

Abstract
The gamma-aminobutyric acid (GABA) metabolite gamma-hydroxybutyric acid (GHB) shows a variety of behavioural effects when administered to animals and humans, including reward/addiction properties and absence seizures. At the cellular level, these actions of GHB are mediated by activation of neuronal GABAB receptors (GABABRs) where it acts as a weak agonist. Because astrocytes respond to endogenous and exogenously applied GABA by activation of both GABAA and GABABRs, here we investigated the action of GHB on astrocytes on the ventral tegmental area (VTA) and the ventrobasal (VB) thalamic nucleus, two brain areas involved in the reward and proepileptic action of GHB, respectively, and compared it with that of the potent GABABR agonist baclofen. We found that GHB and baclofen elicited dose-dependent (ED50: 1.6 mM and 1.3 µM, respectively) transient increases in intracellular Ca2+ in VTA and VB astrocytes of young mice and rats, which were accounted for by activation of their GABABRs and mediated by Ca2+ release from intracellular store release. In contrast, prolonged GHB and baclofen exposure caused a reduction in spontaneous astrocyte activity and glutamate release from VTA astrocytes. These findings have key (patho)physiological implications for our understanding of the addictive and proepileptic actions of GHB.
Keywords: ventral tegmental area, thalamus, baclofen, reward, absence seizures
1. Introduction
Gamma-hydroxybutyric acid (GHB) is an endogenous central nervous system (CNS) substance that results from the metabolism of the neurotransmitter gamma-aminobutyric acid (GABA) [1–3], but can also act as a source of neuronal GABA [4]. In humans, exogenously administered GHB elicits a variety of behavioural responses that at progressively increasing doses include sedation, memory loss, euphoria, behavioural disinhibition, sleep and coma [2,5–8]. GHB has also found some limited clinical use in the treatment of alcohol and opiate withdrawal [9–12] as well as of narcolepsy and cataplexy [13–15]. In the early 1970s, GHB emerged as a recreational drug and still remains one of the most commonly used ‘club drugs’ [16–19], with GHB overdoses accounting for a substantial proportion of the hospital emergencies that are linked to recreational nightlife settings [20].
A neuropharmacological profile similar to that observed in humans characterizes the behaviour of animals injected with GHB. The most extensively studied behavioural actions of GHB are its ability to induce self-administration and absence seizures [3]; indeed, the systemic administration of low doses of GHB has become the most widely used pharmacological model of these non-convulsive seizures [21]. Thus, oral GHB induces self-administration and conditioned place preference in mice and rats [22–25]. In addition, GHB administration leads to a moderate stimulation of the dopaminergic reward system [26], and in vitro low concentration of GHB preferentially inhibits the GABAergic neurons of the ventral tegmental area (VTA), one of the key brain-reward areas [25,27]. Indeed, GHB induces conditioned place preference when injected directly in the VTA [28], but not in the nucleus accumbens, another important brain-reward area. As far as absence seizures are concerned, the ability of GHB to elicit absence seizures is not restricted to its systemic administration but importantly it also occurs when it is injected directly into the ventrobasal (VB) nucleus of the thalamus [29], one of the key areas for the generation of these non-convulsive seizures [30].
In both mice and rats, all behavioural and cellular actions of GHB, including its reward- and seizure-eliciting properties, can be explained by it acting as a weak agonist at GABAB receptors (GABABRs) [31,32], though the existence of a putative GHB receptor site has been suggested [33] (but see [3]). Thus, the pro-absence action of GHB is mimicked by the selective GABABR agonist baclofen and is fully antagonized by different selective GABABR antagonists [3], and in some studies by NCS382, an antagonist of the putative GHB receptor site [34]. Interestingly, micromolar GHB concentrations have recently been suggested to activate recombinant α4βδ GABAARs [35], though the physiological significance of this finding remains obscure as these results could not be reproduced in different brain areas, including thalamus, cerebellum and hippocampus [36] that contain this combination of GABAAR subunits.
It is now well-established that astrocytes respond to different neurotransmitters, including GABA [37,38] and signal back to neurons via the release of gliotransmitters [39,40], thus modulating neuronal excitability and different forms of synaptic plasticity [41]. Thus, because astrocytes respond to GABABR activation [42], and the VTA and VB thalamus contribute to the behavioural actions of GHB [3], we investigated the effect of GHB on this glial cell type in brain slices and compared it with the selective GABABR agonist baclofen. We report that GHB and baclofen elicit a dose-dependent increase of intracellular Ca2+ ([Ca2+]i) in VTA and VB astrocytes which is fully accounted for by activation of their GABABRs and is mediated by Ca2+ release from intracellular stores. In contrast to many other studies which found that transient as well as sustained agonist-induced astrocytic [Ca2+]i elevations result in increased gliotransmitter release, prolonged GHB and baclofen exposure causes a reduction in spontaneous glutamate release from VTA astrocytes.
2. Materials and methods
All experiments were performed in accordance with the Animals (Scientific Procedures) Act 1986, UK, and with approval of local animal welfare and ethical review bodies.
(a). Slice preparation and maintaining solutions
Male and female Wistar rats (8–12 days old), as well as GABAB KO mice and their wild-type littermates (kindly provided by B. Bettler, Basel, Switzerland) [43] were deeply anaesthetized with isoflurane, and the brain quickly removed. Slices of VTA or VB thalamus were prepared as described previously [44,45]. Briefly, the brain was glued with cyanoacrylate adhesive to a metal block and submerged in the bath of a Leica or Microm MV tissue slicer. The bathing solution was of composition (in mM): NaCl 120, NaHCO3 16, KCl 1, KH2PO4 1.25, MgSO4 5, CaCl2 1, 10 glucose, and was maintained at 5°C. Slices (350 µm) were cut in the horizontal plane, and then stored in a 95% O2, 5% CO2-bubbled solution of identical composition at room temperature. Following a 1 h recovery period, experiments were performed in a solution of composition (in mM): NaCl 120, NaHCO3 16 or 25, KCl 2, KH2PO4 1.25, MgSO4 1, CaCl2 2, 10 glucose, at room temperature (20–24°C), unless otherwise stated. Slices were loaded with Fluo-4 AM (Invitrogen, Paisley, UK) by incubating for 40–60 min at 28°C with 5 μM of the indicator dye and 0.01% pluronic acid. TTX (1 µm) was present in the perfusate of all experiments. Agonists (GHB and baclofen) were bath-applied for 2 min with or without GABABR antagonists (unless stated otherwise). Chemicals were purchased from Sigma (St Louis, MO), except CGP65426, MTEP, CNQX, CPCCOEt and baclofen (Tocris, Bristol, UK), D-APV and TTX (Abcam, Cambridge, UK), and GHB (that was kindly donated by Unavera ChemLab GmbH, Mittenwald, Germany).
(b). Fluorescence imaging
The slices were placed in a recording chamber, whereas the patch-electrode headstage micromanipulators were mounted on a movable platform (MP MTP-01, Scientifica, UK). Fluorescence was measured using a Noran Odyssey confocal (Thermo Noran, USA) fitted to a Nikon E600FN upright microscope. Averages of eight whole field images (206 µm × 158 µm) were routinely acquired every 5 s with a 40× objective lens (NA = 0.8). Acquisition and image analyses were performed using Noran Intervision and Metamorph software. Fluorescence values over time for specific regions of interest were exported into sigmaplot (Jandel, USA) for further analysis. The number of responding astrocytes (reported in the text and figures) is expressed as an absolute number of responding astrocytes per imaged slice area. Because the imaged area (see above) was the same in all experiments, and astrocytes are evenly distributed throughout the brain parenchyma [46], using the absolute number of responding astrocytes provides a valid and efficient way of determining agonist and antagonist effects as reported previously [47,48].
Two photon laser scanning microscopy was performed using a Prairie Ultima (Prairie Technologies, Madison, WI) microscope and a Ti : sapphire pulsed laser (Chameleon Ultra II, Coherent, UK) tuned to λ = 810 nm. Image acquisition was controlled using Prairieview software, and laser intensity was modified using a Pockels cell electroacoustic modulator (ConOptics, USA). Slices were imaged using a 40×/0.8 NA objective lens, and fluorescence signals from Fluo-4 indicator were collected in the epicollection mode using multi-alkali photomultiplier tubes (Hamamatsu Photonics, Hamamatsu, Japan).
(c). Electrophysiology
Patch-clamp recordings from VTA neurons were made using pipettes (2–4 MΩ) containing an internal solution of composition (in mM): KMeSO4 120, HEPES 10, EGTA 0.1, Na2ATP 4, GTP 0.5, osmolarity adjusted to 295 mOsm l−1 with KCl. Currents were recorded at −60 mV using a multi-clamp 700B amplifier, digitized with a Digidata 1440A, and acquired and analysed using pClamp (Molecular Devices). Neurons with more than 20% change in access resistance were excluded. Slow inward currents (SICs) were analysed using the event detection protocols in the Clampfit routine of pClamp. Events were accepted as SICs if their amplitude was more than 20 pA and their time to peak was more than 20 ms [47,49]. Data were exported to SigmaPlot (Jandel) for additional analysis and plotting.
(d). Statistics
All quantitative data are expressed in the text as mean ± s.e.m. Statistical test used was unpaired Student's t-test. Dose–response curve fitting was conducted using the fitting procedures of SigmaPlot (Jandel) and Prizm (GraphPad). In the figures, asterisk indicates significance of *p < 0.05, **p < 0.005 and ***p < 0.0005.
3. Results
In the continuous presence of TTX (1 µM), relatively brief (2 min) application of GHB to rat VTA slices bulk-loaded with the Ca2+ indicator Fluo-4 elicited robust and synchronous somatic [Ca2+]i transients in astrocytes (figure 1a, top row and the electronic supplementary material, movie S1), with increasing numbers of cells responding to increasing agonist concentration. The magnitude of GHB-induced elevations was not different in magnitude to those evoked by the selective GABABR agonist baclofen (figure 1a, bottom row). These [Ca2+]i elevations were present in the soma and in the fine astrocytic processes (electronic supplementary material, movie S1), and sometimes seen in processes without an apparent clear somatic response. Moreover, the GHB- and baclofen-elicited [Ca2+]i transients could be observed in the mouse VTA (figure 1c) as well as in the rat nucleus accumbens (not shown) and in the mouse and rat VB thalamus (figure 1c). The astrocytic effect of GHB and baclofen was concentration dependent with an ED50 of 1.6 mM and 1.3 µM, respectively (figure 1b), with 10 mM GHB and 10 µM baclofen evoking [Ca2+]i transients in 6.82 ± 0.39 astrocytes (n = 11 slices) and 7.14 ± 1.14 astrocytes (n = 14 slices), respectively.
Figure 1.

GHB and baclofen elicit [Ca2+]i transients in VTA and thalamic astrocytes. (a) Fluorescence images of Fluo-4-loaded rat VTA slices before (control) and during a brief bath application of 10 mM GHB (top) or 10 µM baclofen (bottom) show robust and synchronous [Ca2+]i transients in response to both drugs. The transients evoked in astrocytes in the two experiments are shown in fluorescence plots on the right. (b) Dose–response curves of the number of astrocytes responding to GHB and baclofen (number of slices is indicated below each data point). (c) Fluorescence plots from different preparations of mouse and rat VTA and VB thalamus show that GABABR activation resulting in [Ca2+]i elevations is a general mechanism, i.e. it can be observed in different species and brain regions. Glutamate (100 µM) applications are also shown for comparison. (Online version in colour.)
To confirm that GABABRs were indeed mediating the astrocytic GHB and baclofen responses described above, experiments were conducted using pharmacological and transgenic interventions. Both GHB and baclofen effects were virtually abolished by two structurally different GABABR antagonists, CGP5426 and SCH50911 (baclofen: p < 1 × 10−5, p < 1 × 10−3, respectively; GHB: p < 1 × 10−7, p < 1 × 10−5, respectively; figure 2a). Interestingly, GHB responses were also markedly inhibited by the putative GHB receptor antagonists NCS382 (GHB: 7.71 ± 0.89 astrocytes, n = 19 slices; GHB + NCS382: 1.5 ± 0.46, n = 8, p < 1 × 10−4; figure 2a), which, however, had no effect on the number of astrocytes responding to baclofen application (baclofen: 8.5 ± 1.28, n = 8 slices; baclofen + NCS382: 10 ± 0.57, n = 3 slices). Finally, astrocytes in VTA slices from wild-type littermate mice (WT, GABABR+/+) readily responded with [Ca2+]i elevations to GHB and baclofen applications, whereas both agonists failed to elicit astrocytic transients in slices from GABABR knockout (GABABR KO,−/−) mice (figure 2b,c). However, astrocytes in GABABR KO and WT littermate mice responded similarly to glutamate (figure 2b,c), thus excluding the possibility that the mechanism underlying [Ca2+]i transients in astrocytes from the GABABR KO mice was compromised. Identical results were obtained in the VB thalamus of GABABR KO and WT littermate mice (figure 2b,c).
Figure 2.

GHB and baclofen elicit astrocytic [Ca2+]i elevations acting via GABABRs. (a) Summary bar graphs showing the number of astrocytes responding to a brief bath-application of 10 mM GHB and 10 µM baclofen alone or together with one of different GABABR antagonists (as indicated). Astrocyte responses to both drugs are abolished by the two GABABR antagonists, CGP65426 and Sch50911, whereas the GHB effect, but not that of baclofen, is also inhibited by the putative GHB receptor antagonist NCS382. Example fluorescence traces are shown on the right. (b) Fluorescence traces from experiments in mouse VTA slices (left column) show that astrocytes from WT littermates respond to baclofen, whereas in GABABR KO mice baclofen responses are absent though astrocytes still respond to a 100 µM glutamate application. Similar results are seen in the VB thalamus (right column), indicating common mechanisms for astrocyte [Ca2+]i elevations in different brain areas. (c) Summary bar graphs of similar experiments as in (b) showing number of responding astrocytes conducted in VTA for WT (left) and GABABR KO (right) astrocytes with GHB and baclofen. (Online version in colour.)
Although all our experiments were conducted in the presence of TTX to block neuronal activity, the possibility might exist that the observed effect could be owing to activation of neuronal GABABRs that resulted in glutamate release and an indirect effect mediated by the activation of metabotropic glutamate receptors (mGluRs), which comprise a major signalling pathway in astrocytes [50]. As shown in figure 3, however, the response of VTA astrocytes to both GHB and baclofen was not affected by APV, NBQX or by the combined application of mGluR5 and mGluR1 antagonists (MTEP and CPCCOEt, respectively).
Figure 3.
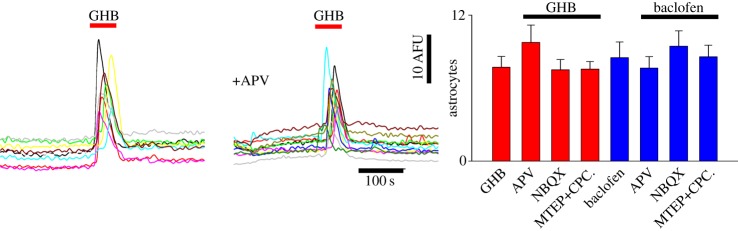
GHB- and baclofen-elicited astrocytic [Ca2+]i transients are unaffected by block of glutamate receptors. Fluorescence traces show responses of astrocytes in a VTA slice to 10 mM GHB in control conditions and in the presence of APV. The summary bar graph on the right quantifies the lack of effect of NMDA, non-NMDA and mGluR antagonists (APV, NBQX and MTEP+ CPCCOEt, respectively) on the number of astrocytes responding to application of 10 mM GHB or 10 µM baclofen. (Online version in colour.)
To investigate the sources of signalling elicited by GHB activation, we manipulated extracellular and intracellular [Ca2+]. To deplete intracellular stores, we used the SERCA pump inhibitor cyclopiazonic acid (CPA; 10 µM), whereas in separate experiments we tested the action of GHB and baclofen in slices bathed with a nominal extracellular Ca2+-free solution. Responses to baclofen were significantly reduced by both interventions (ctrl: 5.75 ± 0.70 astrocytes, n = 12 slices; CPA: 0.3 ± 0.15, n = 12, p < 1 × 10−8; 0 mM Ca2+: 0.22 ± 0.11, n = 12, p < 1 × 10−8) as were those to GHB (ctrl: 5.6 ± 0.56, n = 12; CPA: 0.28 ± 0.88, n = 16, p < 1 × 10−5; 0 mM Ca2+: 0.46 ± 0.13, n = 12, p < 1 × 10−5), showing that the effects of both agonists are dependent on Ca2+ release from intracellular stores and an extracellular Ca2+ supply, as previously described for similar [Ca2+]i transients observed spontaneously and evoked by other neurotransmitter agonists [51,52].
We have previously shown that the sustained stimulation of G-protein coupled mGluRs either by synaptic glutamate release or by agonist exposure results in an increase in the frequency of spontaneous astrocyte [Ca2+]i oscillations which is associated with a [Ca2+]i-dependent increase in the frequency of astrocytic glutamate release events, i.e. neuronal SICs [49,53]. To investigate whether a similar phenomenon resulted from sustained GABABR activation, we applied GHB and baclofen for increasing durations to VTA slices. Although our experiments show that a 2 min baclofen application elicited astrocyte [Ca2+]i elevations (figures 1–3), a 3 min pre-exposure did not lead to an increase in the number of spontaneous astrocytic [Ca2+]i transients, whereas following a 15 min pre-exposure to baclofen spontaneous astrocyte activity was reduced compared with control slices (ctrl: 7.16 ± 0.54 astrocytes, n = 8 slices; baclofen 15 min: 1.78 ± 0.32, n = 12, p < 1 × 10−5; figure 4a). Exposure to GHB also resulted in a reduction that approached statistical significance (ctrl: 6.3 ± 1.12, n = 12, GHB 15 min: 4.0 ± 0.5, n = 12, p = 0.069; figure 4a). To test whether these changes in spontaneous [Ca2+]i signalling were translated to spontaneous astrocyte–neuron glutamatergic signalling recorded as SICs, we therefore treated VTA slices with baclofen or GHB for extended periods of 1–3 h and compared these results with those of untreated slices. Patch-clamp recordings made from VTA neurons in the presence of TTX showed the presence of spontaneous SICs in the VTA, confirming the existence of spontaneous astrocyte–neuron signalling in this brain area (figure 4b). Short (3 min) pre-incubation of the slice with baclofen application, however, did not induce astrocytic glutamate release as measured by SIC recording (ctrl: 0.056 ± 0.016 SICs per min, n = 11 slices; baclofen: 0.086 ± 0.027, n = 11, p = 0.2). Nevertheless, in agreement with the observed decrease in astrocytic [Ca2+]i elevations observed during prolonged GABABR activation (1–3 h), SIC frequency was significantly reduced by sustained pre-exposure to GHB and baclofen (ctrl: 0.066 ± 0.017 SICs per min, n = 28 slices; baclofen: 0.026 ± 0.009, n = 26, p < 0.05; GHB: 0.01 ± 0.008, n = 11, p < 0.05; figure 4c, right plot).
Figure 4.

Time-dependent effect of GHB and baclofen on astrocytic [Ca2+]i signalling and glutamate release. (a) Fluorescence traces show spontaneous astrocytic [Ca2+]i transients in control conditions and in a VTA slice pre-exposed to 10 µM baclofen for 15 min. Bar graphs on the right summarize data from similar experiments where slices were pre-exposed to either 10 µM baclofen or 10 mM GHB for 3 and 15 min. (b) Patch-clamp recording from a VTA neuron on an expanded timebase illustrating spontaneous SICs in control conditions (top trace), and lower trace showing effect of long duration 10 µM baclofen application (SICs highlighted by filled circle). (c) Summary data showing the frequency of SICs in control conditions, and following long (>1 h) exposure to either 10 mM GHB or 10 µM baclofen. (Online version in colour.)
4. Discussion
The main findings of this study are that GHB consistently elicits robust [Ca2+]i transients in astrocytes of both the VTA and the VB thalamus of young rodents, two brain areas that are involved in the reward properties and the pro-absence effect of this drug, respectively. This astrocytic GHB action is mediated by GABABRs and is comparable to that evoked in astrocytes of the same regions by the selective GABABR agonist baclofen. Moreover, prolonged GHB and baclofen exposure causes a reduction in spontaneous glutamate release from VTA astrocytes.
(a). Astrocytic gamma-hydroxybutyric acid effects
The astrocytic response to GHB (and baclofen) was highly reliable and consistent from trial to trial in the age range tested, and thus did not show the variability that has been reported in a previous study [42]. Determination of whether there is a difference in age-dependent GABABR signalling that is also region-dependent will require further studies in the VTA and VB thalamus.
Both GHB- and baclofen-elicited [Ca2+]i transients were (i) abolished by two highly selective but structurally dissimilar GABABR antagonists, (ii) absent in GABABR KO mice and (iii) unaffected by the block of ionotropic and metabotropic glutamate receptors, indicating that in astrocytes, as occurs in neurons, the action of GHB is mediated by GABABRs. Indeed, the 1000-fold difference in the ED50 of the two drugs on astrocytic responses is very similar to that reported for their activation of pre- and post-synaptic neuronal GABABRs, including hyperpolarization of the membrane potential and inhibition of synaptic potentials [54]. However, the astrocytic effect of GHB was also markedly blocked by NCS382, an antagonist of the putative GHB receptor. This represents the first solid evidence of a CNS action of GHB being mediated by its putative receptor [3], and might provide an explanation for the block of GHB-elicited absence seizures observed in some studies [34]. Finally, the lack of action of ionotropic and metabotropic glutamate receptors on GHB-elicited [Ca2+]i transients indicates that these responses are not mediated by activation of astrocytes by glutamate, secondary to neuronal activation, and suggests that they results from direct activation of astrocytic GABABRs.
GHB- (and baclofen)-elicited [Ca2+]i transients were blocked following perfusion of VTA slices with either a nominal extracellular Ca2+-free solution or with CPA. Thus, our results indicate that both GHB and baclofen astrocytic responses in VTA involve intracellular Ca2+ stores and their refilling by extracellular Ca2+. Whether GHB activity requires Gi/o proteins (which are classically associated with neuronal GABABR activation) or Gq proteins (which have been strongly linked to Ca2+ release from intracellular stores) remains to be elucidated, as is indeed the case for the astrocytic response to baclofen in VTA or any other brain region.
Finally, in contrast to the activation of astrocytes, brief application of GHB and baclofen had no effect on spontaneous [Ca2+]i transients (see control grey bar in figure 4a), but decreased their frequency when applied for periods of 15 min or longer. A similar picture was obtained when looking at spontaneous SICs, the neuronal counterpart of astrocytic released glutamate, suggesting that long-term activation of GABABR lead to a reduction in the glutamate-dependent astrocyte to neuron signalling.
(b). Potential (patho)physiological implications of gamma-hydroxybutyric acid activation of astrocytes
Our finding of an astrocytic activation by GHB opens new avenues in the interpretation of the effects of this substance. As far as absence seizures are concerned, our results suggest for the first time that the ability of GHB to induce these seizures might involve an astrocytic component together with a neuronal component. This, together with the already described loss of function of GAT1 in thalamic astrocytes of genetic models of absence seizures [29] and the fact that GABA transporters affect astrocytic [Ca2+]i transients [55], clearly stresses the importance of this glial cell type in the generation of these non-convulsive seizures. Finally, our data showing a block of GHB-elicited astrocytic [Ca2+]i transients by NCS382 provide a potential explanation for the anti-absence effect of this putative GHB receptor antagonist, the action of which had so far been difficult to reconcile with its lack of action on neuronal response in thalamus [3], one of the key regions responsible for the generation of these non-convulsive seizures.
Drugs that cause addiction act in the VTA to increase dopamine release in target areas, notably the nucleus accumbens. It has been suggested that GHB achieves this by inhibiting GABAergic interneuron activity which results in disinhibition of dopaminergic neurons in the VTA, and so increased dopamine release [56,57]. Our observations suggest a possible astrocytic contribution to such a mechanism, if a reduced astrocytic glutamate release which may ordinarily contribute to tonic glutamate has a physiological role in driving GABAergic interneuron activity. Reducing such a drive would therefore be expected to increase the output of dopaminergic neurons. There are reports from many brain areas of a Ca2+-dependent gliotransmitter release increase following the activation of G-protein coupled receptors [58]: these examples are linked to PLC and IP3 production via Gq, whereas GABABRs are coupled to adenylyl cyclase via Gi/o [54]. We found that in astrocytes, GABABR activation elicits [Ca2+]i elevations. The mechanism underlying this increase is unclear but may involve some activation of Gq-coupled pathways, or novel interactions between GABAB auxiliary subunits and intracellular Ca2+ release pathways. Whatever the physiological role of the GABABR-induced [Ca2+]i elevations, it does not seem to invoke glutamate gliotransmitter release. It does, however, provide an experimental reporter of astrocyte GABABR activation, and our observations are consistent with dominant activation of non-Gq pathways, because we found a reduction in spontaneous gliotransmitter glutamate release (i.e. SICs). Interestingly, this observation is analogous to the effect of GABABR activation on inhibiting spontaneous presynaptic neurotransmitter release [59], and indicates the complexity of signalling pathways which may influence the ways that astrocytes interact with neuronal activity in different brain areas.
Acknowledgements
We thank Prof. B. Bettler (Department of Biomedicine, Institute of Physiology, University of Basel, Basel, Switzerland) for the generous gift of GABAB KO mice.
Funding statement
This work was supported by the Wellcome Trust (091882), the MRC (G0900671) and the EU (FP7 HEALTH-F2-2007-202167).
References
- 1.Hechler V, Ratomponirina C, Maitre M. 1997. γ-Hydroxybutyrate conversion into GABA induces displacement of GABAB binding that is blocked by valproate and ethosuximide. J. Pharmacol. Exp. Ther. 281, 753–760. [PubMed] [Google Scholar]
- 2.Wong CG, Gibson KM, Snead OC., III 2004. From the street to the brain: neurobiology of the recreational drug γ-hydroxybutyric acid. Trends Pharmacol. Sci. 25, 29–34. ( 10.1016/j.tips.2003.11.001) [DOI] [PubMed] [Google Scholar]
- 3.Crunelli V, Emri Z, Leresche N. 2006. Unravelling the brain targets of γ-hydroxybutyric acid. Curr. Opin. Pharmacol. 6, 44–52. ( 10.1016/j.coph.2005.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambliss KL, Gibson KM. 1992. Succinic semialdehyde dehydrogenase from mammalian brain: subunit analysis using polyclonal antiserum. Int. J. Biochem. 24, 1493–1499. ( 10.1016/0020-711X(92)90077-E) [DOI] [PubMed] [Google Scholar]
- 5.Laborit H, Buchard F, Laborit G, Kind A, Weber B. 1960. [Use of sodium 4-hydroxybutyrate in anesthesia and resuscitation]. Agressologie: revue internationale de physio-biologie et de pharmacologie appliquees aux effets de l'agression 1, 549–560. [PubMed] [Google Scholar]
- 6.Miotto K, Darakjian J, Basch J, Murray S, Zogg J, Rawson R. 2001. Gamma-hydroxybutyric acid: patterns of use, effects and withdrawal. Am. J. Addict. 10, 232–241. ( 10.1080/105504901750532111) [DOI] [PubMed] [Google Scholar]
- 7.Carter LP, Richards BD, Mintzer MZ, Griffiths RR. 2006. Relative abuse liability of GHB in humans: a comparison of psychomotor, subjective, and cognitive effects of supratherapeutic doses of triazolam, pentobarbital, and GHB. Neuropsychopharmacology 31, 2537–2551. ( 10.1038/sj.npp.1301146) [DOI] [PubMed] [Google Scholar]
- 8.Abanades S, Farre M, Segura M, Pichini S, Pastor A, Pacifici R, Pellegrini M, de la Torre R. 2007. Disposition of γ-hydroxybutyric acid in conventional and nonconventional biologic fluids after single drug administration: issues in methodology and drug monitoring. Ther. Drug Monit. 29, 64–70. ( 10.1097/FTD.0b013e3180307e5e) [DOI] [PubMed] [Google Scholar]
- 9.Gallimberti L, Canton G, Gentile N, Ferri M, Cibin M, Ferrara SD, Fadda F, Gessa GL. 1989. γ-hydroxybutyric acid for treatment of alcohol withdrawal syndrome. Lancet 2, 787–789. ( 10.1016/S0140-6736(89)90842-8) [DOI] [PubMed] [Google Scholar]
- 10.Addolorato G, Balducci G, Capristo E, Attilia ML, Taggi F, Gasbarrini G, Ceccanti M. 1999. γ-Hydroxybutyric acid (GHB) in the treatment of alcohol withdrawal syndrome: a randomized comparative study versus benzodiazepine. Alcohol Clin. Exp. Res. 23, 1596–1604. [PubMed] [Google Scholar]
- 11.Caputo F, Vignoli T, Lorenzini F, Ciuffoli E, Del Re A, Stefanini GF, Addolorato G, Trevisani F, Bernardi M. 2005. Suppression of craving for γ-hydroxybutyric acid by naltrexone administration: three case reports. Clin. Neuropharmacol. 28, 87–89. ( 10.1097/01.wnf.0000159954.49393.76) [DOI] [PubMed] [Google Scholar]
- 12.Nava F, Premi S, Manzato E, Campagnola W, Lucchini A, Gessa GL. 2007. Gamma-hydroxybutyrate reduces both withdrawal syndrome and hypercortisolism in severe abstinent alcoholics: an open study versus diazepam. Am. J. Drug Alcohol Abuse 33, 379–392. ( 10.1080/00952990701315046) [DOI] [PubMed] [Google Scholar]
- 13.Black J, Guilleminault C. 2001. Medications for the treatment of narcolepsy. Expert Opin. Emerg. Drugs 6, 239–247. ( 10.1517/14728214.6.2.239) [DOI] [PubMed] [Google Scholar]
- 14.Tunnicliff G, Raess BU. 2002. γ-Hydroxybutyrate (orphan medical). Curr. Opin. Invest. Drugs 3, 278–283. [PubMed] [Google Scholar]
- 15.Pardi D, Black J. 2006. γ-Hydroxybutyrate/sodium oxybate: neurobiology, and impact on sleep and wakefulness. CNS Drugs 20, 993–1018. ( 10.2165/00023210-200620120-00004) [DOI] [PubMed] [Google Scholar]
- 16.Degenhardt L, Copeland J, Dillon P. 2005. Recent trends in the use of ‘club drugs’: an Australian review. Subst. Use Misuse 40, 1241–1256. ( 10.1081/JA-200066777) [DOI] [PubMed] [Google Scholar]
- 17.Ricaurte GA, McCann UD. 2005. Recognition and management of complications of new recreational drug use. Lancet 365, 2137–2145. ( 10.1016/S0140-6736(05)66737-2) [DOI] [PubMed] [Google Scholar]
- 18.Snead OC, III, Gibson KM. 2005. γ-Hydroxybutyric acid. N. Engl. J. Med. 352, 2721–2732. ( 10.1056/NEJMra044047) [DOI] [PubMed] [Google Scholar]
- 19.Sumnall HR, Woolfall K, Edwards S, Cole JC, Beynon CM. 2008. Use, function, and subjective experiences of γ-hydroxybutyrate (GHB). Drug Alcohol Dependence 92, 286–290. ( 10.1016/j.drugalcdep.2007.07.009) [DOI] [PubMed] [Google Scholar]
- 20.European Monitoring Centre for Drugs and Drug Addiction. 2008. GHB and its precursor GBL: an emerging trend case study . EMCDDA Thematic Paper 320. Lisbon, Portugal: EMCDDA. ( 10.2810/16549) [DOI] [Google Scholar]
- 21.Velazquez JL, Huo JZ, Dominguez LG, Leshchenko Y, Snead OC., III 2007. Typical versus atypical absence seizures: network mechanisms of the spread of paroxysms. Epilepsia 48, 1585–1593. ( 10.1111/j.1528-1167.2007.01120.x) [DOI] [PubMed] [Google Scholar]
- 22.Colombo G, Agabio R, Balaklievskaia N, Diaz G, Lobina C, Reali R, Gessa GL. 1995. Oral self-administration of γ-hydroxybutyric acid in the rat. Eur. J. Pharmacol. 285, 103–107. ( 10.1016/0014-2999(95)00493-5) [DOI] [PubMed] [Google Scholar]
- 23.Martellotta MC, Fattore L, Cossu G, Fratta W. 1997. Rewarding properties of γ-hydroxybutyric acid: an evaluation through place preference paradigm. Psychopharmacology 132, 1–5. ( 10.1007/s002130050312) [DOI] [PubMed] [Google Scholar]
- 24.Itzhak Y, Ali SF. 2002. Repeated administration of γ-hydroxybutyric acid (GHB) to mice: assessment of the sedative and rewarding effects of GHB. Ann. NY Acad. Sci. 965, 451–460. ( 10.1111/j.1749-6632.2002.tb04186.x) [DOI] [PubMed] [Google Scholar]
- 25.Labouebe G, et al. 2007. RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat. Neurosci. 10, 1559–1568. ( 10.1038/nn2006) [DOI] [PubMed] [Google Scholar]
- 26.Pistis M, Muntoni AL, Pillolla G, Perra S, Cignarella G, Melis M, Gessa GL. 2005. γ-Hydroxybutyric acid (GHB) and the mesoaccumbens reward circuit: evidence for GABAB receptor-mediated effects. Neuroscience 131, 465–474. ( 10.1016/j.neuroscience.2004.11.021) [DOI] [PubMed] [Google Scholar]
- 27.Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Luscher C. 2004. Bi-directional effects of GABAB receptor agonists on the mesolimbic dopamine system. Nat. Neurosci. 7, 153–159. ( 10.1038/nn1181) [DOI] [PubMed] [Google Scholar]
- 28.Watson J, et al. 2010. Gamma-hydroxybutyrate does not maintain self-administration but induces conditioned place preference when injected in the ventral tegmental area. Int. J. Neuropsychopharmacol. 13, 143–153. ( 10.1017/S1461145709990186) [DOI] [PubMed] [Google Scholar]
- 29.Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. 2009. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398. ( 10.1038/nm.2058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crunelli V, Leresche N. 2002. Childhood absence epilepsy: genes, channels, neurons and networks. Nat. Rev. Neurosci. 3, 371–382. ( 10.1038/nrn811) [DOI] [PubMed] [Google Scholar]
- 31.Mathivet P, Bernasconi R, De Barry J, Marescaux C, Bittiger H. 1997. Binding characteristics of γ-hydroxybutyric acid as a weak but selective GABAB receptor agonist. Eur. J. Pharmacol. 321, 67–75. ( 10.1016/S0014-2999(96)00916-8) [DOI] [PubMed] [Google Scholar]
- 32.Lingenhoehl K, Brom R, Heid J, Beck P, Froestl W, Kaupmann K, Bettler B, Mosbacher J. 1999. γ-Hydroxybutyrate is a weak agonist at recombinant GABAB receptors. Neuropharmacology 38, 1667–1673. ( 10.1016/S0028-3908(99)00131-8) [DOI] [PubMed] [Google Scholar]
- 33.Maitre M. 1997. The γ-hydroxybutyrate signalling system in brain: organization and functional implications. Prog. Neurobiol. 51, 337–361. ( 10.1016/S0301-0082(96)00064-0) [DOI] [PubMed] [Google Scholar]
- 34.Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. 1998. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog. Neurobiol. 55, 27–57. ( 10.1016/S0301-0082(97)00091-9) [DOI] [PubMed] [Google Scholar]
- 35.Absalom N, et al. 2012. α4βδ GABAA receptors are high-affinity targets for γ-hydroxybutyric acid (GHB). Proc. Natl Acad. Sci. USA 109, 13 404–13 409. ( 10.1073/pnas.1204376109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Connelly WM, Errington AC, Crunelli V. 2013. γ-Hydroxybutyric acid (GHB) is not an agonist of extrasynaptic GABAA receptors. PLoS ONE 8, e79062 ( 10.1371/journal.pone.0079062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraser DD, Mudrick-Donnon LA, MacVicar BA. 1994. Astrocytic GABA receptors. Glia 11, 83–93. ( 10.1002/glia.440110203) [DOI] [PubMed] [Google Scholar]
- 38.Kang J, Jiang L, Goldman SA, Nedergaard M. 1998. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692. ( 10.1038/3684) [DOI] [PubMed] [Google Scholar]
- 39.Volterra A, Meldolesi J. 2005. Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640. ( 10.1038/nrn1722) [DOI] [PubMed] [Google Scholar]
- 40.Zorec R, Araque A, Carmignoto G, Haydon PG, Verkhratsky A, Parpura V. 2012. Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro 4, e00080 ( 10.1042/AN20110061). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perea G, Navarrete M, Araque A. 2009. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. ( 10.1016/j.tins.2009.05.001) [DOI] [PubMed] [Google Scholar]
- 42.Meier SD, Kafitz KW, Rose CR. 2008. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia 56, 1127–1137. ( 10.1002/glia.20684) [DOI] [PubMed] [Google Scholar]
- 43.Schuler V, et al. 2001. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABAB responses in mice lacking GABAB1. Neuron 31, 47–58. ( 10.1016/S0896-6273(01)00345-2) [DOI] [PubMed] [Google Scholar]
- 44.Mueller AL, Brodie MS. 1989. Intracellular recording from putative dopamine-containing neurons in the ventral tegmental area of Tsai in a brain slice preparation. J. Neurosci. Methods 28, 15–22. ( 10.1016/0165-0270(89)90005-8) [DOI] [PubMed] [Google Scholar]
- 45.Parri HR, Crunelli V. 2001. Pacemaker calcium oscillations in thalamic astrocytes in situ. Neuroreport 12, 3897–3900. ( 10.1097/00001756-200112210-00008) [DOI] [PubMed] [Google Scholar]
- 46.Bushong EA, Martone ME, Jones YZ, Ellisman MH. 2002. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parri HR, Gould TM, Crunelli V. 2010. Sensory and cortical activation of distinct glial cell subtypes in the somatosensory thalamus of young rats. Eur. J. Neurosci. 32, 29–40. ( 10.1111/j.1460-9568.2010.07281.x.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, Parri HR. 2013. α7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer's mouse model. PLoS ONE 8, e81828 ( 10.1371/journal.pone.0081828) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pirttimaki TM, Hall SD, Parri HR. 2011. Sustained neuronal activity generated by glial plasticity. J. Neurosci. 31, 7637–7647. ( 10.1523/JNEUROSCI.5783-10.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parpura V, Verkhratsky A. 2013. Astroglial amino acid-based transmitter receptors. Amino Acids 44, 1151–1158. ( 10.1007/s00726-013-1458-4) [DOI] [PubMed] [Google Scholar]
- 51.Porter JT, McCarthy KD. 1997. Astrocytic neurotransmitter receptors in situ and in vivo. Prog. Neurobiol. 51, 439–455. ( 10.1016/S0301-0082(96)00068-8) [DOI] [PubMed] [Google Scholar]
- 52.Parri HR, Crunelli V. 2002. Astrocytes, spontaneity, and the developing thalamus. J. Physiol. Paris 96, 221–230. ( 10.1016/S0928-4257(02)00009-8) [DOI] [PubMed] [Google Scholar]
- 53.Pirttimaki TM, Parri HR. 2012. Glutamatergic input–output properties of thalamic astrocytes. Neuroscience 205, 18–28. ( 10.1016/j.neuroscience.2011.12.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gassmann M, Bettler B. 2012. Regulation of neuronal GABAB receptor functions by subunit composition. Nat. Rev. Neurosci. 13, 380–394. ( 10.1038/nrn3249) [DOI] [PubMed] [Google Scholar]
- 55.Losi G, Mariotti L, Carmignoto G. 2014. GABAergic interneuron to astrocyte signalling: a neglected form of cell communication in the brain. Phil. Trans. R. Soc. B 369, 20130609 ( 10.1098/rstb.2013.0609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carter LP, Wu H, Chen W, Cruz CM, Lamb RJ, Koek W, Coop A, France CP. 2004. Effects of γ-hydroxybutyrate (GHB) on schedule-controlled responding in rats: role of GHB and GABAB receptors. J. Pharmacol. Exp. Ther. 308, 182–188. ( 10.1124/jpet.103.058909) [DOI] [PubMed] [Google Scholar]
- 57.Creed MC, Luscher C. 2013. Drug-evoked synaptic plasticity: beyond metaplasticity. Curr. Opin. Neurobiol. 23, 553–558. ( 10.1016/j.conb.2013.03.005) [DOI] [PubMed] [Google Scholar]
- 58.Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. 2014. Gliotransmitters travel in time and space. Neuron 81, 728–739. ( 10.1016/j.neuron.2014.02.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manzoni OJ, Williams JT. 1999. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J. Neurosci. 19, 6629–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]