

Abstract
Livestock especially cattle are known as a main reservoir of Escherichia coli O157:H7. This bacterium is considered as a pathogenic agent characterized by producing toxins, which are familiarly known as Shiga-like toxin-1 (Stx1) and Stx2. The aim of this work was to analyse the novel sequence of the 16S rRNA gene of strains isolated in this study in order to know the phylogenetic relationships between these sequences and those between the sequences of bacteria available in databanks. The results of this analysis showed that the strains KL-48(2) and SM25(1) that originated from human and cattle feces, respectively, are closely related among them and with respect to E. coli EDL 933, E. coli Sakai, E. coli ATCC 43894, E. coli O111:H-, E. coli O121:H19, E. coli O104:H4, and Shigella sonnei with more than 99% similarity values.
1. Introduction
The identification of pathogenic bacteria was traditionally performed by isolating the organism and studying it phenotypically by means of Gram staining and culture and biochemical methods, which has been the gold standard of bacterial identification [1].
With the invention of polymerase chain reaction (PCR) and automated DNA sequencing, the genome of some bacteria has been sequenced completely. A comparison of the genomic sequences of bacterial species showed that the 16S ribosomal RNA (rRNA) gene is highly conserved within a species and among species of the same genus and, hence, can be used as the new gold standard for the specification of bacteria [2]. To study bacterial phylogeny and taxonomy, the 16S rRNA gene sequences are very useful. With the gene presence in almost all bacteria, often existing as a multigene family, or operons, the function of the 16S rRNA gene over time has not changed, suggesting that random sequence changes are a more accurate measure of time and the 16S rRNA gene (1500 bp) is large enough for informatics purposes [3].
Using 16S rRNA sequences, numerous bacterial genera and species have been reclassified and renamed; classification of uncultivable bacteria has been made possible, phylogenetic relationships have been determined, and the discovery and classification of novel bacterial species have been facilitated [4]. This method has been successful in identifying Enterobacteriaceae species from a bone marrow transplant recipient [2], and the use of this method to identify or discover novel bacteria in clinical microbiology laboratories has successfully been reported also [4, 5].
Escherichia coli O157:H7 as one of enterohemorrhagic Escherichia coli (EHEC) are predominant strains causing infections to human. This disease ranges from simple diarrhea to the more complicated hemorrhagic colitis (HC) and hemolytic uremic syndrome (HUS) [6, 7]. Most infections caused by these bacteria are a result of the consumption of less cooked meat and unpasteurized dairy products and drinking water contaminated with feces [8]. In this study, we report the application of such technique to confirm novel E. coli strainsisolated from feces of human and Bali cattle and thus make the phylogenetic tree in order to know the relationship to each order sequence that is available in the databank. This study also intended to clarify previous study that identified that local isolates of E. coli O157:H7 that originated from animals and humans share genetic similarity coefficients [9, 10].
2. Materials and Methods
2.1. Bacterial Strains
Bacterial strains that were investigated in this study are, namely, SM-25(1) and KL-48(2). These strains were isolated from 80 feces samples of Bali cattle and 76 feces samples of humans suffering renal failure at the Sanglah General Hospital Centre, respectively. Both strains had been identified as serotype E. coli O157:H7 according to their genetic marker covering stx1, stx2, and eae gene [11, 12].
2.2. Extraction of DNA and PCR
DNA was extracted from bacterial strains using QIAamp DNA Mini Kits (Qiagen) according to manufacturer's instructions as described previously [11]. The 16S rRNA gene was amplified using Platinum PCR Supermix kit (Invitrogen) on Thermocycler Eppendorf Mastercycler personal/PTC 100. The PCR program was carried out in 40 μL reaction volumes containing 2 μL DNA template (300 ng/μL), 34 μL PCR Supermix 2x, and 2 μL (20 pmol/μL) of each primer. The primers were used in this study, that is, 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and U1492R (5′-GGTTACCTTGTTACGACTT-3′) [13]. The PCR amplification has initial DNA denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 55°C for 1 min, and elongation at 72°C for 1 min, which was followed by a final extension at 72°C for 5 min. 5 μL PCR product was analyzed by electrophoresis (Bio-Rad) in 1% agarose (Gibco BRL) gel, at 90 volts for 45 min, followed by staining with 1% solution of ethidium bromide (50 μL/L) and destaining with TBE 1x for 10 min. Gel was visualized by UV transillumination and recorded by digital camera FE-270 7.1 megapixels.
2.3. Sequencing and Phylogenetic Analysis
The sequencing of 16S rRNA gene was conducted using genetic analyzer (ABI Prism 3130 and 3130 xl Genetic Analyzer) at Eijkman Institute for Molecular Biology, Jakarta. The sequencing used both primers: Stx2 (F) and Stx2 (R). The sequences were edited to exclude the PCR primer binding sites and manually were corrected using MEGA 5.2 version software. The full gene sequences of strains KL-48(2) and SM-25(1) were compared automatically using the BLAST against the sequences of bacteria available in databanks (http://www.ncbi.nlm.nih.gov/). The phylogenetic analysis was constructed using neighbor-joining algorithm [14, 15].
2.4. Statistical Criteria for Species Identification
Identification of serotype was done through sequence similarity and/or difference nucleotides per total nucleotides. The criteria were determined based on the following: if the different nucleotides between the query and the study strain were 1–1.5% (14–22 bp), 1.5–5.0% (23–72 bp), and 5.0–7.0% (72–98 bp), the query strain should be given to the same species or genus or a different genus, respectively. Confirmation of strains was also determined based on the guidelines recommended by Janda and Abbott [16, 17].
2.5. GenBank Accession Number
The complete sequences (1380 bp) of the 16S rRNA gene of both strains KL-48(2) and SM-25(1) have been deposited in the International Nucleotide Sequence Database (INSD), that is, in the National Center for Biotechnology Information (NCBI).
3. Results and Discussion
The analysis of 16S rRNA gene of Escherichia coli O157:H7 local isolates as an objective to be confirmed in this study has been successfully sequenced. Full sequences (1380 bp) of the 16S rRNA gene of both strains have been registered in GenBank with accession numbers KF768068 and KF768069 for strains SM-25(1) and KL-48(2), respectively. Alignment of the 16S rRNA gene of isolates E. coli SM-25(1) and E. coli KL-48(2) against some of those available in databanks is shown in Figure 1.
Figure 1.

Nucleotides sequence of the 16S rRNA gene of the isolates E. coli SM-25(1) and E. coli KL-48(2) among nucleotides sequence of those available in databanks. Data indicated that the position of nucleotides is different among isolates and identical data for all isolates are not shown.
According to Figure 1, it showed some similarity or difference among nucleotides sequences that were aligned. Isolates E. coli SM-25(1) and E. coli KL-48(2) have tendency to show nucleotides sequence closely with isolates that originated from same species and distinctly for different species or genus. These results are propped by the ribosomal RNA sequencing as a more powerful technique for identification of bacteria, and these results agree with previous study. Patel [3] successfully uses 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. Woo et al. [4] had used 16S rRNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories, and Fattahi et al. [18] had developed the 16S rRNA as a PCR target for detection of E. coli in Rainbow Trout.
Furthermore, Patel [3] reported that the use of 16S rRNA gene sequence to study bacterial taxonomy has been used widely for a number of reasons. These reasons include (i) its presence in almost all bacteria, often existing as a multigene family or operons; (ii) the fact that the function of the 16S rRNA gene over time has not changed, suggesting that random sequence changes are a more accurate measure of time (evolution); and (iii) the fact that the 16S rRNA gene (1,500 bp) is large enough for informatics purposes.
The analysis of similarity or nucleotides different both E. coli SM-25(1) and KL-48(2) strains were studied against some strains of E. coli, that is, E. coli Sakai (BA000007), E. coli EDL 933 (AE005174), E. coli O104:H4 (AFOB02000112), E. coli O111:H-(GU237022), E. coli O121:H19 (JASV01000004), E. coli O26:H11 (AP010953), and E. coli ATCC 43894 as a bacterial control. The similarity analysis was also conducted on some strains of non-E. coli, that is, Aeromonas sp. (FM957460), Vibrio sp. (FM957459), Shigella sonnei (FR870445), Streptomyces sp. (AJ391832), Bacillus sp. (AB851799), and Shigella dysenteriae (CP000034) that are summarized in Table 1.
Table 1.
Similarity analysis and nucleotides different among 16S rRNA genes using PHYDIT program.
| Aeromonas | Vibrio sp. | E. coli EDL 933 W | E. coli Sakai | E. coli ATCC 43894 | E. coli O111:H— | E. coli O104:H4 | E. coli O121:H19 | E. coli SM25(1)∗ | E. coli KL48(2)∗ | Shigella sonnei | Streptomyces sp. | Bacillus sp. | E. coli O26:H11 | Shigella dysenteriae | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aeromonas | — | 158/1500 | 151/1376 | 151/1376 | 150/1376 | 154/1503 | 149/1375 | 153/1529 | 147/1376 | 148/1376 | 161/1529 | 329/1445 | 357/1377 | 817/1451 | 817/1451 |
| Vibrio sp. | 89.47 | — | 131/1380 | 131/1380 | 130/1380 | 134/1501 | 141/1379 | 146/1506 | 140/1380 | 138/1380 | 252/1642 | 326/1418 | 458/1528 | 810/1429 | 809/1429 |
| E. coli EDL 933 W | 89.03 | 90.51 | — | 0/1380 | 1/1380 | 6/1380 | 19/1379 | 13/1379 | 12/1380 | 9/1380 | 16/1380 | 282/1346 | 305/1325 | 739/1340 | 741/1340 |
| E. coli Sakai | 89.03 | 90.51 | 100.00 | — | 1/1380 | 6/1380 | 19/1379 | 13/1379 | 12/1380 | 9/1380 | 16/1380 | 282/1346 | 305/1325 | 739/1340 | 741/1340 |
| E. coli ATCC 43894 | 89.10 | 90.58 | 99.93 | 99.93 | — | 5/1380 | 18/1379 | 12/1379 | 11/1380 | 8/1380 | 15/1380 | 282/1346 | 305/1325 | 739/1340 | 741/1340 |
| E. coli O111:H— | 89.75 | 91.07 | 99.57 | 99.57 | 99.64 | — | 15/1379 | 11/1506 | 14/1380 | 11/1380 | 24/1506 | 301/1420 | 338/1378 | 797/1424 | 797/1424 |
| E. coli O104:H4 | 89.16 | 89.78 | 98.62 | 98.62 | 98.69 | 98.91 | — | 6/1378 | 7/1379 | 12/1379 | 5/1379 | 280/1345 | 305/1324 | 738/1339 | 740/1339 |
| E. coli O121:H19 | 89.99 | 90.31 | 99.06 | 99.06 | 99.13 | 99.27 | 99.56 | — | 5/1379 | 6/1379 | 21/1539 | 309/1447 | 342/1383 | 808/1451 | 808/1451 |
| E. coli SM25(1)∗ | 89.32 | 89.86 | 99.13 | 99.13 | 99.20 | 98.99 | 99.49 | 99.64 | — | 5/1380 | 4/1380 | 284/1346 | 304/1325 | 737/1340 | 739/1340 |
| E. coli KL48(2)∗ | 89.24 | 90.00 | 99.35 | 99.35 | 99.42 | 99.20 | 99.13 | 99.56 | 99.64 | — | 9/1380 | 285/1346 | 307/1325 | 738/1340 | 740/1340 |
| Shigella sonnei | 89.47 | 84.65 | 98.84 | 98.84 | 98.91 | 98.41 | 99.64 | 98.64 | 99.71 | 99.35 | — | 314/1449 | 511/1601 | 813/1457 | 813/1457 |
| Streptomyces sp. | 77.23 | 77.01 | 79.05 | 79.05 | 79.05 | 78.80 | 79.18 | 78.65 | 78.90 | 78.83 | 78.33 | — | 278/1298 | 827/1427 | 829/1427 |
| Bacillus sp. | 74.07 | 70.03 | 76.98 | 76.98 | 76.98 | 75.47 | 76.96 | 75.27 | 77.06 | 76.83 | 68.08 | 78.58 | — | 768/1312 | 770/1312 |
| E. coli O26:H11 | 43.69 | 43.32 | 44.85 | 44.85 | 44.85 | 44.03 | 44.88 | 44.31 | 45.00 | 44.93 | 44.20 | 42.05 | 41.46 | — | 17/1542 |
| Shigella dysenteriae | 43.69 | 43.39 | 44.70 | 44.70 | 44.70 | 44.03 | 44.73 | 44.31 | 44.85 | 44.78 | 44.20 | 41.91 | 41.31 | 98.90 | — |
*Strain in this study.
The data in Table 1 contain percentage of nucleotide similarity (lower-left triangle) and nucleotides difference/total nucleotides (upper-right triangle) of nucleotides analyzed. The summary of the 16S rRNA similarity analysis in Table 1 showed that E. coli KL-48(2) that originated from human feces has nucleotide similarity of 16S rRNA gene closely against some strains. These strains, that is, E. coli SM25(1), E. coli 121:H19, E. coli ATCC 43894, E. coli Sakai, E. coli EDL 933, Shigella sonnei, E. coli O111:H-, and E. coli O104:H4, are as high as 99.64, 99.56, 99.42, 99.35, 99.35, 99.35, 99.20, and 99.13%, respectively. Furthermore, E. coli SM-25(1) that originated from cattle feces also has high nucleotides similarity to the data of 16S rRNA that are available in GenBank also. It has nucleotides similarity to Shigella sonnei, E. coli O121:H19, E. coli O104:H4, E. coli ATCC 43894, E. coli Sakai, and E. coli EDL 933 as high as 99.71, 99.64, 99.49, 99.20, 99.13, and 99.13%, respectively. On the other hand, both strains showed percentage of nucleotide similarity distinctly to Shigella dysentery, E. coli O26:H11, Bacillus sp., Streptomyces sp., Aeromonas, and Vibrio sp.
Table 1 also showed that the number of nucleotides was different per total nucleotides among all isolates compared. Particularly for both isolates that were investigated, isolate KL-48(2) has few different nucleotides per total nucleotides against E. coli SM25(1), E. coli 121:H19, E. coli ATCC 43894, E. coli Sakai, E. coli EDL 933, Shigella sonnei, E. coli O111:H-, and E. coli O104:H4 as many as 5/1380, 6/1379, 8/1380, 9/1380, 9/1380, 9/1380, 11/1380, and 12/1379 nucleotides, respectively. Isolate SM-25(1) also has few different nucleotides per total nucleotides against Shigella sonnei, E. coli O121:H19, E. coli O104:H4, E. coli ATCC 43894, E. coli Sakai, and E. coli EDL 933 as many as 4/1380, 5/1379, 7/1379, 11/1380, 12/1380, and 12/1380 nucleotides, respectively.
Referred to the concept of similarity or nucleotides different between the query nucleotides and those compared, It is recommended when the sequences similarity is more than 90% or the nucleotides different between the query and those compared 1–1.5% (14–22 bp), the query should be categorized as the same species [16]. This assumption is supported by the similarity concept determined by Janda and Abbott [17]. The guideline recommends (i) the length of 16S rRNA gene should be sequenced minimum 500 to 525 bp and ideally 1,300 to 1,500 bp; (ii) criteria for species identification should be minimum >99% sequence similarity and ideally >99.5%. According to this guideline, E. coli SM-25(1) originated from feces of Bali cattle and E. coli KL-48(2) originated from human feces confirmed as the same species. This assumption was supported by the fact that both strains have nucleotides similarity of 99.64% or these strains have different nucleotides as many as 5/1380 nucleotides.
The high nucleotides similarity between 16S rRNA genes of isolates that originated from cattle and human made the conclusion the probability of the strain originated from feces of cattle as a main reservoir and then transmitted to human as a new host obvious occurred. The transmission of this bacterium from animals (cattle) to human can be facilitated by the consumption of meat that is less cooked or unpasteurized dairy products or drinking water contaminated with feces [19]. The results of the study all at once comes as a deep confirmation of previous study which identified that both isolates E. coli SM-25(1) and E. coli KL-48(2) share protein profile more than 70% [10], and the analysis using random amplified polymorphic DNA (RAPD) method indicated that both isolates also share genetic diversity more than 70% [9]. Moreover, analysis of phenogenotype of both isolates also had the same properties characterized. Both isolates genetically positive eae gene and the phenotypic study also showed either E. coli SM-25(1) or E. coli KL-48(2) had been colonize and causes cytophatic effects on vero cell. This study clarified both isolates had potency to colonize at the intestine host and induce attaching-effaching lesions [12].
The high nucleotides similarity (>99%) of both E. coli KL-48(2) and E. coli SM-25(1) strains with those of some nucleotides sequences that are available in GenBank also concludes that the tendency of both strains having virulent capacity as equal as of those, especially to the strains that are compared that is E. coli Sakai, E. coli EDL 933 and E. coli O104:H4, although there are needed to deep confirmations. on the other hand, the high similarity of E. coli SM-25(1) with Shigella sonnei that is 99.71% showed the probability of E. coli SM-25(1) as a novel strain outside of pathogenic E. coli strain especially to Shigella sonnei should be confirmed using the orther markers as a confirmation.
Based on the data in Table 1, a phylogenetic tree of the 16S rRNA gene was performed using Clustal W programme in the MEGA 5.2 software. The phylogenetic tree was constructed using the neighbor-joining algorithm with bootstrap analysis for 1000 replicates (Figure 2).
Figure 2.
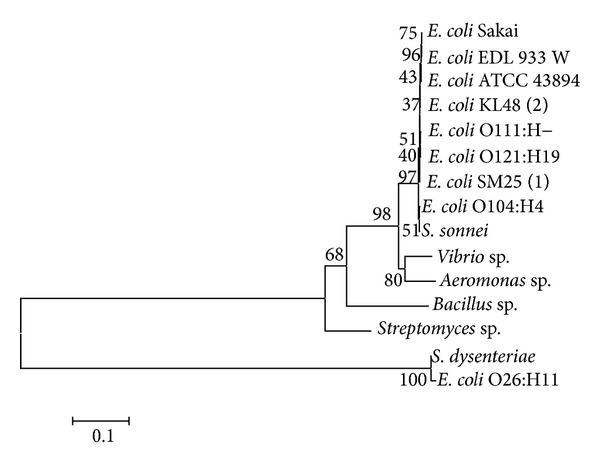
Phylogenetic tree was constructed using neighbor-joining algorithm [14] of nucleotides sequence of 16S rRNA gene. The number in the branch of phylogram indicates bootstrap value (%) by 1000-replication multiple, and scale indicates one per 1000 substitutions of nucleotides sequence of 16S rRNA gene.
Phylogenetic tree in Figure 2 showed that the E. coli KL-48(2) and SM25(1) performed close clade with some strains of pathogenic E. coli except for E. coli O26:H11. On the contrary, both strains also showed distinct clade against some strains that are available in databank. Some of those strains are Streptomyces sp. isolated from Yogyakarta, Bacillus sp. isolated from Jepara, and Vibrio sp.and Aeromonas sp. isolated from Lampung. As a result, both strains are proved to be a strain of pathogenic E. coli which potentially caused a serious outbreak of food borne illness equal to those strains that are characterized by bloody diarrhea and high frequency of serious complications including hemolytic-uremic syndrome (HUS).
4. Conclusion
The novel Escherichia coli strains SM-25(1) and KL-48(2) isolated from cattle feces and human feces, respectively, originated from the same source according to the analysis of 16S rRNA gene. These strains were predicted to have characteristics equal to E. coli Sakai, E. coli EDL 933, E. coli ATCC 43894, E. coli O111:H-, E. coli 121:H19, E. coli O104:H4, and Shigella sonnei.
Acknowledgment
The author gratefully acknowledges the Institute for Research and Community Service at Udayana University for providing financial support in the form of Udayana excellent research grant under Grant no. 175.88/UN 14.2/PNL.01.03.00/2013, dated May 16, 2013.
Conflict of Interests
The author declares that there is no conflict of interests.
References
- 1.O'Hara CM. Manual and automated instrumentation for identification of Enterobacteriaceae and other aerobic gram-negative bacilli. Clinical Microbiology Reviews. 2005;18(1):147–162. doi: 10.1128/CMR.18.1.147-162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo PCY, Leung PKL, Leung KW, Yuen KY. Identification by 16s ribosomal RNA gene sequencing of an Enterobacteriaceae species from a bone marrow transplant recipient. Journal of Clinical Pathology—Molecular Pathology. 2000;53(4):211–215. doi: 10.1136/mp.53.4.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel JB. 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. Molecular Diagnosis. 2001;6(4):313–321. doi: 10.1054/modi.2001.29158. [DOI] [PubMed] [Google Scholar]
- 4.Woo PCY, Lau SKP, Teng JLL, Tse H, Yuen K-Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clinical Microbiology and Infection. 2008;14(10):908–934. doi: 10.1111/j.1469-0691.2008.02070.x. [DOI] [PubMed] [Google Scholar]
- 5.Boudewijns M, Bakkers JM, Sturm PDJ, Melchers WJG. 16S rRNA gene sequencing and the routine clinical microbiology laboratory: a perfect marriage? Journal of Clinical Microbiology. 2006;44(9):3469–3470. doi: 10.1128/JCM.01017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong GL, Hollingsworth J, Morris JG., Jr. Emerging foodborne pathogens: Escherichia coli O157:H7 as a model of entry of a new pathogen into the food supply of the developed world. Epidemiologic Reviews. 1996;18(1):29–51. doi: 10.1093/oxfordjournals.epirev.a017914. [DOI] [PubMed] [Google Scholar]
- 7.Smith SI, Aboaba OO, Odeigha P, et al. Plasmid profile of Escherichia coli 0157:H7 from apparently healthy animals. African Journal of Biotechnology. 2003;2(9):322–324. [Google Scholar]
- 8.Rey J, Sánchez S, Blanco JE, et al. Prevalence, serotypes and virulence genes of Shiga toxin-producing Escherichia coli isolated from ovine and caprine milk and other dairy products in Spain. International Journal of Food Microbiology. 2006;107(2):212–217. doi: 10.1016/j.ijfoodmicro.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 9.Suardana IW, Artama WT, Widiasih DA, Mahardika IGNK. Genetic diversity of Escherichia coli O157:H7 strains using random amplified polymorphic DNA (RAPD) International Research Journal of Microbiology. 2013;4(2):72–78. [Google Scholar]
- 10.Suardana IW, Pinatih KJP, Ratnawati NLKA, Widiasih DA. Protein profile analysis of Escherichia coli O157:H7 from human and animals origin. International Journal of Current Microbiology and Applied Science. 2013;2(6):204–214. [Google Scholar]
- 11.Suardana IW, Artama WT, Asmara W, Daryono BS. Identification of Escherichia coli O157:H7 and detection of Shiga like toxin 1 and 2 genes from animals feces, beef, and human feces. Jurnal Veteriner. 2010;11(4):264–270. [Google Scholar]
- 12.Suardana IW, Artama WT, Asmara W, Daryono BS. Adherence pheno-genotypic of Escherichia coli O157:H7 isolated from beef, feces of cattle, chicken and human. Indonesian Journal of Biotechnology. 2010;16(1):46–52. [Google Scholar]
- 13.James GS. Universal bacterial identification by PCR and DNA sequencing of 16S rRNA gene. In: Schuller M, Sloots TP, Holliday CL, James GS, Carter IWJ, editors. PCR for Clinical Microbiology. New york, NY, USA: Springer; 2010. pp. 209–214. [Google Scholar]
- 14.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 15.Tamura K, Dudley J, Nei M, Kumar S. Mega 4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 16.Bosshard PP, Abels S, Zbinden R, Böttger EC, Altwegg M. Ribosomal DNA sequencing for identification of aerobic gram-positive rods in the clinical laboratory (an 18-month evaluation) Journal of Clinical Microbiology. 2003;41(9):4134–4140. doi: 10.1128/JCM.41.9.4134-4140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 2007;45(9):2761–2764. doi: 10.1128/JCM.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fattahi F, Mirvaghefi A, Farahmand H, Rafiee G, Abdollahi A. Development of 16s rRNA targeted PCR method for the detection of Escherichia coli in rainbow trout (Oncorhynchus mykiss) Iranian Journal of Pathology. 2013;8(1):36–44. [Google Scholar]
- 19.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli . Clinical Microbiology Reviews. 1998;11(1):142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]