phenotype switching in astrocytes and neuroinflammation
Astrocytes are an abundant and diverse subtype of glia. As a critical component of the neurovascular unit, astrocytes ensheath most microvessels via specialized end-foot processes, which help to maintain the integrity of the blood-brain barrier and promote osmotic balance (for review see [22]). Many astrocyte processes are also in close juxtaposition to synapses where they coordinate nutrient exchange to neurons and detoxify the local environment via the uptake of K+, glutamate, and other neurotransmitters [23,24]. Redistribution of imported factors and excitotoxins across numerous neighboring astrocytes is accomplished by an extensive gap junction network, which helps minimize local concentration gradients [25]. By ensheathing synapses, astrocytes play an essential role in establishing and maintaining the structural integrity of nerve terminals and dendritic spines, which, in turn, ensures the fidelity of interneuronal communication. In healthy central nervous system (CNS) tissue, astrocytes effectively carry out all of these functions, plus others. However, with CNS injury, astrocytes often look and appear to behave in very different ways. Hypertrophy of astrocyte somata and processes, with a corresponding increase in the expression of the intermediate filament protein, glial fibrillary acidic protein (GFAP), is a pervasive feature of nearly every form of acute CNS injury as well as most chronic neurodegenerative disorders [22,26,27]. These changes have been extensively documented and are commonly referred to as “astrocyte activation” or “astrocyte reactivity” [28].
Along with activated microglia, astrocyte activation is widely accepted as a hallmark of neuroinflammation, though the functional phenotype of activated astrocytes remains somewhat elusive. Protective roles for activated astrocytes, particularly after acute injury, have been demonstrated by many studies (for a review, see [29]). However, if not properly resolved, astrocyte activation can become a chronic condition with apparently detrimental effects on neuronal function and plasticity [28,30]. Activated astrocytes secrete numerous pro-inflammatory cytokines and other factors that can interfere with synaptic fidelity, impair neuronal viability, and/or maintain the activation state of astrocytes and microglia [31-33]. Activated astrocytes also appear to be compromised in their ability to take up excitotoxins (e.g., K+ and glutamate) from the extracellular milieu, which could, in turn, lead to further synaptic dysfunction, neuronal damage, and neuroinflammation [19,28,33,34]. The appearance of activated astrocytes at very early stages of cognitive impairment in humans [35-37] suggests that astrocyte activation may help initiate and/or drive other pathophysiological changes leading to dementia.
We have hypothesized that CN is a critical mechanism for triggering phenotype switching (i.e., activated vs. non-activated) and neuroinflammatory signaling inherent to astrocytes during neural damage and dysfunction [13,18,33]. In many peripheral tissues, CN is a pivotal regulator of transcriptional programs involved in cellular remodeling [38,39]. Perhaps the best documented example is the role that CN plays in the adaptive immune response through the activation of NFATs (Nuclear Factor of Activated T cells) and NFκB (Nuclear Factor κB) transcription factors. In the resting state, NFATs and NFκB are both sequestered in the cytosol, albeit by different mechanisms. The nuclear translocation signal of NFATs is masked by hyperphosphorylation. When activated, CN directly binds to and dephosphorylates NFATs, exposing the nuclear translocation signal and promoting the accumulation of NFATs in the nucleus [40]. A similar nuclear localization signal is present in NFκB, but is masked not by hyperphosphorylation, but by the binding of subunits called inhibitory κBs (IκBs) [41]. CN helps promote the activation of IκB kinases [42], which phosphorylate and tag IκB for proteasomal degradation, thereby allowing nuclear translocation of NFκB. Once in the nucleus, NFATs and NFκB interact with distinct DNA binding elements to drive the expression of multiple cytokine species that promote (or in some cases suppress) the clonal expansion of T cells.
In astrocytes, CN appears to represent a fundamental link between morphological changes and immune/inflammatory signaling. Forced expression of activated CN in primary mixed neuron/glia cultures was sufficient to cause an increase in the width of astrocyte somata and processes [18], while inhibition of astrocytic CN/NFAT activity in intact amyloid-bearing mice caused a reduction in the surface area of individual hippocampal astrocytes without affecting overall cell number [43]. Multiple extracellular factors that trigger astrocyte hypertrophy and/or neuroinflammation, including many “pro-inflammatory” cytokines (e.g., interleukin 1β (IL-1β), tumor necrosis factor α (TNFα), and interferon γ), glutamate, ATP, thrombin, S100, and Aβ also robustly activate CN in primary astrocyte cultures [19,21,33,44-48]. Once activated, CN helps drive the expression of numerous immune/inflammatory factors in astrocytes [18,33,44,45], many of which are found at elevated levels during injury, aging, and neurodegenerative disease [49-54]. Moreover, we have found that astrocytic CN/NFAT activity can propagate from one astrocyte population to another in an autostimulatory manner [33].
Through its actions on NFAT- and NFκB-dependent transcriptional regulation, CN appears ideally suited to drive the self-perpetuating “cytokine cycles” implicated in chronic neuroinflammation [54,55]. Indeed, similar immune/inflammatory functions of CN have been observed in microglia [56-59], confirming CN’s role as a global mediator of neuroinflammation. Nonetheless, it is probably too simplistic to think of CN strictly as a “pro-inflammatory” mechanism. Extensive work on peripheral immune cells tells us that CN activation can participate in diametrical processes, e.g., cytokine production and clonal expansion under some conditions, and lymphocyte anergy and/or tolerance under different conditions [60]. Such opposing phenotypic characteristics may depend, in part, on the association of CN with a variety of transcription factors. T cell activation, for instance, is largely driven by synergistic actions of NFATs and activator protein 1 (AP1) [40,61] (Figure 1A), while T cell tolerance and/or anergy has been shown to result from interactions between NFATs and forkhead box P3 (FOXP3) [61,62]. Based on these observations in T cells, it is not surprising that CN has been shown to have “anti-“, as well as, “pro-” inflammatory effects in astrocytes. In a series of studies from Torres-Aleman’s group, CN was implicated in both the initiation and resolution of neuroinflammatory signaling in acutely injured mice [45] or mice with progressing amyloid pathology [63]. In the latter study, performed on APP mice expressing a dox-inducible CN fragment in astrocytes, CN was shown to differentially affect neuroinflammation through its interactions with NFκB, peroxisome proliferator-activated receptor γ (PPARγ), and/or forkhead box O3 (FOXO3) transcription factors. These interactions were governed by the presence of specific extracellular factors. TNFα was shown to stimulate interactions between CN, FOXO3, and NFκB leading to increased inflammatory signaling, while insulin-like growth factor (IGF-1) was shown to disrupt the pro-inflammatory interactions between CN, NFκB, and FOXO3 (Figure 1B) in favor of anti-inflammatory interactions between CN, NFκB, and PPARγ (Figure 1C).
Figure 1.
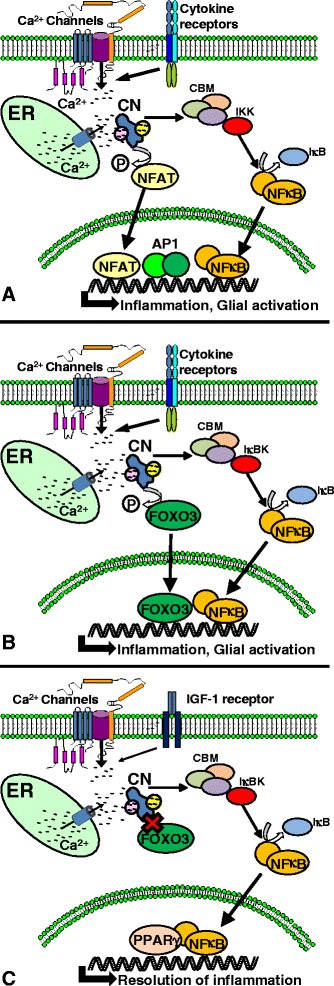
CN pathways can drive or resolve neuroinflammatory signaling in glial cells. (A) Factors that activate glial cells, including multiple cytokine species and Aβ peptides, stimulate Ca2+ release from endoplasmic reticulum (ER) and/or Ca2+ influx across Ca2+ channels in the plasma membrane leading to CN activation. CN directly dephosphorylates and activates NFATs. CN also facilitates the recruitment of IκB kinases (IKK) to the CARMA1-Bcl10-MALT1 (CBM) complex, which, in turn, causes the phosphorylation of IκB and the release (i.e., activation) of NFκB. NFATs and NFκB translocate to the nucleus, and with other transcription factors, such as AP1, drive the expression of numerous cytokines involved in the generation and maintenance of neuroinflammation. (B) CN can also dephosphorylate and activate FOXO3 transcription factors, which can also synergize with NFκB to drive immune/inflammatory signaling in glial cells. (C) Activation of IGF-1 receptors in glial cells also stimulates CN but suppresses its interaction with FOXO3. Simultaneous activation of PPARγ and NFκB by IGF-1 regulates transcriptional programs that reduce or resolve neuroinflammation.
Even the presence of different NFAT isoforms can dramatically alter the functional impact of elevated CN activity. A striking example of this is found in muscle tissue where NFAT4 is selectively activated by Ca2+ elevations in myoblasts, while NFATs 1 and 2 are selectively activated by the same kind of stimulation in myotubes [64]. In skeletal muscle fibers from adult rats, different NFAT isoforms showed varying sensitivities to different stimulation frequencies and regulated distinct genes associated with slow- and fast-twitch transcriptional programs [65]. The idea that individual NFAT isoforms play various roles in astrocyte function has been proposed by our lab and others [13,19,66-68]. We discovered that early stages of cognitive decline were associated with increased nuclear levels of NFAT1 in astrocytes, while NFAT3 was more strongly associated with astrocytes during late stages of AD [19]. Based on the clear association between the NFAT1 isoform and cytokine expression in lymphocytes, as well as the comparatively weak association of NFAT3 with lymphoid tissues [40], we have suggested that NFAT1 is more strongly linked to the neuroinflammatory phenotype of astrocytes [13]. In contrast, NFAT3 has been more closely linked to cell death and degeneration in multiple cell types (e.g., see [69-71]), suggesting that the demise of astrocytes in severe forms of injury/disease results from the selective activation of NFAT3. The other CN-dependent NFAT isoforms (i.e., NFATs 2 and 4) have also been detected in astrocytes and microglia at the mRNA and protein levels [47,56,59,66-68,72-74]. Several recent studies have reported a striking increase in NFAT4 expression in a subgroup of astrocytes following acute CNS trauma [67,68,73], though the subcellular localization of NFAT4 seemed to be limited to the cytosol, with very little expression found in astrocyte nuclei [67]. Other groups have shown that NFAT4 is indeed active in astrocytes and regulates the expression of transcripts involved in Aβ production [66], as discussed in a later section of this review.
In summary, the available literature suggests that CN plays a major role in shaping the neuroinflammatory phenotype of astrocytes. The impact of CN on neuroinflammation appears to be complex and may depend upon the co-activation of other intracellular signaling pathways and/or the association of CN with multiple transcription factors, including different NFAT isoforms, NFκB, AP1, FOXO3, and others. These differential CN interactions likely do not simply promote a global phenotypic change but instead may selectively affect specific glial functions (Figure 1).
CN and astrocyte-mediated glutamate regulation
One of the most important functions of astrocytes is the removal of potentially toxic factors from the extracellular milieu, including K+, glutamate, GABA, and many others. Glutamate is cleared by astrocytes using a variety of excitatory amino acid transporters (EAATs), which energetically couple glutamate uptake to the import of Na+ ions. The EAAT2 isoform (or Glt 1) is expressed predominantly in astrocytes and plays a lead role in glutamate clearance from many brain regions [75]. Loss of EAAT expression and/or function has been linked to a variety of neurologic diseases including AD, amyotrophic lateral sclerosis, Parkinson’s disease, and epilepsy, among others (for reviews, see [24,75,76]). Elevated amyloid levels in human AD brain tissue and in AD animal models is associated with a loss of EAAT expression and/or function [19,77-83]. This loss can occur very early in the progression of cognitive deficits [19], suggesting that changes in glutamate regulation may be key to the initiation/propagation of neuronal degeneration and death. Common phenotypic characteristics of multiple disease models, including increased susceptibility to excitotoxicity, altered synaptic plasticity, and impaired cognition can be recapitulated in otherwise healthy animals by genetically or pharmacologically knocking down EAAT function [84-86]. These observations and many others make EAATs an intensely studied molecular target for preventing and/or limiting neurologic deficits associated with injury and disease.
Changes in EAATs appear to be strongly linked to the activated astrocyte phenotype. Immunohistochemical analyses of postmortem human brain sections revealed an inverse correlation between EAAT2 and GFAP expression levels [87]. Moreover, multiple extracellular factors that trigger profound astrocyte activation (including pro-inflammatory cytokines and Aβ) can cause a reduction in EAAT expression and/or function in cell culture and animal models [19,33,77,83,88-90]. Numerous mechanisms have been proposed for regulation of EAATs at the transcriptional, translational, and post-translational levels. Many of these mechanisms show strong sensitivity to immune/inflammatory signaling factors implicated in astrocyte activation [91]. In regard to transcriptional regulation, binding sites for NFκB and NFAT have been verified in the human EAAT2 promoter [92]. Interestingly, NFκB appears to play critical roles in both the up- and down-regulation of EAAT2 expression levels. Exposure to a variety of protective/reparative factors, including epidermal growth factor and TGF-β, causes an NFκB-mediated increase in EAAT2 expression, while pro-inflammatory factors like TNFα reduce EAAT2 levels in an NFκB-dependent manner [92]. In our work on primary astrocytes [19,33], reductions in EAAT2 levels following treatment with IL-1β or oligomeric Aβ, were strongly inhibited by pretreatment with VIVIT, an NFAT inhibitory peptide. In addition to preserving EAAT levels, VIVIT lowered extracellular glutamate, dampened neuronal hyperactivity, and reduced the appearance of excitotoxic neuronal death. Given the relatively close proximity between the NFκB and NFAT binding sites within the EAAT2 promoter [92], we suggest that the downregulation of EAAT2 during astrocyte activation and/or neuroinflammation is largely attributable to the synergistic actions of CN, NFκB, and NFAT (Figure 2). Additional work will be required to determine the viability of this combinatorial mechanism for EAAT2 regulation, and to identify possible conditions in which NFATs positively regulate EAAT2 expression to promote glutamate uptake.
Figure 2.
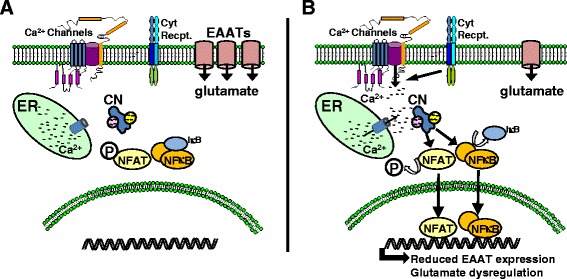
CN-mediated impairment of glutamate uptake in astrocytes. (A) In healthy nervous tissue, astrocytes remove excess glutamate from the extracellular milieu using EAATs. (B) Pro-inflammatory cytokines and Aβ cause hyperactivation of CN in astrocytes leading to the activation of NFAT and NFκB transcription factors. Together, and possibly individually, NFATs and NFκB suppress the transcription of EAATs. Loss of EAAT protein is associated with reduced glutamate uptake, higher extracellular glutamate levels, and increased excitotoxicity.
CN, astrocytes, and gap junctions
Astrocytes are extensively coupled to one another and, to a lesser degree, to other cell types via gap junctions (GJ). The GJ channel consists of a hexamer of specialized proteins called connexins (Cx), which are localized to the cell membrane and directly apposed to a similar Cx hexamer localized to the plasma membrane of another cell [25]. The channel formed from apposing connexin hexamers (i.e., Cx hemichannels) can accommodate the passage of molecules up to ~1.2 kDa and provides a direct cytoplasm-to-cytoplasm pathway for adjacent astrocytes. GJs permit the exchange of numerous small molecules and signaling factors between cells including ions, nucleotides, and amino acids. Passage of critical second messengers across GJs, such as Ca2+ and inositol trisphosphate (IP3), also allow groups of astrocytes to respond in synchrony to highly localized extracellular signaling factors, which, in turn, can influence the plasticity of local synaptic ensembles and/or modulate the tone of the local vascular network [93]. In addition to forming GJs, some Cx hemichannels are unapposed (i.e., not coupled to Cx hemichannels on other cells) and provide a direct path between the astrocyte cytoplasm and the extracellular milieu. Unapposed Cx hemichannels are generally found in a closed channel state, but may become permeable during brain injury and disease [25].
The diffusion of K+ and glutamate across GJ-coupled astrocyte networks during high levels of neuronal activity is one of the fundamental mechanisms for preserving high fidelity interneuronal communication and for preventing excitotoxicity [94]. Transgenic mice lacking Cx43 and/or Cx30 (i.e., the major connexin subtypes found in mature astrocytes) show heightened vulnerability to seizure activity and neuronal death, and may exhibit a variety of neurologic alterations (e.g., altered motor function and impaired cognition) depending on the brain region targeted [95-97]. On the other hand, increased GJ permeability may expose otherwise healthy astrocytes to toxic signals carried from sites of injury or pathology. In this way, GJs have been suggested as a mechanism for spreading pathophysiology [98].
Similar to EAATs, astrocytic GJs are highly sensitive to extracellular factors linked to neuroinflammation and exhibit altered expression and/or function in a variety of different neurologic disorders and diseases [94,99]. Multiple pro-inflammatory cytokines have been shown to either reduce Cx43 expression levels or reduce GJ coupling [100]. The C terminus tail region of Cx43, in particular, contains numerous phosphorylation sites that are regulated by inflammation-sensitive protein kinases [101]. Several research groups have also demonstrated stimulus-evoked dephosphorylation of Cx43 in primary astrocytes, suggesting the dynamic involvement of protein phosphatases [102,103]. Specific dephosphorylation of ser368 in the Cx43 cytoplasmic tail can occur within minutes following a hypoxic insult or after treatment with endogenous factors linked to vascular damage and astrogliosis and is highly sensitive to CN activation [102,103]. Dephosphorylation of ser368 in primary astrocytes following hypoxia or treatment with extracellular factors, such as endothelin-1 and phingosine-1-phosphate, was mostly prevented by pretreatment with commercial CN inhibitors (i.e., cyclosporine A and FK506), supporting a regulatory role for CN in Cx43 dephosphorylation. Additionally, preliminary studies in our laboratory indicate that CN mediates the dephosphorylation of Cx43 ser368 in primary astrocytes exposed to IL-1β or the exogenous Ca2+ mobilizing agents ionomycin and phorbol ester [104]. Parallel investigations of Cx43 in postmortem human hippocampal tissue performed by our group further showed that Cx43 dephosphorylation (at ser368) is increased at early stages of cognitive decline and positively correlated with the levels of a proteolytically active form of CN, suggesting that the CN/Cx43 interaction may have relevance to the progression of neurodegeneration and/or dementia.
Despite these observations, the functional impact of the CN/Cx43 interaction is not presently clear. Using a dye-coupling approach, CN inhibitors were initially shown to prevent the decoupling of GJs in astrocytes following a hypoxic insult [102]. Later studies similarly demonstrated a relationship between GJ inhibition and dephosphorylation of Cx43 ser368, but found that dephosphorylation does not necessarily cause GJ decoupling [103]. Instead, this report suggested that Cx43 dephosphorylation by CN occurs after GJ decoupling and leads to the redistribution of Cx43 to tight junctions. Clearly, additional studies are needed to clarify the role of CN in the regulation of gap junctions and determine whether this interaction significantly impacts astrocyte function in particular, and neurologic function in general (Figure 3).
Figure 3.
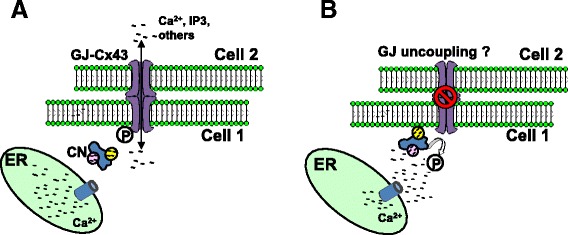
CN dephosphorylates Cx43 and modulates GJ coupling in astrocytes. (A) Cx hexamers on the astrocyte membrane form GJ channels with apposing Cx hexamers in adjacent cells and allow the intercellular passage of small molecules like Ca2+ and IP3. (B) Activation of astrocytes with inflammatory mediators stimulates CN, which dephosphorylates the cytoplasmic tail of Cx43. Dephosphorylation of Cx43 is associated with reduced GJ coupling, though it is unclear whether dephosphorylation is a cause or consequence of decoupling.
CN and astrocytic Ca2+ dysregulation
Similar to neurons, activated astrocytes in models of aging, injury, and disease exhibit numerous signs of Ca2+ dysregulation including elevated expression of a variety of Ca2+ channels and Ca2+ regulated proteins, as well as higher intracellular Ca2+ levels and/or more frequent Ca2+ oscillations [105-107]. While it seems clear how these changes could help set the stage for hyperactive CN signaling, it is also important to note that CN may play an active role in promoting or disrupting Ca2+ homeostasis. At the transcriptional level, CN helps drive the expression of many key Ca2+ signaling mediators in multiple cell types [108-111]. At the post-translational level, CN directly or indirectly regulates plasma membrane Ca2+ channels, intracellular Ca2+ release channels, and Ca2+-dependent enzymes [2]. In astrocytes, inhibition of CN was recently shown to reduce Ca2+ transients evoked by Aβ [21] and to suppress the upregulation of critical proteins involved in Ca2+-induced Ca2+ release, including IP3 receptor 1 and metabotropic glutamate receptor 5 [112]. Other potential targets for CN may be predicted from findings gleaned from neurons and other cell types. For instance, in neurons and cardiomyocytes, CN has been shown to enhance the function of L-type Ca2+ channels, which may, in turn, disrupt cellular activity and viability [113-116]. Though found at very low levels in astrocytes of healthy animals, L-type Ca2+ channels appear to be present at high levels in activated astrocytes following acute injury [117]. This differential pattern of expression in astrocytes is strikingly similar to that of CN. Moreover, we have shown that 50% or more of CN/NFAT activity in primary astrocytes treated with IL-1β is eliminated by co-treatment with the L-type Ca2+ channel blocker nifedipine [33], while others have shown that CN/NFAT signaling is stimulated in astrocytes treated with the L-type Ca2+ channel activator Bayk8644 [44]. These observations suggest that CN/L-type Ca2+ channel interactions may play a critical role in promoting Ca2+ dysregulation in activated astrocytes (Figure 4).
Figure 4.
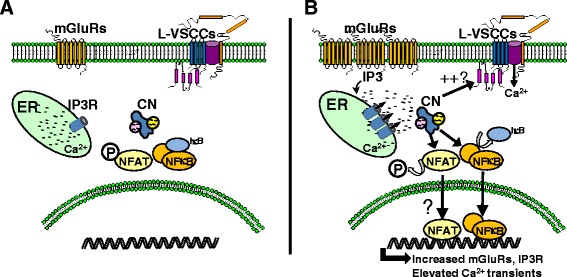
CN is associated with Ca 2+ dysregulation in astrocytes. (A) In healthy nervous tissue, elevations in intracellular Ca2+ are controlled, in part, by Ca2+ release channels (e.g., IP3Rs) located on the endoplasmic reticulum membrane. Levels of several different varieties of plasma membrane Ca2+ channels, including L-type Ca2+ channels are generally low in non-activated astrocytes. (B) Inflammatory mediators, like cytokines and Aβ, cause hyperactivation of CN and its target transcription factors, NFAT and NFκB. These events cause the transcriptional upregulation of IP3 receptors and mGluRs leading to increased Ca2+ transients and/or higher resting levels of cytosolic Ca2+. CN may also increase the function of L-type Ca2+ channels, which, like CN, are found at higher levels in activated astrocytes.
CN and astrocyte-derived Aβ production
In AD, CN expression and activity levels are directly correlated to increasing levels of the Aβ peptide [19]. Aβ deposits in both human and mouse tissue are often surrounded by astrocytes that label very intensely for CN [18-20]. The sensitivity of CN activity to rising Aβ levels has been well-documented by several different research groups using a variety of experimental models. Application of the pathogenic, oligomeric form of Aβ rapidly and robustly activates CN signaling pathways in both neurons [71,118-123] and astrocytes [19,21,66,112] and leads to numerous deleterious neurologic changes including enhanced synaptic depression, impaired synaptic potentiation, glutamate dysregulation, dendritic atrophy, cell death, and cognitive deficits.
Interestingly, APP/PS1 mice treated with FK506 exhibit reductions in Aβ plaque load relative to vehicle-treated APP mice [124], suggesting that CN not only responds to elevated Aβ, but also actively contributes to the accumulation of Aβ pathology. Though the mechanisms through which CN modulates Aβ levels are unsettled, several studies indicate that the CN/NFAT signaling pathway positively regulates the expression of beta-secretases [66,125], the rate-limiting enzymes for formation of Aβ peptides (Figure 5). Jin et al. [66] showed that the NFAT4 isoform binds to the beta-site APP cleaving enzyme 1 (BACE1) promoter in primary astrocytes, which in turn, helps drive Aβ production in response to rising intracellular Ca2+ levels. Similarly, our group showed that inhibition of astrocytic NFAT activity reduces soluble Aβ levels and plaque load in the hippocampus of APP/PS1 mice [43] in parallel with a reduction in BACE1 protein levels. We did not observe any changes in the levels for several key Aβ clearing enzymes, including neprilysin and insulin degrading enzyme. Together, these findings are consistent with the notion that astrocytic NFAT/BACE1 interactions play an important role in amyloid regulation. Although BACE1 expression is very low in astrocytes of intact animals and humans, the sheer abundance of this cell type, relative to other cell types, could provide enough BACE activity to contribute significantly to the production of amyloid peptides and subsequent formation of parenchymal Aβ deposits [126]. Nonetheless, effects of CN/NFAT on other key regulators of Aβ formation and clearance, including gamma secretases, apolipoprotein E, and lipoprotein receptor-related protein 1, among others, are not clear and will require further investigation.
Figure 5.
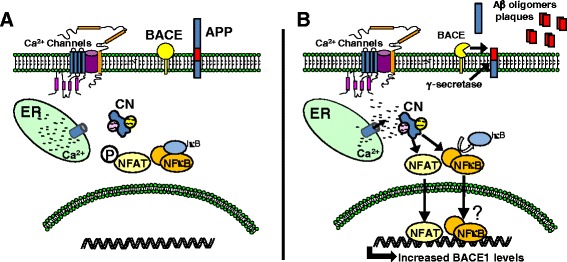
CN regulates Aβ metabolism in astrocytes. (A) In resting astrocytes, BACE expression and activity levels are relatively low. (B) Activation of astrocytes with inflammatory mediators stimulates CN. Nuclear translocation of NFATs and possibly NFκB leads to the transcriptional upregulation of the protease BACE1 (the rate limiting enzyme for production of neurotoxic Aβ peptides). Elevated levels of BACE1 are associated with the increased production of Aβ peptides, which further aggregate and form Aβ plaques.
In addition to directly influencing Aβ production in astrocytes, it is perhaps just as likely that astrocytic CN indirectly influences Aβ production/metabolism in neurons. Indeed, numerous “pro-inflammatory” factors released by glial cells, many of which are sensitive to CN activity, have been shown to stimulate neuronal APP and/or Aβ production, e.g., see [127-131]. Similarly, the loss of any number of protective glial properties during chronic activation/neuroinflammation would be expected to disrupt Ca2+ homeostasis in neurons and/or erode neuronal viability leading to greater Aβ levels [132-136]. Whether direct or indirect, these observations suggest that inhibition of astrocytic CN activity may be an effective strategy for slowing the progression of Aβ pathology.
Impact of astrocytic CN on neurologic function
Numerous studies have shown that commercial CN inhibitors dampen glial activation, impart neuroprotection, and/or improve neurologic function in animal models of aging, injury, and disease [3,12]. Surely, at least some of these beneficial effects are attributable to direct inhibition of deleterious neuronal CN signaling pathways, which have been shown to play important roles in neuronal degeneration and altered synaptic function (e.g., see [105-107]). However, what about the impact of glial CN signaling? This is a difficult question to address in intact animals using basic pharmacologic reagents, given their lack of cellular specificity. To overcome this difficulty, our group recently employed an adeno-associated virus-based approach to selectively express the NFAT inhibitor, VIVIT, in hippocampal astrocytes of AD model mice [43]. Suppression of astrocytic NFAT signaling in pre-symptomatic mice was sufficient for reducing glial activation and Aβ plaque pathology during advanced age. Arguably more important, this knockdown also proved beneficial to neurologic function and plasticity, i.e., VIVIT-treated mice showed improved synaptic strength, increased levels of long-term potentiation, and better avoidance learning relative to AD mice treated with vehicle or control adeno-associated virus vectors. These observations support a detrimental role for astrocytic CN/NFAT signaling in neurologic function and are consistent with cell culture studies that report lower levels of pro-inflammatory cytokines, reduced Aβ production/lower BACE1 activity, reduced Ca2+ transients, lower extracellular glutamate levels, reduced neuronal excitability, and less excitotoxic neuronal death following inhibition of astrocytic CN/NFATs as discussed in preceding sections.
In addition to directly suppressing NFAT activity, overexpression of VIVIT may help to divert CN activity to other substrates that are more protective in nature. For instance, Fernandez et al. [63] reported that overexpression of an activated form of CN in astrocytes of intact APP mice led to the increased association of CN with PPARγ and NFκB, which, in turn, reduced glial activation and amyloid pathology and improved cognition. Together with our findings, it is tempting to speculate that astrocytic CN is generally protective in astrocytes, unless it interacts extensively with NFAT transcription factors, in which case the astrocyte phenotype becomes harmful, marked by increased expression of pro-inflammatory cytokines, impaired glutamate uptake, and other deleterious properties.
Conclusions
We are just beginning to understand the contribution of CN to glial function. While it seems clear that CN is intimately involved in neuroinflammation, much work is still needed to fully assess the specific interactions between CN-dependent and -independent transcription factors, and how these interactions regulate specific inflammatory phenotypes. While this review has focused primarily on the role of CN in astrocytes, other work showing similar roles of CN in microglia [56-58,74] underscores the necessity to characterize the signaling properties of CN in other glial subtypes. Furthermore, the identification of other cellular targets of CN, including glutamate transporters, gap junctions, and BACE, suggests that CN’s impact on glial function may extend well beyond immune/inflammatory signaling. These observations highlight some of the complexities facing future research into the role of glial CN signaling, but also hint at the potential of discovering new molecular targets for treating neural injury and neurodegenerative disease.
Acknowledgements
Work supported by awards from the NIH (AG027297), the Kentucky Spinal Cord and Head Injury Research Trust (12-10A), and The Hazel Embry Research Fund to CMN and an award from the PhRMA Foundation to JLF.
Abbreviations
- Aβ
β-amyloid
- AD
Alzheimer’s disease
- AP1
Activator protein 1
- APP
Amyloid precursor protein
- BACE1
Beta-site APP cleaving enzyme 1
- CaM
Calmodulin
- CN
Calcineurin
- CNS
Central nervous system
- Cx
Connexins
- EAAT
Excitatory amino acid transporter
- FOXO3
Forkhead box O3
- FOXP3
Forkhead box P3
- GJ
Gap junction
- GFAP
Glial fibrillary acidic protein
- IGF-1
Insulin-like growth factor
- IκB
Inhibitory κ B
- IL-1β
Interleukin 1β
- IP3
Inositol triphosphate
- NFATs
Nuclear factor of activated T cells
- NFκB
Nuclear factor κ B
- PPARγ
Peroxisome proliferator-activated receptor
- PS1
Presenilin 1
- TNFα
Tumor necrosis factor α
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JLF and CMN researched and wrote this manuscript. Both authors read and approved the final manuscript.
Contributor Information
Jennifer L Furman, Email: furmanj@neuro.wustl.edu.
Christopher M Norris, Email: cnorr2@uky.edu.
References
- 1.Musson RE, Smit NP. Regulatory mechanisms of calcineurin phosphatase activity. Curr Med Chem. 2011;18:301–315. doi: 10.2174/092986711794088407. [DOI] [PubMed] [Google Scholar]
- 2.Baumgartel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learn Mem. 2012;19:375–384. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- 3.Norris CM. Calpain interactions with the protein phosphatase calcineurin in neurodegeneration. Adv Biochem Health Dis. 2014;8:17–45. [Google Scholar]
- 4.Stemmer PM, Klee CB. Dual calcium ion regulation of calcineurin by calmodulin and calcineurin B. Biochemistry. 1994;33:6859–6866. doi: 10.1021/bi00188a015. [DOI] [PubMed] [Google Scholar]
- 5.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 6.Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res. 1986;397:161–172. doi: 10.1016/0006-8993(86)91381-8. [DOI] [PubMed] [Google Scholar]
- 7.Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. Calcineurin in human brain and its relation to extrapyramidal system. Immunohistochemical study on postmortem human brains. Acta Neuropathol. 1986;72:150–156. doi: 10.1007/BF00685977. [DOI] [PubMed] [Google Scholar]
- 8.Kuno T, Mukai H, Ito A, Chang CD, Kishima K, Saito N, Tanaka C. Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J Neurochem. 1992;58:1643–1651. doi: 10.1111/j.1471-4159.1992.tb10036.x. [DOI] [PubMed] [Google Scholar]
- 9.Polli JW, Billingsley ML, Kincaid RL. Expression of the calmodulin-dependent protein phosphatase, calcineurin, in rat brain: developmental patterns and the role of nigrostriatal innervation. Brain Res Dev Brain Res. 1991;63:105–119. doi: 10.1016/0165-3806(91)90071-P. [DOI] [PubMed] [Google Scholar]
- 10.Halpain S, Girault JA, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 11.Halpain S, Greengard P. Activation of NMDA receptors induces rapid dephosphorylation of the cytoskeletal protein MAP2. Neuron. 1990;5:237–246. doi: 10.1016/0896-6273(90)90161-8. [DOI] [PubMed] [Google Scholar]
- 12.Reese LC, Taglialatela G. A role for calcineurin in Alzheimer’s disease. Curr Neuropharmacol. 2011;9:685–692. doi: 10.2174/157015911798376316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdul HM, Furman JL, Sama MA, Mathis DM, Norris CM. NFATs and Alzheimer’s disease. Mol Cell Pharmacol. 2010;2:7–14. [PMC free article] [PubMed] [Google Scholar]
- 14.Vinade L, Goncalves CA, Wofchuk S, Gottfried C, Rodnight R. Evidence for a role for calcium ions in the dephosphorylation of glial fibrillary acidic protein (GFAP) in immature hippocampal slices and in astrocyte cultures from the rat. Brain Res Dev Brain Res. 1997;104:11–17. doi: 10.1016/S0165-3806(97)00114-4. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda T, Takuma K, Asano S, Kishida Y, Nakamura H, Mori K, Maeda S, Baba A: Involvement of calcineurin in Ca2+paradox-like injury of cultured rat astrocytes.J Neurochem 1998, 70:2004–2011. [DOI] [PubMed]
- 16.Hashimoto T, Kawamata T, Saito N, Sasaki M, Nakai M, Niu S, Taniguchi T, Terashima A, Yasuda M, Maeda K, Tanaka C. Isoform-specific redistribution of calcineurin A alpha and A beta in the hippocampal CA1 region of gerbils after transient ischemia. J Neurochem. 1998;70:1289–1298. doi: 10.1046/j.1471-4159.1998.70031289.x. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari D, Stroh C, Schulze-Osthoff K. P2X7/P2Z purinoreceptor-mediated activation of transcription factor NFAT in microglial cells. J Biol Chem. 1999;274:13205–13210. doi: 10.1074/jbc.274.19.13205. [DOI] [PubMed] [Google Scholar]
- 18.Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celsi F, Svedberg M, Unger C, Cotman CW, Carri MT, Ottersen OP, Nordberg A, Torp R. Beta-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 21.Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia. 2013;61:1134–1145. doi: 10.1002/glia.22502. [DOI] [PubMed] [Google Scholar]
- 22.Verkhratsky A, Sofroniew MV, Messing A, deLanerolle NC, Rempe D, Rodriguez JJ, Nedergaard M. Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro. 2012;4(3):e00082. doi: 10.1042/AN20120010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schousboe A, Waagepetersen HS. Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox Res. 2005;8:221–225. doi: 10.1007/BF03033975. [DOI] [PubMed] [Google Scholar]
- 24.Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 25.Eugenin EA, Basilio D, Saez JC, Orellana JA, Raine CS, Bukauskas F, Bennett MV, Berman JW. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol. 2012;7:499–518. doi: 10.1007/s11481-012-9352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 29.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 31.Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci U S A. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keene SD, Greco TM, Parastatidis I, Lee SH, Hughes EG, Balice-Gordon RJ, Speicher DW, Ischiropoulos H. Mass spectrometric and computational analysis of cytokine-induced alterations in the astrocyte secretome. Proteomics. 2009;9:768–782. doi: 10.1002/pmic.200800385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuller S, Steele M, Munch G. Activated astroglia during chronic inflammation in Alzheimer’s disease–do they neglect their neurosupportive roles? Mutat Res. 2010;690:40–49. doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, Nordberg A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53:37–46. doi: 10.2967/jnumed.110.087031. [DOI] [PubMed] [Google Scholar]
- 36.Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27:252–261. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/S0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 39.Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol. 2002;156:771–774. doi: 10.1083/jcb.200111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 41.Liou HC, Baltimore D. Regulation of the NF-kappa B/rel transcription factor and I kappa B inhibitor system. Curr Opin Cell Biol. 1993;5:477–487. doi: 10.1016/0955-0674(93)90014-H. [DOI] [PubMed] [Google Scholar]
- 42.Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, Marienfeld RB. The Ca2 + −dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J Biol Chem. 2011;286:7522–7534. doi: 10.1074/jbc.M110.155895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canellada A, Ramirez BG, Minami T, Redondo JM, Cano E. Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1-4 and cyclooxygenase-2 as NFAT target genes. Glia. 2008;56:709–722. doi: 10.1002/glia.20647. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci. 2007;27:8745–8756. doi: 10.1523/JNEUROSCI.1002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leal RB, Frizzo JK, Tramontina F, Fieuw-Makaroff S, Bobrovskaya L, Dunkley PR, Goncalves CA. S100B protein stimulates calcineurin activity. Neuroreport. 2004;15:317–320. doi: 10.1097/00001756-200402090-00021. [DOI] [PubMed] [Google Scholar]
- 47.Perez-Ortiz JM, Serrano-Perez MC, Pastor MD, Martin ED, Calvo S, Rincon M, Tranque P. Mechanical lesion activates newly identified NFATc1 in primary astrocytes: implication of ATP and purinergic receptors. Eur J Neurosci. 2008;27:2453–2465. doi: 10.1111/j.1460-9568.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- 48.Furman JL, Artiushin IA, Norris CM. Disparate effects of serum on basal and evoked NFAT activity in primary astrocyte cultures. Neurosci Lett. 2010;469:365–369. doi: 10.1016/j.neulet.2009.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29:1805–1816. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilcock DM. Neuroinflammatory phenotypes and their roles in Alzheimer’s disease. Neurodegener Dis. 2014;13:183–185. doi: 10.1159/000354228. [DOI] [PubMed] [Google Scholar]
- 53.Mrak RE, Griffin WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 2001;22:915–922. doi: 10.1016/S0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- 54.Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda M, Inoue K. Activation of P2X7 receptors induces CCL3 production in microglial cells through transcription factor NFAT. J Neurochem. 2009;108:115–125. doi: 10.1111/j.1471-4159.2008.05744.x. [DOI] [PubMed] [Google Scholar]
- 58.Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem. 2010;114:810–819. doi: 10.1111/j.1471-4159.2010.06809.x. [DOI] [PubMed] [Google Scholar]
- 59.Kim B, Jeong HK, Kim JH, Lee SY, Jou I, Joe EH. Uridine 5′-diphosphate induces chemokine expression in microglia and astrocytes through activation of the P2Y6 receptor. J Immunol. 2011;186:3701–3709. doi: 10.4049/jimmunol.1000212. [DOI] [PubMed] [Google Scholar]
- 60.Im SH, Rao A: Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling.Mol Cells 2004, 18:1–9. [PubMed]
- 61.Rudensky AY, Gavin M, Zheng Y. FOXP3 and NFAT: partners in tolerance. Cell. 2006;126:253–256. doi: 10.1016/j.cell.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez AM, Jimenez S, Mecha M, Davila D, Guaza C, Vitorica J, Torres-Aleman I. Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol Psychiatry. 2012;17:705–718. doi: 10.1038/mp.2011.128. [DOI] [PubMed] [Google Scholar]
- 64.Abbott KL, Friday BB, Thaloor D, Murphy TJ, Pavlath GK. Activation and cellular localization of the cyclosporine A-sensitive transcription factor NF-AT in skeletal muscle cells. Mol Biol Cell. 1998;9:2905–2916. doi: 10.1091/mbc.9.10.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calabria E, Ciciliot S, Moretti I, Garcia M, Picard A, Dyar KA, Pallafacchina G, Tothova J, Schiaffino S, Murgia M. NFAT isoforms control activity-dependent muscle fiber type specification. Proc Natl Acad Sci U S A. 2009;106:13335–13340. doi: 10.1073/pnas.0812911106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin SM, Cho HJ, Kim YW, Hwang JY, Mook-Jung I. Abeta-induced Ca(2+) influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem Biophys Res Commun. 2012;425:649–655. doi: 10.1016/j.bbrc.2012.07.123. [DOI] [PubMed] [Google Scholar]
- 67.Serrano-Perez MC, Martin ED, Vaquero CF, Azcoitia I, Calvo S, Cano E, Tranque P. Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia. 2011;59:94–107. doi: 10.1002/glia.21079. [DOI] [PubMed] [Google Scholar]
- 68.Yan HQ, Shin SS, Ma X, Li Y, Dixon CE. Differential effect of traumatic brain injury on the nuclear factor of activated T Cells C3 and C4 isoforms in the rat hippocampus. Brain Res. 2014;1548:63–72. doi: 10.1016/j.brainres.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shioda N, Han F, Moriguchi S, Fukunaga K. Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J Neurochem. 2007;102:1506–1517. doi: 10.1111/j.1471-4159.2007.04600.x. [DOI] [PubMed] [Google Scholar]
- 71.Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaguchi R, Hosaka M, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, Takeuchi T. Cyclophilin C-associated protein regulation of phagocytic functions via NFAT activation in macrophages. Brain Res. 2011;1397:55–65. doi: 10.1016/j.brainres.2011.03.036. [DOI] [PubMed] [Google Scholar]
- 73.Neria F, del Carmen S-PM, Velasco P, Urso K, Tranque P, Cano E. NFATc3 promotes Ca(2+) -dependent MMP3 expression in astroglial cells. Glia. 2013;61:1052–1066. doi: 10.1002/glia.22494. [DOI] [PubMed] [Google Scholar]
- 74.Rojanathammanee L, Puig KL, Combs CK. Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro and in a transgenic mouse model of Alzheimer disease. J Nutr. 2013;143:597–605. doi: 10.3945/jn.112.169516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15:461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 76.Sultana R, Butterfield DA. Alterations of some membrane transport proteins in Alzheimer’s disease: role of amyloid beta-peptide. Mol Biosyst. 2008;4:36–41. doi: 10.1039/B715278G. [DOI] [PubMed] [Google Scholar]
- 77.Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG. Amyloid-beta1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci. 2013;33:5312–5318. doi: 10.1523/JNEUROSCI.5274-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matos M, Augusto E, Machado NJ, dos Santos-Rodrigues A, Cunha RA, Agostinho P. Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis. 2012;31:555–567. doi: 10.3233/JAD-2012-120469. [DOI] [PubMed] [Google Scholar]
- 79.Schallier A, Smolders I, Van Dam D, Loyens E, De Deyn PP, Michotte A, Michotte Y, Massie A. Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer’s disease. J Alzheimers Dis. 2011;24:287–300. doi: 10.3233/JAD-2011-101005. [DOI] [PubMed] [Google Scholar]
- 80.Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL. Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem. 2010;113:978–989. doi: 10.1111/j.1471-4159.2010.06661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matos M, Augusto E, Oliveira CR, Agostinho P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience. 2008;156:898–910. doi: 10.1016/j.neuroscience.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 82.Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 83.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 84.Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14:547–553. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- 85.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/S0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 86.Mookherjee P, Green PS, Watson GS, Marques MA, Tanaka K, Meeker KD, Meabon JS, Li N, Zhu P, Olson VG, Cook DG. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J Alzheimers Dis. 2011;26:447–455. doi: 10.3233/JAD-2011-110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging. 2010;31(4):578–590. doi: 10.1016/j.neurobiolaging.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 88.Fang J, Han D, Hong J, Tan Q, Tian Y. The chemokine, macrophage inflammatory protein-2gamma, reduces the expression of glutamate transporter-1 on astrocytes and increases neuronal sensitivity to glutamate excitotoxicity. J Neuroinflammation. 2012;9:267. doi: 10.1186/1742-2094-9-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boycott HE, Wilkinson JA, Boyle JP, Pearson HA, Peers C. Differential involvement of TNF alpha in hypoxic suppression of astrocyte glutamate transporters. Glia. 2008;56:998–1004. doi: 10.1002/glia.20673. [DOI] [PubMed] [Google Scholar]
- 90.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271:15303–15306. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- 91.Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- 92.Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2) Proc Natl Acad Sci U S A. 2003;100:1955–1960. doi: 10.1073/pnas.0136555100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Bock M, Decrock E, Wang N, Bol M, Vinken M, Bultynck G, Leybaert L: The dual face of connexin-based astroglial Ca2+communication: a key player in brain physiology and a prime target in pathology.Biochim Biophys Acta 2014, 1843(10):2211–2232. [DOI] [PubMed]
- 94.Kielian T. Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem. 2008;106:1000–1016. doi: 10.1111/j.1471-4159.2008.05405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Doring B, Frisch C, Sohl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 99.Kielian T, Esen N. Effects of neuroinflammation on glia-glia gap junctional intercellular communication: a perspective. Neurochem Int. 2004;45:429–436. doi: 10.1016/j.neuint.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 100.Orellana JA, Martinez AD, Retamal MA. Gap junction channels and hemichannels in the CNS: regulation by signaling molecules. Neuropharmacology. 2013;75:567–582. doi: 10.1016/j.neuropharm.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 101.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li WE, Nagy JI. Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur J Neurosci. 2000;12:2644–2650. doi: 10.1046/j.1460-9568.2000.00162.x. [DOI] [PubMed] [Google Scholar]
- 103.Tence M, Ezan P, Amigou E, Giaume C. Increased interaction of connexin43 with zonula occludens-1 during inhibition of gap junctions by G protein-coupled receptor agonists. Cell Signal. 2012;24:86–98. doi: 10.1016/j.cellsig.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 104.Furman JL, Mohmmad Abdul H, Norris CM. Alterations in connexin 43 phosphorylation during the progression of Alzheimer’s disease: possible role of astrocytic calcineurin. Alzheimers Dement. 2012;8:P300. doi: 10.1016/j.jalz.2012.05.814. [DOI] [Google Scholar]
- 105.Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin DT, Wu J, Holstein D, Upadhyay G, Rourk W, Muller E, Lechleiter JD: Ca2+signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes.Neurobiol Aging 2007, 28:99–111. [DOI] [PubMed]
- 107.Sama DM, Norris CM. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res Rev. 2013;12:982–995. doi: 10.1016/j.arr.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carafoli E, Genazzani A, Geurini D. Calcium controls the transcription of its own transporters and channels in developing neurons. Biochem Biophys Res Comm. 1999;266:624–632. doi: 10.1006/bbrc.1999.1879. [DOI] [PubMed] [Google Scholar]
- 109.Genazzani AA, Carafoli E, Guerini D. Calcineurin controls inositol 1,4,5-trisphosphate type 1 receptor expression in neurons. Proc Natl Acad Sci U S A. 1999;96:5797–5801. doi: 10.1073/pnas.96.10.5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- 111.Groth RD, Coicou LG, Mermelstein PG, Seybold VS. Neurotrophin activation of NFAT-dependent transcription contributes to the regulation of pro-nociceptive genes. J Neurochem. 2007;102:1162–1174. doi: 10.1111/j.1471-4159.2007.04632.x. [DOI] [PubMed] [Google Scholar]
- 112.Grolla AA, Fakhfouri G, Balzaretti G, Marcello E, Gardoni F, Canonico PL, DiLuca M, Genazzani AA, Lim D: Aβ leads to Ca2+signaling alterations and transcriptional changes in glial cells.Neurobiol Aging 2013, 34:511–522. [DOI] [PubMed]
- 113.Norris CM, Blalock EM, Chen KC, Porter NM, Landfield PW: Calcineurin enhances L-type Ca2+channel activity in hippocampal neurons: increased effect with age in culture.Neuroscience 2002, 110:213–225. [DOI] [PMC free article] [PubMed]
- 114.Norris CM, Blalock EM, Chen KC, Porter NM, Thibault O, Kraner SD, Landfield PW: Hippocampal ‘zipper’ slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca2+channels with aging.Neurobiol Aging 2010, 31:328–338. [DOI] [PMC free article] [PubMed]
- 115.Yatani A, Honda R, Tymitz KM, Lalli MJ, Molkentin JD: Enhanced Ca2+channel currents in cardiac hypertrophy induced by activation of calcineurin-dependent pathway.J Mol Cell Cardiol 2001, 33:249–259. [DOI] [PubMed]
- 116.Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA: Physical and functional interaction between calcineurin and the cardiac L-type Ca2+channel.Circ Res 2009, 105:51–60. [DOI] [PMC free article] [PubMed]
- 117.Westenbroek RE, Bausch SB, Lin RC, Franck JE, Noebels JL, Catterall WA: Upregulation of L-type Ca2+channels in reactive astrocytes after brain injury, hypomyelination, and ischemia.J Neurosci 1998, 18:2321–2334. [DOI] [PMC free article] [PubMed]
- 118.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008;7:824–835. doi: 10.1111/j.1474-9726.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 124.Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AbetaPP/PS1 double transgenic mice. J Alzheimers Dis. 2010;22:97–105. doi: 10.3233/JAD-2010-100261. [DOI] [PubMed] [Google Scholar]
- 125.Cho HJ, Jin SM, Youn HD, Huh K, Mook-Jung I. Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell. 2008;7:137–147. doi: 10.1111/j.1474-9726.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- 126.Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Forloni G, Demicheli F, Giorgi S, Bendotti C, Angeretti N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res. 1992;16:128–134. doi: 10.1016/0169-328X(92)90202-M. [DOI] [PubMed] [Google Scholar]
- 128.Li Y, Wang J, Sheng JG, Liu L, Barger SW, Jones RA, Van Eldik LJ, Mrak RE, Griffin WS. S100 beta increases levels of beta-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. doi: 10.1046/j.1471-4159.1998.71041421.x. [DOI] [PubMed] [Google Scholar]
- 129.Sastre Ml Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sutinen EM, Pirttilä T, Anderson G, Salminen A, Ojala JO. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J Neuroinflammation. 2012;9:199. doi: 10.1186/1742-2094-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/S0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pierrot N, Ghisdal P, Caumont AS, Octave JN. Intraneuronal amyloid-beta1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–1150. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- 134.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, Banerjee PK, Lahiri DK. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res. 2010;88:143–154. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marklund N, Farrokhnia N, Hånell A, Vanmechelen E, Enblad P, Zetterberg H, Blennow K, Hillered L. Monitoring of β-amyloid dynamics after human traumatic brain injury. J Neurotrauma. 2014;31:42–55. doi: 10.1089/neu.2013.2964. [DOI] [PubMed] [Google Scholar]