

Abstract
Background
Many previous studies have shown that soybean WRKY transcription factors are involved in the plant response to biotic and abiotic stresses. Phakopsora pachyrhizi is the causal agent of Asian Soybean Rust, one of the most important soybean diseases. There are evidences that WRKYs are involved in the resistance of some soybean genotypes against that fungus. The number of WRKY genes already annotated in soybean genome was underrepresented. In the present study, a genome-wide annotation of the soybean WRKY family was carried out and members involved in the response to P. pachyrhizi were identified.
Results
As a result of a soybean genomic databases search, 182 WRKY-encoding genes were annotated and 33 putative pseudogenes identified. Genes involved in the response to P. pachyrhizi infection were identified using superSAGE, RNA-Seq of microdissected lesions and microarray experiments. Seventy-five genes were differentially expressed during fungal infection. The expression of eight WRKY genes was validated by RT-qPCR. The expression of these genes in a resistant genotype was earlier and/or stronger compared with a susceptible genotype in response to P. pachyrhizi infection. Soybean somatic embryos were transformed in order to overexpress or silence WRKY genes. Embryos overexpressing a WRKY gene were obtained, but they were unable to convert into plants. When infected with P. pachyrhizi, the leaves of the silenced transgenic line showed a higher number of lesions than the wild-type plants.
Conclusions
The present study reports a genome-wide annotation of soybean WRKY family. The participation of some members in response to P. pachyrhizi infection was demonstrated. The results contribute to the elucidation of gene function and suggest the manipulation of WRKYs as a strategy to increase fungal resistance in soybean plants.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-014-0236-0) contains supplementary material, which is available to authorized users.
Keywords: Glycine max, Genetic transformation, Fungus resistance, Transcription factors, Asian Soybean Rust, Functional analysis
Background
Soybean (Glycine max) is one of the most important crops in the world. At present, one of the major diseases affecting soybean production is Asian Soybean Rust (ASR), which results from infection with Phakopsora pachyrhizi [1]. Under conditions that are favorable for fungal propagation, infection results in yield losses ranging from 10 to 80% [2-4].
Three infection types have been described on soybean accessions inoculated with P. pachyrhizi: (1) susceptible reaction characterized by “tan” lesions with many uredinia and prolific sporulation; (2) resistant reaction typified by reddish brown lesions with few uredinia and little to moderate sporulation; and (3) resistant reaction with no visible lesions or uredinia, conferring the immune phenotype [5,3]. Six single dominant genes (Rpp1 to Rpp6) conditioning soybean resistance and/or immunity to P. pachyrhizi have been identified so far [5-14]. The effectiveness of these genes is limited through virulent ASR isolates that are able to overcome the resistance mechanism conferred by each of them [1,15]. For this reason, the most successful method to control fungal spread is the application of fungicides, which are costly and have a negative impact on the environment, favor a selection of pathogen resistance and, in severe cases, are ineffective [16]. In this context, understanding the molecular basis of the soybean defense against fungal infection and growth, identifying genes involved in susceptible or resistant response and characterizing their individual roles are key steps for engineering durable and quantitative disease resistance. Therefore, genetic transformation represents a powerful tool for functional studies.
Many studies have implicated a role for soybean WRKY transcription factors in the response to P. pachyrhizi infection [17-22]. WRKY genes might regulate the expression of defense genes, modulating immediate downstream target genes or activating/repressing other transcriptional factors [23].
WRKY transcription factors comprise one of the largest families of regulatory proteins in plants. Previous studies have identified 72 WRKY-encoding genes in Arabidopsis [24], approximately 100 members in rice [25-28], 104 in poplar [29], 86 in Brachypodium distachyon [30], 80 in grape [31] and 116 and 102 genes in two different species of cotton [32]. A genome-wide analysis in primitive eukaryotes [33] revealed the widespread occurrence of WRKY proteins.
The most prominent feature of these proteins is the WRKY domain, which is a highly conserved 60 amino acid region hallmarked by the heptapeptide WRKYGQK followed by a C2H2- or C2HC zinc-finger motif. As deduced from the results of a nuclear magnetic resonance analysis of a WRKY domain of AtWRKY4, the conserved WRKYGQK sequence is directly involved in DNA binding [34], but the zinc finger motif is also required [35]. Most of the well-characterized WRKY proteins bind to the W-box element (C/T)TGAC(C/T) in the promoter region of the target genes [36]. The specificity of the binding site is partly dependent on the DNA sequences adjacent to the W-box core, and the involvement of WRKY factors in protein complexes might be the major criteria in determining promoter selectivity [37].
The identification of 64 WRKY genes expressed in various soybean tissues and in response to abiotic stress was previously assessed using RT-PCR [38]. However, due to the unavailability of the complete soybean genome sequence at that time, the number of members of this gene family was underrepresented. Yin et al. [39] identified 133 WRKY members in soybean genome. Now a day, several databases for soybean genome analysis are publicly available. PlantTFDB [40] SoyDB [41] and SoyTFKB [42] are transcription factor databases which contain valuable information, including protein sequence, protein domains, predicted tertiary structures and links to external databases. However, despite the usefulness, these databases have performed systematic annotations resulting in different numbers of soybean WRKY transcription factors and some incorrect gene models. So, until now, there is no a comprehensive curate list of soybean WRKY genes. Besides, there is inconsistent nomenclature for soybean WRKY members in the literature. The Phytozome database (http://www.phytozome.org) assigns names from Arabidopsis orthologs, while Zhou et al. [38] identified 64 soybean WRKY genes (deposited in http://www.ncbi.nlm.nih.gov/) and randomly assigned a number to each gene. Moreover, studies of the individual genes [43,44] have assigned numbers different from those proposed by Zhou et al. [38]. The present study reports a genome-wide annotation of the WRKY family in soybean and a functional analysis of some genes involved in response to P. pachyrhizi infection.
Results
Annotation and in silico characterization
In total, 182 potentially WRKY-encoding genes were identified and annotated in the present work (Table 1 and Additional file 1). Additionally, a total of 33 putative WRKY pseudogenes were found (Additional file 2). Some of them were identified in our search and other ones were previously described in the USM data set [45]. Transcripts for 152 annotated WRKY genes were detected on SoyBase EST database (http://soybase.org/) and/or on five global expression experiments: SuperSAGE of soybean leaves 12, 24 and 48 hours after inoculation (hai) of P. pachyrhizi [46], RNA-Seq of microdissected lesions 10 days after inoculation of P. pachyrhizi, two different microarrays of leaves 12 and 120 hai of P. pachyrhizi (available in the current literature) and RNA-Seq expression data of healthy plants in different developmental stages [47], available at SoyBase [48]. The GmWRKY genes were distributed over the 20 soybean chromosomes with protein sequences ranging from 121 to 1,356 amino acids in length (Table 1 and Additional file 1). There was an average of 9.1 WRKY genes per chromosome, with the highest number of genes (15 genes) located on chromosome 6.
Table 1.
Annotation of Glycine max WRKY transcription factors (Choromosome 1 to 3)
| Chr | Gene ID a | Name b | Alternative transcripts | CDS (pb) | Protein (aa) | Groups c | Expression | Soybase | Domain modifications |
|---|---|---|---|---|---|---|---|---|---|
| (Phytozome) | Confirmed d | EST ID | |||||||
| 1 | Glyma01g05050 | GmWRKY3 | 1 | 1530 | 510 | IIb | + | - | |
| 1 | Glyma01g06550 | GmWRKY9 | 1 | 1368 | 456 | I | + | EU019557.1 | |
| 1 | Glyma01g06870 | GmWRKY28 | 2 | 894 | 298 | IIc | + | CA938308.1 | |
| 1 | Glyma01g31921 | GmWRKY5 | 2 | 1524 | 508 | I | + | EU019554.1 | WRKYGQK → WRKYGEK (N-terminal) |
| 1 | Glyma01g39600 | GmWRKY35 | 2 | 966 | 322 | IId | + | BG651351.1 | |
| 1 | Glyma01g43130 | GmWRKY65 | 1 | 738 | 246 | IIe | + | - | CX(N)CX(N)HXH/C → CX(N)CX(N)HXD |
| 1 | Glyma01g43420 | GmWRKY12 | 1 | 969 | 323 | III | + | EU019558.1 | |
| 2 | Glyma02g01031 | GmWRKY66 | 1 | 1455 | 485 | IIb | - | - | |
| 2 | Glyma02g01420 | GmWRKY67 | 1 | 963 | 321 | IIc | + | BT096212.1 | |
| 2 | Glyma02g02430* | GmWRKY68 | 2 | 1443 | 481 | IIb | - | - | |
| 2 | Glyma02g12490 | GmWRKY69 | 1 | 1368 | 456 | I | + | FK022538.1 | |
| 2 | Glyma02g12830 | GmWRKY32 | 1 | 882 | 294 | IIc | + | BM527576.1 | |
| 2 | Glyma02g15920 | GmWRKY22 | 4 | 1068 | 355 | IId | + | AK244154.1 | |
| 2 | Glyma02g36510 | GmWRKY70 | 1 | 1518 | 506 | I | + | FG988660.1 | |
| 2 | Glyma02g39870 | GmWRKY39 | 1 | 1743 | 581 | I | + | BM188894.1 | |
| 2 | Glyma02g45530 | GmWRKY71 | 1 | 1014 | 338 | IIc | + | BE020472.1 | |
| 2 | Glyma02g46280 | GmWRKY72 | 2 | 1206 | 402 | IIb | - | - | |
| 2 | Glyma02g46690 | GmWRKY73 | 2 | 1767 | 589 | I | + | BG789786.1 | |
| 2 | Glyma02g47650 | GmWRKY74 | 1 | 1524 | 508 | I | + | CO984087.1 | |
| 3 | Glyma03g00460 | GmWRKY75 | 1 | 816 | 272 | III | + | BT095645.1 | |
| 3 | Glyma03g05220 | GmWRKY76 | 1 | 1524 | 508 | I | + | EV272592.1 | WRKYGQK → WRKYGEK (N-terminal) |
| 3 | Glyma03g25770 | GmWRKY77 | 1 | 717 | 239 | IIc | + | EV274902.1 | |
| 3 | Glyma03g31630 | GmWRKY15 | 2 | 1026 | 342 | IId | + | CD397604.1 | |
| 3 | Glyma03g33376 | GmWRKY29 | 2 | 1347 | 449 | I | + | EU019569.1 | |
| 3 | Glyma03g37870 | GmWRKY41 | 1 | 762 | 254 | IIe | + | EU019577.1 | |
| 3 | Glyma03g37940 | GmWRKY51 | 1 | 864 | 288 | IIc | + | BT098285.1 | |
| 3 | Glyma03g38360 | GmWRKY78 | 2 | 1626 | 542 | IIb | + | DB956313.1 | |
| 3 | Glyma03g41750 | GmWRKY43 | 1 | 1089 | 363 | III | + | EU019579.1 |
aReannotated genes with original sequences containing wrong start\stop codons are marked with (*).
bThe names GmWRKY1-64 are given according to Zhou et al. [38]; GmWRKY65-182 are given according to the chromosome order.
cThe classification according to Eugelm et al. [24].
dThe expression confirmation according to SoyBase ESTs, RNA-Seq analysis (in silico analysis) and RNA-Seq of ASR lesion microdissection (experimental analysis).
The proteins were assigned to three major groups and subgroups in accordance with Eugelm et al. [24]. Group I, II and III contained 31, 126 and 25 soybean WRKY genes, respectively (Table 1 and Additional file 1). A total of 13, 33, 42, 16 and 22 proteins were assigned to subgroups IIa, IIb, IIc, IId and IIe, respectively.
Although the WRKYGQK signature was highly conserved in the soybean WRKYs, 15 proteins with amino acid substitutions in the signature of the C-terminal domain were identified. These variant proteins were distributed among all groups, except subgroup IId. WRKYGKK was the most common variant and was shared by 11 genes. Other atypical sequences, such as WRKYGEK, WRKYEDK, WKKYGQK, CRKYGQK and WHQYGLK, occurred in single proteins. Nine WRKY proteins contained incomplete and/or amino acid substitutions in the zinc-finger sequence (Table 1 and Additional file 1). Some of these proteins contained patterns of zinc-finger motifs that have not been reported in the literature. Expression was detected for nine genes presenting modifications in the WRKY signature and for six genes with modifications in the zinc-finger motif, indicating that these genes might be functional. Moreover, another highly conserved domain, the zinc cluster, was identified upstream of the WRKY domain in IId gene members.
The phylogenetic approach performed with the WRKY domain sequences confirmed the division of GmWRKY members in the five groups (I, IIa + IIb, IIc, IId + IIe and III) (Figure 1 and Additional file 3). These groups correspond to the WRKY domain classification (groups and subgroups I, IIa, IIb, IIc, IId, IIe and III) that has already been demonstrated in other studies. Genes from Group IIa are closely related with those from Group IIb, while genes from Group IId are closely related with those from Group IIe.
Figure 1.
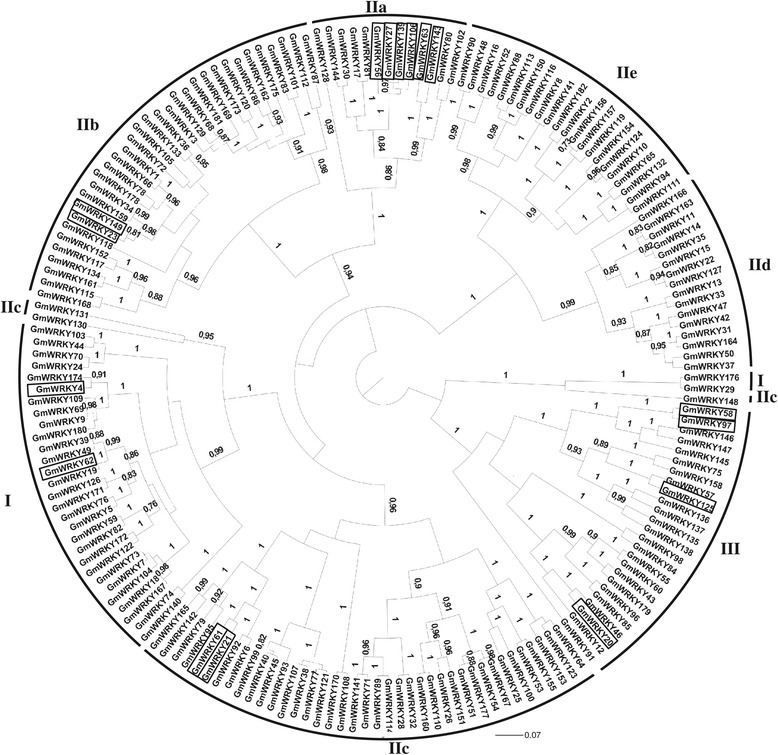
Dendogram representing the relationship among the soybean WRKY proteins. The tree was reconstructed using a Bayesian (BA) method. A total of 182 amino acid sequences from G. max and 65 sites corresponding to WRKY domain were included in the analysis. The posteriori probability values are labeled above the branches and only values higher than 70% are presented. The groups I, IIa, IIb, IIc, IId, IIe and III are indicated. Differentially expressed genes in response to P. pachyrhizi infection are boxed in black.
Gene expression data
An overview of the differential expressed soybean WRKY genes that were modulated in response to P. pachyrhizi infection is presented in Table 2 and Additional file 4. The expression data were obtained from four global expression experiments: SuperSAGE of leaves 12, 24 and 48 hours after inoculation (hai), RNA-Seq of microdissected lesions 10 days after inoculation and two different microarrays of leaves 12 and 120 hai, available in the current literature [17,22]. Seventy-five genes showed differential expression in at least one experiment, whereas 16 genes showed differential expression in more than one experiment. Genes from groups I, II and III responded to this stress condition.
Table 2.
Expression pattern of WRKY encoding-genes under P. pachyrhizi infection a (Group I and IIa)
| Group | Gene ID | SuperSage - LGE | RNA-Seq of lesion LCM b | Microarray – van de Mortel et al. [ 17 ] c | Microarray - Schneider et al. [ 22 ] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Incompatible reaction (PI561356- Rpp1 ) | PI561356 X BRS231 | Incompatible reaction (PI230970- Rpp2 ) | Compatible reaction (Embrapa48) | Compatible reaction (PI462312- Rpp3 X Taiwan 80-2) | Incompatible reaction (PI462312- Rpp3 X Hawaii 94-1) | ||||||
| Inoculated X Mock | Inoculated | Inoculated X Mock | Inoculated X Mock | Inoculated X Mock | Inoculated X Mock | ||||||
| 12, 24, 48 h | 10 days | 12 h | 120 h | 12 h | 120 h | 12 h | 144 h | 12 h | 144 h | ||
| I | Glyma03g05220 | x | x | x | x | ||||||
| I | Glyma01g31921 | x | x | x | x | ||||||
| I | Glyma18g44030 | x | x | x | |||||||
| I | Glyma09g41670 | x | x | x | x | ||||||
| I | Glyma18g06360 | x | x | x | x | x | |||||
| I | Glyma11g29720 | x | x | x | x | ||||||
| I | Glyma14g38010 | x | x | x | x | ||||||
| I | Glyma02g39870 | x | x | x | x | ||||||
| I | Glyma09g38581 | x | |||||||||
| I | Glyma04g12830 | x | x | x | x | ||||||
| I | Glyma06g47880 | x | x | x | x | ||||||
| I | Glyma08g43770 | x | x | x | x | ||||||
| I | Glyma18g09040 | x | x | x | x | ||||||
| I | Glyma07g35381 | x | |||||||||
| I | Glyma18g49830 | x | x | ||||||||
| I | Glyma08g26230 | x | x | x | |||||||
| IIa | Glyma04g06470 | x | |||||||||
| IIa | Glyma17g33920 | x | x | x | x | x | |||||
| IIa | Glyma14g11920 | x | x | x | x | x | x | x | |||
| IIa | Glyma15g00570 | x | x | x | x | x | x | ||||
| IIa | Glyma13g44730 | x | x | x | x | ||||||
| IIa | Glyma08g23380 | x | x | x | x | x | x | ||||
| IIa | Glyma07g02630 | x | x | x | x | x | |||||
| IIa | Glyma17g33891 | x | x | x | x | ||||||
| IIa | Glyma14g11960 | x | x | x | x | x | |||||
aThe expression data were obtained from four global expression experiments: SuperSAGE available at www.lge.ibi.unicamp.br/soja/, RNA-Seq of microdissected lesions and two different microarrays available in the current literature. The x denotes significant differences (p < 0.05). The genes indicated in bold were used in further analyses. The genes were ordered according to the clustering analysis.
bLCM: laser-capture microdissection.
cSome probes hybridized with more than one gene.
Some of the genes that presented differential expression profiles in response to the fungus were randomly selected from each classification group for more detailed analyses. GmWRKY27 (Glyma15g00570) and GmWRKY125 (Glyma09g41050) were differentially expressed in three of the four experiments, while GmWRKY56 (Glyma08g23380), GmWRKY106 (Glyma07g02630) and GmWRKY20 (Glyma08g02580) in the two microarrays. GmWRKY139 (Glyma13g44730), GmWRKY46 (Glyma05g36970), GmWRKY57 (Glyma18g44560) were also analyzed because they were closely related to at least one of the genes evaluated above. Interestingly, none of these genes was expressed in rust infection lesions at ten days after fungus inoculation (RNA-Seq).
The differential expression of these genes was confirmed using RT-qPCR. The transcript levels during the course of fungus infection in a resistant genotype (PI561356) and in a susceptible genotype (Embrapa-48) were compared with those in the mock-inoculated plants (Figure 2).
Figure 2.
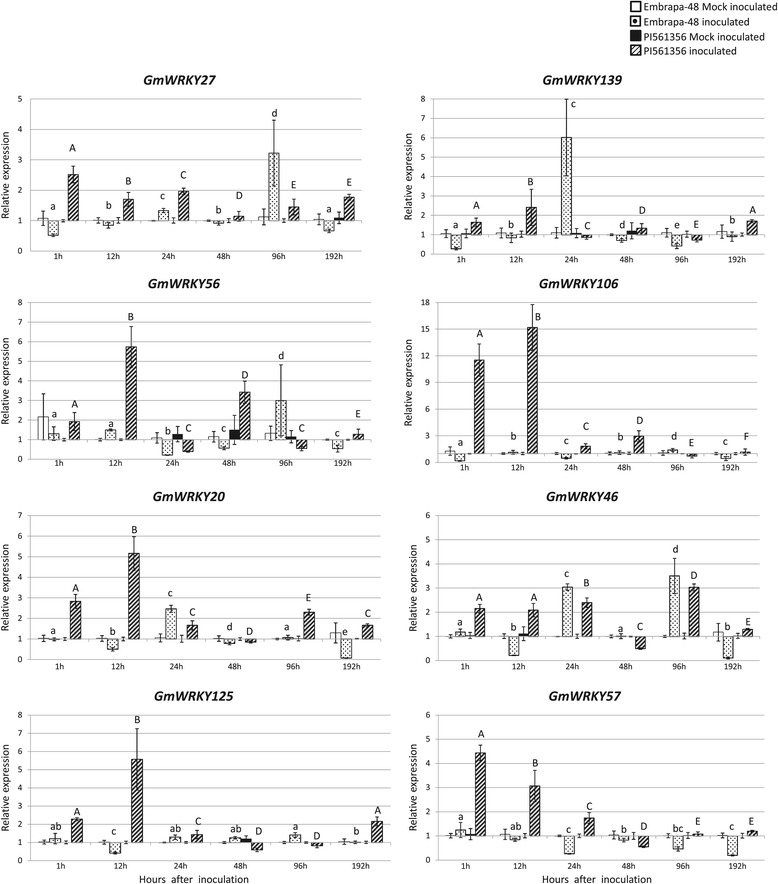
Expression patterns of WRKY genes in leaves of three-week-old soybean plants infected with P. pachyrizi. The gene response in susceptible (Embrapa-48) and resistant (PI 561356) genotypes during P. pachyrizi infection (inoculated) was evaluated using RT-qPCR. Mock-inoculated plants were used as a control. The values (mean ± SD) were calculated based on three biological replicates and four technical replicates. Multifactorial analysis of three factors (genotype, treatment and time) was highly significant: GmWRKY57, GmWRKY27, GmWRKY125, GmWRKY20 and GmWRKY46 p = 0.0001; GmWRKY139 p = 0.0265; GmWRKY56 p = 0.0003. The means indicated with the same letters in the same cultivar and treatment did not differ significantly (Tukey’s multiple comparison test, p < 0.05). Lower case letters were used to identify differences among inoculated Embrapa-48 plants and capital letters were used to identify differences among inoculated PI561356 plants. F-Box protein and metalloprotease reference genes were used as internal controls to normalize the amount of mRNA present in each sample. Transcript levels of WRKY genes present in mock-inoculated plants were used to calculate transcript accumulation in the inoculated plants.
The interaction among the genotypes, time-course and pathogen presence was highly significant (p < 0.0001). In the inoculated plants, the eight genes showed early expression in PI561356 (resistant) compared with Embrapa 48 (susceptible). In the Embrapa 48, the expression peaks were higher at 24 and/or 96 hai, while in PI561356, these peaks varied from one to 24 hai. Furthermore, GmWRKY56, GmWRKY106, GmWRKY20 and GmWRKY125 presented a stronger response in the resistant genotype. Interestingly, the homologous genes (GmWRKY27 and GmWRKY139, GmWRKY125 and GmWRKY57) did not overlap with their expression peaks in the resistant genotype. GmWRKY27 and GmWRKY57 showed higher expression levels at one hai followed by a decrease in expression, whereas GmWRKY139 and GmWRKY125 presented higher transcript levels at 12 hai.
GmWRKY27 overexpression and silencing in soybean plants
GmWRKY27 was selected for further functional characterization because it was one of the genes that showed differential expression in different experiments. Furthermore, it was also shown that this gene is involved in different abiotic stresses [38]. To determine the functional role of the GmWRKY27 in response to P. pachyrhizi infection, soybean somatic embryos were transformed to obtain gene overexpression and silencing. In the overexpression experiments, GFP expression was detected in hygromycin-resistant globular embryos (Additional file 5A and B). The histodifferentiated embryos of nine independent transgenic lines (seven from Biobalistic and two from bombardment/Agrobacterium) were obtained. The presence of the T-DNA in the embryo genomes was confirmed using PCR, and the GmWRKY27 expression was significantly higher in the embryos of the four independent transgenic lines (Additional file 5C). However, the development of transgenic embryos overexpressing GmWRKY27 was not successful. As a consequence, those embryos were not able to develop into plants.
For gene silencing, a vector carrying a 176-bp inverted-repeat fragment sequence from GmWRKY27 was constructed. This fragment shared 83% similarity with the homologous region of GmWRKY139 and 70% and 67% similarity with GmWRKY56 and GmWRKY106 respectively. These data confirm the close relationship among the genes, which was also observed in the phylogenetic analysis (Figure 1). This high sequence similarity suggests that the silencing construct would target the four genes.
A more detailed structural analysis of the four homologous genes showed that the WRKYGQK signature, zinc-finger motif and other residues in the sequences were highly conserved among the four corresponding proteins (Figure 3A). The sequence identity of the complete proteins varied from 66% to 94% (Table 3). The four soybean genes were putative orthologs of AtWRKY40, AtWRKY18 and AtWRKY60 Arabidopsis genes, as shown in the phylogenetic tree (Additional file 3). The gene structure of GmWRKY27, GmWRKY139, GmWRKY56 and GmWRKY106 was similar, with the WRKY domain present in the fourth exon (Figure 3B). Interestingly, GmWRKY56 had four alternative transcripts, and one of the transcripts lacked the WRKY domain.
Figure 3.

Amino acid alignment, conserved residues and structure of the four soybean WRKY genes. (A) Amino acid alignment and identification of conserved residues. The conserved WRKY amino acid signature and the amino acid forming the zinc-finger motif are highlighted in black and gray, respectively. Other conserved amino acids are boxed in black. Multiple sequence alignment was performed using CLUSTAL W 2.1. Highly conserved residues are indicated by (*), strongly similar by (:) and weakly similar by (.). (B) Structure of WRKY-encoding genes. Glyma08gg23380.1, Glyma08gg23380.2, Glyma08gg23380.3 and Glyma08gg23380.4 are alternative transcripts of Glyma08gg23380. The gray boxes represent exons and the black boxes indicate the exons that contain the WRKY domain. The dotted lines represent introns.
Table 3.
Identity percentage (%) among the sequences of the four soybean and three Arabidopsis WRKY
| GmWRKY139 | GmWRKY56 | GmWRKY106 | AtWRKY40 | AtWRKY18 | AtWRKY60 | |
|---|---|---|---|---|---|---|
| GmWRKY27 | 83,6 | 69,7 | 66,7 | 59,4 | 42,2 | 36 |
| GmWRKY139 | 75,72 | 76,8 | 53,4 | 46,77 | 39,16 | |
| GmWRKY56 | 94,6 | 49,52 | 45,05 | 39 | ||
| GmWRKY106 | 48,9 | 46,94 | 37,94 |
Two independent transgenic lines (cultivar BRSMG 68 Vencedora) carrying the silencing construct were obtained. The molecular analysis revealed that one of the repeats (176-bp fragment) was eliminated from the first line. Therefore, the post-transcriptional silencing was not triggered, which was confirmed using RT-qPCR (data not shown). In the second transgenic line (P3-2) the complete cassette was successfully integrated (data not shown). As anticipated, the results from the RT-qPCR analysis showed that the expression of the four homologous genes was significantly reduced (Figure 4). The transgenic line exhibited no major phenotypic alterations.
Figure 4.

Expression levels (RT-qPCR) of the soybean-silenced transgenic line for the four WRKY genes. Expression levels of the four WRKY genes in a wild-type (wt) soybean plants and in a transgenic soybean line P3-2. F-Box protein and metalloprotease reference genes were used as internal controls to normalize the amount of mRNA present in each sample. Transcript levels of WRKY genes present in the wild type were used to calibrate transcript amounts in P3-2. *Means are significantly different in the wild type and P3-2 plants (Student’s t-test, p < 0.05).
The silenced line was shown to be more susceptible to P. pachyrhizi
A detached leaf assay was performed to confirm the involvement of GmWRKY27, GmWRKY139, GmWRKY56 and GmWRKY106 in the soybean response to P. pachyrhizi infection. As previously described, detached leaf and intact plant bioassays revealed a high correlation [49]. In the present study, “tan” lesions could be observed on all detached leaves of both transgenic and wild type samples at 12 days after P. pachyrhizi inoculation. However, the number of lesions was significantly higher in the leaves of the transgenic line (Figure 5). No visible differences were observed concerning the appearance of the lesions and pustule formation or eruption (data not shown).
Figure 5.
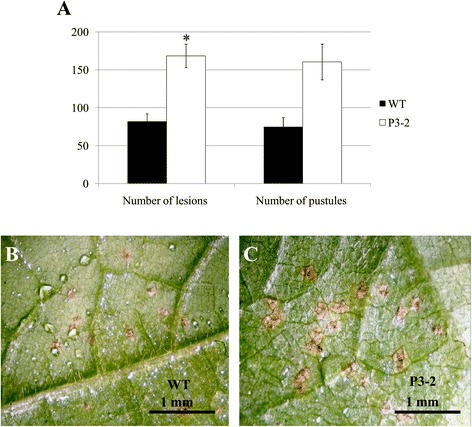
P. pachyrhizi development on the detached leaves at 12 days after inoculation. Three detached leaves of each one transgenic line and two wild-type plants were inoculated with 105/mL spore suspension and incubated at 20°C. (A) Two infection parameters were evaluated: number of lesions and number of pustules. *Means are significantly different in leaves of wild type (wt) and transgenic soybean line P3-2. (Student’s t-test, p < 0.05). (B) Low number of tan-colored lesions and pustules under stereomicroscope in a leaf of wild-type (wt) plant. (C) High number of tan-colored lesions and pustules under stereomicroscope in a leaf of transgenic soybean line P3-2 with suppression of the four WRKYs.
Discussion
Soybean WRKY genes
Whole genome sequencing [50] has facilitated the accurate annotation of soybean gene families. In this study, we present the annotation of 182 WRKY transcription factors in soybean. The transcripts of 152 genes were detected, suggesting they can be expressed at the protein level; however, specific conditions might be necessary for the successful transcription of the remaining genes.
As discussed before, there is inconsistent nomenclature for soybean WRKY members in the literature. To unify the terminology, we proposed a nomenclature based on the previously described WRKY-encoding genes [38], with some modifications. Data from sequence comparisons have shown that GmWRKY18 and GmWRKY35 is the same gene. In addition, GmWRKY3 does not exist in the soybean genome; indeed, this sequence represents a chimeric transcript produced through trans-splicing between N-terminal and C-terminal sequences from Glyma02g46690 and Glyma14g01980, respectively. The remaining 118 genes were numbered according to the order of the chromosomes (Table 1 and Additional file 1).
More WRKY genes have been identified in soybean than in other species, such as rice, Arabidopsis, cotton, grape and B. distachyon [24-28]. The duplication events have been greatly over-retained, specifically in the case of transcription factors [51]. Thus, functional redundancy is a common feature in plant species. However, homologous genes might diverge in function providing a source of evolutionary novelty [52].
The phylogenetic approach used in this study allowed the division of the soybean WRKY genes in the five groups previously reported [26,53,54].
In soybean, the members of group I contained domains with a C2H2-type zinc-finger motif. The same characteristic is observed in Arabidopsis, while in rice, the WRKY domains of group I members include two types of zinc-finger motifs: C2H2 and C2HC [25,27].
Although the WRKYGQK signature was highly conserved among soybean WRKY proteins, as illustrated in Figure 6, variation was identified in 21 genes. Zhou et al. [38] previously showed that GmWRKY6 (Glyma08g15050) and GmWRKY21 (Glyma04g39650) contain the variant WRKYGKK rather than the conserved WRKYGQK motif. Slight variations in this region have also been reported in Arabidopsis, rice, tobacco, barley, canola and sunflower [25,26,55-58]. Compared with Arabidopsis, which contains four WRKYGKK variants, the number of genes with a modified WRKYGQK motif is greater in soybean.
Figure 6.

Conservation analysis of the consensus sequence of the WRKYGQK domain. Analysis of the 182 soybean WRKY genes identified was performed using the MEME suite. The overall height in each stack indicates the sequence conservation at each position. The height of each residue letter is proportional to the relative frequency of the corresponding residue. Amino acids are colored according to their chemical properties: green for polar, non-charged, non-aliphatic residues (NQST), magenta for the most acidic residues (DE), blue for the most hydrophobic residues (A, C, F, I, L, V, W and M), red for positively charged residues (KR), pink for histidine (H), orange for glycine (G), yellow for proline (P) and turquoise for tyrosine (Y).
Some unusual GmWRKY-encoding genes (i.e., containing a modified WRKY signature and/or zinc-finger motif) produced mRNA (Table 2 and Additional file 4). Further analyses are necessary to determine whether these genes function as transcription factors or if they induce post-transcriptional regulation through RNAi, as previously suggested [23]. Variant proteins might have abolished or decreased capacities to bind to the W-box [35,37]. It has been suggested that WRKY proteins without the canonical WRKYGQK motif might have different binding sites [37,56], target genes and possibly divergent roles [57].
Functional analysis
Despite the fact that the identification or prediction of many WRKY genes from different species has been previously achieved, only a small number of these have been functionally characterized. Information concerning the role of soybean genes (Glyma13g00380-GmWRKY13, Glyma04g39650-GmWRKY21, Glyma10g01450-GmWRKY54 and Glyma18g44560-GmWRKY57) during abiotic stress has been based on data obtained from heterologous expression systems [38,43]. The data from expression analyses [17,44] or using transient gene silencing [59] supports a role for the WRKY genes in response to biotic stresses. Studies concerning global expression profiling have demonstrated the importance of WRKY-encoding genes in transcriptional reprogramming during P. pachyrhizi infection in soybean plants [17-22].
To determine which soybean WRKY genes are involved in plant defense against P. pachyrhizi infection, we performed a series of analyses to examine their expression patterns after infection. We initially compared the microarray data available in the literature [17,22] with the results obtained from two additional experiments: SuperSAGE and RNA-Seq. Many genes were differentially expressed in only one library, while a few of them showed differential expression in more than one library. The modulation in the transcript levels of eight genes was validated, showing the reliability of data mining. The similar expression patterns in response to P. pachyrhizi infection was observed among closely related genes (Figure 1), such as GmWRKY61 (Glyma06g15220) and GmWRKY21 (Glyma04g39650), GmWRKY143 (Glyma14g11920) and GmWRKY63 (Glyma17g33920), GmWRKY106 and GmWRKY56, GmWRKY58 (Glyma04g40130) and GmWRKY97 (Glyma06g14720). This similar expression pattern suggests that these genes might share similar functions in disease resistance. The redundant function of GmWRKY genes might be beneficial in protecting the cell or organism under various stress conditions and eliciting multiple pathways that lead to the wide array of physiological responses that occur following pathogen infections [60].
Global expression data have suggested that the timing and the degree of induction of the defense pathway are determinants for the induction of soybean resistance to P. pachyrhizi [17,20,22,60]. In our study, the induced expression of GmWRKY20, GmWRKY27, GmWRKY46, GmWRKY57, GmWRKY56, GmWRKY106, GmWRKY125 and GmWRKY139 in response to P. pachyrhizi was earlier and/or stronger in the resistant genotype. The expression of most genes analyzed peaked at 12 hai in the resistant genotype; therefore, we propose that these genes might be involved in non-specific defense responses. Van de Mortel et al. [17] and Schneider et al. [22] reported that P. pachyrhizi infections induce biphasic global expression. Gene expression initially peaked at 12 hai, which corresponded with the early infection processes of appressoria formation and epidermal cell penetration. The authors suggested that this peak corresponded to a non-specific defense response similar to pathogen-triggered immunity. A second phase of gene expression, which began at 72 hai and continued until 288 hai, is coincident with haustoria formation and effector protein secretion. The authors suggested that this response is consistent with the activation of RPP2 and RPP3-mediated resistance. It has been shown that gene expression is rapid and increased in the incompatible interaction [17,18,22].
The closely related genes GmWRKY27, GmWRKY139, GmWRKY56 and GmWRKY106 are putative orthologues of AtWRKY40, AtWRKY18 and AtWRKY60 Arabidopsis genes. In both species, these genes were classified into group IIa. The three Arabidopsis WRKYs are involved in stress responses, which include resistance against the bacteria Pseudomonas syringae and fungus Botrytis cinerea [61,62]. AtWRKY18 is a salicylic acid-induced gene that positively regulates SAR [63,64] and modulates PR gene expression; AtWRKY18 overexpression increases resistance to P. syringae [65]. AtWRKY40 and AtWRKY60 proteins antagonize AtWRKY18 during P. syringae infection. The gain or loss of gene function in single, double or triple combination mutants resulted in increased susceptibility to B. cinerea [61]. Some rice, barley and Brassica napus WRKY members from group IIa are also involved in the response to fungal and bacterial pathogens, as demonstrated using expression studies. OsWRKY62 and OsWRKY76 are upregulated in Magnaporthe grisea infected-leaves and downregulated in Xanthomonas oryzae-inoculated leaves [66]. HvWRKY1 and HvWRKY2 play an important role in response to Blumeria graminis infection [55], and BnWRKY18 and BnWRKY40 play a role in the response to Sclerotinia sclerotiorum and Alternaria brassicae infections [57].
Most available information concerning soybean gene function is based on data obtained from heterologous expression systems. However, as the activity of many proteins frequently depends on specific interactions that are only found in homologous backgrounds, the present study was based on a homologous expression system. An RNA interference approach was used for the silencing of four soybean homologous genes (GmWRKY27, GmWRKY139, GmWRKY56 and GmWRKY106). The quadruple silencing is an advantage because a single knockout of transcription factors rarely results in altered phenotypes due to functional redundancy among closely related members [65]. The transgenic RNAi line used in this study generated a significant reduction in the transcript levels of the four target genes. When infected with P. pachyrhizi, the transgenic line showed increased susceptibility to the fungus. Taken together, the results strongly suggest that at least one of the four genes might be involved in the soybean resistance phenotype.
Pandey et al. [59] silenced 64 soybean WRKYs individually using virus-induced gene silencing (VIGS) to test their involvement in Rpp2-mediated resistance against P. pachyrhizi infection. Three of these genes (GmWRKY45, GmWRKY40 and GmWRKY36) compromised the resistance phenotype when silenced. Phenotypic alterations were not evidenced when GmWRKY56 and GmWRKY106 genes were individually silenced. However, in the present study, an increased susceptibility to P. pachyrhizi infection was observed in the quadruple-silenced (GmWRKY27, GmWRKY139, GmWRKY56 and GmWRKY106) line, suggesting that this phenotype is a consequence of GmWRKY27 and/or GmWRKY139 silencing. Moreover, the four genes analyzed in this study could also play a synergistic role in the pathogenic defense response.
A previous study showed that GmWRKY27 is also strongly induced under conditions of drought and salt stress in the soybean [38]. Altogether, these data suggest that this gene is probably involved in a non-specific response that occurs upstream of biotic and abiotic stress defense routes, in contrast with the specific Rpp2-response of the genes identified by Pandey et al. [59] in response to the fungal infection.
GmWRKY27 was selected for use in the overexpression study. Histodifferentiated embryos overexpressing this gene were obtained from four independent transformation experiments. However, the plants were not recovered. The most likely explanation is that the constitutive overexpression of the GmWRKY27 might affect the regeneration of plants. The use of constitutive promoters in investigation of genes whose constant overexpression has deleterious effects on the plant is a major limitation [67]. Chen and Chen [65] reported that high levels of AtWRKY18 cause severe abnormalities in plant growth. Even at moderate levels, the individual or combinatorial overexpression of AtWRKY18, AtWRKY40 and AtWRKY60 leads to the development of smaller plants or death shortly after germination [61].
The deleterious effect of the excessive production of these WRKYs during plant growth suggested that the expression of this gene might require proper regulation during the activation of plant defense responses. However, in healthy plants, the expression of these genes is negatively regulated, as demonstrated by Chen and Chen [65] for the AtWRKY18.
To a certain extent, the lethality problems observed in this study could be partially overcome using tissue-specific, developmentally regulated or inducible promoters. Although the number of tissue-specific promoters has increased in recent years, soybean leaf-specific promoters are still unavailable.
Conclusions
In the present study, 182 WRKY transcription factors were annotated in soybean. Seventy-five genes were identified as involved in the soybean response to P. pachyrhizi infection based on transcriptional regulation. The participation of four genes in response to pathogen infection was demonstrated using an RNAi approach. Further investigations are required to provide clues regarding the functions of the individual genes. The results contribute to the elucidation of gene function and suggest the manipulation of WRKYs as a strategy to increase fungal resistance in soybean plants.
Methods
Database search and sequence annotation
To search for Glycine max (Gm) WRKY transcription factor we use two different approaches as follow: first we downloaded soybean proteome from Phytozome (http://www.phytozome.org) and SoyBase (http://soybase.org/) databases to perform a Batch BLAST using BLASTALL software [68]. The WRKY domains previously identified in Arabidopsis [40], poplar [40] and soybean [40-42,45] genomes were checked on the SMART Web Site and were used as queries to perform tblastp (e-value cut off of 10) searches. After doing Batch BLAST searches we checked for soybean WRKY genes in PlantTFDB (http://planttfdb.cbi.pku.edu.cn/) transcription factor database and USM data set [45].
Additionally, we used the coding sequences (CDS) to perform blast searches against the Phytozome database (www.phytozome.org) and PLAZA (http://bioinformatics.psb.ugent.be/plaza/) to retrieve any additional WRKY genes. The Phytozome database was also used to obtain the gene structures. The automated WRKY-predicted gene sequences that contained incorrect gene models (wrong start/stop codons or truncated proteins) were reannotated using GENSCAN [69] and FEGENESH [70] predictors, considering 2, 5 or 10-kb DNA sequences obtained from Gbrowse. The sequences were aligned with ClustalX v2.1 [71], and the domains manually examined. The sequences without conserved WRKYGQK domain signatures were discarded. The degree of conservation of the WRKYGQK and zinc finger domains was analyzed using the MEME suite (http://meme.sdsc.edu/meme/). The annotated genes were classified in groups and subgroups proposed consistent with the methods of Eugelm et al. [24] for Arabidopsis thaliana. A nomenclature for the WRKY-encoding genes identified in this work was adopted, according to the order of the chromosomes. The structures of the four soybean WRKY-encoding genes selected to the functional analysis and their alternative transcripts were analyzed using Fancy Gene v1.4 [72].
Soybean WRKY relationships
In order to classify the soybean WRKY genes identified, a phylogenetic approach was performed with two dataset: the first one contained only soybean WRKY sequences and the second included also Arabidopsis thaliana and Populus trichocarpa WRKY sequences, downloaded from PlantTFDB database. The multiple sequences alignments were performed with MUSCLE software [73], implemented in MEGA5 (Molecular Evolutionary analysis) software [74]. Phylogenetic analyses were conducted with WRKY domain sequences using Bayesian approach implemented in BEAST1.7 software [75]. The best-fit model of protein evolution was determined using ProTest [76], which selected the JTT model for protein matrix substitution. The Yule tree was selected as a tree prior for Bayesian analysis and 30.000.000 generations were performed with Markov Chain Monte Carlo (MCMC) algorithms. The trees were visualized and edited in FigTree v1.3.1 software [77].
Gene expression data mining
The GmWRKY CDSs were searched into RNA-Seq expression data [47] which is available at SoyBase [48]. In addition, the expression profiles of the WRKY genes that were modulated in response to P. pachyrhizi infection were obtained from four different sources. The reaction of soybean plants to rust infection of the first three experiments was assessed by the inoculation of P. pachyrhizi spores collected in the field into plants maintained under greenhouse conditions at Embrapa Soja, Londrina, PR, Brazil. The sources used to obtain the expression profiles of the WRKY genes are described:
a) SuperSAGE: The libraries were constructed using the leaves of a soybean resistant genotype (PI561356), which carries the Rpp1 resistance gene, infected with P. pachyrhizi vs. uninfected leaves (mock inoculation/control) collected at 12, 24 and 48 hours after inoculation (hai). A Plant RNeasy kit (Qiagen) was used for RNA extraction and equal amounts of RNA from each sample were used to construct the RNA pools. The libraries (inoculated and mock) were constructed at GenXPro GmbH (Frankfurt, Germany) using previously described methods [78] and subsequently sequenced using the Illumina Genome Analyzer IIx. The SuperSAGE tags were analyzed using the DiscoverySpace software v.4.01 [79] to identify unique (unitags) and differentially expressed tags (p ≤ 0.05). The libraries were constructed as part of the GenoSoja project (Brazilian Soybean Genome Consortium), and the results are available in the LGE (Laboratório de Genômica e Expressão, UNICAMP) Soybean Genome database [80] for members of the consortium.
b) RNA-Seq of lesion LCM (Laser Capture Microdissection): foliar segments (1 cm2) containing P. pachyrhizi lesions from two soybean resistant (PI561356) and susceptible BRS231 [81] genotypes at the V2 growth stage were collected at 10 days after infection. The leaf segments were immediately fixed on ice in Farmer’s solution [82], dehydrated and embedded on paraffin in accord with the methods of Cai and Lashbrook [83]. Serial sections of 12-μm in thickness were generated using a rotary microtome and transferred to microscope membrane slides. Twenty sections containing a variable number of rust lesions were prepared for each biological replicate/treatment. The PixCell II LCM system (Arcturus) and CapSure Macro LCM (Arcturus) were used to collect the foliar cells within the lesion. Total RNA was extracted using the PicoPure RNA Isolation Kit (Arcturus) from the cells collected at a variable number of infection sites for each biological replicate. The synthesis of cDNA was conducted, and high-performance paired-end (108 bp) sequencing was performed on the Illumina genome analyzer GAAllx. Low-quality RNA-Seq reads were discarded. The reads (a total of 86,301,242) were aligned against the soybean genome, and the corresponding genes were predicted using the TopHat [84] and SOAP2 [85] alignment programs. Gene expression was calculated using the FPKM (fragments per kilobase of exon per million fragments mapped) value [86]. To identify differentially expressed genes, a pair-wise comparison between the FPKM values of both genotypes was performed using a t-test at the 99% confidence level. This library was constructed as part of the Biotecsur Consortium and the results are available [87] for members of the consortium.
c) Microarray [17]: The expression o WRKY genes in the leaves of the soybean resistant genotype (PI970230), which carries the Rpp2 gene, and in the soybean susceptible genotype (Embrapa 48) in response to P. pachyrhizi infection were compared with that of uninfected leaves (mock inoculation). In the present study, the data obtained at 12 and 120 hai were considered because the highest gene expression was exhibited at these time points. Only the 46 probes previously described as WRKYs were examined. The specificity of probes was analyzed using the SoyBase and Phytozome databases. Probes with e-values <0.05 were considered.
d) Microarray [22]: The global expression of the soybean cultivar Ankur (PI462312), which carries the Rpp3 resistance gene, which was inoculated with avirulent (Hawaii 94-1) and virulent (Taiwan 80-2) isolates of P. pachyrhizi, was analyzed. The Affy probe sets were searched using the tools available in the Soybase database. In the present study, only the WRKY probes that hybridized with a single locus in the soybean genome were selected. The data obtained at 12 and 120 hai were considered because the highest gene expression was exhibited at these time points. The genes with a p-value <0.05 were considered as differentially expressed.
P. pachyrhizi bioassay for gene expression analysis
Soybean plants were grown in a pot-based system maintained in greenhouse conditions at 28 ± 1°C under a 16/8 h light/dark cycle with a light intensity of 22.5 μEm-2s-1 in Embrapa Soja, Londrina, PR, Brazil. The Embrapa-48 genotype, which develops a “tan” lesion [17], was used as the susceptible standard, and the PI561356 genotype, which carries the Rpp1 resistance gene [88], was used as the resistant standard. ASR isolated from Brazilian fields was maintained in a susceptible cultivar. Spores harvested from leaves exhibiting sporulating uredinia and diluted in distilled water containing 0.05% Tween-20 to a final concentration of 3 × 105 spores/mL. The spore suspension was sprayed onto plantlets at the V2 developmental stage. The same solution without spores was used for the mock inoculation. Subsequently, the water-misted bags were placed over each pot for one day. One trifoliate leaf from each plant was collected at 1, 12, 24, 48, 96 and 192 hai, frozen in liquid nitrogen, and stored at -80°C. Three biological replicates from each genotype/treatment were analyzed.
Expression pattern analysis using reverse transcription and quantitative real-time PCR (real-time RT-qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) and further treated with DNAse (Promega) according to the manufacturer’s instruction. The first-strand cDNAs were obtained using 2 μg of DNA-free RNA using the M-MLV Reverse Transcriptase System (Invitrogen) with a 24-polyVT primer. The RT-qPCR was conducted using a StepOne Applied Biosystems Real-Time cycler™. The PCR-cycling conditions were implemented as follows: 5 min at 94°C, followed by 40 cycles of 10 s at 94°C, 15 s at 60°C and 15 s at 72°C, and a final step of 2 min at 60°C. A melting curve analysis was performed at the end of the PCR run over a range of 55–99°C, increasing the temperature stepwise by 0.1°C every 1 s. Each 25-μL reaction comprised 12.5 μL of diluted DNA template, 1X PCR buffer (Invitrogen), 2.4 mM of MgCl2, 0.024 mM of dNTPs, 0.1 μM of each primer, 2.5 μL of SYBR Green (1:100000-Molecular Probes Inc.) and 0.03 U of Platinum Taq DNA polymerase (Invitrogen). The cDNA (1:100) templates were evaluated. All PCR reactions were performed in technical quadruplicates. Reactions lacking templates were used as negative controls.
The PCR reactions were performed using gene-specific primers (Table 4). Primer-pairs designed to amplify F-Box proteins and metalloprotease sequences were used to normalize the amount of mRNA present in each sample. These genes were previously confirmed as good reference genes for the experimental conditions used in the present study [89]. The expression analyses were performed after the comparative quantification of amplified products using the 2-ΔΔCt method [90]. The results were statistically compared using variance analysis with three-factor factorial treatments: genotype, time and pathogen presence. The data were transformed using the weighted least squares method. The means were compared using Tukey’s multiple comparison test.
Table 4.
Primer set designed for RT-qPCR
| Target | Orientation | Tm (°C)* | Primer Sequence | PCR product size (pb) |
|---|---|---|---|---|
| GmWRKY20 transcripts | Forward | 60 | 5′-TTGCAAAGTTCAGAAGTATCTTGTC-3′ | 264 |
| Reverse | 60 | 5′-GTGACCTGTTGTAGATCCCATC-3′ | ||
| GmWRKY27 transcripts | Forward | 61.69 | 5′-GATTGTGCATTTGCTAATCATGC-3′ | 105 |
| Reverse | 59.93 | 5′- GCTATAGAAACTTCGCCAGAAC-3′ | ||
| GmWRKY46 transcripts | Reverse | 60 | 5′-CAATGCATCATCAACTTCCG-3′ | 213 |
| Forward | 60 | 5′-CAAGACCACTTTCACAGCTCAC-3′ | ||
| GmWRKY56 transcripts | Forward | 60.06 | 5′-CACCCATCTGCCTCATCAC-3′ | 234 |
| Reverse | 59.17 | 5′-GGAGGCCGAGTCTGTACAAT-3′ | ||
| GmWRKY57 transcripts | Forward | 60 | 5′-TCCCTCAACTTCCTCCAATC-3′ | 170 |
| Reverse | 60 | 5′-GGAAGGGTTCAAAGGCATC-3′ | ||
| GmWRKY106 transcripts | Forward | 59.72 | 5′-GGAAATAAAGTTCCACTAAGGATGAC-3′ | 174 |
| Reverse | 60.57 | 5′ - CCGAGAATGTGTGCTACAACC-3′ | ||
| GmWRKY125 transcripts | Forward | 60 | 5′-TCTCATCTTCCAATAATTTCCCA-3′ | 135 |
| Reverse | 60 | 5′-CATGATGCCTTGGTGAGCTA-3′ | ||
| GmWRKY139 transcripts | Forward | 60.53 | 5′-CAAATCCTTTTGGTGGGAATC-3′ | 145 |
| Reverse | 59.30 | 5′-CTATAGAAATTTCGCAAGAACTTAACC-3′ | ||
| Metalloprotease transcripts | Forward | 60.5 | 5′-ATGAATGACGGTTCCCATGTA-3′ | 114 |
| Reverse | 60.17 | 5′-GGCATTAAGGCAGCTCACTCT-3′ | ||
| FBox transcripts | Forward | 60.25 | 5′-AGATAGGGAAATGTTGCAGGT-3′ | 93 |
| Reverse | 59.84 | 5′-CTAATGGCAATTGCAGCTCTC-3′ |
*Calculated Tm under PCR conditions.
Silencing and overexpression vectors construction
The open reading frame (ORF) of GmWRKY27 (Glyma15g00570), according to Phytozome v1.0, was amplified from the MGBR-46 Conquista soybean cultivar using a high-fidelity Taq DNA Polymerase (Pfu–Fermentas). The Gateway® System (Invitrogen) was used to recombine the PCR product into the overexpression pH7WG2D,1 vector [91]. The T-DNA region of the resulting pH7WG2D,1-GmWRKY27 vector contained the GmWRKY27 gene ORF under the control of the CaMV 35S promoter, the hygromycin-phosphotransferase marker gene (hpt) and the green fluorescent protein reporter gene (gfp) (Figure 7A). A RNAi silencing vector was constructed using pH7GWIWG2(II),0 [91]. The T-DNA region of the resulting pH7GWIWG2(II),0-GmWRKY27 contained inverted repeat fragments (176 bp) from the GmWRKY27 sequence, which were separated by an intron from the Arabidopsis genomic DNA sequence, under the control of the CaMV 35S promoter and the hygromycin-phosphotransferase marker gene (hpt) (Figure 7B). Both constructs were confirmed using DNA sequencing.
Figure 7.
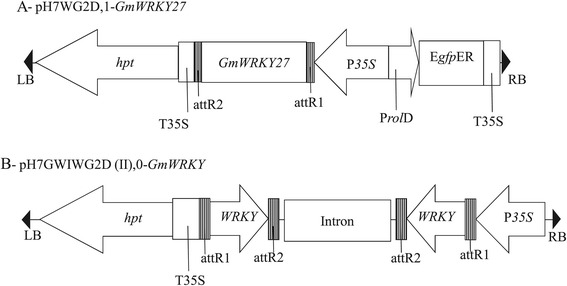
T-DNA region of binary vectors used for Gm WRKY27 overexpression or Gm WRKY silence in soybean. (A) Overexpression construct - pH7WG2D.1-GmWRKY27. The full-length ORF of GmWRKY27 was cloned in the vector. (B) RNAi suppression construct - pH7GWIWG2(II).0-GmWRKY. Inverted repeats of a 176-bp WRKY fragment was cloned into the vector. RB – T-DNA right border, LB – left border, hpt – hygromycin phosphotransferase gene, P35S – Cauliflower mosaic virus (CaMV) 35S promoter, T35S – CaMV 35S terminator, EgfpER – enhanced green fluorescent protein, ProlD – root loci D promoter, WRKY – soybean 176 pb WRKY fragment, attB1 and attB2 – LR reaction site.
Soybean transformation and plant regeneration
Pods containing immature seeds of 3-5 mm in length from soybean cultivars MGBR 46 (Conquista), BRSMG 68 (Vencedora) and IAS5 were harvested from field grown plants. They are all susceptible to P. pachyrhizi. Somatic embryogenesis was induced from immature cotyledons and proliferated using the methods of Droste et al. [92]. Proliferating embryogenic tissues were subjected to transformation through particle bombardment using a particle inflow gun (PIG) [93] following the procedure of Droste et al. [92] or using the combined methods of DNA-free particle bombardment and Agrobacterium transformation [94]. After cultivating for three months in hygromycin-B selection medium, the hygromycin-resistant embryogenic soybean tissues were visually selected and individually cultured for the establishment of lines corresponding to putative independent transformation events.
Embryo histodifferentiation, conversion into plants and acclimation were carried out as previously described [92]. All plants derived from an independent sample of hygromycin-resistant tissue were considered as cloned plants. The plants derived from non-transformed embryogenic tissues submitted to the same culture conditions were recovered and used as controls for molecular characterization and bioassays.
Screening for transgenic embryos and plants
Total DNA was extracted [95] from hygromycin-resistant histodifferentiated embryos and plant leaves. The putative transgenic embryos/plants were PCR-screened for the presence of the complete T-DNA using different primer combinations (Table 5). The PCR mixture consisted of 200 ng of template DNA, 0.4 mM of dNTPs, 0.4 μM of each primer, 2.5 mM of MgCl2, 1X Taq Buffer, 1 U of Taq DNA Polymerase (Invitrogen), and autoclaved distilled water in a final volume of 25 μl. The reactions were initially heated (5 min at 94°C) and subjected to 30 cycles of the following conditions: 45 s at 94°C, 45 s at 58°C and 1 min at 72°C. Subsequent to electrophoresis on a 1% agarose gel containing ethidium bromide (0.01 mg/L), the PCR products were visualized under ultraviolet light.
Table 5.
Primer set designed to gene isolation and transgene detection
| Target | Orientation | Tm (°C) | Primer Sequence | PCR product size (pb) |
|---|---|---|---|---|
| 35S | Forward | 52 | 5′-GGACCCCCACCCACGAGGAG-3′ | 139* |
| GmWRKY27 overexpression | Forward | 58 | 5′- CACCATGGATTATTCATCATGGATTAACA-3′ | 921 |
| Reverse | 5′- TTAATTATTATTGTGCAACATTTTTC-3′ | |||
| WRKY (RNAi) | Forward | 58 | 5′- CACCCTTCCTTGGATCTCAACATTAATCT -3′ | 176 |
| Reverse | 5′- TAACTTCTTGTTTTCTGCACTCACC -3′ | |||
| Intron | Forward | 60 | 5′- TGCCTCTTCTTACGGCTTTCTGTG -3′ | 400 |
| Reverse | 5′ - TGCCGTCTGTGATGGCTTCCA -3′ | |||
| Hpt | Forward | 60 | 5′-GCGATTGCTGATCCCCATGTGTGTAT-3′ | 512 |
| Reverse | 5′-GGTTTCCACTATCGGCGAGTACTT-3′ |
*Fragment length was considered from the beginning of the primer sequence until the end of the promoter sequence.
GFP expression was detected under blue light using an Olympus® fluorescence stereomicroscope equipped with a BP filter set containing a 488 nm excitation filter and a 505-530 nm emission filter. The images were captured using the QCapture Pro™ 6 software (QImaging®).
Gene overexpression or silencing was confirmed using RT-qPCR. The RNA extraction, cDNA synthesis and qPCR analysis were performed as described above.
Fungal bioassay
A detached leaf method was used to evaluate the P. pachyrhizi infection [49]. Three fully expanded leaves from each one transgenic line and two wild-type plants (2-month-old) were collected, rinsed with sterile distilled water and cut in 5 cm × 5 cm pieces. For the inoculation, 1 mL of a uredospore suspension (105 spores/mL) was dripped onto each leaf piece, which was subsequently placed with its abaxial side upwards in a Petri dish covered with wet filter paper. The material was incubated at 20°C under a 12/12 h light/dark cycle. The number of lesions and pustules (uredium) was recorded at 12 days after inoculation. A non-parametric Student’s t-test was conducted to compare the effect of P. pachyrhizi on transgenic and non-transgenic plants. The results with p < 0.05 were considered significant.
Acknowledgments
We thank Dr. Elsa Mundstock and Gilberto P. Mesquita from “Núcleo de Assessoria Estatística” from Universidade Ferderal do Rio Grande do Sul for statistical support; to Dr. Cláudia Godoy for providing fungal isolates and Dr. Emerson Del Ponte, Dr. Cláudia Godoy, Dr. Juliano dos Santos, Larissa Bittecourt and Silvia Richter for their technical assistance. This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico and Consórcio Nacional do Genoma da Soja (CNPq-GENOSOJA) and BIOTECSUR (European Union/MERCOSUL).
Additional files
Annotation of Glycine max WRKY transcription factors (Chromosome 4 to 20).
Pseudogenes list.
Phylogenetic tree representing relationship among WRKY proteins of three species. The tree was reconstructed using a Bayesian (BA) method. A total of 289 amino acid sequences from Glycine max, Arabidopsis thaliana and Populus trichocarpa and 65 sites corresponding to WRKY domain were included in the analysis. The posteriori probability values are labeled above the branches and only values higher than 70% are presented. The groups I, IIa, IIb, IIc, IId, IIe and III are indicated. *Differentially expressed genes in response to P. pachyrhizi infection.
Expression pattern of WRKY encoding-genes under P. pachyrhizi infection (Group IIb to III).
Characterization of soybean transgenic lines overexpressing GmWRKY27 . GFP expression analyses in wild type (A) and hygromycin-resistant embryogenic tissues (B). GFP expression was detected under blue light using a fluorescence stereomicroscope Olympus®, equipped with a BP filter set containing a 488 nm excitation filter and a 505-530 nm emission filter. (C) Expression levels (RT-qPCR) of the GmWRKY27 in wild-type (WT) soybean plants and in histodifferentiated embryos of different transgenic soybean lines. Venc (BRSMG68 Vencedora) P2-1, IAS-5 P1-1, Conq (MGBR-46 Conquista) P1-1 lines were obtained from Biobalistic and IAS-5 P3-1 line from Biobalistic/Agrobacterium transformation experiments. F-Box protein and metalloprotease reference genes were used as internal controls to normalize the amount of mRNA present in each sample. Transcript levels of WRKY genes present in the wt were used to calibrate the transcript amounts in transgenic embryos. *Means are significantly different in the wt and transgenic lines (Student’s t-test, p < 0.05).
Footnotes
Marta Bencke-Malato and Caroline Cabreira contributed equally to this work.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
Conceived and designed the experiments: MB-M, BW-S, LBN, MM-P, MHB-Z, APC. Performed the experiments: MB-M, BW-S, FCM-G, MCCGdeC, RS, MBO. Vectors construction: MSH. Performed data analysis: MB-M, CC, BW-S, MCCGdeC, LBN, EM, GW, ACT-Z, MSH, RLMW. Wrote the paper: MB-M, BW-S, LBN. Revised the paper: MM-P, MHB-Z, RVA, ACT-Z. Supervised and coordinated the study: MHB-Z. All authors read and approved the final manuscript.
Contributor Information
Marta Bencke-Malato, Email: mbencke@gmail.com.
Caroline Cabreira, Email: carol.cabreira@yahoo.com.br.
Beatriz Wiebke-Strohm, Email: strohm@sinos.net.
Lauro Bücker-Neto, Email: laurobucker@gmail.com.
Estefania Mancini, Email: soyestepi@gmail.com.
Marina B Osorio, Email: miaosorio@gmail.com.
Milena S Homrich, Email: mihomrich@yahoo.com.br.
Andreia Carina Turchetto-Zolet, Email: aturchetto@gmail.com.
Mayra CCG De Carvalho, Email: mayragallo@hotmail.com.
Renata Stolf, Email: renatastolf@hotmail.com.
Ricardo LM Weber, Email: rlmweber@yahoo.com.br.
Gastón Westergaard, Email: gastongw@gmail.com.
Atílio P Castagnaro, Email: atilio@eeaoc.org.ar.
Ricardo V Abdelnoor, Email: ricardo@cnpso.embrapa.br.
Francismar C Marcelino-Guimarães, Email: francmg@gmail.com.
Márcia Margis-Pinheiro, Email: marcia.margis@ufrgs.br.
Maria Helena Bodanese-Zanettini, Email: mhbzanettini@yahoo.com.br.
References
- 1.Miles MR, Frederick RD, Hartman GL: Evaluation of soybean germplasm for resistance toPhakopsora pachyrhizi.Plant Health Prog 2006, doi:10.1094/PHP-2006-0104-01-RS.
- 2.Ogle HJ, Byth DE, McLean R. Effect of rust (Phakopsora pachyrhizi) on soybean yield and quality in south-eastern Queensland. Australian J Agric Res. 1979;30:883–893. doi: 10.1071/AR9790883. [DOI] [Google Scholar]
- 3.Bromfield KR. Monograph No. 11. St. Paul, Minnesota: American Phytopathological Society; 1984. Soybean Rust. [Google Scholar]
- 4.Patil VS, Wuike RV, Thakare CS, Chirame BB. Viability of uredospores of Phakopsora pachyrhizi Syd. at different storage conditions. J Maharashtra Agric Universities. 1997;22:260–261. [Google Scholar]
- 5.Bromfield KR, Hartwig EE. Resistance to Soybean Rust and mode of inheritance. Crop Sci. 1980;20:254–255. doi: 10.2135/cropsci1980.0011183X002000020026x. [DOI] [Google Scholar]
- 6.Cheng YW, Chan KL. The breeding of ‘Tainung 3’ soybean. J Taiwan Agric Res. 1968;17:30–35. [Google Scholar]
- 7.Hidayat OO, Somaatmadja S. Screening of soybean breeding lines for resistance to soybean rust (Phakopsora pachyrhizi Sydow) Soybean Rust Newsl. 1977;1:9–22. [Google Scholar]
- 8.Singh BB, Thapliyal PN. Breeding for resistance to Soybean Rust in India. In: Ford RE, Sinclair JB, editors. Rust of Soybean: The Problem and Research Needs. Urbana, IL: College of Agriculture, University of Illinois at Urbana-Champaign; 1977. pp. 62–65. [Google Scholar]
- 9.McLean RJ, Byth DE. Inheritance in resistance to rust (Phakopsora pachyrhizi) in soybeans. Aust J Agric Res. 1980;31:951–956. doi: 10.1071/AR9800951. [DOI] [Google Scholar]
- 10.Hartwig EE, Bromfield KR. Relationships among three genes conferring specific resistance to rust in soybeans. Crop Sci. 1983;23:237–239. doi: 10.2135/cropsci1983.0011183X002300020012x. [DOI] [Google Scholar]
- 11.Hartwig EE. Identification of a fourth major gene conferring resistance to soybean rust. Crop Sci. 1986;26:1135–1136. doi: 10.2135/cropsci1986.0011183X002600060010x. [DOI] [Google Scholar]
- 12.Monteros MJ, Missaoui AM, Phillips DV, Walker DR, Boerma HR. Mapping and confirmation of the ‘Hyuuga’ red-brown lesion resistance gene for Asian Soybean Rust. Crop Sci. 2007;47:829–834. doi: 10.2135/cropsci06.07.0462. [DOI] [Google Scholar]
- 13.Garcia A, Calvo ES, de Souza KRA, Harada A, Hiromoto DM, Vieira LG. Molecular mapping of soybean rust (Phakopsora pachyrhizi) resistance genes: discovery of a novel locus and alleles. Theor Appl Genet. 2008;117:545–553. doi: 10.1007/s00122-008-0798-z. [DOI] [PubMed] [Google Scholar]
- 14.Li S, Smith JR, Ray JD, Frederick RD. Identification of a new soybean rust resistance gene in PI 567102B. Theor Appl Genet. 2012;125(1):133–142. doi: 10.1007/s00122-012-1821-y. [DOI] [PubMed] [Google Scholar]
- 15.Bonde MR, Nester SE, Austin CN, Stone CL, Frederick RD, Hartman GL, Miles MR. Evaluation of virulence of Phakopsora pachyrhizi and P. meibomiae isolates. Plant Dis. 2006;90:708–716. doi: 10.1094/PD-90-0708. [DOI] [PubMed] [Google Scholar]
- 16.Sconyers LE, Kemerait RC, Brock J, Phillips DV, Jost PH, Sikora EJ, Gutierrez-Estrada A, Mueller JD, Marois JJ, Wright DL, Harmon CL: Asian soybean rust development in 2005: A perspective from the Southeastern United States. In APSnet Features 2006. doi:10.1094/APSnetFeatures-2006-0106.
- 17.van de Mortel M, Recknor JC, Graham MA, Nettleton D, Dittman JD, Nelson RT, Godoy CV, Abdelnoor RV, Almeida AMR, Baum TJ, Whitham SA. Distinct biphasic mRNA changes in response to Asian soybean rust infection. Mol Plant Microbe Interact. 2007;20:887–899. doi: 10.1094/MPMI-20-8-0887. [DOI] [PubMed] [Google Scholar]
- 18.Panthee DR, Yuan JS, Wright DL, Marois JJ, Mailhot D, Stewart CN., Jr Gene expression analysis in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) in an early growth stage. Funct Integr Genomics. 2007;7:291–301. doi: 10.1007/s10142-007-0045-8. [DOI] [PubMed] [Google Scholar]
- 19.Panthee DR, Marois JJ, Wright DL, Narvaez D, Yuan JS, Stewart CN., Jr Differential expression of genes in soybean in response to the causal agent of Asian soybean rust (Phakopsora pachyrhizi Sydow) is soybean growth stage-specific. Theor Appl Genet. 2009;118:359–370. doi: 10.1007/s00122-008-0905-1. [DOI] [PubMed] [Google Scholar]
- 20.Choi JJ, Alkharouf NW, Schneider KT, Matthews BF, Frederick RD. Expression patterns in soybean resistant to Phakopsora pachyrhizi reveal the importance of peroxidases and lipoxygenases. Funct Integr Genomics. 2008;8:341–359. doi: 10.1007/s10142-008-0080-0. [DOI] [PubMed] [Google Scholar]
- 21.Tremblay A, Hosseini P, Alkharouf N, Li S, Matthewsa BF. Transcriptome analysis of a compatible response by Glycine max to Phakopsora pachyrhizi infection. Plant Sci. 2010;179:183–193. doi: 10.1016/j.plantsci.2010.04.011. [DOI] [Google Scholar]
- 22.Schneider KT, van de Mortel M, Bancroft TJ, Braun E, Nettleton D, Nelson RT, Frederick RD, Baum TJ, Graham MA, Whitham SA. Biphasic gene expression changes elicited by Phakopsora pachyrhizi in soybean correlate with fungal penetration and haustoria formation. Plant Physiol. 2011;157:355–371. doi: 10.1104/pp.111.181149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pandey SP, Somssich IE. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009;150:1648–1655. doi: 10.1104/pp.109.138990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5:199–206. doi: 10.1016/S1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- 25.Xie Z, Zhang ZL, Zou X, Huang J, Ruas P, Thompson D, Shen QJ. Annotations and Functional Analyses of the Rice WRKY Gene Superfamily Reveal Positive and Negative Regulators of Abscisic Acid Signaling in Aleurone Cells. Plant Physiol. 2005;137:176–189. doi: 10.1104/pp.104.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evolutionary Biol. 2005;5:1–12. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu KL, Guo ZJ, Wang HH, Li J. The WRKY family of transcription factors in rice and arabidopsis and their origins. DNA Res. 2005;12:9–26. doi: 10.1093/dnares/12.1.9. [DOI] [PubMed] [Google Scholar]
- 28.Ross CA, Liu Y, Shen QJ. The WRKY Gene Family in Rice (Oryza sativa) J Integr Plant Biol. 2007;49:827–842. doi: 10.1111/j.1744-7909.2007.00504.x. [DOI] [Google Scholar]
- 29.He HS, Dong Q, Shao YH, Jiang HY, Zhu SW, Cheng B, Xiang Y: Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa.Plant Cell Rep 2012, doi:10.1007/s00299–012–1241–0.
- 30.Wen F, Zhu H, Li P, Jiang M, Mao W, Ong C, Chu Z: Genome-wide evolutionary characterization and expression analyses of wrky family genes in brachypodium distachyon.DNA Res 2014, 1–13. doi:10.1093/dnares/dst060. [DOI] [PMC free article] [PubMed]
- 31.Zhang Y, Feng J. Identification and characterization of the grape WRKY family. BioMed Research International. 2014;Article ID 787680:14. doi: 10.1155/2014/787680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dou L, Zhang X, Pang C, Song M, Wei H, Fan S, Yu S: Genome‑wide analysis of the WRKY gene family in cotton.Mol Genet Genomics doi:10.1007/s00438-014-0872-y. [DOI] [PubMed]
- 33.Ülker B, Somssich IE. WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004;7:491–498. doi: 10.1016/j.pbi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Hayami N, Terada T, Shirouzu M, Tanaka A, Seki M, Shinozaki K, Yokoyama S. Solution structure of an arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17:944–956. doi: 10.1105/tpc.104.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeo K, Hayashi S, Kojima-Suzuki H, Morikami A, Nakamura K. Role of conserved residues of the WRKY domain in the DNA-binding of tobacco WRKY family proteins. Biosci Biotech Biochem. 2001;65:2428–2436. doi: 10.1271/bbb.65.2428. [DOI] [PubMed] [Google Scholar]
- 36.Eulgem T, Somssich IE. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10:366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 37.Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol Biol. 2008;68:81–92. doi: 10.1007/s11103-008-9353-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Q, Tian A, Zou H, Xie Z, Lei G, Huang J, Wang C, Wang H, Zhang J, Chen S. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J. 2008;6:486–503. doi: 10.1111/j.1467-7652.2008.00336.x. [DOI] [PubMed] [Google Scholar]
- 39.Huang S, Gao Y, Liu J, Peng X, Niu X, Fei Z, Cao S, Liu Y. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol Genet Genomics. 2012;287:495–513. doi: 10.1007/s00438-012-0696-6. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Jin JP, Tang L, Zhao Y, Gu XC, Gao G, Luo JC. PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011;39:1114–1117. doi: 10.1093/nar/gkq1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Libault M, Joshi T, Valliyodan B, Nguyen H, Xu D, Stacey G, Cheng J. SoyDB: A Knowledge Database of Soybean Transcription Factors. BMC Plant Biol. 2010;10:14–26. doi: 10.1186/1471-2229-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soybean trancription factor knowledge base. [http://www.igece.org/Soybean_TF/]
- 43.Zhang L, Wang X, Bi Y, Zhang C, Fan Y, Lei W. Isolation and functional analysis of transcription factor GmWRKY57b from soybean. Chin Sci Bulletin. 2008;53:3538–3545. doi: 10.1007/s11434-008-0483-2. [DOI] [Google Scholar]
- 44.Kang SG, Park E, Do KS. Identification of a pathogen-induced Glycine max transcription factor GmWRKY1. Plant Pathol J. 2009;25:381–388. doi: 10.5423/PPJ.2009.25.4.381. [DOI] [Google Scholar]
- 45.IGECE: http://systemsbiology.usm.edu/BrachyWRKY/WRKY/Soybean.html.
- 46.LGE genômica e expressão. [http://www.lge.ibi.unicamp.br/soja/]
- 47.Severin AJ, Woody JL, Bolon Y, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptoma. BMC Plant Biol. 2010;10:160–176. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grant D, Nelson R, Cannon S, Shoemaker R. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2010;38(Database issue):D843–846. doi: 10.1093/nar/gkp798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Twizeyimana M, Bandyopadhyay R, Ojiambo P, Paul C, Hartman GL. Proceedings of 2006 National Soybean rust symposium, Saint Louis. 2006. A detached leaf method to evaluate soybean for resistance to rust National Soybean. Rust symposium. [Google Scholar]
- 50.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–184. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 51.Freeling M. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annual Rev Plant Biol. 2009;60:433–453. doi: 10.1146/annurev.arplant.043008.092122. [DOI] [PubMed] [Google Scholar]
- 52.Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martinez-Garcia JF, Bilbao-Castro JR, Robertson DL. Genome wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss and algae. Plant Physiol. 2010;153:1398–1412. doi: 10.1104/pp.110.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rushton PJ, Bokowiec MT, Han S, Zhang H, Brannock JF, Chen X, Laudeman TW, Timko MP. Tobacco transcription factors: novel insights into transcriptional regulation in the solanaceae. Plant Physiol. 2008;147:280–295. doi: 10.1104/pp.107.114041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15:247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Mangelsen E, Kilian J, Berendzen KW, Kolukisaoglu UH, Harter K, Jansson C, Wanke D. Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots. BMC Genomics. 2008;9:194–211. doi: 10.1186/1471-2164-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Verk MC, Pappaioannou D, Neeleman L, Bol JF, Linthorst HJM. A novel WRKY transcription factor is required for induction of PR-1a gene expression by salicylic acid and bacterial elicitors. Plant Physiol. 2008;146:1983–1995. doi: 10.1104/pp.107.112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang B, Jiang Y, Rahman MH, Deyholos MK, Kav NNV. Identification and expression analysis of WRKY transcription factor in canola (Brassica napus L.) in response to fungal pathogens and hormone treatments. BMC Plant Biol. 2009;9:68–87. doi: 10.1186/1471-2229-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giacomelli JI, Ribichich KF, Dezar CA, Chan RL. Expression analyses indicate the involvement of sunflower WRKY transcription factors in stress responses, and phylogenetic reconstructions reveal the existence of a novel clade in the Asteraceae. Plant Sci. 2010;178:398–410. doi: 10.1016/j.plantsci.2010.02.008. [DOI] [Google Scholar]
- 59.Pandey AK, Yang C, Zhang C, Graham MA, Horstman HD, Lee Y, Zabotina OA, Hill JH, Pedley KF, Whitham SA. Functional Analysis of the Asian Soybean Rust Resistance Pathway Mediated by Rpp2. Mol Plant Microbe Interact. 2011;24:194–206. doi: 10.1094/MPMI-08-10-0187. [DOI] [PubMed] [Google Scholar]
- 60.Goellner K, Loehrer M, Langenbach C, Conrath U, Koch E, Schaffrath U. Phakopsora pachyrhizi, the causal agent of Asian soybean rust. Mol Plant Pathol. 2010;11:169–177. doi: 10.1111/j.1364-3703.2009.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18:1310–1326. doi: 10.1105/tpc.105.037523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen HC, Lai Z, Shi J, Xiao Y, Chen Z, Xu X. Roles of arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010;10:281–296. doi: 10.1186/1471-2229-10-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu D, Chen C, Chen Z. Evidence for an Important Role of WRKY DNA Binding Proteins in the Regulation of NPR1 Gene Expression. Plant Cell. 2001;13:1527–1539. doi: 10.1105/tpc.13.7.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang D, Amornsiripanitch N, Dong X. A Genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. Plos Pathog. 2006;2:1042–1050. doi: 10.1371/journal.ppat.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen C, Chen Z. potentiation of developmentally regulated plant defense response by atwrky18, a pathogen-induced arabidopsis transcription factor. Plant Physiol. 2002;129:706–716. doi: 10.1104/pp.001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryu H, Han M, Lee S, Cho JI, Sunggi HNR, Lee YH, Bhoo SH, Wang GL, Hahn TR, Jeon JS. A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 2006;25:836–847. doi: 10.1007/s00299-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 67.Zuo J, Chua N. Chemical-inducible systems for regulated expression of plant genes. Curr Opin Biotechnol. 2000;11:146–151. doi: 10.1016/S0958-1669(00)00073-2. [DOI] [PubMed] [Google Scholar]
- 68.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 69.The GENSCAN Web Server at MIT. [http://genes.mit.edu/GENSCAN.html]
- 70.FEGENESH. [http://linux1.softberry.com/]
- 71.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.FancyGENE. [http://bio.ieo.eu/fancygene/]
- 73.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinforma. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 77.Molecular Evolution, Phylogenetics and epidemiology - FigTree. [http://tree.bio.ed.ac.uk/software/figtree/]
- 78.Matsumura H, Krueger DH, Kahl G, Terauchi R. SuperSAGE: A Modern Platform for Genome-Wide Quantitative Transcript Profiling. Curr Pharm Biotechnol. 2008;9:368–374. doi: 10.2174/138920108785915157. [DOI] [PubMed] [Google Scholar]
- 79.Robertson N, Oveisi-Fordorei M, Zuyderduyn SD, Varhol RJ, Fjell C, Marra M, Jones C, Siddiqui A. DiscoverySpace: an interactive data analysis application. Genome Biol. 2007;8:R6. doi: 10.1186/gb-2007-8-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.LGE Genômica e Expressão. [http://www.lge.ibi.unicamp.br/soja/]
- 81.Ribeiro AS, Moreira JUV, Pierozzi PHB, Rachid BF, Toledo JFF, Arias CAA, Soares RM, Godoy CV. Genetic control of Asian rust in soybean. Euphytica. 2007;157:15–25. doi: 10.1007/s10681-007-9404-8. [DOI] [Google Scholar]
- 82.Kerk NM, Ceserani T, Tausta SL, Sussex IM, Nelson TM, Ceserani T. Laser capture microdissection of cells from plant tissues. Plant Physiol. 2003;132:27–35. doi: 10.1104/pp.102.018127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai S, Lashbrook CC. Laser capture microdissection of plant cells from tape-transferred paraffin sections promotes recovery of structurally intact RNA for global gene profiling. Plant J. 2006;48:628–637. doi: 10.1111/j.1365-313X.2006.02886.x. [DOI] [PubMed] [Google Scholar]
- 84.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinforma. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinforma. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 86.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 87.Biotecsur. [http://bioinfo.cnpso.embrapa.br/biotecsoja/sistema_autenticacao/login.php]
- 88.Kim KS, Unfried JR, Hyten DL, Frederick RD, Hartman GL, Nelson RL, Song Q, Diers BW. Molecular mapping of soybean rust resistance in soybean accession PI 561356 and SNP haplotype analysis of the Rpp1 region in diverse germplasm. Theor Appl Genet. 2012;125:1339–1352. doi: 10.1007/s00122-012-1932-5. [DOI] [PubMed] [Google Scholar]
- 89.Libault M, Thibivilliers S, Bilgin DD, Radwan O, Benitez M, Clough SJ, Stacey G. Identification of four soybean reference genes for gene expression normalization. Plant Genome. 2008;1:44–54. doi: 10.3835/plantgenome2008.02.0091. [DOI] [Google Scholar]
- 90.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 91.Karimi M, Inze D, Depicker A. GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002;7:193–195. doi: 10.1016/S1360-1385(02)02251-3. [DOI] [PubMed] [Google Scholar]
- 92.Droste A, Pasquali G, Bodanese-Zanettini MH. Transgenic fertile plants of soybean (Glycine max (L.) Merrill) obtained from bombarded embryogenic tissue. Euphytica. 2002;127:367–376. doi: 10.1023/A:1020370913140. [DOI] [Google Scholar]
- 93.Finer JJ, Vain P, Jones MW, McMullen MD. Development of the particle inflow gun for DNA delivery to plant cells. Plant Cell Rep. 1992;11:323–328. doi: 10.1007/BF00233358. [DOI] [PubMed] [Google Scholar]
- 94.Wiebke-Strohm B, Droste A, Pasquali G, Osório MB, Bucker-Neto L, Passaglia LMP, Bencke M, Homrich MS, Margis-Pinheiro M, Bodanese-Zanettini MH. Transgenic fertile soybean plants derived from somatic embryos transformed via the combined DNA-free particle bombardment and Agrobacterium system. Euphytica. 2011;177:343–354. doi: 10.1007/s10681-010-0249-1. [DOI] [Google Scholar]
- 95.Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]