

Abstract
ACE-536, a recombinant protein containing a modified activin receptor type IIB, is being developed for the treatment of anemias caused by ineffective erythropoiesis, such as thalassemias and myelodysplastic syndromes. ACE-536 acts through a mechanism distinct from erythropoiesis-stimulating agents to promote late-stage erythroid differentiation by binding to transforming growth factor-β superfamily ligands and inhibiting signaling through transcription factors Smad 2/3. The goal of this Phase 1 study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamic effects of ascending dose levels of ACE-536 in healthy volunteers. Thirty-two postmenopausal women were randomized in sequential cohorts of eight subjects each to receive up to two doses of either ACE-536 (0.0625–0.25 mg/kg) or placebo (3:1 randomization) given subcutaneously every 2 weeks. Mean baseline age was 59.4 years, and hemoglobin was 13.2 g/dL. ACE-536 was well tolerated at dose levels up to 0.25 mg/kg over the 1-month treatment period. There were no serious or severe adverse events, nor clinically meaningful changes in safety laboratory measures or vital signs. Mean ACE-536 AUC0–14d and Cmax increased proportionally after first dose; mean t½ was 15–16 days. Dose-dependent increases in hemoglobin concentration were observed, beginning 7 days after initiation of treatment and maintained for several weeks following treatment. The proportion of subjects with a hemoglobin increase ≥1.0 g/dL increased in a dose-dependent manner to 83.3% of subjects in the highest dose group, 0.25 mg/kg. ACE-536 was well tolerated and resulted in sustained increases in hemoglobin levels in healthy postmenopausal women. Am. J. Hematol. 89:766–770, 2014. © 2014 Wiley Periodicals, Inc.
Introduction
Erythropoiesis is a multistep process by which erythroid precursor cells proliferate and differentiate into mature red blood cells (RBCs) to maintain hemoglobin concentrations in the blood within a narrow range. Under the influence of erythropoietin (EPO), committed progenitor cells divide and differentiate into proerythroblasts [1]. These cells further mature through a series of normoblast stages before undergoing enucleation to form the reticulocyte and mature RBC. Ligands of the transforming growth factor-β superfamily, including certain growth and differentiation factors (GDFs), their respective “activin” receptors, and the intracellular signaling proteins Smad 2/3, have been shown to be negative modulators of late-stage erythropoiesis [2,3]. This mechanism of action is thus distinct from EPO, which stimulates proliferation of RBC progenitor cells during early stages of erythropoiesis.
Diseases such as the thalassemias [4,5] and myelodysplastic syndromes (MDS) [6] are characterized by ineffective erythropoiesis, which can lead to overproduction of EPO, erythroid hyperplasia, and bone marrow expansion, as well as dysregulated iron metabolism, including suppressed hepcidin production. Increased Smad 2 phosphorylation and signaling may play a role in conditions of ineffective erythropoiesis, as has been shown in bone marrow biopsies of patients with MDS [7]. In β-thalassemia, defective β-globin chain synthesis and increased free heme in developing normoblasts, along with other factors, lead to an excess of α-globin chains, which precipitate and form hemichromes, resulting in ineffective erythropoiesis and cell death [4].
ACE-536, a recombinant protein comprised of a modified extracellular domain of the human activin receptor type IIB (ActRIIB) linked to the Fc region of human immunoglobulin IgG1 (modified ActRIIB-huIgG1), is being developed for the treatment of conditions with anemia caused by ineffective erythropoiesis. ACE-536 binds to ligands in the transforming growth factor-β superfamily that regulate late-stage erythropoiesis, including the GDFs GDF11 [8,9] and GDF8, and thus functions as a ligand trap to block the interaction of these ligands with activin receptors on developing normoblasts and consequently reduce Smad 2/3 signaling. Nonclinical pharmacology studies have demonstrated that treatment with ACE-536 or its murine version RAP-536 (containing murine IgG Fc in place of human IgG Fc) can increase RBC, hematocrit, and hemoglobin levels in a dose-dependent manner [10]. ACE-536 or RAP-536 treatment increases RBC parameters in multiple species, and prevented or reduced anemia in different murine anemia models, including MDS, due to abortive erythroid maturation [10].
This article reports the results from a first-in-human, Phase 1, ascending-dose, randomized, double-blind, placebo-controlled clinical trial of ACE-536 in healthy, postmenopausal women to evaluate its safety, tolerability, pharmacokinetics (PK), and pharmacodynamic (PD) effects.
Methods
Standard protocol approvals, registrations, and subject consents
The study, identified by the sponsor as Study A536-02, was entered in the http://ClinicalTrials.gov registry with Identifier NCT01432717. It was approved by the local institutional review board and conducted in accordance with applicable national and local regulations and requirements for good clinical practices. All subjects were required to provide written informed consent before screening.
Study design
This completed Phase 1, randomized, double-blind, placebo-controlled, dose escalation trial was conducted between September 2011 and September 2012 at a single clinical research center (Celerion, Tempe, AZ) in 32 healthy postmenopausal women. The primary objective was to evaluate the safety and tolerability of multiple ascending dose levels of ACE-536 administered subcutaneously (SC). Secondary objectives included the evaluation of PK and PD effects of ACE-536 in healthy subjects.
Subjects were randomized into cohorts of eight subjects each (six active and two placebo control subjects per cohort). Subjects were evaluated for study inclusion and exclusion criteria during the screening period (within 28 days prior to the first dose), and those who met the study entry criteria were enrolled and treated with study drug (ACE-536 or placebo) beginning on Day 1. Subjects were to receive a total of two doses of study drug administered via SC injection on Day 1 and Day 15.
ACE-536 was tested at dose levels of 0.0625, 0.125, and 0.25 mg/kg in four cohorts as follows: Cohort 1, 0.0625 mg/kg for two doses; Cohort 2, 0.125 mg/kg for one dose (due to study interruption; see Results); Cohort 3, 0.125 mg/kg for two doses; and Cohort 4, 0.25 mg/kg for two doses. Subjects were evaluated for safety and PD effects through Day 57, with follow-up safety visits at approximately 2 and 10 weeks after the Day 57 visit.
Prior to administering the second dose of study drug on Day 15 (Cohorts 1, 3, and 4), adverse events (AEs), blood pressure, and hemoglobin levels were evaluated for each subject. The second dose was not given to a subject if a Grade 3 AE occurred, if the Day 15 hemoglobin was >15.5 g/dL or had increased ≥1.0 g/dL at any time after Day 1, or if the Day 15 blood pressure was elevated (systolic > 140 mmHg and/or diastolic > 90 mmHg as an average of two readings at least 15 min apart).
Safety was evaluated by a Safety Review Team, which was comprised at minimum of the principal investigator (Dr. Allison), the medical monitor (Dr. Attie), and an independent hematologist (Dr. David Kuter). Escalation to the next dose level proceeded only after the Safety Review Team reviewed cumulative, blinded safety data when at least six subjects completed Day 29 in the previous cohort. Dose escalation was to be halted if a dose-limiting toxicity (e.g., serious AE, Grade ≥ 3 AE, or Grade ≥ 2 AE of bleeding or hypertension) occurred in two or more subjects or if a hemoglobin increase of ≥1.5 g/dL occurred in two or more subjects within 28 days of first dose.
Subject eligibility
Eligible subjects were healthy, postmenopausal women who understood and signed written informed consent, aged 45–75 years, with a body mass index (BMI) of 20–32 kg/m2. Postmenopausal women were chosen because of the potential risk of ACE-536 binding to activin(s) and inhibiting their activity in the pituitary gland and/or gonads and subsequent potential effects on fertility. Screening and Day 1 hemoglobin had to be ≥11.0 g/dL and ≤14.5 g/dL.
Subjects who had a platelet count <100,000/µL or donated or lost ≥500 mL of whole blood within 2 months prior to Day 1 were excluded. Subjects who had received an erythropoiesis-stimulating agent (ESA), systemic glucocorticoid therapy, or anticoagulant therapy within 6 months prior to Day 1 were also excluded.
Randomization and blinding
A computer-generated randomization scheme was prepared by the statistics group at Celerion using SAS v9.1.3. The investigators, all other site personnel (except for the pharmacist preparing study drug), and all subjects remained blinded to subject treatment assignments throughout the study.
Study drug
ACE-536 drug substance was manufactured by Acceleron Pharma Inc. (Cambridge, MA). ACE-536 drug product was provided as a frozen, sterile, liquid formulation at a nominal concentration of 50 mg/mL in Tris-buffered saline for SC injection. The placebo used in this study was sterile normal saline (0.9% sodium chloride for injection) for SC injection.
Assessments and endpoints
Safety
Safety endpoints were assessed at baseline and regularly throughout the study (eight visits during the first 8 weeks following the first dose, and 2 additional visits during the 10-week follow-up period). They included evaluation of AEs, physical examination, vital signs, electrocardiogram, hematology, chemistry, urinalysis, coagulation profile, troponin-I, B-type natriuretic peptide, follicle-stimulating hormone (FSH), and antidrug antibodies. AEs which occurred after signing of the consent form and through Day 71 were recorded regardless of whether they were considered to be study drug related. Only events that were newly occurring or increasing in severity or frequency during or after study drug administration were analyzed. AEs were graded as mild, moderate, severe, or life-threatening by National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. Serious AEs were defined in accordance with International Conference on Harmonisation Good Clinical Practice guidelines.
Serum samples for anti-ACE-536 antibody analysis were collected at baseline and Days 29, 57, 71, and 127, and assessed using a validated, qualitative, immunoassay performed at ICON Development Solutions (Whitesboro, NY). The assay used was an electrochemiluminescent immunoassay with ACE-536-coated assay plates followed by detection with biotinylated ACE-536. The sensitivity of the assay was 286 ng/mL, and the interbatch coefficient of variation was ≤26.9%.
Pharmacokinetics
Serum samples for the determination of ACE-536 concentration were collected on study days 1 (at 0, 3, and 6 hr), 3, 5, 8, 15 (at 0, 3, and 6 hr), 22, 29, 43, 57, and 71. ACE-536 was assessed using a validated, sandwich enzyme-linked immunosorbent assay performed at ICON Development Solutions. The assay used an anti-ACE-536 antibody for capture followed by an anti-human IgG1 antibody for detection. The assay range was 50.0–600 ng/mL. The inter-batch coefficient of variation was ≤12.0%. The following PK parameters were calculated, using standard noncompartmental or compartmental methods as implemented in WinNonlin, Version 6.3 (Pharsight, Mountain View, CA): area under the concentration–time curve (AUC), peak concentration (Cmax), time to peak concentration (Tmax), elimination half-life (t1/2), clearance (Cl/F), and volume of distribution (Vz/F).
Pharmacodynamics
Protocol-specified PD assessments included RBC count, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, peripheral blood smear (for RBC morphology), serum iron, total iron-binding capacity, unsaturated iron-binding capacity, transferrin, ferritin, and transferrin saturation (calculated as serum iron/total iron-binding capacity), hemoglobin electrophoresis, hepcidin, EPO, bone-specific alkaline phosphatase, and C-terminal telopeptide.
Statistical methods
The sample size for this Phase 1 dose-escalation study was sufficient to evaluate safety, tolerability, and PK based on clinical considerations. Although PD endpoints were also evaluated, the study was not powered for efficacy. Baseline value was defined as the last observation prior to study drug administration. Data from subjects who received placebo were pooled across all four cohorts.
Demographics and safety evaluations were analyzed by descriptive statistics using data from all subjects who received ACE-536 or placebo. AEs were coded using the Medical Dictionary for Regulatory Activities, version 14.1.
PK and PD parameters were evaluated by descriptive statistics using data from all subjects who received at least one dose of study drug and had at least one post-dose measurement. Statistical analysis results were considered significant at P < 0.05 when using a two sample Student's t-test of mean change from baseline vs. placebo. No adjustments were made for multiple comparisons.
Results
Subject disposition and characteristics
Thirty-two postmenopausal women were enrolled, including 24 subjects randomized to active treatment and eight randomized to placebo. All subjects were treated as they were randomized and completed all protocol-specified visits. The study was temporarily interrupted due to a regulatory hold related to a contract manufacturer. This interruption occurred following the first dose of 0.125 mg/kg in Cohort 2, and, as a result, the second dose was not administered to these eight subjects. The 0.125 mg/kg dose level was repeated in Cohort 3 with administration of both doses to an additional eight subjects. The study was ended by the Sponsor following completion of the 0.25 mg/kg dose level based upon protocol-defined dose escalation stopping rules related to increases in hemoglobin levels.
A total of 24 subjects received ACE-536, and eight subjects received placebo. Six subjects received two doses of ACE-536 at 0.0625 mg/kg. A total of seven subjects received one dose of ACE-536 at 0.125 mg/kg, including six subjects from Cohort 2 and one subject from Cohort 3 who did not receive the second dose per protocol-specified rules due to elevated blood pressure on Day 15 prior to scheduled dosing (not reported as an AE and not associated with increased hemoglobin). The remaining five subjects in Cohort 3 received two doses of ACE-536 at 0.125 mg/kg. One subject received two doses, and five subjects received only one dose of ACE-536 at 0.25 mg/kg; four subjects did not receive the second dose due to increase in hemoglobin level (per protocol) and one subject did not receive the second dose due to an AE of rash.
Demographics and baseline characteristics are summarized in Supporting Information Table 1. The mean subject age was 59.4 years (range: 49–71 years), mean weight was 68.8 kg (52.9–84.1 kg), and mean BMI was 26.1 kg/m2 (20.9–31.6 kg/m2), overall. Mean hemoglobin at screening was 13.3 g/dL (range: 11.6–14.3 g/dL) and on Day 1 was 13.2 g/dL (range: 12.0–14.2 g/dL). No notable differences were seen among the four active treatment groups and the placebo group for baseline age, weight, height, BMI, hemoglobin level, or race. Hispanic or Latino ethnicity ranged from 0% of subjects in two of the dose groups to 50% of subjects in the 0.125 mg/kg × 1 dose group, and was 22% overall.
Safety and tolerability
ACE-536 administered SC at doses up to and including 0.25 mg/kg was generally well tolerated. No serious, severe, or life-threatening AEs or deaths were reported. The incidence of AEs was comparable across treatment groups, including placebo. The majority of AEs (59 of 71 events, 83%) was mild in severity and resolved without sequelae.
The most frequently reported AE was headache (four active-treated patients, three placebo-treated, all not related). The majority of events were considered by the investigator to be unrelated to the study treatment. Among the 24 subjects treated with ACE-536, seven (29%) experienced at least one AE considered at least possibly related to the study drug (Supporting Information Table II). Adverse events that were considered at least possibly drug-related across ACE-536 dose groups included injection-site hemorrhage (three subjects), injection-site macule (two subjects), and dry skin, macule, hyperesthesia, muscle spasms, myalgia, generalized pruritus, and papular rash (one subject each). Injection-site pain was reported in one placebo-treated subject.
No clinically relevant abnormalities were observed in physical examination, vital signs, electrocardiogram, or urinalysis. There were no dose-dependent changes in mean QTc interval by electrocardiogram. Overall, mean values for safety laboratory parameters generally remained within the normal range, and there were no clinically meaningful abnormalities. In particular, there were no hemoglobin levels that exceeded the normal range, no clinically significant changes in blood pressure or coagulation profile including PT/INR, PTT, and adenosine diphosphate and arachidonic acid aggregation platelet function assays, and no aspirin or antiplatelet concomitant medications reported. No antidrug antibodies were detected. Mean FSH levels decreased slightly in the 0.25 mg/kg group, but remained within normal limits and returned to baseline levels by Day 57.
Pharmacokinetics
PK parameters were estimated by noncompartmental or single compartmental (for Cmax and Tmax) analysis of ACE-536 serum concentration data. Mean AUC0–14d and Cmax after the first dose increased proportionally with ACE-536 dose level from 0.0625 to 0.25 mg/kg (Supporting Information Table III). At 0.25 mg/kg, mean Cmax was 1.9 µg/mL after the first dose. ACE-536 absorption and elimination were largely independent of dose level, with mean Tmax of 7–10 days and mean t1/2 of 15–16 days after the first dose (Supporting Information Table III and Fig. 1). Every 2-week dosing resulted in increased Cmax and AUC after the second dose, with mean accumulation ratios ranging from 1.5 to 2.2. Estimates for clearance (Cl), and volume of distribution (Vz) are presented in Supporting Information Table III.
Figure 1.
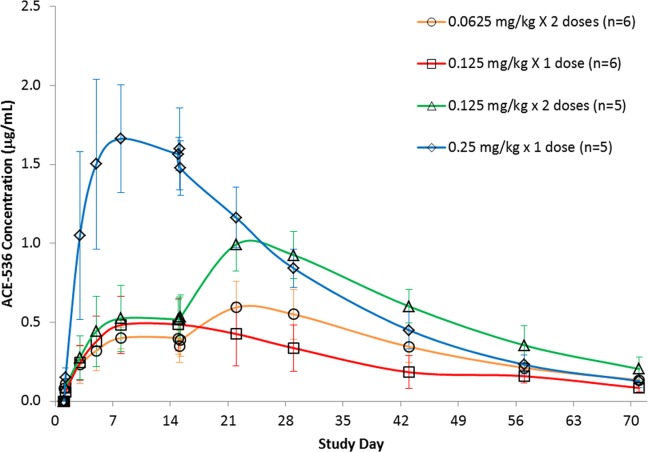
Pharmacokinetic profile of ACE-536 concentrations following one or two subcutaneous doses in healthy postmenopausal women treated with ACE-536 (mean ± SD).
Pharmacodynamic effects
Sustained increases in mean RBC, hematocrit, and hemoglobin levels were observed in the ACE-536-treated groups, compared with placebo. Mean levels in the 0.25 mg/kg group were increased from Day 8 through Day 57 (Fig. 2). The mean maximum increase in hemoglobin (using maximum value at any time point minus baseline value) in the 0.25 mg/kg group (n = 6) was 1.3 g/dL (P < 0.01 vs. placebo group [n = 8]). Mean levels in the placebo group were decreased through Day 43, possibly due in part to blood collection.
Figure 2.
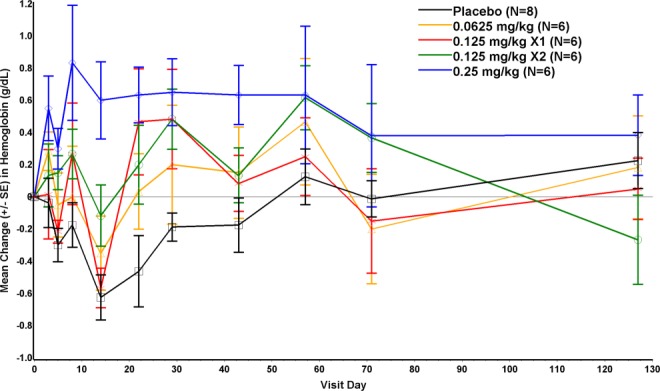
Change from baseline in hemoglobin concentrations (g/dL, mean ± SE); baseline hemoglobin was last nonmissing value prior to dosing.
The proportion of subjects with hemoglobin increase ≥1.0 g/dL showed a dose-dependent increase (Fig. 3). In the 0.25 mg/kg group, five of six (83.3%) had an increase ≥1.0 g/dL, despite five of six subjects only receiving one dose. The proportion achieving this response was 41.6% in the two groups treated with 0.125 mg/kg, 16.7% in the 0.0625 mg/kg group, and 12.5% in the placebo group. The mean duration of hemoglobin increase ≥1.0 g/dL in the 0.125 and 0.25 mg/kg groups was approximately 14 days in responders who received one dose (n = 7) and approximately 21 days in responders who received two doses (n = 3).
Figure 3.
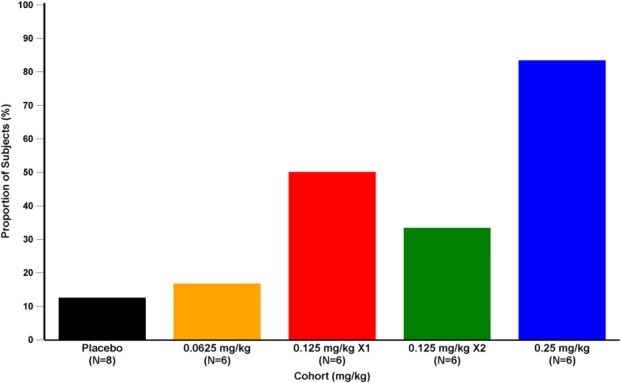
Proportion of subjects (%) with hemoglobin increase ≥1.0 g/dL following dosing with ACE-536 or placebo.
Changes from baseline to Day 15 for RBC parameters and indices are shown in Table I. Increases in hemoglobin were significantly different from placebo in the 0.25 mg/kg group, and increases in RBC were significantly different from placebo in the 0.125 ×2 doses and 0.25 mg/kg groups. Increases from baseline in mean reticulocyte count were significantly different from placebo in the 0.125 ×1 dose and 0.25 mg/kg groups. Minimal increases from baseline were also observed for mean EPO levels for some of the ACE-536 groups; the majority of individual values remained within normal limits. There were no significant changes in serum iron or ferritin levels relative to placebo. No significant changes in hepcidin (Table I) or hemoglobin electrophoresis results were observed. No significant changes in the bone formation biomarker bone-specific alkaline phosphatase and the bone resorption biomarker C-terminal telopeptide were seen (Table I).
TABLE I.
Pharmacodynamic Effects of ACE-536 by Treatment Group (Change from Baseline, Mean (SD))
| ACE-536 treatment (mg/kg) | |||||
|---|---|---|---|---|---|
| Placebo (N = 8) | 0.0625 × 2 (N = 6) | 0.125 × 1 (N = 6) | 0.125 × 2 (N = 6) | 0.25 × 2 (N = 6) | |
| Change from baseline to Day 15 | |||||
| RBC (millions/µL) | −0.27 (0.14) | −0.11 (0.16) | −0.16 (0.11) | −0.04a (0.17) | 0.14a (0.19) |
| Hemoglobin (g/dL) | −0.6 (0.4) | −0.4 (0.6) | −0.6 (0.3) | −0.1 (0.5) | 0.6a (0.6) |
| MCV (fL) | −0.3 (0.9) | −0.2 (1.2) | −0.2 (0.8) | 0.3 (0.5) | −0.3 (0.5) |
| MCHC (g/dL) | 0.2 (0.3) | 0.0 (0.5) | −0.2 (0.2) | −0.1 (0.1) | 0.6 (0.8) |
| Reticulocytes (%) | −0.06 (0.12) | −0.05 (0.15) | 0.15a (0.19) | 0.12b (0.08) | 0.22a (0.15) |
| Change from baseline to Day 29 | |||||
| Hepcidin (ng/mL) | −0.4 (4.5) | 0.0 (4.7) | −0.7 (5.0) | 1.2 (4.2) | 1.6 (6.9) |
| Erythropoietin (U/L) | −1.4 (10.4) | 3.0 (2.4) | −1.2 (2.9) | 5.8b (5.6) | 4.2 (3.9) |
| FSH (IU/L) | 1.1 (5.4) | 2.5 (6.9) | 1.5 (6.5) | 6.3 (8.0) | −13.2b (17.5) |
| BSAP (µg/L) | −0.7 (3.0) [n = 4] | ND | ND | 1.6 (1.2) | 0.7 (0.9) |
| CTX (pg/mL) | 52.3 (87.2) [n = 4] | ND | ND | −44.2 (122.8) | −29.0 (127.3) |
P < 0.01
P < 0.05 vs. placebo. If not shown, P-value was not significant.
ND, not done (these tests were performed only for Cohorts 3 and 4).
RBC: red blood cell; MCV: mean corpuscular volume; MCHC: mean corpuscular hemoglobin concentration; FSH: follicle-stimulating hormone; BSAP: bone-specific alkaline phosphatase; CTX: C-terminal telopeptide.
Discussion
ACE-536 is a recombinant protein composed of a modified extracellular domain of the ActRIIB linked to the Fc region of human immunoglobulin IgG1. It is being developed as a treatment of anemia in conditions characterized by ineffective erythropoiesis, such as in β-thalassemia and MDS, where often the anemia is either unresponsive or becomes refractory to ESAs. In this study in healthy volunteers, ACE-536 was found to be safe and well-tolerated at dose levels up to 0.25 mg/kg. The most common AE was headache (in 22% of subjects overall), and injection-site reactions were minimal. Clinically significant AEs described for ESAs, such as hypertension, stroke, and other cardiovascular effects, were not observed in this study as events related to ACE-536. One patient had transiently elevated blood pressure on Day 15 that was not associated with increased hemoglobin. Effects of ACE-536 on blood pressure continue to be monitored in ongoing trials.
The PK profile of ACE-536 demonstrated a long-acting pattern of exposure with subcutaneous dosing, as was expected for this fusion protein. Cmax after the first dose and AUC0–14d were generally dose-proportional, whereas Tmax and t½ were not, and were approximately 7–10 days and 15–16 days, respectively. Every 2-week dosing used in this study resulted in notable Cmax and AUC accumulation. These results suggest less frequent SC dosing of ACE-536, for example, every 3 weeks may be appropriate for future studies.
The substantial increases in hemoglobin seen after even a single dose of ACE-536 at 0.25 mg/kg were consistent with results from mechanistic and animal pharmacology studies [10]. The rapid effects on erythropoiesis, including small increases in reticulocyte counts, support the purported mechanism of action of ACE-536 on late-stage differentiation. This is different from the more delayed effects of ESAs, which target earlier proliferative stages [1]. Although minimal increases in EPO levels were also observed in this study and may have contributed to the rise in hemoglobin levels, this may have been a secondary response to the increased maturation process. The effects of ACE-536 on RBCs and hemoglobin levels in healthy volunteers was expected based on nonclinical studies in healthy animals and suggests that the GDF11-Smad 2/3 signaling pathway is constitutively active in regulating terminal erythroid differentiation and maturation. ACE-536 is thus believed to act through a distinct mechanism from that of ESAs and could potentially be used to treat anemias resistant or refractory to ESAs, or in combination with ESAs in certain conditions.
A similar investigative product, sotatercept (ACE-011), based on a soluble activin receptor type IIA, demonstrated increases in both erythropoietic parameters and markers of bone formation, as well as decreases in FSH, in single-dose and multiple-dose studies in healthy volunteers [11,12], and its mouse version has been recently shown to correct ineffective erythropoiesis in a preclinical model of β-thalassemia [13]. In comparison, ACE-536 had minimal effects on FSH levels or markers of bone formation or resorption, suggesting reduced binding of ACE-536 to activins at the dose levels tested. This was an intended result for this modified receptor. Thus, the mechanism of action of ACE-536 may be via binding to other ligands, including GDF11 [8–10], resulting in reduced Smad 2/3 signaling [10,14].
ACE-536 is a novel regulator of erythroid differentiation that acts via a mechanism distinct from ESAs. ACE-536, or its murine version RAP-536, has been shown to be active in mouse models of conditions in which endogenous EPO levels are elevated and exogenous ESAs are either ineffective, such as in β-thalassemia, or face resistance or refractoriness, such as in MDS. RAP-536 treatment for 2 months in a thalassemia mouse model increased RBC, hematocrit, and hemoglobin levels, with decreased RBC inclusion bodies and hemolysis [15]. In a murine MDS model, RAP-536 increased erythropoietic parameters and decreased bone marrow erythroid hyperplasia [10].
In conclusion, results from this Phase 1 study indicate that ACE-536 can actively stimulate RBC production and increase hemoglobin levels in healthy adult female subjects. The treatment was generally safe and well tolerated at effective dose levels, administered over a 1-month course. These findings support the ongoing Phase 2 clinical trials of ACE-536 in patients with MDS and β-thalassemia, to treat conditions characterized by anemia due to ineffective erythropoiesis.
Acknowledgments
The authors thank the following individuals for their contributions to the study: Sasha Bowden, Yijun Yang, Christopher Rovaldi, John Oram, Ravi Kumar, James Desiderio, and Steven Ertel (Acceleron Pharma); Sharon Wagner, Theresa Sullivan, Sara Azzam, Kate Reese, and Anthea Cromie, PhD (Celerion); Angie Aldridge and Pamela Henderson (study monitors); and ICON Development Solutions (LLC Whitesboro, NY). They also thank David Kuter, MD, DPhil (Director, Center for Hematology, Massachusetts General Hospital Cancer Center) for his role as an independent hematologist and member of the Safety Review Team. Celgene Corporation and Acceleron are jointly developing ACE-536.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Tables.
References
- 1.Doshi S, Krzyzanski W, Yue S, et al. Clinical pharmacokinetics and pharmacodynamics of erythropoiesis-stimulating agents. Clin Pharmacokinet. 2013;52:1063–1083. doi: 10.1007/s40262-013-0098-x. [DOI] [PubMed] [Google Scholar]
- 2.Söderberg SS, Karlsson G, Karlsson S. Complex and context dependent regulation of hematopoiesis by TGF-β superfamily signaling. Ann NY Acad Sci. 2009;1176:55–69. doi: 10.1111/j.1749-6632.2009.04569.x. [DOI] [PubMed] [Google Scholar]
- 3.Iancu-Rubin C, Mosoyan G, Wang J, et al. Stromal cell-mediated inhibition of erythropoiesis can be attenuated by sotatercept (ACE-011), an activin receptor type II ligand trap. Exp Hematol. 2013;41:155–166. doi: 10.1016/j.exphem.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Ginzburg Y, Rivella S. β-thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118:4321–4330. doi: 10.1182/blood-2011-03-283614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in β-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109:5027–5035. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santini V, Girelli D, Sanna A, et al. Hepcidin levels and their determinants in different types of myelodysplastic syndromes. PLoS One. 2011;6:e23109. doi: 10.1371/journal.pone.0023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou L, Nguyen AN, Sohal D, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112:3434–3443. doi: 10.1182/blood-2008-02-139824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh SP, Yeo CY, Lee Y, et al. Activin type IIA and IIB receptors mediate GDF11 signaling in axial vertebral patterning. Gene Dev. 2002;16:2749–2754. doi: 10.1101/gad.1021802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22:260. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- 10.Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408–414. doi: 10.1038/nm.3512. [DOI] [PubMed] [Google Scholar]
- 11.Ruckle J, Jacobs M, Kramer W, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res. 2009;24:744–752. doi: 10.1359/jbmr.081208. [DOI] [PubMed] [Google Scholar]
- 12.Sherman ML, Borgstein NG, Mook L, et al. Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy postmenopausal women. J Clin Pharmacol. 2013;53:1121–1130. doi: 10.1002/jcph.160. [DOI] [PubMed] [Google Scholar]
- 13.Dussiot M, Taciel TT, Fricot A, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat Med. 2014;20:398–407. doi: 10.1038/nm.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sako D, Grinberg AV, Liu J, et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J Biol Chem. 2010;285:21037–21048. doi: 10.1074/jbc.M110.114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suragani RN, Li R, Cawley S, et al. ACE-536 improves ineffective erythropoiesis, anemia and co-morbidities in β-thalassemia. Blood. 2012;120:248. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Tables.