

Abstract
Objective
Drug–drug interactions (DDIs) are an important consideration in both drug development and clinical application, especially for co-administered medications. While it is necessary to identify all possible DDIs during clinical trials, DDIs are frequently reported after the drugs are approved for clinical use, and they are a common cause of adverse drug reactions (ADR) and increasing healthcare costs. Computational prediction may assist in identifying potential DDIs during clinical trials.
Methods
Here we propose a heterogeneous network-assisted inference (HNAI) framework to assist with the prediction of DDIs. First, we constructed a comprehensive DDI network that contained 6946 unique DDI pairs connecting 721 approved drugs based on DrugBank data. Next, we calculated drug–drug pair similarities using four features: phenotypic similarity based on a comprehensive drug–ADR network, therapeutic similarity based on the drug Anatomical Therapeutic Chemical classification system, chemical structural similarity from SMILES data, and genomic similarity based on a large drug–target interaction network built using the DrugBank and Therapeutic Target Database. Finally, we applied five predictive models in the HNAI framework: naive Bayes, decision tree, k-nearest neighbor, logistic regression, and support vector machine, respectively.
Results
The area under the receiver operating characteristic curve of the HNAI models is 0.67 as evaluated using fivefold cross-validation. Using antipsychotic drugs as an example, several HNAI-predicted DDIs that involve weight gain and cytochrome P450 inhibition were supported by literature resources.
Conclusions
Through machine learning-based integration of drug phenotypic, therapeutic, structural, and genomic similarities, we demonstrated that HNAI is promising for uncovering DDIs in drug development and postmarketing surveillance.
Introduction
Drug–drug interactions (DDIs) occur during the co-administration of medications. They are a common cause of adverse drug reactions (ADRs) and lead to increasing healthcare costs.1–3 Many DDIs are not identified during the clinical trial phase and are reported after the drugs are approved for clinical use. Such DDIs often lead to patient morbidity and mortality, accounting for 3–5% of all inpatient medication errors.4 Clinical DDIs can also cause serious social and economic problems. Thus, there is an urgent need to detect or determine DDIs before medications are approved or administered.
Currently, DDI prediction focuses on testing metabolic profiles, for instance for cytochrome P450 (CYP450)5–7 or transporter-associated8 pharmacokinetic interactions. However, the limited ability to identify DDIs using experimental approaches is a major obstacle during drug development.9 Due to the lack of comprehensive experimental data, high study cost, long experimental duration, and animal welfare considerations, the use of computational prediction and assessment of DDIs has been encouraged.10 11 During the past decade, several methods have been designed and made available for the prediction of potential DDIs.12–21 Duke et al12 combined a literature discovery approach with analyses of a large electronic medical record database to predict and evaluate new DDIs. Their method enables the detection of clinically significant DDIs and also evaluates the possible molecular mechanisms of the predicted DDIs. Huang et al13 developed a metric S-score method with 82% accuracy and a 62% recall rate to predict pharmacodynamic DDIs. Tari et al14 proposed a method that integrated text mining and automated reasoning to predict DDIs, and found that 81.3% (256/315) of the interactions were correctly predicted. Gottlieb et al15 proposed the inferring drug interactions (INDI) method, which infers both pharmacokinetic and CYP450-associated DDIs as well as pharmacodynamic DDIs. High specificity and sensitivity levels were found in cross-validation when INDI was used. Cami et al18 presented a predictive pharmacointeraction networks (PPIN) method to predict DDIs by utilizing the network topological structure of all known DDIs as well as other intrinsic and taxonomic properties of ADRs. A 48% sensitivity and 90% specificity were found with the PPIN model. Recently, network pharmacology approaches, such as a network-based drug development strategy, have created a novel paradigm for drug discovery.16 22–25 Therefore, development of a machine learning-based model using multi-dimensional drug properties might be a promising strategy to predict unknown DDIs.
In this study, we propose a heterogeneous network-assisted inference (HNAI) framework (figure 1) for large-scale prediction of ligand–receptor DDIs that may occur at previously identified drug receptor sites. First, we constructed a comprehensive DDI network which contained 6946 high-quality, unique DDI pairs connecting 721 approved drugs from the DrugBank database. Next, we calculated four types of drug–drug similarities as the features of each drug–drug pair. Three of these types of drug similarities (phenotypic, therapeutic, and structural similarities) were calculated using a previous method.25 Additionally, we introduced a new type, the drug's genomic similarity, based on a large drug–target interaction (DTI) network built from DrugBank26 and the Therapeutic Target Database (TTD).27 Finally, we applied five machine learning algorithms to serve as predictive models in the HNAI framework: naive Bayes (NB), decision tree (DT), k-nearest neighbors (k-NN), logistic regression (LR), and support vector machine (SVM). Using fivefold cross-validation, we demonstrated that HNAI yielded high performance. In our previous work,25 we have shown the potential value of the integration of a drug's phenotypic, therapeutic, and structural similarities for DTI prediction. Here, we extended this method to predict DDIs using machine learning-based integration of drug phenotypic, therapeutic, structural, and genomic similarities. There are three improvements in this study compared with the previous work25: (i) we introduced an additional important drug similarity type (genomic similarity) and built a predictive HNAI framework by combining the drug genomic similarity and three previously reported drug similarities25 for DDI prediction; (ii) we systematically evaluated five machine learning algorithms and built the predictive models in the HNAI framework; and (iii) although we used the existing network construction method25 to calculate drug phenotypic similarity, the previous drug–ADR network was only built using data from MetaADEDB.28 In this study, we built a more comprehensive drug–ADR network by integrating data from MetaADEDB and the FDA Adverse Events Reporting System (FAERS) created by the US Food and Drug Administration (US FDA). Collectively, our work could provide an alternative tool and may have the potential to improve ligand–receptor DDI prediction in drug development and postmarketing surveillance.
Figure 1.

The heterogeneous network-assisted inference (HNAI) framework for predicting drug–drug interactions (DDI). (A) Collection of a comprehensive gold standard DDI dataset from the DrugBank database and construction of a DDI network. (B) Calculation of four drug–drug pair similarities. Phenotypic similarity is based on a comprehensive drug–adverse drug reaction network, therapeutic similarity is based on the drug Anatomical Therapeutic Chemical (ATC) classification system, structural similarity is derived from chemical structural data, and genomic similarity is based on a large drug–target interaction network from DrugBank and the Therapeutic Target Database. The drug phenotypic, therapeutic, and structural similarities were calculated using a previously published method.25 (C) Construction and evaluation of machine learning-based HNAI models.
Methods
Data collection
DDI network
We collected DDI data from the DrugBank database26 (V.3.0; http://www.DrugBank.ca/). More than 16 453 DDI pairs exist for approved and experimental drugs. These DDIs can be categorized into three types: pharmaceutical, pharmacokinetic, and pharmacodynamic interactions. We excluded the following drugs that cannot be used to calculate four types of similarities: antibody drugs, inorganic salts, and drugs that do not have Anatomical Therapeutic Chemical (ATC) classification system codes or known target information or known ADR information. Thus, we focused on predicting the pharmacokinetic and pharmacodynamic interactions (ligand–receptor DDIs) that may occur at previously identified ligand–receptor sites. In total, 6946 high-quality, unique ligand–receptor DDI pairs connecting 721 approved drugs are compiled for model construction and validation. We retrieved SMILES data for drugs from the DrugBank database, and then converted them into canonical SMILES using Open Babel29 (V.2.3.1).
Drug–ADR associations and their network
We prepared two datasets. The first drug–ADR dataset was downloaded from a recently published database, MetaADEDB.28 MetaADEDB is a comprehensive database of ADRs that annotates more than 520 000 drug–ADR associations covering 3059 unique compounds (including 1330 drugs) and a total of 13 200 ADR items. These annotations are based on data from the integration of three resources: CTD,30 SIDER31 (V.2.0), and OFFSIDES.32 Here, we only retrieved data with clinically reported evidence. The second dataset was obtained from FAERS, which was created by the US FDA. We retrieved data from the first quarter of 2009 through the fourth quarter of 2012 (zipped file names: aers_ascii_20xxqx.zip, web site: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm083765.htm). In the zip files, each ADR report was assigned an Individual Safety Report (ISR) ID. We retrieved details of the reports from several separate files using each ISR ID. In each file, the ADR suspect drugs are classified into the four categories: primary suspect drug (PS), secondary suspect drug (SS), concomitant (C), and interacting (I). Here, we only used drugs labeled PS or SS to collect drug–ADR associations according to the work by Takarabe et al.33 We annotated all ADR terms and drugs using the Unified Medical Language System (UMLS) and Medical Subject Headings (MeSH) vocabularies. Finally, we obtained the unique drug-ADR associations by removing the duplicated drug-ADR associations between FAERS and MetaADEDB for drug phenotypic similarity calculation using a previous method.25
DTIs and their network
We collected DTI data from two databases, DrugBank26 and TTD.27 In total, we obtained 2912 DTI pairs connecting 674 unique target proteins and 721 approved drugs. We annotated the detailed therapeutic information for all drugs based on their ATC codes from the DrugBank database.
Measurement of four types of similarities
The phenotypic similarity 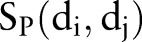 , therapeutic similarity
, therapeutic similarity 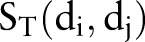 , and chemical structural similarity
, and chemical structural similarity 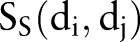 were calculated according to the previous work.25 Additionally, we introduced a genomic similarity
were calculated according to the previous work.25 Additionally, we introduced a genomic similarity 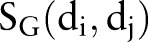 to describe a drug–drug pair in this study. For genomic similarity, each drug is coded using target protein bit vectors (figure 1C). Each bit represents one target protein. If a target protein is associated with a drug in the DTI network, the corresponding bit will be set to ‘1’, otherwise ‘0’. Then, the
to describe a drug–drug pair in this study. For genomic similarity, each drug is coded using target protein bit vectors (figure 1C). Each bit represents one target protein. If a target protein is associated with a drug in the DTI network, the corresponding bit will be set to ‘1’, otherwise ‘0’. Then, the 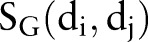 between drug di and dj is calculated using the Tanimoto coefficient34 by inputting the drug's target protein bit vectors.
between drug di and dj is calculated using the Tanimoto coefficient34 by inputting the drug's target protein bit vectors.
Development of predictive models using HNAI
Figure 1 illustrates the entire computational framework of HNAI. We collected 6946 high-quality, unique drug–drug pairs as the positive DDI set. Here, the positive DDI pairs are clinically reported DDIs. We then employed the same number (6946) of non-DDI pairs that were randomly selected from the 721 approved drugs to serve as a negative DDI set based on previous work15 (see online supplementary table S1). Each drug–drug pair is represented by four different similarities: 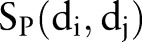 ,
, 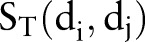 ,
,  , and
, and 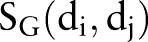 . Finally, we implemented the predictive models in HNAI using five machine learning algorithms: NB, DT, k-NN, LR, and SVM, respectively. Four of these algorithms (NB, DT, k-NN, and LR) are implemented in Orange Canvas (V.2.0b; http://www.ailab.si/orange/). We built the SVM model using the LIBSVM package (V.3.1).35 We briefly describe these algorithms below.
. Finally, we implemented the predictive models in HNAI using five machine learning algorithms: NB, DT, k-NN, LR, and SVM, respectively. Four of these algorithms (NB, DT, k-NN, and LR) are implemented in Orange Canvas (V.2.0b; http://www.ailab.si/orange/). We built the SVM model using the LIBSVM package (V.3.1).35 We briefly describe these algorithms below.
Naive Bayes
Bayesian algorithms classify instances in a dataset using the equal and independent contributions of their attributes.36 Thus, the NB classifier estimates the posterior probability using the following equation:
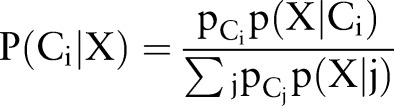 |
1 |
C4.5 decision tree
C4.5 DT constructs a DT using the same steps as Iterative Dichotomiser 3 (ID3) using a set of training data. The elements of the tree generated by ID3 and C4.5 DT include leaf and decision nodes. The leaf denotes the class (eg, DDI and non-DDI) and the decision node specifies the test to be implemented on four drug–drug pair similarities; there is one branch and one sub-tree for each possible result of the test.37
k-Nearest neighbors
The k-NN algorithm categorizes drug–drug pairs using the closest training examples in the four drug–drug pair similarity spaces. Here, we used a hamming distance matrix to measure a nearness and implemented the standard protocol of 3-NN using three steps: (i) to calculate the distances between an unknown drug–drug pair and all drug–drug pairs in the training set; (ii) to select three drug–drug pairs that are most similar to the drug–drug pair y from the training set based on the calculated hamming distances; and (iii) to categorize drug–drug pair y into the group (eg, DDI or non-DDI) to which the majority of the three drug–drug pairs belong.
Logistic regression
The LR algorithm is used to estimate empirical values of the parameters in a qualitative response model using a logistic function.38 In a binary LR, the outcome is set as either ‘positive (DDI)’ or ‘negative (non-DDI)’.
Support vector machine
Cortes and Vapnik39 developed the SVM algorithm for pattern recognition in order to minimize the structural risk under the frame of the Vapnik-Chervonenkis (VC) theory. In this study, each drug–drug pair is represented by an Eigenvector t, which has four components using the four similarities: 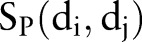 ,
, 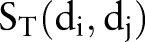 ,
, 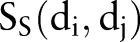 , and
, and 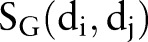 (figure 1C). The category label y was added in SVM training. The ith molecule in the dataset is defined as Mi=(ti, yi), where yi=1 for the ‘DDI’ category and yi=−1 for the ‘non-DDI’ category. SVM builds a classifier according to the following equation:
(figure 1C). The category label y was added in SVM training. The ith molecule in the dataset is defined as Mi=(ti, yi), where yi=1 for the ‘DDI’ category and yi=−1 for the ‘non-DDI’ category. SVM builds a classifier according to the following equation:
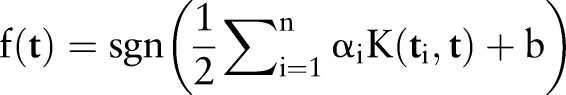 |
2 |
In equation 2, αi is the coefficient to be trained for molecule i and K is a kernel function. Parameter αi is trained by maximizing the Lagrangian expression, as below:
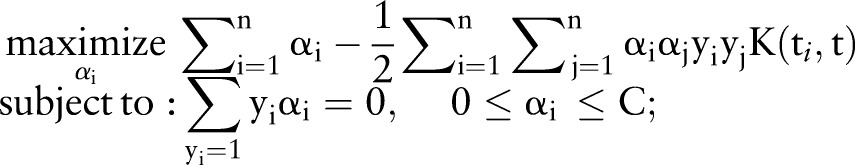 |
3 |
Here, we used the Gaussian radial basis function. We trained the different kernel parameter γ and penalty parameter C based on the training set using a grid search strategy by fivefold cross-validation to obtain an optimal SVM model. Additionally, we used a Bayesian approach for the SVM model to generate a probability estimation for each drug–drug pair class in a binary classifier as described in a previous work.6
Evaluation of HNAI models
In this study, we used the fivefold cross-validation techniques to evaluate the performance of all models. In a fivefold cross-validation, the entire dataset is divided equally into five cross-validation splits. Within each cross-validation step, the model is trained on a set of four cross-validation splits. The fifth sub-sample set is then used as an internal validation set (test set). We then used the receiver operating characteristic (ROC) curve40 to evaluate the performance of each model.
Statistical analysis and network visualization
We calculated network topological parameters (eg, degree) and built the network graph using Cytoscape (V.2.8).41 We performed the statistical analysis (eg, Fisher's exact test and Wilcoxon's test) using the R platform (V.3.01; http://www.r-project.org/).
Results
Construction of a network with known DDI
We compiled a large ligand–receptor DDI dataset from the DrugBank database to serve as the gold standard for model evaluation in the HNAI. This dataset included 6946 DDI pairs connecting 721 US FDA-approved small molecular drugs (see online supplementary table S1). In figure 2, drugs are grouped by first-level ATC codes. We found that most DDIs occur in more than one class of drugs based on first-level ATC codes. For example, we found 487 drugs with degrees larger than 5 in the DDI network. In total, 106 of the 487 drugs were grouped within the cardiovascular system (green, p=2.8×10−5, Fisher's exact test). In addition, the average degree of 173 nervous system drugs (purple) is 25.8, which is significantly higher than the 17.2 found for 548 non-nervous system drugs (p=3.3×10−4, Wilcoxon's test). Collectively, cardiovascular system and nervous system drugs have a high risk of DDIs.
Figure 2.
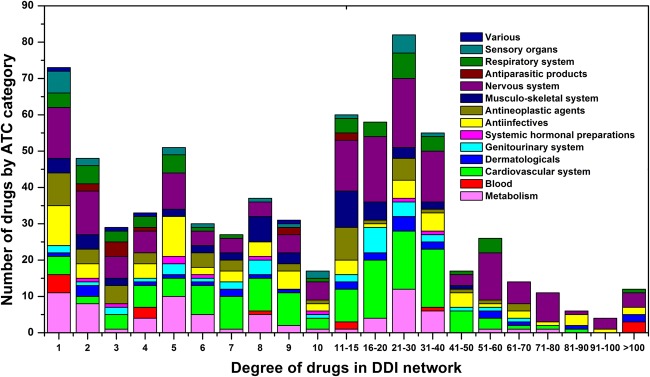
The degree distribution of drugs in the drug–drug interaction (DDI) network categorized by the first level of the drug Anatomical Therapeutic Chemical (ATC) classification system. Drugs are grouped by their ATC classification system. We investigated how specific DDIs occur within ATC classes. We found that most DDIs occurred in more than one class of drugs based on first-level ATC codes. 487 drugs with degrees above 5 were found in the DDI network. Among 487 drugs, 106 drugs were grouped within the cardiovascular system (green, p=2.8×10−5, Fisher's exact test). In addition, the average degree of 173 nervous system drugs (purple) is 25.8, which is significantly higher than the 17.2 found for 548 non-nervous system drugs (p=3.3×10−4, Wilcoxon's test).
The DDI network is represented graphically in figure 3 using the grid layout from Cytoscape. It includes 6946 high-quality DDI pairs connecting 721 approved drugs. In figure 3, a node represents a drug and an edge denotes an association between two drugs that have clinically reported or literature curated DDIs. Each node is color-coded based on the first-level ATC codes. The average drug degree in this network was 19.3, and the degree distribution is a power-law distribution (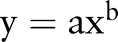 , a=94.0, b=−0.19). The nine most connected drugs in the DDI network were voriconazole (degree=203), triprolidine (164), warfarin (137), treprostinil (126), acenocoumarol (116), tramadol (108), ritonavir (86), cimetidine (80), and ergonovine (30) (figure 3). In order to interpret this network, we investigated the distribution of four similarities in positive DDI pairs and random DDI pairs (see online supplementary table S1). The positive DDI pairs are clinically reported or literature curated interactions. We found that the similarities of positive DDI pairs are significantly higher than those of random DDI pairs (see online supplementary figure S1). For structural similarity, DDI pairs are more enriched with high structural similarity than random DDI pairs (p=1.4×10−109, Fisher's exact test; see online supplementary figure S1A). Moreover, the average similarities (phenotypic, therapeutic, and genomic) of positive DDI pairs are significantly higher than those of random DDI pairs (p<2.2×10−16, Wilcoxon's test; see online supplementary figure S1). Collectively, these investigations confirm the hypothesis that drugs that are similar (ie, measured by phenotypic, therapeutic, structural, and genomic features) to each other tend to have a high risk of DDIs.
, a=94.0, b=−0.19). The nine most connected drugs in the DDI network were voriconazole (degree=203), triprolidine (164), warfarin (137), treprostinil (126), acenocoumarol (116), tramadol (108), ritonavir (86), cimetidine (80), and ergonovine (30) (figure 3). In order to interpret this network, we investigated the distribution of four similarities in positive DDI pairs and random DDI pairs (see online supplementary table S1). The positive DDI pairs are clinically reported or literature curated interactions. We found that the similarities of positive DDI pairs are significantly higher than those of random DDI pairs (see online supplementary figure S1). For structural similarity, DDI pairs are more enriched with high structural similarity than random DDI pairs (p=1.4×10−109, Fisher's exact test; see online supplementary figure S1A). Moreover, the average similarities (phenotypic, therapeutic, and genomic) of positive DDI pairs are significantly higher than those of random DDI pairs (p<2.2×10−16, Wilcoxon's test; see online supplementary figure S1). Collectively, these investigations confirm the hypothesis that drugs that are similar (ie, measured by phenotypic, therapeutic, structural, and genomic features) to each other tend to have a high risk of DDIs.
Figure 3.

The drug–drug interaction (DDI) network. The network contains data collected from the DrugBank database, which included 6946 DDI pairs connecting 721 approved drugs. Drug nodes are colored according to the first-level of their Anatomical Therapeutic Chemical classification. The size of a drug node reflects the degree of the drug in the network. The distribution of four similarities in positive DDI pairs and random DDI pairs is provided in online supplementary figure S1. We found that the similarities among positive DDI pairs were significantly stronger than those of random DDI pairs.
Performance of HNAI models
HNAI implements several predictive models. The HNAI model hypothesis asserts that if two drugs have similar chemical structures, share a similar target protein and similar ADRs, and have similar therapeutic purposes, they have a high probability of DDIs (see online supplementary figure S1). We compiled 6946 high-quality clinically reported DDIs to serve as positive DDI pairs. We then constructed the balanced non-DDI pairs (named negative DDI pairs; see online supplementary table S1) to build predictive machine learning models. The area under the ROC curve (AUC) ranged from 0.565 to 0.666 using the five different HNAI models and the fivefold cross-validation evaluation in figure 4. Because the NB, SVM, and LR algorithms utilize the prior of the positive and negative DDIs during model building, the ratio of positive to negative classes might affect the performance of the model. In order to investigate the influence on performance of the unbalanced positive and negative classes, we rebuilt the models using two unbalanced datasets. As shown in online supplementary figure S2, we found that the performance of a balanced dataset is marginally higher than that of an unbalanced dataset. Of the five different machine learning algorithms, SVM had the best performance (figure 4). This evaluation indicated that SVM might be a good choice for DDI prediction, which coincides with the previous work.6 39 42 The SVM algorithm typically uses a portion of the training set as support vectors to build the classification hyper-plane. If data bias occurs within the non-support vectors, it will not affect the performance of the SVM model. In contrast, k-NN uses the entire training set for model building. Any error or data bias in the training set will influence the model's performance. Although NB and DT had the worst performances, their advantages include the low calculation cost and simplicity in their practical applications.
Figure 4.
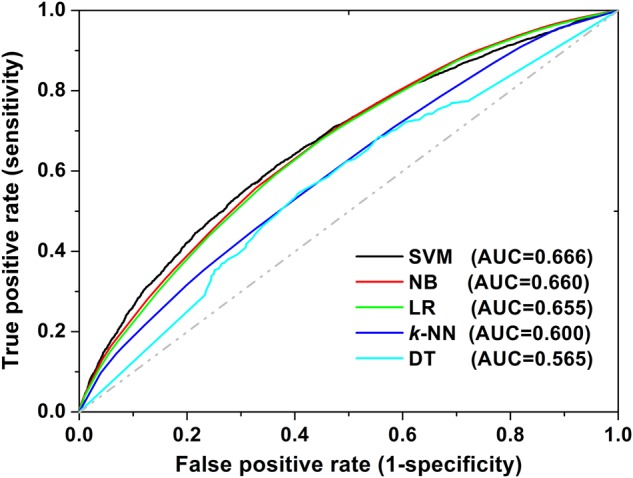
The receiver operating characteristic (ROC) curves of fivefold cross-validation using five models in heterogeneous network-assisted inference. The models were built using naive Bayes (NB, red curve), decision tree (DT, cyan curve), k-nearest neighbors (k-NN, blue curve), logistic regression (LR, green curve), and support vector machine (SVM, black curve) algorithms. The AUC is the area under the ROC curve.
Case study: discovery of potential DDIs for antipsychotic drugs
To predict novel potential DDIs, we employed a global training set that included 6946 positive DDI pairs and an equally sized, randomly generated set of negative DDI pairs that are not known to interact (see online supplementary table S1). A global HNAI model was built using the SVM algorithm to predict the possible drug–drug pairs, including drugs without known DDIs. We predicted 81 580 new possible DDIs connecting 729 approved drugs (see online supplementary table S2) using the default probability score of 0.5 based on the previous work.6 7 Here, we used the higher probability score of 0.8 to define the high-confidence predicted DDIs. Among 81 580 new predicted DDIs, 559 predicted DDIs have a probability score of greater than 0.8, suggesting that they are potential DDI candidates that could be further validated using clinical data or electronic health records (EHRs). Here, we explored some features of the predicted DDIs using antipsychotic drugs as examples.
For antipsychotic drugs, we predicted 36 DDIs with probability scores above 0.8 using the global SVM-based HNAI model (see online supplementary table S3). In addition, we collected 798 clinically reported DDI pairs for 43 antipsychotic drugs from DrugBank. To further explore the molecular mechanisms of antipsychotic DDIs, we collected 332 experimentally validated DTI pairs from DrugBank. Those DTI pairs connected 44 target proteins and 43 antipsychotic drugs (see online supplementary table S3). We constructed a DDI and DTI network for these 43 antipsychotic drugs as shown in figure 5. We then examined the possible molecular mechanisms of antipsychotic DDIs using this network. In DrugBank and TTD, the average number of target proteins for 721 approved drugs was 4.0 (2912/721). The average number of target proteins for 43 antipsychotic drugs was 7.7 (332/43), which is significantly higher than the average number of 3.8 for non-antipsychotic drugs in DrugBank and TTD (p=3.1×10−6, Wilcoxon's test). Thus, antipsychotic drugs bind with multiple proteins, for example, polypharmacology. The effects of antipsychotic drugs are characterized by their antagonistic profiles and high affinity for dopamine receptors, serotonin, histamine receptor H1, muscarinic receptors, and adrenergic receptors.43 44 The polypharmacology of antipsychotic agents may help explain the occurrence of particular DDIs or ADRs. High binding affinity at muscarinic receptors is linked to several ADRs, including dry mouth, constipation, and blurred vision, whereas high binding affinity at the histamine H1 receptor is linked to weight gain.43 44
Figure 5.

Drug–drug and drug–target interaction network for antipsychotic drugs. This network has: (i) 332 drug–target interaction pairs (gold edges) connecting 43 antipsychotic drugs (pink circles) and 44 target proteins (gold squares); (ii) 798 clinically reported drug–drug interactions (DDIs; gray edges) connecting 238 drugs; and (iii) 36 predicted DDIs (magenta edges) with probability scores of greater than 0.8 that connect 28 drugs (see details in online supplementary table S3). Drug nodes are colored according to the first level of their Anatomical Therapeutic Chemical classification in figure 3. The size of a node reflects the degree of the drug or target protein in the network.
In this work, clozapine (DrugBank ID DB00363) was predicted to interact with olanzapine (see online supplementary table S3). Clozapine and olanzapine are often co-prescribed during clinical treatment for clozapine-resistant schizophrenia.45 46 To validate the possible DDIs between clozapine and olanzapine, we systematically examined their DTI profiles. The binding affinities of clozapine and olanzapine to the histamine H1 receptor ranged from 1 to 10 nmol.43 Thus, the co-administration of clozapine and olanzapine increases the synergistic effects of the histamine H1 receptor that is linked to weight gain. Previous studies found that patients whose treatments included co-administered clozapine and olanzapine had the greatest weight gain.43 47
Ziprasidone (DrugBank ID DB00246) was the fifth atypical antipsychotic drug approved by the US FDA in 2001. Ziprasidone has a modest effect on the electrical activity of the heart, which results in an increased risk of arrhythmias due to additive QTc-prolonging effects.26 Thus, a drug that affects the QT interval could further increase the QTc-prolonging effects of ziprasidone. Chintalgattu et al48 found that a multi-target receptor tyrosine kinase inhibitor, sunitinib, has a high risk of causing cardiac dysfunction. Furthermore, the co-administration of sunitinib and ziprasidone will increase the risk of cardiotoxicity. Herein, ziprasidone was predicted to have a DDI with citalopram (DrugBank ID DB00215) according to the global SVM model in HNAI. To validate possible drug interactions between ziprasidone and citalopram, we surveyed CYP450-related metabolic profiles. Ziprasidone was reported to be an inhibitor of CYP2D6, 2C19, 2C9, and 3A4. Citalopram was reported as a substrate of CYP2C19, 2D6, and 3A4.49 Therefore, ziprasidone and citalopram have a high risk of causing CYP450-associated DDIs when they are co-administered. In addition, we examined predicted DDIs among drugs from different families. As shown in online supplementary table S3, ziprasidone was predicted to have a DDI with aspirin. The previous study revealed that co-administration of ziprasidone and aspirin causes a risk of severe cardiotoxicity in elderly patients.50 Collectively, our HNAI framework might predict possible DDIs among drugs from different families.
Discussion
Understanding DDIs is an essential step in drug development and drug co-administration. Currently, the US FDA and pharmaceutical companies are interested in the development and application of computational prediction and assessment of DDIs.11 In this work, we developed a HNAI framework and employed five predictive machine learning models for DDI prediction (figure 1). During the past decade, several computational methods have been reported for computational DDI prediction.12–21 Compared to previous methods, one advantage of our HNAI is that its models are built only by utilizing four similarities as features of each drug–drug pair. Although several reported models have higher performance than our method, those models are constructed using high-dimensional features, thereby increasing the complexity and ‘black-box’ of their models. The method in Vilar et al20 was based on molecular structural similarity information derived from fingerprint-based modeling. Using the data from the earlier version of the DrugBank database for evaluation, they reported an overall sensitivity of 0.68, specificity of 0.96, and precision of 0.26. Tatonetti et al19 built eight LR models to predict DDIs involved in eight types of ADRs: high cholesterol, renal impairment, diabetes, liver dysfunction, hepatotoxicity, depression, and suicide. The AUC value ranged from 0.51 to 0.71 for eight models as evaluated using one validation set from the Veterans Affairs Hospital in Arizona. In this work, we built the HNAI framework with a satisfactory performance for DDI prediction through machine learning-based integration of four types of drug similarity properties. In a previous work,25 we reported the potential value of the integration of drug phenotypic, therapeutic, and structural similarities for DTI prediction. Building on DTI prediction, in this work we expanded the approach (ie, machine learning-based integration of four types of drug similarity properties: phenotypic, therapeutic, structural, and genomic similarities) for DDI prediction. The HNAI hypothesis asserts that if two drugs have high drug phenotypic, therapeutic, structural, or genomic similarity, they have a high probability of DDIs. We found that the structural similarity of positive DDI pairs is significantly higher than that of random DDI pairs (see online supplementary figure S1A). This observation is in agreement with Vilar's work20 showing that drug structural similarity information can be used for DDI prediction. Furthermore, we found that the systematic machine learning-based integration of various data sources, including drug ADRs, drug ATC codes, drug chemical structure, and drug genomic information, can improve the performance of DDI prediction compared to using only individual data sources (see online supplementary figure S3). Collectively, this study presents a simple and promising strategy to predict unknown DDIs using network pharmacology data and machine learning approaches.
Limitations and future work
There are several limitations in the HNAI framework. First, it lacks a ‘gold standard’ non-DDI dataset to serve as negative DDIs for model building. We cannot definitively confirm that two drugs in a non-DDI dataset do not interact, so the collection and reporting of non-DDIs in open source databases, EHRs, and other clinical documents should be encouraged. Second, there is a limitation to the accuracy of the four drug similarities. In order to evaluate the relative importance of each similarity in the overall HNAI predictive model, we rebuilt the machine learning models by using the combinations of the three different similarities. As shown in online supplementary figure S3, we found that the models had poor performance when structural or phenotypic features were removed. Thus, structural and phenotypic features are important for model performance compared to the other two features. Third, data incompleteness is a limitation. Although we used two large networks, a drug–ADR association network and a DTI, to measure drug phenotypic and genomic similarities, both networks are incomplete. However, the availability of drug information has increased with the reporting of DTIs and ADRs from hospitals and pharmaceutical companies, and the availability of dozens of databases covering drug mechanisms.11 GlaxoSmithKline recently announced its intention to release patient-level raw data from clinical trials of approved and failed investigational compounds.51 These efforts will provide new resources to predict DDIs or ADRs in drug development and clinical applications. Finally, there may be several additional limitations associated with each similarity. Although chemical two-dimensional (2D) structural similarities calculated using MACCS keys have been successfully used for DDI or ADR assessment,6 20 2D structurally similar molecules can have very different shapes in 3D structure. Furthermore, 2D structurally dissimilar compounds may have very similar shapes in 3D that might play a crucial role in ligand–receptor binding.52 Here, genomic similarity was calculated using the broad DTI data from the DrugBank and TTD. However, detailed drug target information was lost in the current HNAI framework due to lack of access to data on mode-of-actions (eg, competitive reversible binding, non-competitive irreversible binding), biochemical effects (agonist-activator, partial agonist, antagonist effects), and binding location (active site, and allosteric sites).
However, we predicted thousands of possible DDIs using the HNAI framework. In the future, we plan to validate more predicted DDIs using Vanderbilt's EHRs from the Synthetic Derivative (SD) database (https://starbrite.vanderbilt.edu/biovu/sddata.html). We also intend to: (i) improve the HNAI framework through global integration of more useful features from drug 3D shape similarity, comprehensive DTI networks (eg, ChEMBL53), and EHRs from Vanderbilt's SD database; and (ii) address the molecular mechanisms and genetic profiles of DDIs by integrating drug response data and genome-wide genotyping data from Vanderbilt's DNA bank (https://starbrite.vanderbilt.edu/biovu/).
Conclusion
We have presented a HNAI framework to predict DDIs by utilizing the drug phenotypic, therapeutic, structural, and genomic similarities. We applied five machine learning-based predictive models on a large DDI dataset from the DrugBank database. The SVM model had an AUC value of 0.67 based on fivefold cross-validation. In our exploration of several novel predicted DDIs involving antipsychotic drugs, we demonstrated the potential utility of the HNAI framework for the identification of antipsychotic DDIs. In summary, we showed that a machine learning-based integration of drug phenotypic, therapeutic, structural, and genomic similarities using a systems pharmacology approach is a simple yet efficient strategy to predict unknown DDIs.
Supplementary Material
Acknowledgments
We thank Ms Rebecca Hiller Posey for polishing an earlier draft of the manuscript.
Footnotes
Correction notice: This article has been corrected since it was published Online First. Equation 3 has been corrected.
Contributors: ZZ and FC conceived and designed the study. FC carried out the experiments and analyzed the data. FC and ZZ interpreted the results and wrote the manuscript.
Funding: This work was partially supported by National Institutes of Health grants (R01LM011177, P50CA095103, and P30CA068485), The Robert J Kleberg, Jr and Helen C Kleberg Foundation, and Ingram Professorship Funds to Dr Zhao. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discovery 2005;4:825–33 [DOI] [PubMed] [Google Scholar]
- 2.Juurlink DN, Mamdani M, Kopp A, et al. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA 2003;289:1652–8 [DOI] [PubMed] [Google Scholar]
- 3.Edwards IR, Aronson JK. Adverse drug reactions: definitions, diagnosis, and management. Lancet 2000;356:1255–9 [DOI] [PubMed] [Google Scholar]
- 4.Leape LL, Bates DW, Cullen DJ, et al. Systems analysis of adverse drug events. ADE Prevention Study Group. JAMA 1995;274:35–43 [PubMed] [Google Scholar]
- 5.Veith H, Southall N, Huang R, et al. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat Biotechnol 2009;27:1050–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng F, Yu Y, Shen J, et al. Classification of cytochrome P450 inhibitors and non-inhibitors using combined classifiers. J Chem Inf Model 2011;51:996–1011 [DOI] [PubMed] [Google Scholar]
- 7.Cheng F, Yu Y, Zhou Y, et al. Insights into molecular basis of cytochrome p450 inhibitory promiscuity of compounds. J Chem Inf Model 2011;51:2482–95 [DOI] [PubMed] [Google Scholar]
- 8.Huang SM, Temple R, Throckmorton DC, et al. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther 2007;81:298–304 [DOI] [PubMed] [Google Scholar]
- 9.Nemeroff CB, Preskorn SH, Devane CL. Antidepressant drug-drug interactions: clinical relevance and risk management. CNS Spectrums 2007;12:1–1317514084 [Google Scholar]
- 10.Cheng F, Li W, Liu G, et al. In silico ADMET prediction: recent advances, current challenges and future trends. Curr Top Med Chem 2013;13:1273–89 [DOI] [PubMed] [Google Scholar]
- 11.Percha B, Altman RB. Informatics confronts drug-drug interactions. Trends Pharmacol Sci 2013;34:178–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duke JD, Han X, Wang Z, et al. Literature based drug interaction prediction with clinical assessment using electronic medical records: novel myopathy associated drug interactions. PLoS Comput Biol 2012;8:e1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Niu C, Green CD, et al. Systematic prediction of pharmacodynamic drug-drug interactions through protein-protein-interaction network. PLoS Comput Biol 2013;9:e1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tari L, Anwar S, Liang S, et al. Discovering drug-drug interactions: a text-mining and reasoning approach based on properties of drug metabolism. Bioinformatics 2010;26:i547–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottlieb A, Stein GY, Oron Y, et al. INDI: a computational framework for inferring drug interactions and their associated recommendations. Mol Syst Biol 2012;8:592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Csermely P, Korcsmaros T, Kiss HJ, et al. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther 2013;138:333–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, Wu Y, Xu H, et al. DTome: a web-based tool for drug-target interactome construction. BMC Bioinformatics 2012;13(Suppl 9):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cami A, Manzi S, Arnold A, et al. Pharmacointeraction network models predict unknown drug-drug interactions. PLoS ONE 2013;8:e61468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatonetti NP, Fernald GH, Altman RB. A novel signal detection algorithm for identifying hidden drug-drug interactions in adverse event reports. J Am Med Inform Assoc 2012;19:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilar S, Harpaz R, Uriarte E, et al. Drug-drug interaction through molecular structure similarity analysis. J Am Med Inform Assoc 2012;19:1066–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M, Wu Y, Chen Y, et al. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J Am Med Inform Assoc 2012;19:e28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng F, Liu C, Jiang J, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol 2012;8:e1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng F, Zhou Y, Li W, et al. Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS ONE 2012;7:e41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng F, Zhou Y, Li J, et al. Prediction of chemical-protein interactions: multitarget-QSAR versus computational chemogenomic methods. Mol Biosyst 2012;8:2373–84 [DOI] [PubMed] [Google Scholar]
- 25.Cheng F, Li W, Wu Z, et al. Prediction of polypharmacological profiles of drugs by the integration of chemical, side effect, and therapeutic space. J Chem Inf Model 2013;53:753–62 [DOI] [PubMed] [Google Scholar]
- 26.Knox C, Law V, Jewison T, et al. DrugBank 3.0: a comprehensive resource for 'omics’ research on drugs. Nucleic Acids Res 2011;39:D1035–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu F, Shi Z, Qin C, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res 2012;40:D1128–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng F, Li W, Wang X, et al. Adverse drug events: database construction and in silico prediction. J Chem Inf Model 2013;53:744–52 [DOI] [PubMed] [Google Scholar]
- 29.O'Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminform 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis AP, King BL, Mockus S, et al. The comparative toxicogenomics database: update 2011. Nucleic Acids Res 2011;39:D1067–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn M, Campillos M, Letunic I, et al. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 2010;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatonetti NP, Ye PP, Daneshjou R, et al. Data-driven prediction of drug effects and interactions. Sci Transl Med 2012;4:125ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takarabe M, Kotera M, Nishimura Y, et al. Drug target prediction using adverse event report systems: a pharmacogenomic approach. Bioinformatics 2012;28:i611–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discov Today 2006;11:1046–53 [DOI] [PubMed] [Google Scholar]
- 35.Chang CC, Lin C-J. LIBSVM : a library for support vector machines. http://www.csie.ntu.edu.tw/~cjlin/libsvm (accessed 18 Jan 2010)
- 36.Watson P. Naive Bayes classification using 2D pharmacophore feature triplet vectors. J Chem Inf Model 2008;48:166–78 [DOI] [PubMed] [Google Scholar]
- 37.Quinlan JR. C4.5: Programs for machine learning. Morgan Kaufmann Publishers, 1993 [Google Scholar]
- 38.Hosmer DW, Lemeshow S. Applied logistic regression. 2nd edn Wiley, 2000 [Google Scholar]
- 39.Cortes C, Vapnik V. Support-vector networks. Mach Learn 1995; 20:273–97 [Google Scholar]
- 40.Baldi P, Brunak S, Chauvin Y, et al. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics 2000;16:412–24 [DOI] [PubMed] [Google Scholar]
- 41.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng F, Shen J, Yu Y, et al. In silico prediction of Tetrahymena pyriformis toxicity for diverse industrial chemicals with substructure pattern recognition and machine learning methods. Chemosphere 2011;82:1636–43 [DOI] [PubMed] [Google Scholar]
- 43.Nasrallah HA. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiat 2008;13:27–35 [DOI] [PubMed] [Google Scholar]
- 44.Stephen MS. Describing an atypical antipsychotic: receptor binding and its role in pathophysiology. J Clin Psychiat 2003;5:9–13 [Google Scholar]
- 45.Gupta S, Sonnenberg SJ, Frank B. Olanzapine augmentation of clozapine. Ann Clin Psychiat 1998;10:113–15 [DOI] [PubMed] [Google Scholar]
- 46.Kerwin WR, Bolonna A. Management of clozapine-resistant schizophrenia. Adv Psychiat Treatment 2005;11:101–6 [Google Scholar]
- 47.Allison DB, Mentore JL, Heo M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Pychiat 1999;156:1686–96 [DOI] [PubMed] [Google Scholar]
- 48.Chintalgattu V, Rees ML, Culver JC, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 2013;5:187ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Preissner S, Kroll K, Dunkel M, et al. SuperCYP: a comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res 2010;38:D237–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee TW, Tsai SJ, Hwang JP. Severe cardiovascular side effects of olanzapine in an elderly patient: case report. Int J Psychiat Med 2003;33:399–401 [DOI] [PubMed] [Google Scholar]
- 51.Harrison C. GlaxoSmithKline opens the door on clinical data sharing. Nat Rev Drug Discov 2012;11:891–2 [DOI] [PubMed] [Google Scholar]
- 52.Hu G, Kuang G, Xiao W, et al. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J Chem Inf Model 2012;52:1103–13 [DOI] [PubMed] [Google Scholar]
- 53.Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014;42:D1083–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.