

Abstract
Background
Respiratory viral infections are the most common trigger of acute exacerbations in patients with allergic asthma. The anti-viral response of airway epithelial cells (AEC) may be impaired in asthmatics, while cytokines produced by AEC may drive the inflammatory response. We investigated whether AEC cultured in the presence of Th2 cytokines associated with an allergic environment exhibited altered responses to double-stranded RNA, a virus-like stimulus.
Methods
We undertook preliminary studies using the MLE-12 cell line derived from mouse distal respiratory epithelial cells, then confirmed and extended our findings using low-passage human AEC. Cells were cultured in the absence or presence of the Th2 cytokines IL-4 and IL-13 for 48 hours, then stimulated with poly I:C for 4 hours. Expression of relevant anti-viral response and cytokine genes was assessed by quantitative real-time PCR. Secretion of cytokine proteins was assessed by immunoassay.
Results
Following stimulation with poly I:C, MLE-12 cells pre-treated with Th2 cytokines exhibited significantly higher levels of expression of mRNA for the cytokine genes Cxcl10 and Cxcl11, as well as a trend towards increased expression of Cxcl9 and Il6. Expression of anti-viral response genes was mostly unchanged, although Stat1, Ifit1 and Ifitm3 were significantly increased in Th2 cytokine pre-treated cells. Human AEC pre-treated with IL-4 and IL-13, then stimulated with poly I:C, similarly exhibited significantly higher expression of IL8, CXCL9, CXCL10, CXCL11 and CCL5 genes. In parallel, there was significantly increased secretion of CXCL8 and CCL5, as well as a trend towards increased secretion of CXCL10 and IL-6. Again, expression of anti-viral response genes was not decreased. Rather, there was significantly enhanced expression of mRNA for type III interferons, RNA helicases and other interferon-stimulated genes.
Conclusion
The Th2 cytokine environment appears to promote increased production of pro-inflammatory chemokines by AEC in response to double-stranded RNA, which could help explain the exaggerated inflammatory response to respiratory viral infection in allergic asthmatics. However, any impairment of anti-viral host defences in asthmatics appears unlikely to be a consequence of Th2 cytokine-induced downregulation of the expression of viral response genes by AEC.
Electronic supplementary material
The online version of this article (doi:10.1186/s40247-014-0011-6) contains supplementary material, which is available to authorized users.
Keywords: Airway epithelium, Innate interferons, Anti-viral response, Th2 cytokines
Background
Acute exacerbations of asthma are associated with worsening clinical manifestations requiring a change in treatment strategy [1]. They are the main reason for hospitalisation and the major source of health care costs in asthma [2]. Exacerbations are frequently related to respiratory viral infections, most commonly with human rhinovirus (RV) [3]. Furthermore, asthmatics may develop more severe and longer-lasting RV infections [4],[5].
The airway epithelium is a key player in acute exacerbations of asthma. Not only is it the target of most respiratory viral infections, but it is also an important source of pro-inflammatory cytokines [6]. Several investigators have suggested that one reason for the strong link between exacerbations of asthma and viral infections is that in allergic asthmatics, innate responses to viral infection are impaired. In vitro, there is considerable evidence of decreased production of interferon (IFN)-α2, IFN-β1 and IFN-λ2/3 by airway epithelial cells (AEC) from asthmatics, in response to stimulation with double-stranded RNA (dsRNA) or with RV [7]-[11]. This has been related to impaired toll-like receptor (TLR) and helicase signalling [12]. It has also been suggested that similar impairment is demonstrable in atopic individuals even without asthma [13], although this has not been confirmed.
However, whether the impaired anti-viral cytokine responses translate as increased viral replication in cultures of AEC from allergic asthmatics is much less clear. Although various studies do suggest this [8],[9],[13], others have disagreed [14],[15]. Experimentally, Th2 cytokine pre-treatment of AEC has been reported to increase susceptibility to infection [16],[17] suggested to be related to mucous metaplasia. Again, however, this is controversial, as recent reports have demonstrated either no effect [18] or even that pre-treatment of human AEC with interleukin (IL)-4 and IL-13 was associated with resistance to infection, related to decreased numbers of ciliated cells, with equivalent effect on AEC from asthmatics or non-asthmatics [19].
Another possible reason for the association between viral infections and exacerbations of allergic asthma might be that asthmatic AEC exhibit enhanced expression of pro-inflammatory cytokines in response to viral infection. This has been demonstrated by experimental stimulation with dsRNA, as well by direct infection with viruses including RV [20]-[22]. Furthermore, when stimulated with dsRNA, both asthmatic AEC and normal AEC pre-treated with IL-4 have also been reported to exhibit relatively increased expression of thymic stromal lymphopoietin (TSLP) [10],[23], a cytokine that can induce and amplify Th2 responses.
Overall, however, there remains uncertainty about the nature of the altered responses of AEC to respiratory viral infection in allergic asthmatics, or what might be the mechanism underlying such changes. To further investigate this, we cultured mouse and human AEC in the presence of Th2 cytokines and stimulated them with dsRNA, which is a TLR3 agonist that is also recognised by the RNA helicase IFIH1 and mimics viral infection [24],[25]. We examined the effect of pre-treatment with Th2 cytokines on the expression of innate and interferon-stimulated anti-viral response genes, as well as of a range of pro-inflammatory cytokines. Our results suggest that a Th2 cytokine environment may promote increased production of pro-inflammatory chemokines by AEC in response to respiratory viral infection, but is unlikely to be responsible for any impairment of anti-viral host defences in asthmatics.
Methods
Culture of MLE-12 cells
Preliminary experiments used an SV40-transformed mouse-derived AEC line designated MLE-12 (American Type Culture Collection, Manassas, VA, USA). These cells retain key morphological and functional characteristics of distal airway epithelium [26]. MLE-12 cells were grown in a 50:50 mix of Dulbecco’s Modified Eagle Medium:Ham’s F-12 with 2% heat-inactivated fetal bovine serum and other relevant supplements (L-glutamine, transferrin, sodium selenite, hydrocortisone, β-estradiol, insulin-like growth factor-1 and antibiotics) at 37°C in an atmosphere of 5% CO2. Cells were used between passage 2 and 8. To assess responses to poly I:C and the effects of Th2 cytokine pre-treatment, MLE-12 cells were cultured in 25 cm2 flasks at 5×105/flask, in media either with or without 20 ng/mL of mouse IL-4 and IL-13 (R&D Systems, Minneapolis, MN, USA) for 48 hours, of which the last 16 hours were in serum-free medium. Cells were then stimulated with 10 μg/mL of poly I:C (Invivogen, San Diego, CA, USA) for 4 hours and total RNA was extracted using TriReagent (Sigma-Aldrich) and stored at −80°C. Five independent experiments were performed.
Culture of human bronchial epithelial AEC
Approval of all experiments with human lung tissues was provided by the Ethics Review Committee of the South West Sydney Area Health Service, Royal Prince Alfred Hospital and the University of Sydney Human Research Ethics Committee. Bronchial epithelial layers were isolated from 4th-6th order bronchi from lung tissue obtained from 5 patients undergoing lung resection or transplantation (3 with interstitial lung disease, 1 with emphysema, 1 with a neoplasm). Cells were maintained and expanded in Ham’s F-12 with growth supplements as previously described [27]. All experiments were performed with cells at passage 2. AEC were seeded in 6-well plates at a density of 2×105/well in 2 ml BEGM (Lonza, Basel, Switzerland) and incubated at 37°C in an atmosphere of 5% CO2. After 16 hours, the medium was changed and cells were cultured either with or without 20 ng/ml of human IL-4 (R&D Systems) and IL-13 (Peprotech, Rocky Hill, NJ) for 48 hours. AEC were then stimulated with 10 μg/ml poly I:C (Sigma-Aldrich) for 4 hours. Culture supernatants were collected and stored at −20°C, while cells were lysed in TriReagent and RNA stored at −80°C.
Expression of mRNA for cytokines
Quantitative real-time PCR was used to assess the expression of relevant genes, with detection of amplified products using SYBR green (BioLine, Tauton, MA, USA). Primers were designed in-house or sourced from published articles. Reactions were performed using a Roche LightCycler 480 (Roche Diagnostics, Indianapolis, IN, USA), with gene expression normalised to the housekeeping-gene hypoxanthine-guanine phosphoribosyltransferase (HPRT). Each sample was assessed in triplicate.
Protein immunoassays
For a limited subset of cytokines (CXCL8, CXCL10, CCL5 and IL-6) the concentrations of protein in the supernatants were determined using enzyme-linked immunoassays (R&D Systems) according to the manufacturer’s instructions. Each sample was assessed in duplicate.
Statistical analysis
Data are presented either as arithmetic means ± s.e.m. (MLE-12 cells) or as before-after plots for individual samples (human AEC). To compare the response of Th2 cytokine pre-treated cells, both unstimulated and following stimulation with poly I:C, changes were assessed by a ratio paired t-test, to cater for baseline variability. The software package GraphPad Prism 6.03 (GraphPad Software, San Diego, CA, USA) was used for data analysis and preparation of graphs.
Results
MLE-12 cells
Preliminary experiments using these cells revealed that mRNA expression for the chemokine genes Cxcl10 and Cxcl11 was significantly increased in cells that had been pre-treated with Th2 cytokines and then stimulated with poly I:C (Table 1). There was also a trend towards increased expression of Cxcl9 and of the pro-inflammatory cytokine Il6. In contrast, levels of expression of the Th2-promoting cytokine Il33 were significantly decreased in cells that had been pre-treated with Th2 cytokines and then stimulated with poly I:C, while those of Tslp were unchanged. Unexpectedly, levels of expression of major anti-viral response genes, including the RNA helicases Ddx58 (also known as RIG-I), Ddx60 and Ifih1 (also known as MDA-5) were mostly unchanged, while the interferon-induced genes Stat1, Ifit1 and Ifitm3 were significantly increased in cells pre-treated with Th2 cytokines.
Table 1.
Relative expression by MLE-12 cells of mRNA for chemokine, cytokine and interferon-stimulated genes
| Medium + Poly I:C | Th2 pre-treatment + Poly I:C | |
|---|---|---|
| Cxcl1 | 2.3 ± 0.3 | 2.1 ± 0.4 |
| Cxcl9 | 99.0 ± 27.7 | 178.9 ± 52.7+ |
| Cxcl10 | 46.2 ± 29.8 | 210.5 ± 61.0* |
| Cxcl11 | 8.6 ± 2.2 | 61.2 ± 10.8** |
| Ccl5 | 18.7 ± 2.0 | 26.8 ± 10.3 |
| Il6 | 1.0 ± 0.4 | 2.1 ± 0.2+ |
| Il33 | 2.3 ± 0.3 | 1.2 ± 0.2* |
| Tslp | 0.5 ± 0.2 | 0.9 ± 0.4 |
| Ddx58 | 1.2 ± 0.4 | 1.9 ± 0.7 |
| Ddx60 | 3.5 ± 0.8 | 5.4 ± 1.2 |
| Ifih1 | 2.8 ± 0.7 | 3.5 ± 1.7 |
| Oasl1 | 10.4 ± 3.1 | 9.6 ± 3.8 |
| Stat1 | 3.2 ± 1.9 | 139.8 ± 30.0** |
| Stat2 | 1.2 ± 0.5 | 1.9 ± 0.8 |
| Ifit1 | 4.3 ± 0.8 | 20.4 ± 7.2* |
| Ifitm3 | 1.0 ± 0.5 | 5.6 ± 1.3* |
MLE-12 cells stimulated with poly I:C for 4 hours following culture for 48 hours in either medium alone or medium containing IL-4 and IL-13. mRNA expression shown as stimulation ratio (mean ± s.e.m.) relative to cells cultured in medium alone. + 0.05 < p < 0.1; *p < 0.05; **p < 0.01 by ratio paired t-test, n = 5 separate experiments.
Human AEC
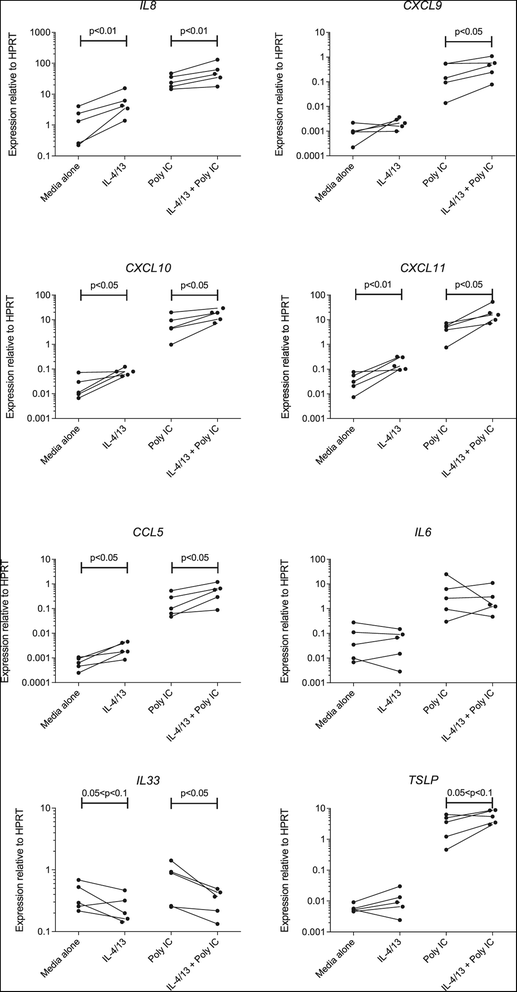
To confirm and extend these findings, we undertook a comprehensive assessment of the expression of relevant innate interferons, interferon-stimulated anti-viral response genes and pro-inflammatory cytokines by human AEC. As a first step, we showed that cells cultured in the presence of IL-4 and IL-13 exhibited a 2.5-fold increase in expression of mRNA for periostin (expression relative to HPRT 0.61 ± 0.14 in media vs. 1.56 ± 0.28 in the presence of IL-4/13, p < 0.05, unpaired t- test), establishing that these cells exhibited a phenotypic change typical of a Th2 environment [28]. Next, we examined the expression of a variety of chemokines and pro-inflammatory cytokines, some of which are known to be interferon-stimulated genes [29]. As shown in Figure 1, baseline levels of expression of the chemokines IL8, CXCL10, CXCL11 and CCL5 were all significantly higher in cells that had been pre-treated with Th2 cytokines. Furthermore, there was significantly increased expression of IL8, CXCL9, CXCL10, CXCL11 and CCL5 in cells that were then stimulated with poly I:C. However, no such increases were observed for IL6. Expression of the Th2-promoting cytokine IL33 was significantly decreased, while there was a trend towards increased expression of TSLP.
Figure 1.

Before-and-after plots showing effects of prior exposure to Th2 cytokines on the expression of mRNA for chemokine and cytokine genes by human AEC at baseline (left) or following stimulation with poly I:C (right). Data are mean values for individual patients, showing expression relative to the housekeeping gene HPRT. Note the logarithmic y-axis. p values for significant differences between cells cultured in media IL-4 and IL-13 were assessed by ratio paired t-test.
For a limited subset of cytokines, results were confirmed by assessing cytokine protein in culture supernatants, as shown in Figure 2. Interestingly, not only were levels of CXCL8 and CCL5 protein significantly increased, together with a trend towards an increase in levels of CXCL10, but in addition there was also a trend towards elevated levels of IL-6 protein.
Figure 2.

Before-and-after plots showing effects of prior exposure to Th2 cytokines on the secretion of chemokine and cytokine proteins by human AEC at baseline (left) or following stimulation with poly I:C (right). Data are mean values for individual patients. p values for differences between cells cultured in media with or without IL-4 and IL-13 were assessed by ratio paired t-test.
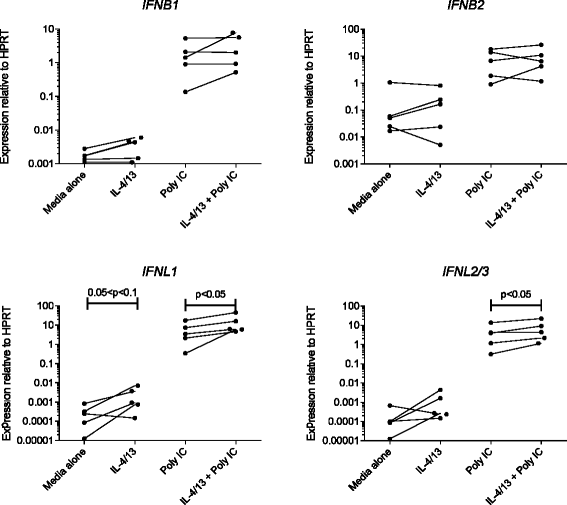
We then examined the expression of innate interferons known to be associated with an anti-viral response. Figure 3 demonstrates that expression of IFNB1 and IFNB2 by AEC in response to poly I:C was unchanged in cells that had been pre-treated with Th2 cytokines. However, there was a modest but statistically significant increase in the expression of both IFNL1 and IFNL2/3.
Figure 3.

Before-and-after plots showing effects of prior exposure to Th2 cytokines on the expression of mRNA for type I and type III interferon genes by human AEC at baseline (left) or following stimulation with poly I:C (right). Data are mean values for individual patients, showing expression relative to the housekeeping gene HPRT. p values for significant differences between cells cultured in media with or without IL-4 and IL-13 were assessed by ratio paired t-test.
Expression of a range of interferon-stimulated anti-viral response genes in cells at baseline or after stimulation with poly I:C is presented in Figure 4. The RNA helicases DDX58, DDX60 and IFIH1 were all significantly up-regulated in cells that had been pre-treated with Th2 cytokines and stimulated with poly I:C, while DDX58 and IFIH1 was also significantly increased at baseline. In addition, there was a trend towards increased expression of the anti-viral transmembrane protein IFITM3. Expression of the transcription factors STAT1 and STAT2 was significantly higher, and there was a trend towards increased expression of the transcription factor regulator OASL1. However, there was no change in expression of the transcription factor IRF3.
Figure 4.
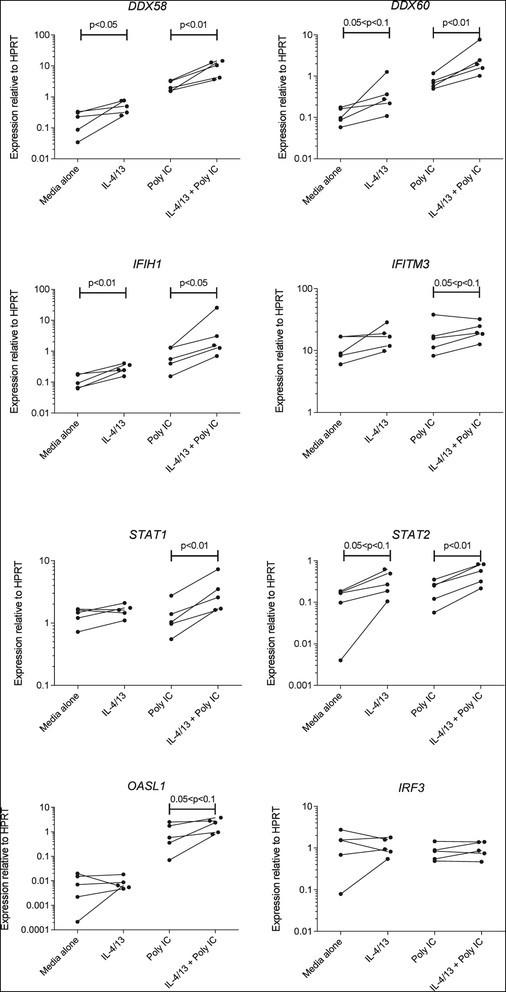
Before-and-after plots showing effects of prior exposure to Th2 cytokines on the expression of mRNA for anti-viral response genes by human AEC at baseline (left) or following stimulation with poly I:C (right). Data are mean values for individual patients, showing expression relative to the housekeeping gene HPRT. p values for differences between cells cultured in media with or without IL-4 and IL-13 were assessed by ratio paired t-test.
Discussion
In this study, we investigated aspects of the relationship between respiratory viral infections and acute exacerbations of allergic asthma. Using exposure to dsRNA as a surrogate for viral infection, we assessed the effects of prior exposure to Th2 cytokines on the expression by AEC of anti-viral host defence genes including RNA helicases and interferons; signalling pathways that are up-regulated by innate interferons; and various cytokines able to promote an inflammatory response or amplify a Th2 response. In preliminary work using mouse MLE-12 cells, an immortalised line derived from distal AEC, we showed that expression of several chemokines and pro-inflammatory cytokines was significantly up-regulated in cells that had been pre-treated with Th2 cytokines and then stimulated with poly I:C, while expression of major anti-viral response genes was either unchanged or was also significantly increased. This was unexpected and we therefore undertook further work using low-passage human bronchial epithelial cells.
The primary response of AEC to viral infection is the production of interferons, mostly interferon-β1 and the various type III interferons (IFN-λ1/2/3) [30]. Because the magnitude of induction of interferons in AEC is relatively low compared to blood leucocytes [30], detection of secreted interferon proteins is difficult, so we assessed expression of these genes by quantitative real-time PCR. We found that in human AEC which had been pre-treated with Th2 cytokines, expression of β interferons was unchanged, while λ interferons exhibited modest but statistically significant up-regulation.
The innate interferons in turn stimulate expression of numerous other genes [29],[31], including not only anti-viral response genes but also chemokines and other pro-inflammatory cytokines, which are secreted at levels that readily permit detection by enzyme immunoassay. Thus we were able to assess the latter in terms of both mRNA expression and protein concentrations in supernatants of AEC in culture. We noted increased expression and secretion of various chemokines, including the neutrophil chemoattractant CXCL8, the T cell chemoattractants CXCL9, CXCL10 and CXCL11, as well as the T cell/eosinophil chemoattractant CCL5. These results were largely similar to the data for MLE-12 cells. Although we observed no change in expression of the IL6 gene, which is consistent with previously reported data [7], there was some increase in levels of IL-6 protein, possibly indicating secretion of pre-formed cytokine. Interestingly, we observed decreased expression of mRNA for the Th2-promoting cytokine IL-33, again analogous to the finding in MLE-12 cells, while expression of TSLP was increased.
Some of the increases in cytokine protein concentrations were not statistically significant, which may have been because culture supernatants were collected at 4 hours after stimulation, a relatively early time point for assessment of secretion of cytokine proteins. Ideally, we would have wished to perform parallel experiments in which cells were collected at 4 hours after stimulation for assessment of mRNA and at 16–24 hours for assessment of protein, but this was not feasible because of the limited availability of human AEC.
With respect to other genes involved in anti-viral defence, we demonstrated up-regulation of the expression of RNA helicases and of the transcription factors STAT1 and STAT2, as well as of other interferon-stimulated genes. However, it was noteworthy that there was no change in the expression of IRF3, even though this transcription factor is believed to be critically involved in the anti-viral response and regulates IFNB, CXCL9, CXCL10 and CCL5[32].
The relationship between respiratory viral infections and asthma is complex, and the underlying mechanisms of cause and effect remain incompletely defined and controversial. For example, there is little doubt that wheezing lower respiratory viral infections in early life are associated with the development of allergic asthma in childhood [33],[34], but it has been suggested that whereas allergic sensitisation increases the risk of wheezing, the converse is not true [35]. Alternatively, some investigators have speculated that development of severe respiratory viral infections is simply an indicator of a genetic predisposition to asthma [36]. Similarly, there is agreement that exacerbations of allergic asthma are most commonly a consequence of viral infections, especially with RV [37]-[39]. However, there is considerable debate about the extent to which an impaired host response might contribute to the development of these infections, or to the severity of infections, or whether the inflammatory response to infection might be significantly different in asthmatics [40].
Our finding of enhanced expression and secretion of a variety of chemokines by AEC pre-treated with Th2 cytokines is consistent with the notion that the allergic environment promotes increased inflammation in response to respiratory viral infection. Our results are concordant with a very recently published study of the response of human AEC to RV, which also demonstrated that cells pre-treated with Th2 cytokines expressed higher levels of the chemokines CXCL8 and CXCL10, independent of any change in viral replication [18]. Increased production of the major neutrophil chemoattractant CXCL8 might help to explain the neutrophilic response to respiratory viral infection observed in the sputum of asthmatics [41],[42]. Increased production of other chemokines might amplify the recruitment of other cell types as well. In this context, it is noteworthy that CXCL10 could be an important pro-inflammatory mediator in asthmatic exacerbations, as it is relatively resistant to suppression by glucocorticosteroids [43].
With respect to epithelial cell-derived Th2-promoting cytokines, the demonstration of a trend towards increased expression of the TSLP gene is consistent with earlier evidence that pre-treatment of AEC with IL-4 induces enhanced production of TSLP following exposure to dsRNA [23]. In contrast, decreased expression of IL-33 in AEC pre-treated with Th2 cytokines is somewhat surprising. IL-33 is potentially important in the pathogenesis of exacerbations of asthma [44],[45]. Moreover, it could be released from AEC in response to virus-induced injury (together with other Th2-promoting cytokines such as IL-25 and TSLP) and might thus help to drive airway inflammation in acute exacerbations of allergic asthma [46]. In this setting, because IL-33 behaves in many respects like a damage-associated molecule or alarmin [47], it may be regulated primarily via altered cytokine release, rather than altered expression of mRNA.
Our observation that there was no diminution in the expression of interferons and indeed an increase in the expression of type III interferons contrasts with another in vitro study, which indicated that treatment with IL-13 suppressed production of type III interferons in response to dsRNA by a human AEC line [48]. This issue is pertinent, especially in the context of evidence that asthmatics are more susceptible to develop lower respiratory viral infections [4] and that their infections are of greater severity [49]. Infections in asthmatics have also been reported to persist for longer, although this is controversial and the increase in RV-related illness may instead be a result of re-infection [4],[50]-[53]. Various studies have suggested that impaired production of interferons by AEC from asthmatics, and especially of type III interferons in those with severe asthma, may be an important predisposing factor and may influence the outcome of infection [7]-[10]. Moreover, a deficient type III interferon response has been suggested to play a key role in determining the severity of asthma exacerbations [8]. However, the evidence that interferon production by AEC from asthmatics is impaired is by no means clear-cut [40],[54]. Indeed, it has been suggested that increased levels of type III interferons may play a role in driving virus-induced exacerbations of asthma [55]. Consistent with this, there is no evidence of an increased viral load associated with exacerbations [55],[56].
Our results indicate that any impairment of interferon-mediated defences of airway epithelium in asthmatics is unlikely to be a direct effect of Th2 cytokines on AEC. However, additional factors may operate in vivo. For example, AEC recovered from severe asthmatics have inevitably been exposed to combinations of therapeutic drugs [9] which are recognised to have suppressive effects on host anti-viral and inflammatory responses [57],[58]. Nevertheless, a recent study in an animal model of chronic asthma suggests that long-term allergen challenge may be associated with a decrease in expression of type I and type II interferons, as well as with borderline changes in type III interferons [59]. Intriguingly, these authors also reported decreased production of other pro-inflammatory cytokines, such as IL-1β and IL-12, in response to RV infection.
We recognise the inherent weaknesses of in vitro studies. Furthermore, our experiments utilised undifferentiated immersion cultures of AEC rather than differentiated air-liquid interface cultures. Notwithstanding these limitations, however, we believe that our data shed new light on the complex interplay between respiratory viral infections, the host cytokine response, and acute inflammation of the airways in exacerbations of allergic asthma.
Conclusions
Collectively, our results suggest that the Th2 cytokine environment which prevails in allergic asthma could promote increased production of pro-inflammatory mediators by AEC in response to respiratory viral infection, but is unlikely to play a role in any impairment of anti-viral host defences in asthmatics.
Acknowledgements
Work in the authors’ laboratories is supported by grants from NHMRC Australia. The funding agency had no role in the collection, analysis, and interpretation of data; in the writing of the manuscript; or in the decision to submit the manuscript for publication.
Abbreviations
- AEC
Airway epithelial cells
- dsRNA
Double-stranded RNA
- HPRT
Hypoxanthine-guanine phosphoribosyltransferase
- IFN
Interferon
- IL
Interleukin
- RV
Rhinovirus(es)
- TLR
Toll-like receptor
- TSLP
Thymic stromal lymphopoietin
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CH supervised the studies on MLE-12 cells and the molecular biological studies on human AEC. Q-XZ performed the cell culture and enzyme immunoassays for human AEC. RS performed the cell culture and most of the molecular biological studies on MLE-12 cells. LG performed the molecular biological studies on human AEC. BO supervised most of the human AEC studies. RKK conceived the study, participated in its design and co-ordination, and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Cristan Herbert, Email: c.herbert@unsw.edu.au.
Qing-Xiang Zeng, Email: qingxiangqie@163.com.
Ramesh Shanmugasundaram, Email: ramesh_112@hotmail.com.
Linda Garthwaite, Email: l.garthwaite@unsw.edu.au.
Brian G Oliver, Email: brian.oliver@sydney.edu.au.
Rakesh K Kumar, Email: R.Kumar@unsw.edu.au.
References
- 1.Reddel HK, Taylor DR, Bateman ED, Boulet LP, Boushey HA, Busse WW, Casale TB, Chanez P, Enright PL, Gibson PG, de Jongste JC, Kerstjens HA, Lazarus SC, Levy ML, O'Byrne PM, Partridge MR, Pavord ID, Sears MR, Sterk PJ, Stoloff SW, Sullivan SD, Szefler SJ, Thomas MD, Wenzel SE. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med. 2009;180:59–99. doi: 10.1164/rccm.200801-060ST. [DOI] [PubMed] [Google Scholar]
- 2.Bahadori K, Doyle-Waters MM, Marra C, Lynd L, Alasaly K, Swiston J, FitzGerald JM. Economic burden of asthma: a systematic review. BMC Pulm Med. 2009;9:24. doi: 10.1186/1471-2466-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol. 2010;125:1178–1187. doi: 10.1016/j.jaci.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corne JM, Marshall C, Smith S, Schreiber J, Sanderson G, Holgate ST, Johnston SL. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359:831–834. doi: 10.1016/S0140-6736(02)07953-9. [DOI] [PubMed] [Google Scholar]
- 5.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, Contoli M, Sanderson G, Kon OM, Papi A, Jeffery PK, Stanciu LA, Johnston SL. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci U S A. 2008;105:13562–13567. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loxham M, Davies DE, Blume C. Clin Exp Allergy. 2014. [DOI] [PubMed] [Google Scholar]
- 7.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, Holgate ST, Davies DE. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, Slater L, Lewis-Antes A, Kon OM, Holgate ST, Davies DE, Kotenko SV, Papi A, Johnston SL. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 9.Edwards MR, Regamey N, Vareille M, Kieninger E, Gupta A, Shoemark A, Saglani S, Sykes A, Macintyre J, Davies J, Bossley C, Bush A, Johnston SL. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013;6:797–806. doi: 10.1038/mi.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, Howarth PH, Holgate ST, Davies DE. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–632. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- 11.Wagener AH, Zwinderman AH, Luiten S, Fokkens WJ, Bel EH, Sterk PJ, van Drunen CM. dsRNA-induced changes in gene expression profiles of primary nasal and bronchial epithelial cells from patients with asthma, rhinitis and controls. Respir Res. 2014;15:9. doi: 10.1186/1465-9921-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons KS, Hsu AC, Wark PA. TLR3 and MDA5 signalling, although not expression, is impaired in asthmatic epithelial cells in response to rhinovirus infection. Clin Exp Allergy. 2014;44:91–101. doi: 10.1111/cea.12218. [DOI] [PubMed] [Google Scholar]
- 13.Baraldo S, Contoli M, Bazzan E, Turato G, Padovani A, Marku B, Calabrese F, Caramori G, Ballarin A, Snijders D, Barbato A, Saetta M, Papi A. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J Allergy Clin Immunol. 2012;130:1307–1314. doi: 10.1016/j.jaci.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Bochkov YA, Hanson KM, Keles S, Brockman-Schneider RA, Jarjour NN, Gern JE. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Souza N, Favoreto S, Wong H, Ward T, Yagi S, Schnurr D, Finkbeiner WE, Dolganov GM, Widdicombe JH, Boushey HA, Avila PC. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J Allergy Clin Immunol. 2009;123:1384–1390. doi: 10.1016/j.jaci.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xatzipsalti M, Psarros F, Konstantinou G, Gaga M, Gourgiotis D, Saxoni-Papageorgiou P, Papadopoulos NG. Modulation of the epithelial inflammatory response to rhinovirus in an atopic environment. Clin Exp Allergy. 2008;38:466–472. doi: 10.1111/j.1365-2222.2007.02906.x. [DOI] [PubMed] [Google Scholar]
- 17.Lachowicz-Scroggins ME, Boushey HA, Finkbeiner WE, Widdicombe JH. Interleukin-13-induced mucous metaplasia increases susceptibility of human airway epithelium to rhinovirus infection. Am J Respir Cell Mol Biol. 2010;43:652–661. doi: 10.1165/rcmb.2009-0244OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cakebread JA, Haitchi HM, Xu Y, Holgate ST, Roberts G, Davies DE. Rhinovirus-16 induced release of IP-10 and IL-8 is augmented by Th2 cytokines in a pediatric bronchial epithelial cell model. PLoS One. 2014;9:e94010. doi: 10.1371/journal.pone.0094010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakiela B, Gielicz A, Plutecka H, Hubalewska-Mazgaj M, Mastalerz L, Bochenek G, Soja J, Januszek R, Aab A, Musial J, Akdis M, Akdis CA, Sanak M. Th2-type cytokine induced mucous metaplasia decreases susceptibility of human bronchial epithelium to rhinovirus infection. Am J Respir Cell Mol Biol. 2014;51:229–241. doi: 10.1165/rcmb.2013-0395OC. [DOI] [PubMed] [Google Scholar]
- 20.Freishtat RJ, Watson AM, Benton AS, Iqbal SF, Pillai DK, Rose MC, Hoffman EP. Asthmatic airway epithelium is intrinsically inflammatory and mitotically dyssynchronous. Am J Respir Cell Mol Biol. 2011;44:863–869. doi: 10.1165/rcmb.2010-0029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG, Van Eeden S, Bai TR, Dorscheid DR, Knight DA. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to RSV and air pollution. Am J Respir Cell Mol Biol. 2011;45:1090–1100. doi: 10.1165/rcmb.2011-0031OC. [DOI] [PubMed] [Google Scholar]
- 22.Rohde G, Message SD, Haas JJ, Kebadze T, Parker H, Laza-Stanca V, Khaitov MR, Kon OM, Stanciu LA, Mallia P, Edwards MR, Johnston SL. CXC-chemokines and antimicrobial peptides in rhinovirus-induced experimental asthma exacerbations. Clin Exp Allergy. 2014;44:930–939. doi: 10.1111/cea.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 25.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 26.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11029–11033. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge Q, Moir LM, Black JL, Oliver BG, Burgess JK. TGFbeta1 induces IL-6 and inhibits IL-8 release in human bronchial epithelial cells: the role of Smad2/3. J Cell Physiol. 2010;225:846–854. doi: 10.1002/jcp.22295. [DOI] [PubMed] [Google Scholar]
- 28.Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A. 2012;109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khaitov MR, Laza-Stanca V, Edwards MR, Walton RP, Rohde G, Contoli M, Papi A, Stanciu LA, Kotenko SV, Johnston SL. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 31.Proud D, Turner RB, Winther B, Wiehler S, Tiesman JP, Reichling TD, Juhlin KD, Fulmer AW, Ho BY, Walanski AA, Poore CL, Mizoguchi H, Jump L, Moore ML, Zukowski CK, Clymer JW. Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am J Respir Crit Care Med. 2008;178:962–968. doi: 10.1164/rccm.200805-670OC. [DOI] [PubMed] [Google Scholar]
- 32.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 33.Holt PG, Sly PD. Prevention of allergic respiratory disease in infants: current aspects and future perspectives. Curr Opin Allergy Clin Immunol. 2007;7:547–555. doi: 10.1097/ACI.0b013e3282f14a17. [DOI] [PubMed] [Google Scholar]
- 34.Sly PD, Kusel M, Holt PG. Do early-life viral infections cause asthma? J Allergy Clin Immunol. 2010;125:1202–1205. doi: 10.1016/j.jaci.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 35.Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Lee WM, Gern JE, Lemanske RF., Jr Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in Early Life. Am J Respir Crit Care Med. 2012;185:281–285. doi: 10.1164/rccm.201104-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomsen SF, van der Sluis S, Stensballe LG, Posthuma D, Skytthe A, Kyvik KO, Duffy DL, Backer V, Bisgaard H. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry-based twin study. Am J Respir Crit Care Med. 2009;179:1091–1097. doi: 10.1164/rccm.200809-1471OC. [DOI] [PubMed] [Google Scholar]
- 37.Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, Symington P, O'Toole S, Myint SH, Tyrrell DA. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray CS, Simpson A, Custovic A. Allergens, viruses, and asthma exacerbations. Proc Am Thorac Soc. 2004;1:99–104. doi: 10.1513/pats.2306027. [DOI] [PubMed] [Google Scholar]
- 39.Wark PA, Gibson PG. Asthma exacerbations. 3: Pathogenesis. Thorax. 2006;61:909–915. doi: 10.1136/thx.2005.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gavala ML, Bashir H, Gern JE. Virus/allergen interactions in asthma. Curr Allergy Asthma Rep. 2013;13:298–307. doi: 10.1007/s11882-013-0344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wark PA, Johnston SL, Moric I, Simpson JL, Hensley MJ, Gibson PG. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J. 2002;19:68–75. doi: 10.1183/09031936.02.00226302. [DOI] [PubMed] [Google Scholar]
- 42.Grissell TV, Powell H, Shafren DR, Boyle MJ, Hensley MJ, Jones PD, Whitehead BF, Gibson PG. Interleukin-10 gene expression in acute virus-induced asthma. Am J Respir Crit Care Med. 2005;172:433–439. doi: 10.1164/rccm.200412-1621OC. [DOI] [PubMed] [Google Scholar]
- 43.Wark PA, Bucchieri F, Johnston SL, Gibson PG, Hamilton L, Mimica J, Zummo G, Holgate ST, Attia J, Thakkinstian A, Davies DE. IFN-gamma-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbations. J Allergy Clin Immunol. 2007;120:586–593. doi: 10.1016/j.jaci.2007.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG, Hamid Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 45.Bunting MM, Shadie AM, Flesher RP, Nikiforova V, Garthwaite L, Tedla N, Herbert C, Kumar RK. Interleukin-33 drives activation of alveolar macrophages and airway inflammation in a mouse model of acute exacerbation of chronic asthma. Biomed Res Int. 2013;2013:250938. doi: 10.1155/2013/250938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar RK, Foster PS, Rosenberg HF. Respiratory viral infection, epithelial cytokines, and innate lymphoid cells in asthma exacerbations. J Leukoc Biol. 2014;96:391–396. doi: 10.1189/jlb.3RI0314-129R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriwaki A, Matsumoto K, Matsunaga Y, Fukuyama S, Matsumoto T, Kan-o K, Noda N, Asai Y, Nakanishi Y, Inoue H. IL-13 suppresses double-stranded RNA-induced IFN-lambda production in lung cells. Biochem Biophys Res Commun. 2011;404:922–927. doi: 10.1016/j.bbrc.2010.12.082. [DOI] [PubMed] [Google Scholar]
- 49.Olenec JP, Kim WK, Lee WM, Vang F, Pappas TE, Salazar LE, Evans MD, Bork J, Roberg K, Lemanske RF, Jr, Gern JE. Weekly monitoring of children with asthma for infections and illness during common cold seasons. J Allergy Clin Immunol. 2010;125:1001–1006. doi: 10.1016/j.jaci.2010.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kling S, Donninger H, Williams Z, Vermeulen J, Weinberg E, Latiff K, Ghildyal R, Bardin P. Persistence of rhinovirus RNA after asthma exacerbation in children. Clin Exp Allergy. 2005;35:672–678. doi: 10.1111/j.1365-2222.2005.02244.x. [DOI] [PubMed] [Google Scholar]
- 51.Wos M, Sanak M, Soja J, Olechnowicz H, Busse WW, Szczeklik A. The presence of rhinovirus in lower airways of patients with bronchial asthma. Am J Respir Crit Care Med. 2008;177:1082–1089. doi: 10.1164/rccm.200607-973OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wood LG, Powell H, Grissell TV, Davies B, Shafren DR, Whitehead BF, Hensley MJ, Gibson PG. Persistence of rhinovirus RNA and IP-10 gene expression after acute asthma. Respirology. 2011;16:291–299. doi: 10.1111/j.1440-1843.2010.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Engelmann I, Mordacq C, Gosset P, Tillie-Leblond I, Dewilde A, Thumerelle C, Pouessel G, Deschildre A. Rhinovirus and asthma: reinfection, not persistence. Am J Respir Crit Care Med. 2013;188:1165–1167. doi: 10.1164/rccm.201303-0585LE. [DOI] [PubMed] [Google Scholar]
- 54.Sykes A, Macintyre J, Edwards MR, Del Rosario A, Haas J, Gielen V, Kon OM, McHale M, Johnston SL. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. 2014;69:240–246. doi: 10.1136/thoraxjnl-2012-202909. [DOI] [PubMed] [Google Scholar]
- 55.Miller EK, Hernandez JZ, Wimmenauer V, Shepherd BE, Hijano D, Libster R, Serra ME, Bhat N, Batalle JP, Mohamed Y, Reynaldi A, Rodriguez A, Otello M, Pisapia N, Bugna J, Bellabarba M, Kraft D, Coviello S, Ferolla FM, Chen A, London SJ, Siberry GK, Williams JV, Polack FP. A mechanistic role for type III IFN-lambda1 in asthma exacerbations mediated by human rhinoviruses. Am J Respir Crit Care Med. 2012;185:508–516. doi: 10.1164/rccm.201108-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kennedy JL, Shaker M, McMeen V, Gern J, Carper H, Murphy D, Lee WM, Bochkov YA, Vrtis RF, Platts-Mills T, Patrie J, Borish L, Steinke JW, Woods WA, Heymann P. Comparison of viral load in individuals with and without asthma during infections with rhinovirus. Am J Respir Crit Care Med. 2014;189:532–539. doi: 10.1164/rccm.201310-1767OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skevaki CL, Christodoulou I, Spyridaki IS, Tiniakou I, Georgiou V, Xepapadaki P, Kafetzis DA, Papadopoulos NG. Budesonide and formoterol inhibit inflammatory mediator production by bronchial epithelial cells infected with rhinovirus. Clin Exp Allergy. 2009;39:1700–1710. doi: 10.1111/j.1365-2222.2009.03307.x. [DOI] [PubMed] [Google Scholar]
- 58.Davies JM, Carroll ML, Li H, Poh AM, Kirkegard D, Towers M, Upham JW. Budesonide and formoterol reduce early innate anti-viral immune responses in vitro. PLoS One. 2011;6:e27898. doi: 10.1371/journal.pone.0027898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rochlitzer S, Hoymann HG, Muller M, Braun A. No exacerbation but impaired anti-viral mechanisms in a rhinovirus-chronic allergic asthma mouse model. Clin Sci (Lond) 2014;126:55–65. doi: 10.1042/CS20130174. [DOI] [PubMed] [Google Scholar]