

ABSTRACT
A severe, sometimes fatal respiratory disease has been observed in captive ball pythons (Python regius) since the late 1990s. In order to better understand this disease and its etiology, we collected case and control samples and performed pathological and diagnostic analyses. Electron micrographs revealed filamentous virus-like particles in lung epithelial cells of sick animals. Diagnostic testing for known pathogens did not identify an etiologic agent, so unbiased metagenomic sequencing was performed. Abundant nidovirus-like sequences were identified in cases and were used to assemble the genome of a previously unknown virus in the order Nidovirales. The nidoviruses, which were not previously known to infect nonavian reptiles, are a diverse order that includes important human and veterinary pathogens. The presence of the viral RNA was confirmed in all diseased animals (n = 8) but was not detected in healthy pythons or other snakes (n = 57). Viral RNA levels were generally highest in the lung and other respiratory tract tissues. The 33.5-kb viral genome is the largest RNA genome yet described and shares canonical characteristics with other nidovirus genomes, although several features distinguish this from related viruses. This virus, which we named ball python nidovirus (BPNV), will likely establish a new genus in Torovirinae subfamily. The identification of a novel nidovirus in reptiles contributes to our understanding of the biology and evolution of related viruses, and its association with lung disease in pythons is a promising step toward elucidating an etiology for this long-standing veterinary disease.
IMPORTANCE
Ball pythons are popular pets because of their diverse coloration, generally nonaggressive behavior, and relatively small size. Since the 1990s, veterinarians have been aware of an infectious respiratory disease of unknown cause in ball pythons that can be fatal. We used unbiased shotgun sequencing to discover a novel virus in the order Nidovirales that was present in cases but not controls. While nidoviruses are known to infect a variety of animals, this is the first report of a nidovirus recovered from any reptile. This report will enable diagnostics that will assist in determining the role of this virus in the causation of disease, which would allow control of the disease in zoos and private collections. Given its evolutionary divergence from known nidoviruses and its unique host, the study of reptile nidoviruses may further our understanding of related diseases and the viruses that cause them in humans and other animals.
INTRODUCTION
The order Nidovirales encompasses a diverse group of viruses that includes significant veterinary and human pathogens (1–6). These viruses cause a variety of diseases that range from mild enteric infection to severe respiratory disease or hemorrhagic fever (7, 8). Examples of disease-causing nidoviruses include the severe acute respiratory syndrome (SARS) coronavirus, a number of other coronaviruses that cause typically mild respiratory disease in humans, and agriculturally important animal pathogens, such as equine arteritis virus, porcine reproductive and respiratory syndrome virus, and yellow head virus. Nidoviruses are characterized by their overall genome architecture, distinct pattern of gene expression, and presence of a conserved set of functional domains in their nonstructural polyproteins. The nidoviruses cluster into five major groups, which have been taxonomically categorized into four families: Arteriviridae, Roniviridae, Mesoniviridae, and Coronaviridae. Viruses in the Coronaviridae family (subfamilies Torovirinae and Coronavirinae) have the largest known RNA genomes, an attribute thought possible because of a virally encoded proofreading exonuclease (ExoN) that increases replication fidelity (9–12). Although nidoviruses are known to infect mammals, birds, fish, and crustaceans, no nonavian reptile nidovirus has been previously described.
Ball pythons (Python regius) have become one of the most popular types of reptiles sold and kept as pets (13). Native to West Africa, these snakes make popular pets because of their relatively modest size (≤1.5 m), docile behavior, and ease of care. Selective captive breeding has resulted in a tremendous variety of colors and patterns (morphs), many of which command high prices. Since the late 1990s, veterinarians have been aware of respiratory tract disease as a common syndrome affecting ball pythons. This syndrome is often characterized by pharyngitis, sinusitis, stomatitis, tracheitis, and a proliferative interstitial pneumonia. The clinical and epidemiological characteristics suggested an infectious etiology.
In this study, we investigated the pathology and etiology of this disease. We obtained case samples from 7 collections around the United States, performed necropsies, and collected multiple tissues for light microscopy and samples of lung for transmission electron microscopy (TEM). Although TEM of the lung suggested a viral etiology, traditional molecular diagnostic methods did not identify an agent. Metagenomic sequencing was used to identify and assemble the genome of a novel virus in the order Nidovirales. Here we describe clinical and pathological manifestations of this disease, ultrastructural findings, tissue tropism, disease association, and subgenomic RNA expression and analyze the genome of this virus in the context of related viruses.
RESULTS
Pathological findings.
Fixed and frozen samples from affected snakes from collections in Wisconsin, Texas, Florida, Oklahoma, and Pennsylvania were collected between 2006 and 2013 (see Materials and Methods and Table 1). Necropsies were performed on 9 snakes with clinical signs of respiratory disease, and multiple tissues were collected for histopathology; samples of lung from two snakes were collected for TEM.
TABLE 1 .
Lesions identified in ball pythons with respiratory tract disease
| BP no. |
Collection | Age/sex | Sinusitis/ rhinitis |
Stomatitis/ pharyngitis |
Tracheitis | Mucous or caseous material in air passageways |
Thickened lung |
Pulmonary hemorrhage |
Pneumonia |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Juvenile UDSa | NEb | No | No | No | No | No | IPPc; moderate |
| 2 | 1 | Juvenile UDS | NE | Moderate-severe; subacute |
Moderate | Moderate to severe |
Moderate to severe |
No | IPP; severe |
| 5 | 3 | Adult female | NE | No | Mild to moderate hyperplasia |
No | No | Yes | IPP; moderate- severe |
| 6 | 3 | Adult female | Mild; subacute | Mild; subacute | Mild to moderate hyperplasia |
No | No | Yes | IPP; moderate- severe |
| 7 | 3 | Adult female | Mild; subacute | Mild; subacute | Mild to moderate hyperplasia |
No | No | Yes | IPP; moderate- severe |
| 8 | 4 | Juvenile female | Moderate to severe; subacute |
Moderate with epithelial hyperplasia |
Moderate to severe; subacute |
No | No | No | IPP; mild |
| 9 | 5 | Juvenile male | No | No | Moderate to severe with epithelial hyperplasia |
Severe | Moderate | No | IPP; mild |
| 10 | 6 | Adult female | No | No | Moderate | Moderate to severe |
Moderate | No | IPP; severe |
| 11 | 7 | Adult male | NE | No | Necrotizing; severe |
No | Moderate | No | IPP; severe |
UDS, undetermined sex.
NE, not examined.
IPP, interstitial proliferative pneumonia.
Lesions were evident in the upper and lower respiratory tracts of all diseased animals (Table 1 and Fig. 1). Four snakes had stomatitis/pharyngitis, with one having caseous material in the choanae (sinusitis) (Fig. 1A). Three snakes had pulmonary hemorrhage, three had either mucoid or caseous material in air passageways, and four snakes had moderately to severely thickened lungs (Fig. 1B). At a microscopic level, four snakes had mild to severe stomatitis (see Fig. S1A in the supplemental material). Eight of nine snakes had tracheitis, with multifocal mild to severe changes, including infiltrates of heterophils, macrophages, and lymphocytes in the lamina propria, and in four of these cases, there was hyperplasia in the tracheal epithelium (see Fig. S1B).
FIG 1 .

Representative macroscopic lesions. (A) Common wild-type ball python (no. 1), Python regius. The palatine mucosa is both thickened and necrotic, and there is an accumulation of caseous material in the internal choanae. (B) Mojave variant ball python (no. 11), Python regius. The lung is thickened and edematous with abundant mucoid to caseous material (arrow) in air passageways.
In comparison to the histology of normal snake lung, all nine snakes had a proliferative interstitial pneumonia of various severities (Table 1). In some cases, inflammatory cells were primarily around the terminal smooth muscle bundle of each septum that projected into the central lumen of the lung (Fig. 2A and B). Hyperplastic alveolar cells completely proliferated over capillary beds that are normally distributed at the surface of the primitive faveoli (Fig. 2C and D). Epithelial cells were tall and columnar with abundant apical mucus production (mucous metaplasia). Periodic acid-Schiff (PAS) and alcian blue staining were positive, consistent with the presence of mucopolysaccharides. Heterophils, lymphocytes, and in some cases macrophages infiltrated the interstitium of the pulmonary septa (Fig. 2D) and often were seen in air passageways along with sloughed epithelial cells. In deeper areas of the faveolar spaces, the lung was histologically normal. In more severe cases, the proliferative changes occurred diffusely throughout the lung and were seen from the proximal to distal surfaces lining faveolar structures.
FIG 2 .
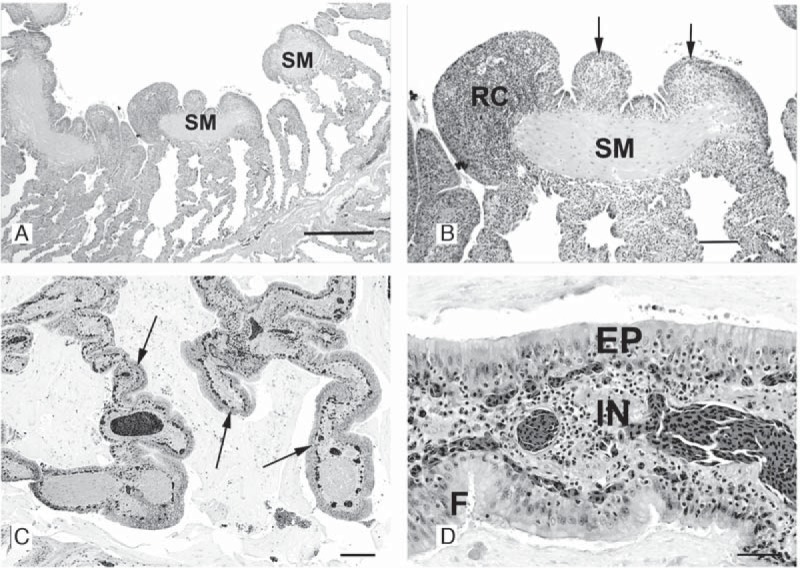
Representative photomicrographs of diseased lung section. (A) Ball python 1 (BP1). Photomicrograph of a cross section of the lung. The smooth muscle (SM) bundle at the luminal end of each septum is hyperplastic and surrounded by inflammatory cells. As the septa approach the base of each faveolus, the septal wall between adjacent faveoli is of normal thickness. No inflammatory cells are seen in air passageways. H&E stain; bar = 500 µm. (B) BP1. Photomicrograph of a cross section of lung. At a higher power, numerous round cells (RC) have infiltrated the submucosa and overlying epithelial cells are hyperplastic (arrows). H&E stain; bar = 100 µm. (C) BP11. Photomicrograph of a cross section of lung. Severe proliferative interstitial pneumonia. Inflammation and epithelial hyperplasia (arrows) markedly thicken the trabecular septa and faveolar walls. Abundant mucinous exudate fills the lumina. H&E stain; bar = 200 µm. (D) BP11. Photomicrograph of a cross section of the lung at a higher magnification. Faveolar septum, severe proliferative interstitial pneumonia. The epithelium (EP) is hypercellular, thick, and densely packed with pseudostratified columnar cells, the majority of which produce mucus. Macrophages, lymphocytes, and granulocytes infiltrate (IN) the epithelium and submucosa. Faveolar capillaries are below the epithelial layer. H&E stain; bar = 50 µm.
Three snakes had mild to marked bronchial epithelial hyperplasia, with mild to moderate smooth muscle hypertrophy of the terminal muscle bundles. In these cases, the bronchus-associated lymphoid tissue was mildly to moderately hyperplastic. Sections of lungs of two cases were stained with Warthin-Starry stain and Brown-Hopps Gram stain; there was no positive staining for bacteria between cilia and microvilli of pneumocytes overlying muscle bundles. Three of five nasal cavities that were examined revealed mild subacute rhinitis/sinusitis. The vomeronasal organ in one snake was almost obliterated by foamy macrophages. Ziehl-Neelsen acid-fast staining was negative for acid-fast bacteria, and methenamine silver staining was negative for yeast and hyphae. Three cases had mild to moderate stomatitis/pharyngitis. The changes seen in the respiratory tract were consistent with a viral infection.
Of the 9 necropsied snakes, lesions were also observed in other areas of the body. These included mild nonsuppurative encephalitis characterized by periventricular perivascular lymphocytic cuffing (in 1 of 9 snakes examined), mild to moderate acute nephritis (1/9), mild nephrosis (1/9), mild to moderate multifocal subacute dermatitis (1/9), mild acute salpingitis (1/9), and hepatic lipidosis (5/9). In the eye of one snake, there was moderate necrotizing conjunctivitis, with diffuse corneal ulceration, mild superficial keratitis, and mild inflammation and necrosis of the inner layer of the spectacle. One snake had mild bacterial colitis. It is unclear whether these other lesions were related to the observed lung pathology.
Ultrastructure of virus morphogenesis.
Two cases with moderate to severe diffuse proliferative pneumonia were selected for ultrastructural examination by TEM (snakes 2 and 6) (Table 1) (14). Virus-like particles were observed within pneumocytes lining the faveoli of both snakes. These particles were observed within ciliated and mucous epithelial cells overlying smooth muscle bundles at the tips of faveolar septa and alveolar type II cells lining faveolar spaces of both snakes (Fig. 3A). The particles corresponded to stages of a virus with both circular and bacillary (elongated rod-shaped) nucleocapsids and were seen within electron-lucent areas of the cytoplasm. At a higher magnification, bacillary nucleocapsids contained a lucent core that was surrounded by fine granular cytoplasmic material (Fig. 3B). The uncoated intracellular capsids measured 10 to 12 nm. Nucleocapsids lined up along membranes of cytoplasmic vesicles of uncertain origin, and as they progressed into a vesicle, a membrane coat was acquired (Fig. 3C). Cross sections of mature virions having a lipid envelope and surface spikes were also identified in cytoplasmic vesicles (Fig. 3D). Bacillary virions were observed within vesicles subadjacent to epithelial cell membranes on the free cell surface (Fig. 3E) and extracellularly between cilia and microvilli projecting into the air passageway (Fig. 3F). The mature particles measured approximately 50 by 180 nm. In some sections, filamentous bacteria (200 nm by 200 µm) were occasionally observed between cilia and closely associated with the cytoplasmic membrane (Fig. 3F).
FIG 3 .

Transmission electron photomicrographs of pulmonary epithelial cells of ball pythons, Python regius, with proliferative pneumonia. (A) BP6. Multiple bacilliform (arrows) and spherical nucleocapsid cores of virions are seen within the cytoplasm. Bar = 200 nm. (B) BP6. At a higher magnification, bacillary nucleocapsids are surrounded by granular cytoplasmic material (white arrows), presumed to be a component of the envelope. Bar = 100 nm. (C) BP6. Progression of immature forms in the cytoplasmic matrix to more mature forms (arrow) within a cytoplasmic vesicle. Bar = 200 nm. (D) BP2. Mature enveloped spherical virions (arrows) within a cytoplasmic vesicle. Spikes can be seen on the surface of several virions (arrowhead). Bar = 200 nm. (E) BP6. Spherical and filamentous mature virions (arrows) can be seen within cytoplasmic vesicles that are subjacent to microvilli projecting from the cell membrane Bar = 200 nm. (F) BP6. Spherical and bacillary forms of mature virions (arrows) can be seen in between microvilli and cilia of a pulmonary epithelial cell. Bacillary bacteria lacking a trilaminar cell wall (arrowheads) are also present. Scale bar corresponds to 1 µm.
Molecular diagnostics.
Pathology and EM findings were consistent with a viral etiology, so reverse transcription-PCR (RT-PCR) was used to attempt to identify an etiologic agent. Consensus primers targeted viruses in the families Paramyxoviridae (genus Ferlavirus), Rhabdoviridae, Reoviridae (genera Orthoreovirus and Aquareovirus), and Filoviridae (15–19). RNA was extracted from frozen lung tissue from animals 1 and 6 and used as a template for reactions, which were uniformly negative, though positive-control primers targeting conserved host sequences were positive.
Metagenomic sequencing and genome assembly.
Because consensus PCR approaches did not identify any candidate etiologic agents, we pursued an unbiased metagenomic shotgun sequencing strategy. Suitable frozen tissue was available for 8 of 9 snakes, and total RNA was extracted and converted into sequencing libraries (see Materials and Methods). The libraries were sequenced on an Illumina HiSeq 2500 instrument to an average depth of 2.9 million pairs of 100-nucleotide (nt) sequences per sample (see Table S2 in the supplemental material). We then used a stepwise analysis pipeline to efficiently identify possible pathogen-derived sequences. First, we removed low-quality and low-complexity sequences and trimmed adapter sequences from reads. For each library, identical reads were collapsed to yield sets of unique sequences. Next, we filtered out snake ribosomal sequences and sequences aligning to the Burmese python and boa constrictor genomes (20, 21). After these operations, approximately 2% of each data set remained.
We then searched for possible pathogen-derived sequences in what remained of the data sets. We performed BLASTx alignments using the remaining reads to search a database of virus protein sequences. In case samples but not in control samples, we observed sequences with similarity to viruses in the Nidovirales order. The data set from snake 2 had the most nidovirus-like sequences: 92 read pairs with BLASTx alignments to nidovirus protein sequences with an expect (E) value of ≤10−3 (see Table S2 in the supplemental material). These sequences were used to seed targeted de novo genome assembly using the PRICE metagenomic assembler (22), which produced a draft assembly of what appeared to be the complete viral genome of 34.5 kb (Fig. 4A). Sanger sequencing of the entire genome and rapid amplification of cDNA ends (RACE) were used to validate the assembly and to confirm that it contained the proper end sequences (Fig. 4B) (23–25). We used the Bowtie2 software alignment tool to retrospectively map paired-end reads to the draft genome assembly and found it to be well supported by deep sequencing data (26, 27). Coverage levels were even across the genome, and read pairs generally mapped concordantly (Fig. 4C and D). We found that the deep-sequencing-based assembly represented nearly the entire genome, except for 165 bases of 3′ untranslated region (UTR) sequence, which were determined by 3′ RACE. This genome sequence has been deposited in GenBank (see below).
FIG 4 .

Ball python nidovirus genome and replicase polyprotein organization. (A) A cartoon showing the genome organization of ball python nidovirus. Major open reading frames are indicated. RFS, position of putative ribosomal frameshift “slippery” sequence. (B) Position of Sanger sequencing PCR and RACE amplicons used to validate the NGS-based assembly (C) Concordance plot of read pairs mapping to assembly. Read pairs from snake no. 1 data set were mapped to the genome assembly using the Bowtie2 aligner. The inferred size of each library molecule is plotted. (D) Read coverage. Reads were aligned as in panel C, and the average number of bases supporting each position in 50-nt sliding windows of the alignment is indicated. The dip at approximately nt 5000 corresponds to a repeat-containing region with decreased mappability. (E) Conserved domains present in the replicase polyproteins (pp1ab). Domains were identified by searching the PFam database using the HMMER3 tool as described in the text and Table 2. Scale bars are indicated.
Comparative analysis.
We next performed comparative and phylogenetic analyses to better understand this virus sequence in the context of related species. The genome of this virus is presumed to be positive-sense single-stranded RNA (ssRNA), and is 33,452 nt long. This is 1,766 nt longer than the genome of the beluga whale coronavirus SW1 (accession no. NC_010646.1), which was previously the longest described RNA genome, at 31,686 nt (28). The genome contains 8 positive-sense open reading frames (ORFs) longer than 400 nt (Fig. 4A). There are also 6 ORFs longer than 400 nt internal to larger ORFs, which are of unknown significance. The overall pattern of ORFs matches the pattern observed for related viruses (1–3, 9, 10, 29). There are 2 large (17,412 and 6,966 nt) 5′ ORFs that are predicted to encode nonstructural proteins (see below), designated ORF1a and ORF1b. The next ORF, ORF2, is predicted to encode the “S” or spike glycoprotein. Then, there are 5 “3′ ORFs,” designated ORF3 to ORF6, that are predicted to encode additional structural proteins (see below). The 5′ and 3′ UTRs are 1,017 and 916 nt, respectively, and RACE analysis corroborated the prediction that the 3′ end of the genome is polyadenylated.
We used several tools to study the predicted proteome of this ball python virus. We used the BLASTp alignment tool to search for similar sequences in the NCBI nonredundant protein database (nr) (30). We also used the more sensitive HMMER3 and HHPRED hidden Markov model-based alignment and structure prediction software tools to detect more distant homologies and identify functional domains within the replicase polyprotein (31, 32) (http://hmmer.org). We used TMHMM to predict transmembrane domains and the NetNGlyc and NetOGlyc tools to predict N- and O-linked glycosylation sites in protein sequences (33, 34) (http://www.cbs.dtu.dk/services/NetNGlyc/).
In other nidoviruses, the replicase gene, comprised of ORF1a and ORF1b, encodes nonstructural proteins involved in viral genome replication and modulation of host cell activities (1–3, 9, 10, 29). ORF1a is translated to produce the pp1a polyprotein. A fraction of translating ribosomes undergo a −1 frameshift near the end of ORF1a, resulting in the continuation of translation though ORF1b and the production of the pp1ab polyprotein. This virus is likely to utilize a similar mechanism of translation. ORF1a and ORF1b are offset by a −1 frame difference and overlap by 52 nt. This overlapping region contains a putative “slippery” sequence (AAAAAAC) that may be the site of frameshifting (Fig. 4A; see also Fig. S2 in the supplemental material) (35, 36). In this virus, pp1a is predicted to be a 5,803-amino-acid (aa) protein with a molecular mass of 663 kDa, and pp1ab is predicted to be 8,108 residues long with a mass of 925 kDa (Fig. 4E). As is the case for other nidoviruses, it is likely that these polyproteins are proteolytically cleaved into multiple functional domains (37).
Nidoviruses are characterized in part by the presence and organization of a set of functional subunits within their pp1ab replicase polyproteins (1, 5, 9, 10, 38, 39). We first queried the snake virus pp1ab sequence against the NCBI nr database using the BLASTp tool. The best alignments produced were to pp1ab sequences from viruses in the Torovirinae subfamily, with bafinivirus sequences producing slightly higher scoring alignments than did torovirus sequences. The best alignment was to the pp1ab sequence of fathead minnow nidovirus (FHMNV) (see Fig. S3 in the supplemental material) (40). This alignment covered nearly the entire second half of pp1ab in 2 contiguous pieces from residues 3973 to 5060 and 5429 to 8029 of snake virus pp1ab. In these regions, the polyproteins shared 22% and 29% pairwise identities, respectively. Thus, the overall organization and domain content of the second half of the snake virus’s pp1ab protein resembled most closely those of bafinivirus replicase polyproteins (see Fig. S3).
We next used the HMMER3 sequence analysis tool (http://hmmer.org) to search the PFam database for particular pp1ab domains, and for the most part, these domains were present and organized as expected given the overall similarity to bafinivirus replicase polyproteins (Fig. 4E and Table 2; see also Fig. S3 in the supplemental material) (1, 9, 32, 41, 42). The expected domains present in snake virus pp1ab included a 3C-like protease located between two predicted transmembrane regions near the end of pp1a (TM2-Mpro-TM-3), an RNA-dependent RNA polymerase (RdRp), a helicase (Zn-Hel), a 3′ to 5′ exoribonuclease (ExoN), a nidoviral uridylate-specific endoribunuclease (NendoU), and a ribose-2′-O-methyltransferase found at the carboxy terminus of pp1ab (O-MT) (Fig. 4E and Table 2) (12, 37, 38, 43, 44). The replicase polyproteins of some taxa of nidoviruses encode a guanine-N7-methyltransferase (1, 9, 43, 45). As is the case for viruses in the Torovirinae subfamily, snake virus pp1ab appears to lack this enzyme. Functional experiments will be necessary to validate the cleavage patterns, biochemical activities, and roles in the virus life cycle of these predicted replicase proteins.
TABLE 2 .
Identification of functional domains in replicase polyprotein
| Domaina | Description | Position in replicase polyprotein (AA)b |
Basis for assignmentc | Alignment supportd |
|---|---|---|---|---|
| ADRP | ADP-ribose-1′′-phosphatase (macrodomain) | Not detected | HMMER/Pfam Macro | |
| PKinase | Protein kinase | 2340–2494 | HMMER/PFam PKinase | 2 × 10−6 |
| TM2 | Transmembrane domain | 4628–4737 | TMHMM | |
| Mpro | 3C-like main protease | 4864–4997 | HMMER/PFamPeptidase_S39 | 6 × 10−5 |
| TM-3 | Transmembrane domain | 5121–5296 | TMHMM | |
| RdRp | RNA-dependent RNA polymerase | 6200–6513 | HMMER/PFam RdRP_1 | 1 × 10−12 |
| Zn-Hel | Zn-finger/5′-to-3′ helicase | 6899–7178 | HMMER/PFam Viral_helicase1 | 8 × 10−14 |
| ExoN | 3′-to-5′ exoribonuclease | 7262–7423 | HMMER/PFam NSP11e | 1 × 10−7 |
| N7 | Guanine-N7-methyltransferase | Not detected | HMMER/PFam NSP11e | |
| NendoU | Nidoviral uridylate-specific endoribonuclease | 7709–7822 | HMMER/PFam EndoU_bacteria | 8 × 10−3 |
| O-MT | Ribose-2′-O-methyltransferase | 7841–8081 | HMMER/PFam NSP13 | 3 × 10−28 |
Domain nomenclature according to reference 1.
Coordinates in amino acids of region within the BPNV pp1a or pp1ab polyprotein as defined by alignment boundaries. Closely juxtaposed TMHelix domains were considered to be part of a single TM domain.
The particular Pfam domain used to identify (or to exclude the presence of) each domain is indicated. Note that Pfam domain nomenclature does not necessarily correspond to current preferred nidovirus nomenclature.
The alignment’s E value from an HMMER3 search of the Pfam database.
The Pfam “NSP11” domain corresponds to the coronavirus nsp14 protein, which contains the ExoN and guanine-N7-MT domains.
One distinguishing feature of the snake virus replicase polyprotein is the apparent lack of an ADP-ribose binding “macro” domain that is present in nidoviruses in the family Coronaviridae (Fig. 4E and Table 2) (46–48). The activity of this domain is not essential for replication in vitro but may help counteract the innate immune response in vivo (49, 50). At approximately the same position of snake virus pp1a is a predicted protein kinase (PFam PKinase domain) that is not present in any other nidovirus as determined by HMMER analysis (HMMER3 E value, 1.5 × 10−6) (Fig. 4B and Table 2). This prediction is also supported by BLASTp analysis, with the best alignments from a search of the NCBI nr database with pp1ab residues 2300 to 2500 being to cellular serine/threonine protein kinases (lowest E value, 3 × 10−5).
In nidoviruses, the ORFs 3′ to the replicase genes encode structural and accessory proteins. The number and organization of these genes vary widely across nidovirus taxa, and in many cases there is little or no detectable similarity between sequences of more distantly related species (1, 9).
Nidovirus S proteins are involved in receptor binding and membrane fusion via their N- and C-terminal S1 and S2 subunits (1, 2, 51, 52). The best alignment from a BLASTp search of the snake virus protein against the nr database was to thrush coronavirus HKU12-600 spike glycoprotein (YP_002308497 [53]). This alignment had 18% pairwise identity over 333 aa in the C-terminal third of the protein, i.e., in the S2 membrane fusion domain (52). The putative S1 receptor-binding domain of this protein possesses no clear similarity to other proteins by BLAST or HMMER analysis (residues 25 to 625 queried). Consistent with its putative identity as a spike glycoprotein, this protein is predicted to contain several transmembrane (TM) domains, including one characteristically near the C terminus, and is predicted to be glycosylated (see Fig. S4 in the supplemental material).
The lipid bilayers of nidoviruses contain one or more proteins in addition to S, including the membrane (M) protein present in some nidoviruses (1, 2). The protein encoded by ORF4 is likely to be a homolog of the M protein of other large nidoviruses. This prediction is based on several attributes of the deduced protein sequence. First, the size of the snake virus protein (215 aa) is just under the range for other Coronaviridae M proteins (216 to 268). Second, the protein contains 3 predicted TM domains in the N-terminal half, a characteristic feature (see Fig. S4 in the supplemental material). Third, the protein possesses sequence similarity to the putative M protein of the white bream virus nidovirus that is detectable by BLASTp analysis of the nr database. The sequences are 21% identical over 116 aa, and the E value of this alignment is slightly lower than those of alignments to various bacterial protein sequences. Whether this putative M protein performs the same functional role as its distant relatives remains to be validated experimentally.
The 152-aa protein predicted to be encoded by ORF5a is similar to nucleocapsids (N) from other nidoviruses. This similarity is detectable by BLASTp, with the best such alignments from a search of the NCBI nr database being to porcine reproductive and respiratory syndrome virus (PRRSV) N proteins (22% pairwise identity over 125 aa) and by HMMER3 analysis, which identifies the Arteri_nucleo PFam domain in this sequence (E value, 5.6 × 10−9). Although the best local alignments from a BLASTp search of nr are to arterivirus N sequences, by pairwise global alignments the ORF5a-encoded N sequence is more similar to bafinivirus and torovirus N sequences (e.g., 24% pairwise identity to white bream virus N) than to arterivirus N sequences (e.g., 18% pairwise identity to PRRSV N). This protein is predicted to be basic, with an isoelectric point of 10.6, and to contain no transmembrane domains, consistent with a putative function as an RNA-binding nucleocapsid protein.
ORF3, ORF5b, and ORF6 encode proteins with no clear homology to other nidovirus proteins or to other viral or nonviral proteins. These proteins are all predicted to contain transmembrane domains and to contain multiple glycosylation sites (see Fig. S4 in the supplemental material). Along with S and M, these proteins are likely additional structural components of the virus particle. In some phyla of large nidoviruses, one of the 3′ ORFs encodes a hemagglutinin-esterase (HE) protein. The snake virus does not appear to encode an HE homolog. Similarly, no putative homolog of the small envelope (E) protein encoded by some nidoviruses was identified for this virus. E homologs have been identified in coronaviruses and arteriviruses but not in other nidovirus lineages (1).
Molecular characterization of subgenomic RNAs.
Nidoviruses generate subgenomic mRNA species (sgRNAs) that are competent for the translation of the downstream 3′ ORFs. In most of these viruses, these subgenomic mRNAs share 5′ and 3′ terminal sequences with the genomic RNA and are transcribed from minus-strand templates that are produced by a process termed discontinuous RNA synthesis (1, 2, 54). In fact, the prefix “nido” refers to the nested nature of these RNA species. In our deep-sequencing data sets, we detected pairs of reads where the first read mapped to the 5′ UTR and the second read mapped near the beginning of the downstream ORFs, suggesting that this virus might also generate sgRNA species by a similar process.
In order to characterize these RNAs, we performed PCR using primer pairs separated by more than 23 kb of genomic sequence but that would amplify putative sgRNAs (Fig. 5A) (55). PCR and sequencing of amplicons revealed sgRNA species for each of the predicted 3′ ORFs, except for ORF5a and -5b, which appear to share a single mRNA. ORF5a and -5b overlap by 251 nt, and their start codons are separated by 208 nt (Fig. 5B and C). The sgRNAs share a 5′-terminal “leader” sequence (approximately nt 1 to 170 of genome) (Fig. 5C). Short regions of homology are evident at the junction points of the sgRNAs (Fig. 5C). These data are consistent with this virus using a strategy of discontinuous RNA synthesis and translation similar to that employed by other nidoviruses (54).
FIG 5 .

Analysis of subgenomic RNA species. (A) A cartoon showing the location of primers used to amplify regions of sgRNAs. Twenty-three kilobases have been removed for clarity. (B) Agarose gel electrophoresis analysis of PCR products obtained from reactions using the indicated primer pairs. Positions of molecular size standards (in bp) are indicated. (C) Sequences of PCR products spanning the junctions between upstream ORF sequences and 5′ UTR (leader) sequence. The top sequence in each triplet is the sequence of the genomic RNA (gRNA) beginning at base 150; the middle sequence is from the amplicons in panel B, corresponding to the indicated subgenomic mRNAs; the bottom sequence is that of the genomic RNA beginning at the indicated base. Regions of homology and putative start codons are underlined. For the ORF5a/b triplet, the start codons of both ORFs are indicated, and 180 nt of intervening sequence has been removed.
Disease association and tissue tropism.
Using this complete genome sequence as a reference, we assessed whether other case and control samples contained sequences from similar viruses as determined by BLASTn (30). Nearly identical sequences were present in the fully filtered data sets of all 8 sequenced diseased snakes (see Table S2 in the supplemental material). The read coverage in the other data sets was not sufficient to generate complete genome assemblies, but the average pairwise nucleotide identity of reads aligning was ≥95% in all cases, suggesting that other diseased snakes were infected by closely related strains of the virus (see Table S2). Conversely, we never observed similar sequences in 57 data sets from tissues from unaffected ball pythons and from other snakes that were collected for other studies. These control data sets corresponded to various tissues (mainly not lung) from a number of snake species, including ball pythons (n = 2), other python species (n = 3), boa constrictors (n = 39), other boa species (n = 10), black-tailed rattlesnakes (n = 1), and California king snakes (n = 2).
To independently confirm the metagenomic sequencing results, we used quantitative RT-PCR (qRT-PCR) to measure viral RNA levels in these samples. Viral RNA was detected in all affected animals but not in control animals (Fig. 6A). Thus, detection of viral RNA by both metagenomic sequencing and qRT-PCR perfectly correlated with disease status, though larger studies will be necessary to confirm this association.
FIG 6 .

Detection of ball python nidovirus RNA is associated with clinical manifestation. (A) Relative levels of viral RNA were measured by qRT-PCR in case and control lung samples. Primers targeted ORF1a or ORF2 (S) sequences. Each row represents values from a single case or control animal of the indicated species. Levels were normalized to levels of cellular GAPDH mRNA and are represented as a heat map. Corallus annulatus and boa constrictor samples have been described previously (59). A polymorphism in the S sequence primer-binding site of snake 8 decreased amplification efficiency. (B) Relative levels of viral RNA in different tissues from infected snakes. For each snake, values were calculated and quantified as in panel A. Lung from a healthy control ball python served as a negative control.
Data sets did not contain consistent evidence of other possible bacterial or viral pathogens. Data sets contained retrovirus-like sequences most similar to that of Python molurus endogenous retrovirus (ERV) sequences (56). These were present in nearly every case and control Python regius data set and most likely derived from ball python ERVs. Also, as with all metagenomic data sets from nonsterile sites, bacterial sequences were present. However, no particular bacterial taxon was associated with cases, in contrast to the nidovirus sequences, which were perfectly associated. Nevertheless, from these sequencing data sets alone, it is not possible to formally rule out a role in disease causality by other viral or bacterial pathogens.
We next sought to determine the distribution of viral load within the tissues of individual infected snakes. Frozen samples from multiple tissues were available for 4 snakes. We extracted RNA from these tissues and used qRT-PCR to quantify viral RNA relative to expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (Fig. 6B). Consistent with the pathology observed, viral load was consistently highest in respiratory tract tissue. Viral RNA levels were at approximately the same level in intestine-derived RNA from the single snake for which this tissue was available (no. 11), a finding consistent with respiratory/enteric tropism of many viruses in the Coronaviridae family (7, 8). Viral RNA was also detectable in other tissues (liver, kidney, heart, spleen, and brain) at levels that were 3 to 5 orders of magnitude lower than in the lung. While these results support the notion that the respiratory tract is the primary location of viral replication, consistent with the disease pathology, additional systematic collection and analysis of samples from diseased animals will be necessary to definitively characterize tissue tropism for this virus.
Phylogenetic analysis.
We performed phylogenetic analyses to understand the evolutionary relationship between this and other nidoviruses. We obtained protein sequences from 33 representative species, and created multiple sequence alignments of the relatively conserved protease, RdRp, and helicase domains of pp1ab (see Table S3 and Fig. S5 in the supplemental material) (42). Bayesian and maximum-likelihood (ML) phylogenies based on individual domain and concatenated alignments had nearly identical topologies (Fig. 7; see also Fig. S6). In these phylogenies, the 5 major groups of nidoviruses (Torovirinae, Coronavirinae, Roniviridae, Mesoniviridae, and Arteriviridae) formed well-separated branches. Ball python nidovirus (BPNV) formed a monophyletic clade with the viruses in the Torovirinae subfamily of the Coronaviridae family. The snake virus is approximately equally distant from the two genera in the subfamily, the toroviruses, which infect mammals, and the bafiniviruses, which infect ray-finned fish (Fig. 7; see also Fig. S6). We noted in the protease alignment that two putative active site motifs previously identified by Ulferts et al. (57), one containing a histidine and a GX[S/C] G motif, aligned in all sequences. A third putative Asp/Glu-containing motif shifted drastically in alignments, even within Coronavirinae. Nevertheless, the topology of the tree generated using the protease alignment was in close agreement with other phylogenies. This analysis did not find a monophyletic grouping of the Torovirinae and Coronavirinae subfamilies of the family Coronaviridae. This paraphyly was found to be significant (P < 0.01) by log likelihood (Shimodaira-Hasegawa) testing (58).
FIG 7 .
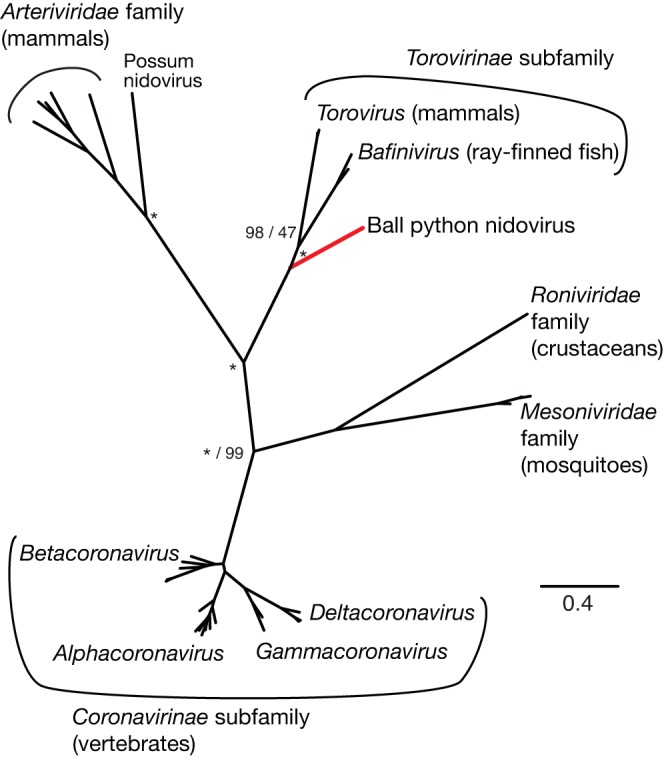
Phylogeny of conserved replicase subunits of representative nidoviruses. Unrooted Bayesian phylogeny based on concatenated multiple sequence alignments of conserved regions of RdRp and helicase domains of replicase polyproteins. At select nodes, Bayesian posterior probabilities and ML bootstrap values for clusters are given to the left and right of forward slashes. Nodes with 100% support are marked with asterisks. The five major nidovirus families and subfamilies are indicated, as are select genera and the currently uncategorized BPNV and possum nidovirus taxa. Known host phyla are indicated in parentheses. The branch leading to BPNV is highlighted red for emphasis.
Attempted isolation of virus in culture.
We attempted to isolate the virus in several snake cell lines, including the boa constrictor JK cell line and the viper VSW and VH-2 lines (59, 60). We prepared an inoculum from frozen infected tissue and applied it to these lines. Viral RNA was detectable in the inoculum but disappeared from the culture supernatants as media were replaced over the course of 14 days, and no cytopathic effects were observed. It is possible that different cells, alternate culture conditions, or inoculum from freshly harvested infected tissue would permit replication.
DISCUSSION
In this report, we describe a severe respiratory disease affecting ball pythons, and we have identified a nidovirus as a candidate etiologic agent by means of association. We collected cases from around the United States and performed a variety of pathological and diagnostic analyses. Respiratory tract pathology was consistent with a viral etiology, as was the presence of virus-like particles in lung epithelium of affected snakes. Using unbiased deep sequencing, we identified and assembled the genome of a previously uncharacterized nidovirus. This virus shares several attributes with other nidoviruses but has several distinguishing characteristics, including its record-setting genome size and reptile host.
The findings presented here raise a number of questions related to the biology of this virus and its relationship to disease. Foremost among these is whether this virus causes the observed respiratory pathology. Several observations suggest a causal relationship. First, detection of viral RNA correlates with clinical signs. Second, the virus load is most profound in the respiratory tract, the site of disease. Third, other animal nidoviruses cause severe respiratory disease. Nevertheless, formal evidence of disease causality remains to be demonstrated. Although virus isolation attempts have yet to succeed, experimental infections using material prepared directly from infected tissues could fulfill Koch’s postulates.
The evolutionary relationship between ball python nidovirus and other nidoviruses is partially clear. In analyses based on the highly conserved replicase subunits, this virus clearly clusters with viruses in the subfamily Torovirinae, the toroviruses found in mammals and the bafiniviruses found in ray-finned fish, with 100% Bayesian posterior probability and ML bootstrap support (Fig. 7; see also Fig. S6 in the supplemental material). This suggests that at least for the core replicase gene, the snake virus shares a common evolutionary origin with these mammal and fish viruses. Although it has not been proven that the virus sequence corresponds to the particles observed in electron micrographs, the ultrastructural appearance and size of the observed virus are notably similar to those of virions of white bream virus in the genus Bafinivirus (61, 62). We propose that ball python nidovirus establish a new genus in the Torovirinae named Barnivirus, for bacilliform reptile nidovirus, a naming convention borrowed from the genus Bafinivirus.
While some studies have found support for the monophyly of Torovirinae and Coronavirinae, in accordance with the current International Committee on Taxonomy of Viruses (ICTV) status of Coronaviridae, our analysis did not find phylogenetic support for this grouping, in agreement with other recent analyses (1, 40, 42, 63). The paraphyly of Coronaviridae was supported by log likelihood testing. The availability of a more complete representation of existing species for comparison results in greater phylogenetic resolution (64). Significant errors can occur in phylogenetic analyses due to incomplete taxum sampling, even if very large sequence length is assessed (65). This further emphasizes the need for exploration of viral diversity and increased sampling of the Nidovirales.
Current standards in herpetoculture encourage disease transmission and may foster evolution of increased pathogen virulence. Captive snake breeding operations typically operate at high stocking densities, and breeders commonly attend trade shows, where animals from different sources are juxtaposed. In addition, animals from geographically and ecologically diverse areas are commonly imported and mixed with minimal quarantine. These practices increase pathogen exposure and lower barriers to transmission. The outcome is evident in the case of farmed turtles, which typically have very high Salmonella carriage rates, in contrast to the low prevalence in wild turtle populations (66–68). It is also possible that these practices select for increased virulence, as has been the case for feline calicivirus (FCV), which has independently evolved multiple times from a pathogen that causes a relatively benign upper respiratory disease to one that causes hemorrhagic disease with high mortality (69). This increase in FCV virulence has been documented only in high-density environments, such as shelters. Surveillance of wild ball pythons in Africa will shed light on whether a similar phenomenon has occurred here. Further investigation of the pathogens of reptiles and improved biosecurity practices and disease surveillance of the herpetoculture industry are indicated.
In addition to disease causality, a number of questions related to the biology of ball python nidovirus and to the epidemiology and natural history of this disease remain open. The precise host range of this virus remains undefined. It is not clear that ball pythons are the primary natural host for this virus, nor whether it can replicate or cause disease in other snakes, other reptiles, or other animals. The routes of transmission for this virus remain unknown. Additionally, the prevalence of this disease is yet to be determined, although it is apparently widespread, since cases were collected from geographically disparate locations in the United States from 2006 to 2013. Additional studies will help to answer these central questions and uncover additional details about the biology of this putative pathogen. The current study is another example where the application of genomic techniques to the study of infectious disease in reptiles and other less well studied organisms has identified new and unexpected relatives of human pathogens (59, 70).
MATERIALS AND METHODS
Collections, animals, and pathological evaluations.
Carcasses and tissues of 9 ball pythons (BP) with clinical signs of respiratory tract disease (stomatitis, pharyngitis, rhinitis, glossitis, tracheitis, bronchitis, and pneumonia) from seven collections in Florida, Oklahoma, Pennsylvania, Texas, and Wisconsin were submitted for necropsy and light microscopic evaluation. Transmission electron microscopy (TEM) and/or molecular diagnostics were performed on selected cases.
(i) Collection 1.
Two snakes from a private collection in Texas (BP1 and -2) were submitted in 2007 for pathological evaluations. The owners reported that affected snakes exhibited acute signs of respiratory disease characterized by audible wheezing along with a clear mucoid nasal discharge, notable “cough-like” forced expiration, and excessive amounts of foamy mucus in the oral cavity. BP1 was euthanatized and immediately necropsied. BP2 was found dead and was refrigerated for 2 days prior to necropsy. Lung was collected from both snakes, and portions were frozen or placed in neutral buffered 10% formalin (NBF) and processed for light microscopy. Lung was sectioned at 6 µm and stained with hematoxylin and eosin (H&E). A portion of lung of BP1 that was fixed in NBF was cut into 1-mm cubes and transferred to 4% paraformaldehyde–2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.24, and processed for TEM (see below). Molecular diagnostics were performed on frozen tissues from two snakes (snakes 3 and 4) from a second collection in Texas (collection 2), but they were negative and are not discussed further in this report.
(ii) Collection 3.
Three ball pythons (5, 6, and 7) from a private collection in Wisconsin were submitted to a veterinarian due to signs of respiratory disease. Ball python 5 was found dead and submitted for necropsy. The following tissues were collected, fixed in NBF, and processed for light microscopy: lungs, kidney, liver, stomach, spleen, and ovary with oviduct. Ball pythons 6 and 7 had acute onset of respiratory signs over a 2- to 3-day period characterized by an increase in respiratory rate and effort. There also was elevation of the cranial third of the snake’s body (from the ground), ultimately resulting in opisthotonos (stargazing). A decision was made to euthanatize and necropsy BP6 and -7. The following tissues were collected from each snake, fixed in NBF, and subsequently embedded in paraffin: trachea, lungs, heart, esophagus, stomach, intestines, liver, pancreas, kidneys, oviduct, brain, spinal cord, nasal sinuses, vomeronasal organ, and skin. The paraffin-embedded tissues were sectioned at 6 µm, processed routinely for light microscopy, and stained with hematoxylin and eosin. Sections of the vomeronasal organ of one case were stained with Ziehl-Neelsen acid-fast stain, which was negative for acid-fast bacteria; methenamine silver staining was negative for yeast and hyphae. A portion of lung from BP6 was fixed in 4% paraformaldehyde–2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.24, and processed for TEM (see below).
(iii) Collection 4.
A full necropsy and pathological evaluation were performed on a 1-year-old female ball python (BP8) from a private collection in Texas that died after a history of respiratory disease for 1 week and respiratory distress for 2 days. The snake was from a group of four snakes. There was a history of respiratory disease in the group, and previously one other snake had died. The snake exhibited marked dyspnea with crackles and wheezes audible without a stethoscope. Other clinical findings included hypersalivation and a swollen, erythematous glottis. Culture of a tracheal swab isolated Shewanella putrefaciens. No response to treatment with the antimicrobial enrofloxacin was noted. The following tissues were analyzed: lungs, kidney, liver, stomach, small intestine, large intestine, heart, brain, oral cavity, pharynx, nasal cavity, trachea, eye, spleen, and ovary with oviduct were placed in NBF and processed routinely for light microscopy. Six-micrometer sections of paraffin-embedded tissues were stained with hematoxylin and eosin.
(iv) Collections 5 and 6.
Two BPs, one from private collection 5 in Florida (BP9) and the other from private collection 6 in Oklahoma (BP10), were submitted to an exotic animal practice for evaluation due to signs of respiratory disease and anorexia. BP9 was part of a small breeding collection where very few sick animals were previously observed. BP9 was euthanized, and the following tissues were collected during necropsy, fixed in NBF, and processed for light microscopy: trachea, lung, kidney, salivary gland, liver, brain, and small intestine. Portions of trachea, liver, and lung were collected, frozen at −20°C, and subsequently submitted for molecular diagnostics. Six-micrometer sections of paraffin-embedded tissues were stained with hematoxylin and eosin.
The second case (BP10) was a 5-year-old female common BP from a large collection (500-plus) of ball pythons in Oklahoma. Prior to 2009, no significant health issues were recognized in this collection. Starting in 2009 several ball pythons showed signs of respiratory disease, and from 2010 to early 2011, three ball pythons died with clinical signs of respiratory disease. Ball python 10 had signs of respiratory disease and was euthanized for pathological evaluation. The following tissues were collected, fixed in NBF, and processed for light microscopy: brain, kidney, large intestine, liver, lung, small intestine, and trachea. Paraffin-embedded tissues were sectioned at 6 µm and were stained with hematoxylin and eosin. Portions of lung were collected, frozen at −20°C, and subsequently submitted for molecular diagnostics.
(v) Collection 7.
Ball python 11 was a 6-year-old adult from a private collection in Pennsylvania. The owner had about 74 snakes in total, all of which were ball pythons. Seven snakes in close proximity to one another from his collection became sequentially ill with clinical signs of respiratory disease, including dyspnea and open-mouth breathing, All ill snakes were immediately quarantined in a separate room from the general population. Of the 7 snakes that became ill, 6 died within a few days of first manifesting signs of respiratory disease. One of the dead snakes was submitted to a private practitioner for necropsy. Portions of trachea, lung, kidney, liver, integument, and small intestine were fixed in NBF and submitted to a pathology service processed for light microscopy. Paraffin-embedded tissues were sectioned at 6 µm and were stained with hematoxylin and eosin, alcian blue, and PAS. Portions of lung, liver, and kidney were collected, frozen at −80°C, and subsequently submitted for molecular diagnostics.
Ethics statement.
No live animals were used in this study. Tissues and carcasses were from animals from private collections. Snakes either died or were euthanized by the attending veterinarian due to the severity of clinical signs and poor prognosis as described above. Tissues or carcasses were collected during necropsy by attending veterinarians and submitted for the purposes of this study, with the owners’ consent. The acquisition of fresh, frozen, and formalin-fixed tissue samples was authorized under University of Florida Institutional Animal Care and Use Committee Protocol A116.
Transmission electron microscopy.
Formalin-fixed lung samples from BP1 and BP6 were cut into 1-mm cubes and transferred to 4% paraformaldehyde–2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.24. Fresh lung from BP6 was also cut into small cubes and fixed directly in 4% paraformaldehyde–2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.24. Fixed tissues were processed with the aid of a Pelco BioWave laboratory microwave (Ted Pella, Redding, CA, USA). The samples were washed in 0.1 M sodium cacodylate buffer, pH 7.24, postfixed with buffered 2% OsO4, water washed, and dehydrated in a graded ethanol series, 25%, 50%, 75%, 95%, and 100%, followed by 100% acetone. Dehydrated samples were infiltrated in graded acetone-Spurrs epoxy resin (Electron Microscopy Sciences, Hatfield, PA) 30%, 50%, 70%, and 100% and cured at 60°C. Cured resin blocks were trimmed, thin sectioned (70 nm), and collected on Formvar-coated copper grids, poststained with 2% aqueous uranyl acetate and Reynolds lead citrate. Sections were examined with a Hitachi H-7000 TEM (Hitachi High Technologies America, Inc., Schaumburg, IL), and digital images were acquired using a Veleta 2k by 2k camera and iTEM software (Olympus Soft-Imaging Solutions Corp., Lakewood, CO).
Clinical molecular diagnostics.
Portions of fresh frozen lung specimens were selected from several of the above cases for identification of nucleic acid sequences for several viruses implicated as causative agents of respiratory tract disease of snakes. RNA was extracted from lung of BP1 and tested by using previously described consensus PCR methods for the genus Ferlavirus in the Paramyxoviridae, the Rhabdoviridae, and the genus Orthoreovirus in the Reoviridae (71–73). Additionally, RNA was extracted from portions of lungs of BP6 and -7 and tested for gene sequences for members of the family Rhabdoviridae and Filoviridae. For molecular diagnostic detection of filoviruses (Marburg viruses and Ebola viruses), RNA was sent to the Viral Special Pathogens Branch, Centers for Disease Control and Prevention. Two independent panfilovirus RT-PCR assays were utilized as previously described (74, 75) using SuperScript III reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. Briefly, RNA was reverse transcribed for 30 min at either 45°C or 50°C, respectively, and then denatured for 2 min at 94 to 95°C. For RNAs amplified according to the protocol described elsewhere (74), reaction mixtures were thermocycled 40 times at 94°C for 15 s, followed by successive incubations at 45°C and 68°C for 30 s each. For RNAs amplified according to the previously described protocol (75), reaction mixtures were treated identically except that the annealing and extension temperatures were 53°C and 72°C degrees, respectively. Positive- and negative-control reactions were carried out in parallel to validate the performance of the testing.
Library preparation and sequencing.
Total RNA was extracted from frozen tissues for sequencing library generation and molecular analysis. RNA was extracted as previously described (59). Two hundred fifty nanograms of RNA was added to 20 µl RT reaction mixtures containing 200 pmol oligonucleotide MDS-286 (random hexamer), 1× reaction buffer, 5 mM dithiothreitol, 1.25 mM (each) deoxynucleoside triphosphates (dNTPs), and 100 U SuperScript III (Life Technologies). The sequences of all oligonucleotides are listed in Table S1 in the supplemental material. Reaction mixtures were incubated for 60 min at 42°C and then for 15 min at 70°C. Then, 1 U RNase H (NEB) diluted in 5 µl 1× RT buffer with 100 pmol MDS-286 was added to reaction mixtures, which were incubated for 30 min at 37°C and then 94°C for 2 min. Then, primer extension was used to convert single-stranded cDNA to double-stranded DNA (dsDNA): 2.5 U Klenow DNA polymerase (3′ to 5′ exo-; NEB) diluted in 5 µl 1× RT buffer with 2 mM (each) dNTP was added to reaction mixtures, which were incubated at 37°C for 15 min. DNA was purified using DNA clean and concentrator columns (CC-5; Zymo Research) according to the manufacturer’s protocol and using a 3:1 ratio of binding buffer/sample. The DNA concentration was determined fluorometrically, and 10 ng was used as a template in 10 µl Nextera transposon reactions, which contained 1× buffer and 0.5 µl transposase mix (Illumina), which were incubated at 55°C for 10 min and then placed on ice. Tagmented DNA was cleaned with CC-5 columns (4:1 buffer ratio), eluted in 20 µl H2O, and used as a template in PCRs that added full-length bar-coded adapters to library molecules. PCR mixtures contained 1× Kapa real-time library amplification PCR mix (Kapa Biosystems), 0.33 µM (each) primers MDS-143 and -445 and 0.017 µM (each) adapter1 and adapter2 bar-coded primers, and 5-µl library template. Thermocycling conditions were 72°C for 3 min, followed by 95°C for 30 s, and then 5 cycles of 98°C for 10 s, 63°C for 30 s, and 72°C for 3 min. Relative concentrations of libraries were then quantified in quantitative PCR (qPCR) reaction mixtures containing 10 mM Tris, pH 8.6, 50 mM KCl, 1.5 mM MgCl2, 5% glycerol, 0.08% IGEPAL CA-630, 0.05% Tween 20, 0.2 mM (each) dNTP, 1× Sybr green (Life Technologies), and 0.5 U Taq polymerase, and 0.1 µM (each) primers MDS-143 and -445. Equivalent amounts of each sample were then pooled, and the pool was cleaned with a DNA CC-5 column according to the manufacturer’s protocol. The pooled libraries were then size selected (350 to 450 nt) using a LabChip XT device (Caliper), and purified again using a CC-5 column. Size-selected pooled libraries were subjected to a final round of amplification in PCR mixtures containing 1× Kapa real-time library amplification mix, 200 pmol each MDS-143 and -445, and 5 µl library template. Reaction conditions were 98°C for 1 min and 16 cycles of 98°C for 15 s, 63°C for 30 s, and 72°C for 3 min. The pooled libraries were finally purified using a DNA CC-5 column and quantified spectroscopically and by using the Illumina library quantification kit (Kapa Biosystems). Sequencing was performed on an Illumina HiSeq 2500 instrument.
Sequence analysis.
The sequence analysis pipeline was similar to that employed previously (59, 76). Low-quality sequences and low-complexity sequences (defined as having a ratio of the length of the Lempel-Ziv-Welch compressed sequence to the uncompressed length of less than 0.46) were removed from further analysis. The first 5 bases of each sequence were trimmed, as was the last base. The CD-HIT sequence clustering tool was then used to collapse reads with >98% global pairwise identity (77). Host-derived sequences were then filtered, first by using the BLASTn alignment tool (version 2.2.25+ [30]) to query a database of snake ribosomal and mitochondrial sequences and then by using the Bowtie2 alignment tool (version 2.0.0-beta7 [26]) to query databases composed of draft assemblies of the Burmese python and boa constrictor genomes (20, 21). Sequences aligning with an expect value of less than 10−12 (BLASTn) or with –local mode alignment scores greater than 86 (Bowtie) were filtered. Similarly, sequences that aligned to the Illumina adapter sequences (see Table S1 in the supplemental material) or to PhiX-174 control sequence were removed. This filtering removed on average 98% of sequences (see Table S2). The remaining sequences were searched against custom databases of viral protein sequences using the BLASTx alignment tool. Sequences aligning to any viral protein sequence with an expect value of less than 0.25 were further examined. False positives were reduced by using BLAST to align putative viral sequences to the NCBI nonredundant nucleotide (nt) and protein (nr) databases. Only sequences whose best hit and whose pair’s best hit were still to viral sequences were retained. The PRICE de novo targeted genome assembler was used to generate initial contiguous virus sequences (22). The reference assembly (NCBI accession no. KJ541759) was assembled using reads from a single snake (no. 2); other animals lacked sufficient coverage to assemble the complete genome. To validate this assembly and generate coverage and pairwise identity data, reads were aligned to Sanger validated assemblies using the BLASTn algorithm. Sequencing data have been deposited in the UCSF Integrated Data Repository.
Sanger sequencing and RACE.
The sequencing-based virus genome assembly was validated using RACE and Sanger sequencing. PCR mixtures contained 1× iProof High-Fidelity DNA polymerase mix (Bio-Rad), 0.5 µM (each) primer, and 2 µl diluted cDNA, prepared as described above. Thermocycling conditions were 98°C for 30 s and then 40 cycles of 98°C for 10 s, 60°C for 20 s, and 72°C for 1 min, with a final 5-min 72°C extension. Primers were designed to tile across the assembly in overlapping ~1,200-bp amplicons (Fig. 4B) (78). Amplicons were verified on agarose gels and sequenced directly. In some cases, amplicons were gel purified with the Zymoclean kit (Zymo Research), cloned into the pCRBlunt vector (Invitrogen) according to the manufacturer’s protocols, and sequenced. 5′ and 3′ RACE was used to validate end sequences, essentially as described previously (24, 25), with primers listed in Table S1 in the supplemental material. Multiple RACE amplicons were cloned and sequenced as described above. The complete and validated genome sequence has been deposited with the NCBI (see below). Subgenomic RNA junctions were amplified and sequenced similarly.
Quantitative PCR.
To measure relative levels of viral RNA in samples, RNA was extracted and reverse transcribed as described above for library preparation. cDNA was diluted 1:10 in 10 mM Tris–0.1 mM EDTA, pH 8. qPCR reaction mixtures contained 10 mM Tris, pH 8.6, 50 mM KCl, 1.5 mM MgCl2, 5% glycerol, 0.08% IGEPAL CA-630, 0.05% Tween 20, 0.2 mM (each) dNTP, 1× Sybr green (Life Technologies), 0.5 U Taq polymerase, and 0.25 µM (each) primer (see Table S1 in the supplemental material). Viral RNA levels were normalized to levels of cellular GAPDH (see Table S1). Reaction efficiencies were determined using serial dilutions of templates (79).
Tissue culture and virus isolation.
Cells were cultured as described previously (59). To prepare inocula, frozen tissues were thawed on ice, and 1-g portions were minced with scalpels and disrupted by Dounce homogenization in ice-cold minimum essential medium (MEM) (Gibco) supplemented with 25 mM HEPES buffer, pH 7.4. Homogenate was clarified by centrifugation at 10,000 × g for 1 min and filtered through a 0.45-µm filter. Filtrate was diluted 1:10 in MEM plus HEPES and added to cultures of 80% confluent cells. Culture medium was harvested and replaced after 20 h and every 2 to 3 days thereafter for an additional 14 days and stored at −80°C until further processing. RNA was isolated from clarified culture supernatant using the ZR viral RNA kit (Zymo Research), according to the manufacturer’s protocol. RNA was reverse transcribed and analyzed by qRT-PCR as described above.
Comparative and phylogenetic analyses.
ORFs in the viral genome were identified using the Geneious software program (version 6.1; BioMatters). Homologs of predicted protein sequences were detected using the BLASTp tool (version 2.2.25+) to search the NCBI nonredundant protein database (nr) (30), and by using the HMMER3 alignment tool (version 3.1b1) to search the PFam database (http://hmmer.org) (32). For HMMER analysis of replicase polyprotein, sequence was split into 400-aa sliding windows offset by 60 aa. For sequences with no detectable similarity by those measures, the HHpred homology detection and structure prediction tool (version 2.0) was also used (31). TMHMM (version 2.0) was used to predict transmembrane domains. The NetNGlyc (1.0) and NetOGlyc (4.0) tools were used to predict N- and O-linked glycosylation sites in protein sequences (33, 34) (http://www.cbs.dtu.dk/services/NetNGlyc/). In cartoons depicting sequence features, all features are to scale and accurately positioned.
Sequences from a set of 33 representative nidoviruses based on a previously published analysis were downloaded (see Table S3 in the supplemental material [42]). Conserved protease, RdRp, and helicase domains in replicase polyproteins were identified using HMMER3 and BLAST alignments. Multiple sequence alignments of these domains were created using MAFFT version 7 with default parameters (see Fig. S5) (80). Amino acid substitution models were evaluated for each region using corrected Aikake information criteria in the software program ProtTest 3.2.2 (81). LG was identified as the best-fit model (82, 83). Bayesian analyses of each alignment were performed using the software program MrBayes 3.2.2 (84) on the CIPRES server (85), with gamma-distributed rate variation and a proportion of invariant sites, and amino acid models were also assessed by model jumping. The first 10% of 1,000,000 iterations were discarded as a burn-in, based on examination of trends of the log probability versus generation. Two independent Bayesian analyses were run to avoid entrapment on local optima.
Maximum-likelihood (ML) analysis was performed using the RAxML software tool on the CIPRES server (86), with gamma-distributed rate variation and a proportion of invariant sites. The amino acid substitution model with the best fit using ProtTest was selected. Gill-associated virus was used as an outgroup. Bootstrap analysis was used to test the strength of the tree topology (1,000 resamplings) (87). Numbers of bootstrap replicates were determined using the stopping criteria of Pattengale et al. (88).
To test for paraphyly of the Coronaviridae, likelihood ratio testing was conducted (58). Trees were constrained to Coronaviridae monophyly (including ball python nidovirus with the Torovirinae) and compared by the Shimodaira-Hasegawa test to the best unconstrained tree identified in RAxML.
Nucleotide sequence accession number.
The complete and validated sequence assembled using reads from snake no. 2 has been deposited with the NCBI under accession no. KJ541759.
SUPPLEMENTAL MATERIAL
Oligonucleotides used in this study
High-throughput sequencing summary
NCBI accession numbers of sequences of representative nidoviruses used for phylogenetic analyses
Representative microscopic lesions in upper respiratory tract. (A) Common wild-type ball python (no. 7), Python regius. Photomicrograph of the oral mucosa, revealing a mild to moderate stomatitis. There is a focal area of necrosis (arrows) and heterophilic inflammation in oral mucosa. H&E stain; bar = 100 µm. (B) Cinnamon variant ball python (no. 8). Photomicrograph of the oral mucosa. There is diffuse necrosis of the oropharyngeal mucosa with numerous lymphocytes and fewer plasma cells and macrophages infiltrating the submucosa. H&E stain; bar = 75 µm. (C) Common wild-type ball python (no. 7). Photomicrograph of a nasal cavity revealing mild heterophilic and lymphocytic inflammation in sinus mucosa. (D) Cinnamon variant ball python (no. 8). Photomicrograph of the tracheal mucosa, revealing areas of hyperplasia and necrosis (arrow). The submucosa (SM) is severely and diffusely thickened, primarily with round cells. Cartilage (CA) surrounds the submucosa. H&E stain; bar = 75 µm. Download
The putative AAAAAAC “slippery sequence” in the region of ORF1a/b overlap is well supported by Illumina and Sanger sequencing. The 55-nt region of overlap between ORF1a and ORF1b is shown, as are Sanger and Illumina sequences mapping to this region. Bases are numbered according to the overall genomic position. Sanger sequences are from directly sequenced PCR products. Twenty randomly selected Illumina reads (from 85× average coverage) are depicted, and disagreements to the consensus sequence are highlighted. Download
Dot plot showing regions of similarity between BPNV and FHMNV replicase polyproteins. Lines in the dot plot represent 100-amino-acid windows with pairwise global alignment scores of ≥40 (BLOSUM50 matrix). A cartoon of BPNV pp1ab with predicted functional domains highlighted (see the text), with highlighting extended into the dot plot, is shown. Blue and orange colors correspond to the ORF1a and ORF1b encoded domains. Download
Transmembrane domains and glycosylation sites in proteins predicted to be encoded by ORF2 to ORF6 of snake nidovirus. Domains and glycosylation sites were predicted as described in the text. Predicted protein lengths are indicated. Download
Multiple alignment of conserved domain of replicase polyproteins of indicated representative nidoviruses: RdRp domain (A), helicase domain (B), and Mpro domain (C). Columns with >75% identity are highlighted. Sequence accession numbers are indicated. Abbreviations are defined in Table S3. Download
Phylogenies of conserved replicase subunits of representative nidoviruses. Unrooted Bayesian phylogenies based on multiple sequence alignments of indicated conserved regions of replicase polyproteins. At select nodes, Bayesian posterior probabilities are indicated in percentages. The five major nidovirus families and subfamilies are indicated, as are select genera and the currently uncategorized BPNV and possum nidovirus taxa. Known host phyla are indicated in parentheses. The branch leading to BPNV is highlighted red for emphasis. Download
ACKNOWLEDGMENTS
We thank April Childress and Shelley Campbell for technical assistance, Jennifer Mann for logistical assistance, Jessica Lund, Eric Chow, and the Center for Advanced Technology at UCSF for assistance with sequencing, and Chris Sanders, Drury Reavill, and Freeland Dunker for negative-control samples.
J.L.D. is supported by the Howard Hughes Medical Institute. M.D.S. is supported by NIH grant 5T32HL007185-34 and the Pacific Southwest Regional Center of Excellence (PSWRCE) (NIH grant U54 AI065359).
Footnotes
Citation Stenglein MD, Jacobson ER, Wozniak EJ, Wellehan JFX, Kincaid A, Gordon M, Porter BF, Baumgartner W, Stahl S, Kelley K, Towner JS, DeRisi JL. 2014. Ball python nidovirus: a candidate etiologic agent for severe respiratory disease in Python regius. mBio 5(5):e01484-14. doi:10.1128/mBio.01484-14.
ADDENDUM IN PROOF
While this article was in press, two studies describing similar viral sequences in pythons with respiratory disease were published:
L. Uccellini, R. J. Ossiboff, R. E. C. de Matos, J. K. Morrisey, A. Petrosov, I. Navarrete-Macias, K. Jain, A. L. Hicks, E. L. Buckles, R. Tokarz, D. McAloose, W. I. Lipkin, Virol. J. 11:144, 2014, doi:10.1186/1743-422X-11-144.
R. Bodewes, C. Lempp, A. Schürch, A. Habierski, K. Hahn, M. Lamers, K. von Dörnberg, P. Wohlsein, J. F. Drexler, B. Haagmans, S. L. Smits, W. Baumgärtner, A. D. M. E. Osterhaus, J. Gen. Virol. pii:vir.0.068700-0, 2014, doi:10.1099/vir.0.068700-0.
REFERENCES
- 1. De Groot RJ, Cowley JA, Enjuanes L, Faaberg KS, Perlman S, Rottier PJM, Snijder EJ, Ziebuhr J, Gorbalenya AE. 2012. Nidovirales, p 785–795 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses, 1st ed. Academic Press, Waltham, MA [Google Scholar]
- 2. Lai MMC, Perlman S, Anderson LJ. 2007. Coronaviridae, p 1305–1335 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3. Snijder EJ, Spaan WJM. 2007. Arteriviruses, p 1337–1355 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4. Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 94:2141–2163. 10.1099/vir.0.056341-0 [DOI] [PubMed] [Google Scholar]
- 5. Lauber C, Ziebuhr J, Junglen S, Drosten C, Zirkel F, Nga PT, Morita K, Snijder EJ, Gorbalenya AE. 2012. Mesoniviridae: a proposed new family in the order Nidovirales formed by a single species of mosquito-borne viruses. Arch. Virol. 157:1623–1628. 10.1007/s00705-012-1295-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Graham RL, Donaldson EF, Baric RS. 2013. A decade after SARS: strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 11:836–848. 10.1038/nrmicro3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450. 10.1038/nrmicro2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weiss SR, Navas-Martin S. 2005. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69:635–664. 10.1128/MMBR.69.4.635-664.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lauber C, Goeman JJ, del Carmen Parquet M, Nga PT, Snijder EJ, Morita K, Gorbalenya AE. 2013. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 9:e1003500. 10.1371/journal.ppat.1003500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ. 2006. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117:17–37. 10.1016/j.virusres.2006.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith EC, Blanc H, Vignuzzi M, Denison MR. 2013. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 9:e1003565. 10.1371/journal.ppat.1003565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, Ziebuhr J. 2006. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 103:5108–5113. 10.1073/pnas.0508200103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barker DG, Barker 2006. Ball pythons: the history, natural history, care and breeding. VPI Library, Boerne, TX [Google Scholar]
- 14. Goldsmith CS, Ksiazek TG, Rollin PE, Comer JA, Nicholson WL, Peret TC, Erdman DD, Bellini WJ, Harcourt BH, Rota PA, Bhatnagar J, Bowen MD, Erickson BR, McMullan LK, Nichol ST, Shieh WJ, Paddock CD, Zaki SR. 2013. Cell culture and electron microscopy for identifying viruses in diseases of unknown cause. Emerg. Infect. Dis. 19:886–891. 10.3201/eid1906.130173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacobson ER. 2007. Infectious diseases and pathology of reptiles: color atlas and text. CRC Press/Taylor & Francis, Boca Raton, FL [Google Scholar]
- 16. Orós J, Sicilia J, Torrent A, Castro P, Déniz S, Arencibia A, Jacobson ER, Homer BL. 2001. Immunohistochemical detection of ophidian paramyxovirus in snakes in the Canary Islands. Vet. Rec. 149:21–23. 10.1136/vr.149.1.21 [DOI] [PubMed] [Google Scholar]
- 17. Ahne W, Thomsen I, Winton J. 1987. Isolation of a reovirus from the snake, Python regius. Brief report. Arch. Virol. 94:135–139. 10.1007/BF01313731 [DOI] [PubMed] [Google Scholar]
- 18. Lamirande EW, Nichols DK, Owens JW, Gaskin JM, Jacobson ER. 1999. Isolation and experimental transmission of a reovirus pathogenic in ratsnakes (Elaphe species). Virus Res. 63:135–141. 10.1016/S0168-1702(99)00067-2 [DOI] [PubMed] [Google Scholar]
- 19. Latney LV, Wellehan J. 2013. Selected emerging infectious diseases of Squamata. Vet. Clin. North Am. Exot. Anim. Pract. 16:319–338. 10.1016/j.cvex.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 20. Castoe TA, de Koning APJ, Hall KT, Card DC, Schield DR, Fujita MK, Ruggiero RP, Degner JF, Daza JM, Gu W, Reyes-Velasco J, Shaney KJ, Castoe JM, Fox SE, Poole AW, Polanco D, Dobry J, Vandewege MW, Li Q, Schott RK, Kapusta A, Minx P, Feschotte C, Uetz P, Ray DA, Hoffmann FG, Bogden R, Smith EN, Chang BSW, Vonk FJ, Casewell NR, Henkel CV, Richardson MK, Mackessy SP, Bronikowski AM, Yandell M, Warren WC, Secor SM, Pollock DD. 2013. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. U. S. A. 110:20645–20650. 10.1073/pnas.1314475110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman JA, Chapuis G, Chikhi R, Chitsaz H, Chou WC, Corbeil J, Del Fabbro C, Docking TR, Durbin R, Earl D, Emrich S, Fedotov P, Fonseca NA, Ganapathy G, Gibbs RA, Gnerre S, Godzaridis É, Goldstein S, Haimel M, Hall G, Haussler D, Hiatt JB, Ho IY, Howard J, Hunt M, Jackman SD, Jaffe DB, Jarvis ED, Jiang H, Kazakov S, Kersey PJ, Kitzman JO, Knight JR, Koren S, Lam T-W, Lavenier D, Laviolette F, Li Y, Li Z, Liu B, Liu Y, Luo R, MacCallum I, MacManes MD, Maillet N, Melnikov S, Naquin D, Ning Z, Otto TD, Paten B, Paulo OS, Phillippy AM, Pina-Martins F, Place M, Przybylski D, Qin X, Qu C, Ribeiro FJ, Richards S, Rokhsar DS, Ruby JG, Scalabrin S, Schatz MC, Schwartz DC, Sergushichev A, Sharpe T, Shaw TI, Shendure J, Shi Y, Simpson JT, Song H, Tsarev F, Vezzi F, Vicedomini R, Vieira BM, Wang J, Worley KC, Yin S, Yiu S-M, Yuan J, Zhang G, Zhang H, Zhou S, Korf IF. 2013. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2:10. 10.1186/2047-217X-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruby JG, Bellare P, DeRisi JL. 2013. PRICE: software for the targeted assembly of components of (meta)genomic sequence data. G3 3:865–880. 10.1534/g3.113.005967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467. 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scotto-Lavino E, Du G, Frohman MA. 2006. 3′ end cDNA amplification using classic RACE. Nat. Protoc. 1:2742–2745. 10.1038/nprot.2006.481 [DOI] [PubMed] [Google Scholar]
- 25. Scotto-Lavino E, Du G, Frohman MA. 2006. 5′ end cDNA amplification using classic RACE. Nat. Protoc. 1:2555–2562. 10.1038/nprot.2006.480 [DOI] [PubMed] [Google Scholar]
- 26. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mihindukulasuriya KA, Wu G, St Leger J, Nordhausen RW, Wang D. 2008. Identification of a novel coronavirus from a Beluga whale by using a panviral microarray. J. Virol. 82:5084–5088. 10.1128/JVI.02722-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brian DA, Baric RS. 2005. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 287:1–30. 10.1007/3-540-26765-4_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33:W244–W248. 10.1093/nar/gki162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 34. Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, Gupta R, Bennett EP, Mandel U, Brunak S, Wandall HH, Levery SB, Clausen H. 2013. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32:1478–1488. 10.1038/emboj.2013.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plant EP, Dinman JD. 2008. The role of programmed-1 ribosomal frameshifting in coronavirus propagation. Front. Biosci. 13:4873–4881. 10.2741/3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dinman JD. 2012. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip. Rev. RNA 3:661–673. 10.1002/wrna.1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ziebuhr J, Snijder EJ, Gorbalenya AE. 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81:853–879 [DOI] [PubMed] [Google Scholar]
- 38. Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, Guan Y, Rozanov M, Spaan WJM, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991–1004. 10.1016/S0022-2836(03)00865-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ziebuhr J. 2005. The coronavirus replicase. Curr. Top. Microbiol. Immunol. 287:57–94. 10.1007/3-540-26765-4_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Batts WN, Goodwin AE, Winton JR. 2012. Genetic analysis of a novel nidovirus from fathead minnows. J. Gen. Virol. 93:1247–1252. 10.1099/vir.0.041210-0 [DOI] [PubMed] [Google Scholar]
- 41. Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222. 10.1093/nar/gkp985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nga PT, del Carmen Parquet M, Lauber C, Parida M, Nabeshima T, Yu F, Thuy NT, Inoue S, Ito T, Okamoto K, Ichinose A, Snijder EJ, Morita K, Gorbalenya AE. 2011. Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes. PLoS Pathog. 7:e1002215. 10.1371/journal.ppat.1002215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bouvet M, Debarnot C, Imbert I, Selisko B, Snijder EJ, Canard B, Decroly E. 2010. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 6:e1000863. 10.1371/journal.ppat.1000863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seybert A, Hegyi A, Siddell SG, Ziebuhr J. 2000. The human coronavirus 229E superfamily 1 helicase has RNA and DNA duplex-unwinding activities with 5′-to-3′ polarity. RNA 6:1056–1068. 10.1017/S1355838200000728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y, Cai H, Pan J, Xiang N, Tien P, Ahola T, Guo D. 2009. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 106:3484–3489. 10.1073/pnas.0808790106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. 2005. The macro domain is an ADP-ribose binding module. EMBO J. 24:1911–1920. 10.1038/sj.emboj.7600664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. 2005. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure 13:1665–1675. 10.1016/j.str.2005.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y, Cong L, Chen C, Wei L, Zhao Q, Xu X, Ma Y, Bartlam M, Rao Z. 2009. Crystal structures of two coronavirus ADP-ribose-1′′-monophosphatases and their complexes with ADP-ribose: a systematic structural analysis of the viral ADRP domain. J. Virol. 83:1083–1092. 10.1128/JVI.01862-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Putics Á, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. 2005. ADP-ribose-1′′ monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 79:12721–12731. 10.1128/JVI.79.20.12721-12731.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuri T, Eriksson KK, Putics A, Züst R, Snijder EJ, Davidson AD, Siddell SG, Thiel V, Ziebuhr J, Weber F. 2011. The ADP-ribose-1′′-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J. Gen. Virol. 92:1899–1905. 10.1099/vir.0.031856-0 [DOI] [PubMed] [Google Scholar]
- 51. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. 10.1038/nsmb.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801–8811. 10.1128/JVI.77.16.8801-8811.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Woo PC, Lau SK, Lam CS, Lai KK, Huang Y, Lee P, Luk GS, Dyrting KC, Chan KH, Yuen KY. 2009. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 83:908–917. 10.1128/JVI.01977-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pasternak AO, Spaan WJ, Snijder EJ. 2006. Nidovirus transcription: how to make sense…? J. Gen. Virol. 87:1403–1421. 10.1099/vir.0.81611-0 [DOI] [PubMed] [Google Scholar]
- 55. Zirkel F, Kurth A, Quan PL, Briese T, Ellerbrok H, Pauli G, Leendertz FH, Lipkin WI, Ziebuhr J, Drosten C, Junglen S. 2011. An insect nidovirus emerging from a primary tropical rainforest. mBio 2(3):e00077-11. 10.1128/mBio.00077-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huder JB, Böni J, Hatt JM, Soldati G, Lutz H, Schüpbach J. 2002. Identification and characterization of two closely related unclassifiable endogenous retroviruses in pythons (Python molurus and Python curtus). J. Virol. 76:7607–7615. 10.1128/JVI.76.15.7607-7615.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ulferts R, Mettenleiter TC, Ziebuhr J. 2011. Characterization of bafinivirus main protease autoprocessing activities. J. Virol. 85:1348–1359. 10.1128/JVI.01716-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shimodaira H, Hasegawa M. 1999. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16:1114. 10.1093/oxfordjournals.molbev.a026201 [DOI] [Google Scholar]
- 59. Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, Dunker F, Derisi JL. 2012. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio 3(4):e00180-12. 10.1128/mBio.00180-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clark HF, Cohen MM, Lunger PD. 1973. Comparative characterization of a C-type virus-producing cell line (VSW) and a virus-free cell line (VH2) from Vipera russelli. J. Natl. Cancer Inst. 51:645–657 [PubMed] [Google Scholar]
- 61. Schütze H, Ulferts R, Schelle B, Bayer S, Granzow H, Hoffmann B, Mettenleiter TC, Ziebuhr J. 2006. Characterization of white bream virus reveals a novel genetic cluster of nidoviruses. J. Virol. 80:11598–11609. 10.1128/JVI.01758-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Granzow H, Weiland F, Fichtner D, Schütze H, Karger A, Mundt E, Dresenkamp B, Martin P, Mettenleiter TC. 2001. Identification and ultrastructural characterization of a novel virus from fish. J. Gen. Virol. 82:2849–2859 [DOI] [PubMed] [Google Scholar]
- 63. González JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L. 2003. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 148:2207–2235. 10.1007/s00705-003-0162-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Flynn JJ, Finarelli JA, Zehr S, Hsu J, Nedbal MA. 2005. Molecular phylogeny of the Carnivora (Mammalia): assessing the impact of increased sampling on resolving enigmatic relationships. Syst. Biol. 54:317–337. 10.1080/10635150590923326 [DOI] [PubMed] [Google Scholar]
- 65. Lunter G. 2007. Dog as an outgroup to human and mouse. PLoS Comput. Biol. 3:e74. 10.1371/journal.pcbi.0030074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Izadjoo MJ, Pantoja CO, Siebeling RJ. 1987. Acquisition of Salmonella flora by turtle hatchlings on commercial turtle farms. Can. J. Microbiol. 33:718–724. 10.1139/m87-125 [DOI] [PubMed] [Google Scholar]
- 67. Richards JM, Brown JD, Kelly TR, Fountain AL, Sleeman JM. 2004. Absence of detectable Salmonella cloacal shedding in free-living reptiles on admission to the wildlife center of Virginia. J. Zoo Wildl. Med. 35:562–563. 10.1638/03-070 [DOI] [PubMed] [Google Scholar]
- 68. Saelinger CA, Lewbart GA, Christian LS, Lemons CL. 2006. Prevalence of Salmonella spp. in cloacal, fecal, and gastrointestinal mucosal samples from wild North American turtles. J. Am. Vet. Med. Assoc. 229:266–268. 10.2460/javma.229.2.266 [DOI] [PubMed] [Google Scholar]
- 69. Coyne KP, Jones BR, Kipar A, Chantrey J, Porter CJ, Barber PJ, Dawson S, Gaskell RM, Radford AD. 2006. Lethal outbreak of disease associated with feline calicivirus infection in cats. Vet. Rec. 158:544–550. 10.1136/vr.158.16.544 [DOI] [PubMed] [Google Scholar]
- 70. Hyndman TH, Marschang RE, Wellehan JF, Jr, Nicholls PK. 2012. Isolation and molecular identification of Sunshine virus, a novel paramyxovirus found in Australian snakes. Infect. Genet. Evol. 12:1436–1446. 10.1016/j.meegid.2012.04.022 [DOI] [PubMed] [Google Scholar]
- 71. Marschang RE, Papp T, Frost JW. 2009. Comparison of paramyxovirus isolates from snakes, lizards and a tortoise. Virus Res. 144:272–279. 10.1016/j.virusres.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 72. Wellehan JF, Jr, Pessier AP, Archer LL, Childress AL, Jacobson ER, Tesh RB. 2012. Initial sequence characterization of the rhabdoviruses of squamate reptiles, including a novel rhabdovirus from a caiman lizard (Dracaena guianensis). Vet. Microbiol. 158:274–279. 10.1016/j.vetmic.2012.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wellehan JF, Jr, Childress AL, Marschang RE, Johnson AJ, Lamirande EW, Roberts JF, Vickers ML, Gaskin JM, Jacobson ER. 2009. Consensus nested PCR amplification and sequencing of diverse reptilian, avian, and mammalian orthoreoviruses. Vet. Microbiol. 133:34–42. 10.1016/j.vetmic.2008.06.011 [DOI] [PubMed] [Google Scholar]
- 74. Sanchez A, Ksiazek TG, Rollin PE, Miranda ME, Trappier SG, Khan AS, Peters CJ, Nichol ST. 1999. Detection and molecular characterization of Ebola viruses causing disease in human and nonhuman primates. J. Infect. Dis. 179(Suppl 1):S164–S169. 10.1086/514282 [DOI] [PubMed] [Google Scholar]
- 75. Ogawa H, Miyamoto H, Ebihara H, Ito K, Morikawa S, Feldmann H, Takada A. 2011. Detection of all known filovirus species by reverse transcription-polymerase chain reaction using a primer set specific for the viral nucleoprotein gene. J. Virol. Methods 171:310–313. 10.1016/j.jviromet.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yozwiak NL, Skewes-Cox P, Stenglein MD, Balmaseda A, Harris E, DeRisi JL. 2012. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl. Trop Dis. 6:e1485. 10.1371/journal.pntd.0001485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics Oxf. Engl. 22:1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 78. Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386 [DOI] [PubMed] [Google Scholar]
- 79. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9:286–298. 10.1093/bib/bbn013 [DOI] [PubMed] [Google Scholar]
- 81. Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. 10.1093/bioinformatics/btr088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25:1307–1320. 10.1093/molbev/msn067 [DOI] [PubMed] [Google Scholar]
- 83. Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. 10.1093/bioinformatics/bti263 [DOI] [PubMed] [Google Scholar]
- 84. Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- 85. Miller M, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees, p 1–8 In Proceedings of the Gateway Computing Environments Workshop (GCE). Institute of Electrical and Electronics Engineers, Piscataway, NJ. 10.1109/GCE.2010.5676129 [DOI] [Google Scholar]
- 86. Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57:758–771. 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 87. Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783. 10.2307/2408678 [DOI] [PubMed] [Google Scholar]
- 88. Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A. 2010. How many bootstrap replicates are necessary? J. Comput. Biol. 17:337–354. 10.1089/cmb.2009.0179 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides used in this study
High-throughput sequencing summary
NCBI accession numbers of sequences of representative nidoviruses used for phylogenetic analyses
Representative microscopic lesions in upper respiratory tract. (A) Common wild-type ball python (no. 7), Python regius. Photomicrograph of the oral mucosa, revealing a mild to moderate stomatitis. There is a focal area of necrosis (arrows) and heterophilic inflammation in oral mucosa. H&E stain; bar = 100 µm. (B) Cinnamon variant ball python (no. 8). Photomicrograph of the oral mucosa. There is diffuse necrosis of the oropharyngeal mucosa with numerous lymphocytes and fewer plasma cells and macrophages infiltrating the submucosa. H&E stain; bar = 75 µm. (C) Common wild-type ball python (no. 7). Photomicrograph of a nasal cavity revealing mild heterophilic and lymphocytic inflammation in sinus mucosa. (D) Cinnamon variant ball python (no. 8). Photomicrograph of the tracheal mucosa, revealing areas of hyperplasia and necrosis (arrow). The submucosa (SM) is severely and diffusely thickened, primarily with round cells. Cartilage (CA) surrounds the submucosa. H&E stain; bar = 75 µm. Download
The putative AAAAAAC “slippery sequence” in the region of ORF1a/b overlap is well supported by Illumina and Sanger sequencing. The 55-nt region of overlap between ORF1a and ORF1b is shown, as are Sanger and Illumina sequences mapping to this region. Bases are numbered according to the overall genomic position. Sanger sequences are from directly sequenced PCR products. Twenty randomly selected Illumina reads (from 85× average coverage) are depicted, and disagreements to the consensus sequence are highlighted. Download
Dot plot showing regions of similarity between BPNV and FHMNV replicase polyproteins. Lines in the dot plot represent 100-amino-acid windows with pairwise global alignment scores of ≥40 (BLOSUM50 matrix). A cartoon of BPNV pp1ab with predicted functional domains highlighted (see the text), with highlighting extended into the dot plot, is shown. Blue and orange colors correspond to the ORF1a and ORF1b encoded domains. Download
Transmembrane domains and glycosylation sites in proteins predicted to be encoded by ORF2 to ORF6 of snake nidovirus. Domains and glycosylation sites were predicted as described in the text. Predicted protein lengths are indicated. Download
Multiple alignment of conserved domain of replicase polyproteins of indicated representative nidoviruses: RdRp domain (A), helicase domain (B), and Mpro domain (C). Columns with >75% identity are highlighted. Sequence accession numbers are indicated. Abbreviations are defined in Table S3. Download
Phylogenies of conserved replicase subunits of representative nidoviruses. Unrooted Bayesian phylogenies based on multiple sequence alignments of indicated conserved regions of replicase polyproteins. At select nodes, Bayesian posterior probabilities are indicated in percentages. The five major nidovirus families and subfamilies are indicated, as are select genera and the currently uncategorized BPNV and possum nidovirus taxa. Known host phyla are indicated in parentheses. The branch leading to BPNV is highlighted red for emphasis. Download