

ABSTRACT
Staphylococcus aureus is a leading cause of both community- and hospital-acquired infections that are increasingly antibiotic resistant. The emergence of S. aureus resistance to even last-line antibiotics heightens the need for the development of new drugs with novel targets. We generated a highly saturated transposon insertion mutant library in the genome of S. aureus and used Tn-seq analysis to probe the entire genome, with unprecedented resolution and sensitivity, for genes of importance in infection. We further identified genes contributing to fitness in various infected compartments (blood and ocular fluids) and compared them to genes required for growth in rich medium. This resulted in the identification of 426 genes that were important for S. aureus fitness during growth in infection models, including 71 genes that could be considered essential for survival specifically during infection. These findings highlight novel as well as previously known genes encoding virulence traits and metabolic pathways important for S. aureus proliferation at sites of infection, which may represent new therapeutic targets.
IMPORTANCE
Staphylococcus aureus continues to be a leading cause of antibiotic-resistant community and nosocomial infection. With the bacterium’s acquisition of resistance to methicillin and, more recently, vancomycin, the need for the development of new drugs with novel targets is urgent. Applying a highly saturated Tn-seq mutant library to analyze fitness and growth requirements in a murine abscess and in various infection-relevant fluids, we identified S. aureus traits that enable it to survive and proliferate during infection. This identifies potential new targeting opportunities for the development of novel therapeutics.
INTRODUCTION
Staphylococcus aureus is routinely isolated as a commensal organism of skin, nares, and mucosal surfaces; however, it causes over half a million infections and over 10,000 deaths per year in the United States alone (1, 2). These include skin and soft tissue infections, pneumonia, bloodstream infections, infective endocarditis, and osteomyelitis (1) and infections of the ocular surface associated with contact lens wear, as well as intraocular infections associated with cataract surgery and other globe-penetrating trauma (3). Throughout the history of antibiotic use, S. aureus has demonstrated the ability to rapidly adapt and develop resistance. Reports arising from the United Kingdom in the early 1960s described methicillin-resistant isolates of S. aureus (MRSA), which subsequently proliferated, mainly in hospitals (4). Intermediate and high-level resistance (5) to vancomycin, a key last-line bactericidal drug for treating MRSA, has now emerged in S. aureus (6–9).
Several methods have been used to identify genes that are essential for S. aureus growth (10–15). The development of next-generation DNA sequencing technology makes possible the simultaneous identification and quantification of individual members in a very large pool of mutants, allowing the comparative assessment of all genes in the S. aureus genome to fitness in infection and in other ecological conditions with unparalleled resolution. It was therefore of interest to use this approach, termed Tn-seq (10, 16–29) or TraDIS (26), to assess the many traits encoded within the S. aureus genome for genes that encode properties that make measurable contributions to the ability of S. aureus to survive and proliferate in infection. In the process, we identified genes of importance in abscess formation, growth, and proliferation in blood and ocular fluids and compared those to a comprehensively identified set of genes contributing to fitness in nutrient-rich medium.
RESULTS
Characterization of a high-density transposon library of S. aureus.
The initial complexity of the HG003 mariner insertion library was found to consist of 71,700 unique insertions in the HG003 chromosome (Fig. 1A, blue track). The location of the insertions showed an even distribution around the chromosome (Fig. 1A and B). Comparison of insertion sites within predicted open reading frames (ORFs) in the S. aureus NCTC8325 genome indicated that 57,191 insertions occurred within coding regions (~93% of all annotated protein ORFs), whereas 14,509 insertions occurred within intergenic regions. The average distance between transposon insertions is 39 bp.
FIG 1 .

Characterization of the S. aureus transposon mutant library. (A) Tn-seq mapping of the 71,700 transposon insertions (blue track; because of density, individual hash marks appear nearly solid). Analysis of initial mutant population recovered on BHI agar. (B) Cumulative number of unique transposon (Tn) insertions over the length of the 2.8-Mb NCTC8325 genome, illustrating absence of hot spots/large gaps in transposon insertion. (C) The number of unique transposon insertions per ORF (left), the transposon density of each ORF (middle), and the ORF dval (right) are shown for nonessential (red) and essential (blue) genes.
The S. aureus NCTC8325 genome encodes 269,583 possible mariner TA insertion sites, with the longest stretch lacking a TA dinucleotide being 331 nucleotides (positions 757836 to 758166). This stretch of nucleotides falls within a gene where there are multiple TA dinucleotides 5′ and 3′ to that sequence, so that there are no genes that lack the potential for transposon insertion.
Among ORF-containing genes, 319 were found to either completely lack transposon insertions (216 ORFs) or possess fewer insertions (>100-fold less) than expected based on gene size (103 ORFs), following growth on brain heart infusion (BHI) agar plates (see Table S1 in the supplemental material). We classified genes as “essential” for growth under a given condition if they had <1 insertion/kb of coding sequence. Essential and nonessential genes formed well-resolved populations (Fig. 1C). An additional advantage of Tn-seq is the ability to quantify gradations in the impact on fitness of individual insertions (16). ORFs possessing comparatively few transposon insertions (1 to 10% of the expected number based on the size of that ORF [or dval of 0.01 to 0.1 as defined in Text S1 in the supplemental material]) identified genes that contributed to fitness in the tested environment but did not meet our definition of essential under that condition (that is, fitness compromised but not essential [see Table S1]). Tn-seq analysis identified 106 additional genes that contributed to fitness, in that transposon insertion compromised fitness following growth on agar plates (see Table S1).
Genes essential for competitive planktonic growth in rich medium.
When cells are plated at a low density, individual colony growth on agar is relatively noncompetitive, allowing mutants with variable growth rates to reach relatively similar colony size/population levels given sufficient incubation time. Therefore, during the initial mutant selection on solid agar as discussed above, colonies were allowed to form for 48 h at 42°C (to select for transposon integrants into the chromosome), irrespective of the growth rate of an individual mutant, to maximize the diversity of mutants recovered. In contrast, to identify genes that contribute to fitness in a competitive growth environment, we inoculated 108 CFU of the mutant library (to ensure complete representation of the initial complexity) recovered from agar plates and subcultured them for approximately 10 generations overnight in BHI (BHI input pool) (see Fig. S1 in the supplemental material).
Analysis of the diversity of transposon insertions in the initial BHI culture identified 420 genes that fulfilled the criteria as being operationally essential for growth (i.e., unable to withstand transposon insertion, resulting in insertions within these genes constituting fewer than 1% of the sequence reads), including 312 identified as being essential for growth on agar (see Table S1 in the supplemental material). Of the 108 new essential genes, approximately half (30 genes) were previously scored as fitness compromised for growth on agar (median dval, 0.048 ± 0.021) (see Table S1). Insertion in an additional 154 genes resulted in fitness compromise when grown competitively in liquid BHI (see Table S1).
Of the 420 genes identified as essential for overnight growth in BHI (see Table S2 in the supplemental material), 284 genes (67.6%) had previously been identified as essential for the growth of S. aureus in culture using other approaches (11) (Fig. 2). The additional 136 newly identified putative essential genes identified by Tn-seq are listed in Table S2.
FIG 2 .
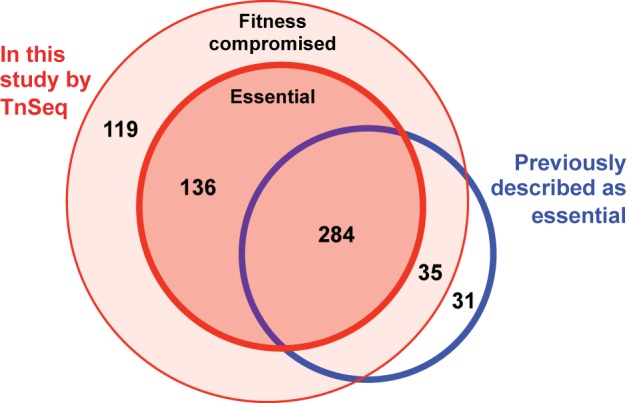
Gene essentiality under laboratory conditions. Agreement between Tn-seq results for 24-h growth in BHI and previous identification of essential genes tested under similar conditions (11).
To validate these results and increase the sensitivity of detection of mutations resulting in fitness compromise, the culture was successively passaged to allow ~10 additional generations of selection (1:1,000 dilution). Following this second overnight passage, quantification of transposon insertions identified 506 genes as essential. Of those, 411 of the 420 genes previously identified as essential in the initial round of cultivation were confirmed, showing the methodological precision. Of the 9 genes not confirmed as essential, 6 were now found to be fitness compromised (see Table S1 in the supplemental material), indicating that they were on the boundary of the definition between essential and fitness compromised in the first analysis. A further 3 genes were found to be neither fitness compromised nor essential—a false-positive rate of 0.7%. Of the 95 genes that now fulfilled the criteria for encoding essential functions upon the second passage (Table S1), 73 were categorized as fitness compromised in the initial 24-h culture (average dval, 0.03 ± 0.03) (Table S1). In addition to the essential genes, from the second BHI passage we identified insertions in 140 genes that fulfilled the definition of having compromised fitness (Table S1), 59 of which exhibited fitness defects in the first broth culture (Table S1).
To standardize comparisons, the initial 24-h culture in BHI broth was used as the inoculum for the infection-related selections described below. Population structures from outgrowth in infection-related environments were then compared to the second broth subculture control, to identify genes of importance only in various infection contexts.
Independent antisense RNA validation.
Of genes identified to be putatively essential for growth in BHI and not previously identified as being essential using other approaches (11), 8 were selected for independent validation using a completely unrelated approach—inducible antisense gene expression (13) (see Table S2 in the supplemental material). These included five genes annotated as encoding hypothetical proteins and three genes that were suggested by others to be essential but were not included in the cumulative essential list of Chaudhuri et al. (11). To deplete transcripts of putatively essential genes, a counter transcript of the gene of interest was expressed from a xylose-inducible promoter during growth in BHI. As a positive control, we generated a construct from which an antisense RNA for fabI, a previously known essential gene (31), was expressed (Fig. 3).
FIG 3 .

Antisense RNA validation of contribution to fitness. Exponential cultures carrying xylose-inducible antisense constructs were diluted into Luria Broth supplemented with 0.2% glucose containing 4% xylose, and optical density at 600 nm (OD600) was monitored. (A to I) Induced control strain with the pEPSA5 vector lacking insert (red squares) compared to induced strain carrying antisense construct (purple X’s). Antisense clones of fragments from genes fabI (A), SAOUHSC_00486 (B), SAOUHSC_00889 (C), SAOUHSC_01263 (D), SAOUHSC_01203 (E), SAOUHSC_01857 (F), SAOUHSC_02383 (G), SAOUHSC_00022 (H), and SAOUHSC_01239 (I). Error bars show the deviations from the means for at least 9 replicates in 3 separate experiments.
Of the eight genes targeted for antisense ablation, clones expressing counter transcripts for three (SAOUHSC_00486, SAOUHSC_00889, and SAOUHSC_01263) displayed no measurable growth within 20 h of inducer being added (Fig. 3). Expression of antisense RNA targeting four genes (SAOUHSC_01203, SAOUHSC_1857, SAOUHSC_2383, and SAOUHSC_00022) resulted in significantly delayed growth compared to the control strain (Fig. 3). One of the eight genes assayed by antisense targeting (SAOUHSC_01239) was not affected by xylose induction (Fig. 3). The basis for the lack of growth inhibition in this strain is the subject of ongoing study and may represent a limitation of the method or a false positive identified by Tn-seq. Independent validation using antisense RNA knockdown readily confirmed the contribution of 87% of the putatively essential genes tested as contributing to S. aureus fitness, validating the method.
Genes contributing to survival and growth in infection-related environments.
We next asked whether S. aureus possesses additional genes that contribute to fitness only in infection-relevant biological fluids, such as whole blood, where host defense factors are present, or fluids from immune privileged ocular tissues (see Fig. S1 in the supplemental material). Each is an important environment for S. aureus infection that varies in nutritional content and defense factors (32–36). Each fluid was inoculated ex vivo using the initial overnight BHI culture (Fig. S1), under conditions that led to approximately 10 additional generations of outgrowth. To determine the reproducibility of the method, a second biological replicate was analyzed and independent dval calculations were made and compared by regression analysis (see Fig. S2; median Pearson coefficient r2 = 0.96 ± 0.14). This again showed the robust reproducibility of the approach and indicated that there were no population bottlenecks, random fluctuations in PCR amplification, or other sources of irreproducible variation as tested.
Following 24-h growth in freshly isolated peripheral whole blood, analysis of the mutant population structure by Tn-seq identified a total of 503 ORFs fulfilling the criteria established for being essential (see Table S1 in the supplemental material). Of these, 31 were new and not detected as essential in the BHI outgrowth control (Table S1). However, mutations in 27 of the 31 newly essential genes identified for growth in blood exhibited some level of growth defect and were categorized as compromised in the BHI outgrowth control (median dval, 0.03 ± 0.04) (Table S1). In addition to essential genes, an additional 120 genes were identified in which transposon insertion resulted in a detectable fitness compromise in blood (Table S1).
Analysis of the Tn-seq data following outgrowth of the transposon mutant pool in ocular fluids indicated that 518 ORFs were essential: 486 essential genes in aqueous fluid and 493 essential genes in vitreous fluid, with 461 essential genes being shared between them (see Table S1 in the supplemental material). Of the 518 identified as essential in one or the other ocular fluid, 488 genes were also essential in the BHI outgrowth control (Table S1). Of the 30 identified as uniquely essential in one or the other of the ocular fluids, 24 encode hypothetical proteins (Table S1). Of the new genes determined to be essential in ocular fluids, 24 were also found to be poorly tolerant of transposon insertion when tested in the BHI outgrowth control (median dval, 0.033 ± 0.06) (Table S1). Since the aqueous and vitreous fluids of the eye share some fluid exchange but have some differences in composition, it was of interest to determine whether differences in genes that are essential for fitness in these two fluids could be detected. Of the 30 genes identified as newly essential for growth in either ocular fluid and not essential in BHI control, 10 were shared between aqueous and vitreous fluids, including 7 annotated as hypothetical. In addition, we identified insertions in an additional 146 genes and 160 genes that resulted in fitness compromise in vitreous and aqueous fluids, respectively, 90 of which were compromised in fitness in both fluids (Table S1), mirroring their related composition.
Genes encoding functions important for survival and proliferation in infection in vivo.
S. aureus is an important cause of wound infections, including abscesses (37). The subcutaneous lesion (abscess) model mimics many parameters of human infection (37) and has demonstrated utility in identifying S. aureus factors contributing to virulence (38). The ultimate goal of this work was to identify genes essential for survival and proliferation of S. aureus in infection—in the presence of an intact immune system and with nutrient limitations that occur in vivo.
Abscesses were harvested and analyzed 24 and 48 h after infection, to identify changes in the population structure of the mutant pool during the course of abscess maturation. Following outgrowth in the abscess of the transposon mutant pool for 48 h, a total of 646 genes met the criteria for encoding functions essential for S. aureus in infection (see Table S1 in the supplemental material). Of these essential genes, 153 were unique to the abscess compared to the BHI outgrowth control (Table S1), and 84 were found to be essential at 24 h as well. Of the newly identified 153 essential genes, 82 are annotated as hypothetical and 55 represent genes for which mutants exhibited fitness compromise in the BHI outgrowth control (Table S1). Additionally, 43 fulfilled the criteria as being essential after the 48-h outgrowth but not at the earlier time point; however, mutations in each of these genes impaired fitness at 24 h (average dval, 0.05 ± 0.12). Interestingly, 26 genes were assessed as being essential for growth at 24 h but not at 48 h. For these 26 genes, an average dval of 0.012 ± 0.01 was seen at 24 h, whereas an average dval of 0.162 ± 0.22 was seen at 48 h, suggesting that that their growth disadvantage may have been overcome by adaptation to the environment with time. In addition, insertions in an additional 356 genes were found to compromise fitness at 24 h, and those in 329 additional genes compromised fitness after 48 h in murine abscesses (Table S1), with 182 of these genes being in common.
Genes not encoding essential functions but differentially important during infection.
While some genes were considered by our criteria to be essential for survival in infection-related environments (i.e., exhibiting a dval of <0.01), other genes exhibited a change in dval ratio (e.g., dval in infection/dval in outgrowth control) while in neither case becoming essential, reflecting selective pressures on a given gene under a specific growth condition. We therefore identified genes for which the dval ratio consistently changed by at least 4-fold in an infection-related environment compared to the BHI control. By this criterion, insertions in 327 genes were identified with decreased frequency (underrepresented) and those in 103 genes were identified with increased frequency (supergrowers) following growth in one or more infection-related environments (see Table S3 in the supplemental material).
Transposon insertions in 28 genes were underrepresented in mutant pools collected from the abscess and ex vivo physiological fluids, including genes with putative roles in metabolism, such as citrate cycle, carbohydrate metabolism, and purine metabolism genes (see Table S3 in the supplemental material). Among the genes found to be underrepresented under all infection-relevant conditions were those for adenylosuccinate synthetase and lyase (SAOUHSC_00019 and SAOUHSC_02126, respectively). The pool of mutants recovered from the abscess was found to have the greatest number of genes (322 genes) for which representation of transposon insertions decreased at least 4-fold compared to BHI outgrowth control. Most notable among these were genes with known roles in S. aureus virulence, such as agrA, agrB, ccpA, codY, vraR, and rot (39, 40). Additionally, 68 genes with ascribed roles in metabolism were found, with strong selection observed on a number of genes that encode proteins involved in carbon (12/68 genes) and nucleotide (21/68 genes) biosynthetic pathways.
When recovery rates of supergrower mutants were compared between ex vivo fluids and abscess, little overlap was found between growth environments. Sixteen genes were found to be supergrowers in one or more ex vivo physiological fluids (see Table S3 in the supplemental material). Transposon insertions in genes involved in purine nucleotide catabolism were recovered with increased abundance in blood and vitreous fluid (SAOUHSC_00372 and SAOUHSC_00373) compared to the BHI outgrowth control. Mutants with transposon insertions in the gene encoding phosphoglyceromutase (SAOUHSC_00798), involved in glycolysis, were more abundant following outgrowth only in ocular fluids. Sixty-five genes were identified with an increased dval ratio specifically following growth in the abscess. Interestingly, insertion mutants in several known pathogenicity genes were found to be enriched following growth in the abscess, including vraFG/graSR, a system that controls resistance to cationic antimicrobial peptides (41, 42), and staphylococcal accessory regulator A (sarA) involved in virulence and biofilm formation (43).
DISCUSSION
We applied Tn-seq to globally identify genes contributing to S. aureus fitness in infection. Most genes found to be essential were, not surprisingly, essential under all growth conditions. Among essential genes, however, were 71 that were defined as essential (exhibited a fitness dval of 0 < 0.1) under one or more infection conditions while being dispensable for outgrowth in BHI under the conditions tested (see Table S1 in the supplemental material). By looking at all the genes that contribute to S. aureus fitness in an abscess in parallel using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (44), we found that certain metabolic pathways are particularly important (Fig. 4). Nucleotide biosynthesis was found to be important for abscess formation and, to a lesser extent, for growth in ex vivo fluids. This suggests that in an abscess, free nucleotides become limiting. The entire pathway involved in metabolizing aspartate to uridylic acid in pyrimidine synthesis (SAOUHSC_01168-01172 and SAOUHSC_2909) and 11 of the first 13 genes in the de novo purine synthesis pathway (SAOUHSC_00101, SAOUHSC_01008, SAOUHSC_01010, and SAOUHSC_01012-01018) were found to be essential (Fig. 5). Several studies previously showed the importance of nucleotide biosynthesis for pathogens during infection (45–49), and mutants in purA, purN, purH, and purD have previously been reported to limit virulence in an animal model (50), consistent with a link between adenosine synthesis and host immune evasion (51).
FIG 4 .

Metabolic genes contributing to S. aureus survival and proliferation in an abscess model.
FIG 5 .

Nucleotide biosynthesis pathways of importance in abscess at 48 h. Pyrimidine (A) and purine (B) biosynthesis pathways. Genes that are critical for survival in abscess at 48 h but not in rich medium outgrowth control are highlighted in yellow.
Another metabolic pathway that was selected for during abscess growth was the conversion of asparagine to aspartate by the enzyme l-asparaginase (SAOUHSC_01497, Fig. 5). This gene showed a remarkable drop in dval (from a dval of 1 in the BHI outgrowth control to undetectable in the abscess) but to our knowledge has not previously been implicated in S. aureus pathogenicity. The conversion of l-asparagine to l-aspartate can provide substrate for pyrimidine biosynthesis, and so this might also be representative of the importance of nucleotide biosynthesis. Interestingly, asparagine assimilation was recently shown to play an important role in Francisella virulence (52). Additionally, mutants in pyruvate carboxylase (SAOUHSC_01064) and aspartate aminotransferase (SAOUHSC_02158), involved in production of aspartate from pyruvate (53), were observed to be less abundant under all infection conditions tested, indicating a critical role for aspartate during infection. These two genes were previously found to be important contributors to virulence in systemic and abscess murine models, using a small-scale transposon screen (53). The parallel pathway of aspartate biosynthesis, mediated by l-asparaginase, was inferred to be unimportant (53), whereas we find this gene to be crucial for growth in an abscess infection.
Among genes that were found to be crucial for infection, we identified many of the genes previously reported to be important for S. aureus pathogenicity, including toxins and transcription factors. This provides an internal control for the method. Importantly, we discovered many new genes, which to our knowledge have not previously been implicated as being important in S. aureus infection. Of the 426 genes contributing to infection (see Table S1 in the supplemental material), over half (246 genes, 57.7%) have unknown functional roles, representing genes which present intriguing targets for new antimicrobials. Bioinformatic interrogation of these hypothetical genes reveals that many are highly conserved in other S. aureus genomes and among related bacteria. Several of the genes annotated as hypothetical are small ORFs (<200 bp) that are identified in only a few S. aureus genomes in the NCBI database. An intriguing possibility is that rather than encoding proteins, these essential regions of the genome may encode regulatory small RNAs or other important signals governing expression of nearby genes with roles in virulence (12, 18, 54). Other hypotheticals appear to possess motifs related to transporter functions or involvement in signal transduction and gene expression, for which roles in infection had not previously been identified.
In addition to identifying genes with critical roles in fitness, Tn-seq also identified genes whose disruption leads to increased fitness in a specific ecology. These genes, termed supergrowers, appear with greater frequency following growth under a given condition compared to the outgrowth control, suggesting that deletion of the respective gene provided a fitness advantage in the given niche. Surprisingly, we found such mutations in genes that have previously been shown, in monoculture, to be required for virulence and pathogenicity in animal models, for example, the pleotropic regulator of virulence gene expression sarA (SAOUHSC_00620) (55). The virulence proteins expressed under SarA control are largely extracellular factors important for the survival of the entire population, also known as “community goods” (30). Mutants within the population that do not make these extracellular products experience a fitness advantage, by benefiting from these products without the fitness cost of producing them. Similarly, extracellular complementation of community goods may mask the impact on fitness of mutations in genes encoding those products. Mutations in these genes will likely not be captured as important by this approach, meaning that the findings reported here represent a conservative estimation of genes contributing to S. aureus fitness in the ecologies tested.
The supergrowers that we have identified in this study, such as sarA, may behave similarly to mutant phenotypes that arise during natural infection, also known as “bacterial cheaters” (30). These findings suggest that Tn-seq may have utility for predicting genes that could produce bacterial cheater phenotypes during infections.
While others have used similar methods to define essential genes in S. aureus, using transposon mutants, either in isolation (56) or in pools (11), or by transcript ablation (13), these studies were largely conducted with comparatively small sets of mutants under laboratory growth conditions, although one also assessed virulence in a Caenorhabditis elegans model (10). The vast majority of previously identified essential genes were also found to be essential in our BHI input pool despite analytical differences between approaches. Indeed, of the 350 genes recently described to be essential for growth under laboratory conditions (11), 284 were found here to be associated with a dval of <0.1 and were considered essential in our study (Fig. 2; see also Table S2 in the supplemental material). When fitness-compromising genes found here are included in the comparison, we capture 91.1% of the previously described essential genes. The remaining 8.9% of genes found by others to be essential were found to have a median dval of 0.373, ranging from 0.107 to 4.100, with only a few falling just above our curation cutoff (6 genes with a dval range of 0.107 to 0.188). Others were clearly nonessential and were well covered both with insertions and with reads in our study. The majority of these inconsistencies are likely attributable to the massively parallel and internally controlled nature of the Tn-seq approach for interrogating fitness, whereas others may be due to subtle differences in methods.
Importantly, we found 136 genes that fulfilled our criteria for essentiality that were not described by Chaudhuri et al. or others (10, 11, 13, 14, 57, 58), constituting a pool of potential new targets that could be exploited in the development of novel therapeutic interventions (59). This included a wealth of previously unidentified and hypothetical genes that appear to contribute to fitness in infection-relevant environments. Uncovering the functions of these genes will shed new light on S. aureus pathogenicity and host interaction.
MATERIALS AND METHODS
Staphylococcus aureus strain HG003 was chosen as the background strain to generate the Tn-seq library. Inducible antisense constructs were initially generated in Escherichia coli DH5α and subsequently tested in S. aureus strain RN4220. The mariner-based bursa aurealis transposon insertion system (60) was used to construct a mutagenized library in HG003 essentially as described by Bae et al. (57). An overview of the experimental strategy to determine genes essential for fitness in competitive, infection-relevant contexts is illustrated in Fig. S1 in the supplemental material (see also Text S1 for further description of methods). Briefly, a 10-µl aliquot of this culture, containing 107 CFU, was then used to inoculate 10 ml of fresh BHI (control) and 10 ml of bovine aqueous or vitreous fluids buffered with 25 mM morpholinepropanesulfonic acid (MOPS). Peripheral blood was freshly isolated from healthy volunteers into heparinized Vacutainer collection tubes (BD, Franklin Lakes, NJ) according to the Massachusetts Eye and Ear Infirmary Institutional Review Board-approved protocol 11-093H. Preliminary studies examining culture of these mutants found a decrease in viable cells following inoculation specifically in blood; therefore, a 100-µl inoculum representing 108 CFU, into 10 ml, was adopted to overcome potential population bottlenecks in the library pool. Bacterial growth in physiological fluids occurred at 37°C with shaking at 225 rpm. Biological replicates were conducted for each growth condition. Bacteria were grown for 24 h, and 9 to 10 generations of outgrowth occurred under each condition, prior to harvest and analyses (see Text S1 in the supplemental material for further description of methods). The competitive ability of various mariner insertion mutants of S. aureus to proliferate at the site of infection was tested using a murine abscess model, essentially as described elsewhere (38) (Fig. S3; see also Text S1 in the supplemental material for further description of methods).
Briefly, a 100-µl aliquot of the input library freezer stock, representing 108 CFU, was inoculated into 100 ml of BHI without antibiotic selection and was cultured overnight at 37°C. Following overnight growth and 1:1,000 dilution into fresh medium, a 1-ml sample of exponential-growth-phase cells was harvested, centrifuged, resuspended in 2 ml sterile phosphate-buffered saline (PBS), diluted 1:10 to achieve 107 CFU/ml in 1 ml sterile PBS, and mixed with an equal volume of autoclaved Cytodex-1 beads (60 to 87 µm; Sigma, St. Louis, MO). A 200-µl aliquot containing 5 × 106 CFU was injected subcutaneously into each shaved hind flank of a 4- to 5-week-old Swiss Webster male mouse, which was anesthetized with ketamine and xylazine and allowed to form abscesses for 24 or 48 h before harvest and analyses. Methods for DNA sequencing and Tn-seq bioinformatic analyses are described in the work of Klein et al. (61) and in Text S1 in the supplemental material. Inducible antisense testing and expanded methods for strain selection and library generation are described in Text S1.
SUPPLEMENTAL MATERIAL
Supplemental materials and methods. Download
Experimental strategy to determine genes essential for fitness in competitive, infection-relevant contexts. The transposon mutant pool (a) was first cultured in nutrient broth (b) to generate a reproducible inoculum for subsequent experiments. To identify genes contributing to fitness in infection, bacteria were inoculated in duplicate ex vivo into aqueous fluid, vitreous fluid, and whole blood (d). Additionally, genes contributing to persistence and proliferation in an abscess in vivo (e) were identified. Each output was sequenced and compared to nutrient broth outgrowth control (c). Download
Reproducibility of biological replicates. Regression statistics for independent biological replicates. Transposon density is the number of transposon insertions per kilobase, calculated independently for each ORF. dval is the number of transposon insertions found in a given ORF versus the number expected based on the size of the ORF. Download
Analysis of mariner mutant population changes in murine abscess by Tn-seq. (A) Flow chart of experimental strategy. (B) Analyses performed (abscesses generated on hind flank illustrated in yellow). (C) Representative abscesses at 24 h (a) and 48 h (b) prior to excision. Download
Genes contributing to S. aureus fitness across various environments.
Comparison of genes curated as essential after growth in BHI in this study compared to previous study of essentiality in S. aureus.
Genes which are either 4-fold underrepresented or 4-fold overrepresented following growth in infection compared to BHI outgrowth control.
Primers used in this study.
ACKNOWLEDGMENTS
This work was supported by NIH/NIAID grant AI107248 and the Harvard-wide Program on Antibiotic Resistance AI083214. M.D.V. was supported in part by the Harvard Medical School/Massachusetts Eye and Ear Infirmary Molecular Bases of Eye Disease fellowship training grant (T32-EY007145).
We thank R. Losick, in whose lab L.F.’s contribution to this work was carried out. We thank F. Lebreton and M. Ramsey for helpful comments during manuscript preparation.
Footnotes
Citation Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Santa Maria J, Jr, Lazinski DW, Camilli A, Walker S, Hooper DC, Gilmore MS. 2014. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 5(5):e01729-14. doi:10.1128/mBio.01729-14.
REFERENCES
- 1. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. 10.1001/jama.298.15.1763 [DOI] [PubMed] [Google Scholar]
- 2. Otto M. 2010. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 64:143–162. 10.1146/annurev.micro.112408.134309 [DOI] [PubMed] [Google Scholar]
- 3. Callegan MC, Jett BD, Hancock LE, Gilmore MS. 1999. Role of hemolysin BL in the pathogenesis of extraintestinal Bacillus cereus infection assessed in an endophthalmitis model. Infect. Immun. 67:3357–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. 2002. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. U. S. A. 99:7687–7692. 10.1073/pnas.122108599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention (CDC) 2002. Staphylococcus aureus resistant to vancomycin—United States, 2002. JAMA 288:824–825. 10.1001/jama.288.7.824 [DOI] [PubMed] [Google Scholar]
- 6. Centers for Disease Control and Prevention (CDC) 2002. Staphylococcus aureus resistant to vancomycin—United States, 2002. MMWR Morb. Mortal. Wkly. Rep. 51:565–567 [PubMed] [Google Scholar]
- 7. Kos VN, Desjardins CA, Griggs A, Cerqueira G, Van Tonder A, Holden MT, Godfrey P, Palmer KL, Bodi K, Mongodin EF, Wortman J, Feldgarden M, Lawley T, Gill SR, Haas BJ, Birren B, Gilmore MS. 2012. Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with methicillin-resistant S. aureus hospital-acquired infection in the United States. mBio 3(3):e00112-12. 10.1128/mBio.00112-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rong SL, Leonard SN. 2010. Heterogeneous vancomycin resistance in Staphylococcus aureus: a review of epidemiology, diagnosis, and clinical significance. Ann. Pharmacother. 44:844–850. 10.1345/aph.1M526 [DOI] [PubMed] [Google Scholar]
- 9. Jarvis WR, Sinkowitz-Cochran RL. 2001. Emerging healthcare-associated problem pathogens in the United States. Postgrad. Med. 109(2 Suppl):3–9. 10.3810/pgm.02.2001.suppl12.60 [DOI] [PubMed] [Google Scholar]
- 10. Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. U. S. A. 101:12312–12317. 10.1073/pnas.0404728101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, Burgis TA, Lockyer M, Garcia-Lara J, Foster SJ, Pleasance SJ, Peters SE, Maskell DJ, Charles IG. 2009. Comprehensive identification of essential Staphylococcus aureus genes using transposon-mediated differential hybridisation (TMDH). BMC Genomics 10:291. 10.1186/1471-2164-10-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L. 2011. The essential genome of a bacterium. Mol. Syst. Biol. 7:528. 10.1038/msb.2011.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu Z-Y, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 43:1387–1400. 10.1046/j.1365-2958.2002.02832.x [DOI] [PubMed] [Google Scholar]
- 14. Ji Y, Zhang B, Van SF, Horn, Warren P, Woodnutt G, Burnham MK, Rosenberg M. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 293:2266–2269. 10.1126/science.1063566 [DOI] [PubMed] [Google Scholar]
- 15. Xu HH, Trawick JD, Haselbeck RJ, Forsyth RA, Yamamoto RT, Archer R, Patterson J, Allen M, Froelich JM, Taylor I, Nakaji D, Maile R, Kedar GC, Pilcher M, Brown-Driver V, McCarthy M, Files A, Robbins D, King P, Sillaots S, Malone C, Zamudio CS, Roemer T, Wang L, Youngman PJ, Wall D. 2010. Staphylococcus aureus TargetArray: comprehensive differential essential gene expression as a mechanistic tool to profile antibacterials. Antimicrob. Agents Chemother. 54:3659–3670. 10.1128/AAC.00308-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6:767–772. 10.1038/nmeth.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Opijnen T, Camilli A. 2012. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 22:2541–2551. 10.1101/gr.137430.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, Rosch JW. 2012. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog. 8:e1002788. 10.1371/journal.ppat.1002788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. 2009. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. U. S. A. 106:16422–16427. 10.1073/pnas.0906627106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sassetti CM, Boyd DH, Rubin EJ. 2001. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 98:12712–12717. 10.1073/pnas.231275498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84. 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 22. Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7:e1002251. 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. 2012. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 8:e1002946. 10.1371/journal.ppat.1002946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6:279–289. 10.1016/j.chom.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, McIver KS. 2013. Genome-wide identification of genes required for fitness of group A Streptococcus in human blood. Infect. Immun. 81:862–875. 10.1128/IAI.00837-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, Wain J, Parkhill J, Turner AK, Turner AK. 2009. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19:2308–2316. 10.1101/gr.097097.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallagher LA, Shendure J, Manoil C. 2011. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio 2(1):e00315-10. 10.1128/mBio.00315-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 100:14339–14344. 10.1073/pnas.2036282100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJ, Willems RJ, van Schaik W. 2012. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet. 8:e1002804. 10.1371/journal.pgen.1002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Popat R, Crusz SA, Messina M, Williams P, West SA, Diggle SP. 2012. Quorum-sensing and cheating in bacterial biofilms. Proc. Biol. Sci. 279:4765–4771. 10.1098/rspb.2012.1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ji Y, Yin D, Fox B, Holmes DJ, Payne D, Rosenberg M. 2004. Validation of antibacterial mechanism of action using regulated antisense RNA expression in Staphylococcus aureus. FEMS Microbiol. Lett. 231:177–184. 10.1016/S0378-1097(03)00931-5 [DOI] [PubMed] [Google Scholar]
- 32. Cassat JE, Skaar EP. 2012. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin. Immunopathol. 34:215–235. 10.1007/s00281-011-0294-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haley KP, Skaar EP. 2012. A battle for iron: host sequestration and Staphylococcus aureus acquisition. Microbes Infect. 14:217–227. 10.1016/j.micinf.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bullen JJ. 1981. The significance of iron in infection. Rev. Infect. Dis. 3:1127–1138. 10.1093/clinids/3.6.1127 [DOI] [PubMed] [Google Scholar]
- 35. Cederlund A, Gudmundsson GH, Agerberth B. 2011. Antimicrobial peptides important in innate immunity. FEBS J. 278:3942–3951. 10.1111/j.1742-4658.2011.08302.x [DOI] [PubMed] [Google Scholar]
- 36. Garreis F, Gottschalt M, Paulsen FP. 2010. Antimicrobial peptides as a major part of the innate immune defense at the ocular surface. Dev. Ophthalmol. 45:16–22. 10.1159/000315016 [DOI] [PubMed] [Google Scholar]
- 37. Bunce C, Wheeler L, Reed G, Musser J, Barg N. 1992. Murine model of cutaneous infection with gram-positive cocci. Infect. Immun. 60:2636–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding Y, Onodera Y, Lee JC, Hooper DC. 2008. NorB, an efflux pump in Staphylococcus aureus strain MW2, contributes to bacterial fitness in abscesses. J. Bacteriol. 190:7123–7129. 10.1128/JB.00655-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Camargo IL, Gilmore MS. 2008. Staphylococcus aureus—probing for host weakness? J. Bacteriol. 190:2253–2256. 10.1128/JB.00043-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Foster TJ, Geoghegan JA, Ganesh VK, Höök M. 2014. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12:49–62. 10.1038/nrmicro3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Falord M, Karimova G, Hiron A, Msadek T. 2012. GraXSR proteins interact with the VraFG ABC transporter to form a five-component system required for cationic antimicrobial peptide sensing and resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 56:1047–1058. 10.1128/AAC.05054-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Falord M, Mäder U, Hiron A, Débarbouillé M, Msadek T. 2011. Investigation of the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress response and cell wall signal transduction pathways. PLoS One 6:e21323. 10.1371/journal.pone.0021323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS. 2004. Global gene expression in Staphylococcus aureus biofilms. J. Bacteriol. 186:4665–4684. 10.1128/JB.186.14.4665-4684.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27:29–34. 10.1093/nar/27.20.e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kirsch DR, Whitney RR. 1991. Pathogenicity of Candida albicans auxotrophic mutants in experimental infections. Infect. Immun. 59:3297–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McFarland WC, Stocker BA. 1987. Effect of different purine auxotrophic mutations on mouse-virulence of a Vi-positive strain of Salmonella Dublin and of two strains of Salmonella typhimurium. Microb. Pathog. 3:129–141. 10.1016/0882-4010(87)90071-4 [DOI] [PubMed] [Google Scholar]
- 47. Mei JM, Nourbakhsh F, Ford CW, Holden DW. 1997. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature-tagged mutagenesis. Mol. Microbiol. 26:399–407. 10.1046/j.1365-2958.1997.5911966.x [DOI] [PubMed] [Google Scholar]
- 48. Polissi A, Pontiggia A, Feger G, Altieri M, Mottl H, Ferrari L, Simon D. 1998. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 66:5620–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. 2008. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 4:e37. 10.1371/journal.ppat.0040037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lan L, Cheng A, Dunman PM, Missiakas D, He C. 2010. Golden pigment production and virulence gene expression are affected by metabolisms in Staphylococcus aureus. J. Bacteriol. 192:3068–3077. 10.1128/JB.00928-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thammavongsa V, Kern JW, Missiakas DM, Schneewind O. 2009. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J. Exp. Med. 206:2417–2427. 10.1084/jem.20090097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gesbert G, Ramond E, Rigard M, Frapy E, Dupuis M, Dubail I, Barel M, Henry T, Meibom K, Charbit A. 2014. Asparagine assimilation is critical for intracellular replication and dissemination of Francisella. Cell. Microbiol. 16:434–449. 10.1111/cmi.12227 [DOI] [PubMed] [Google Scholar]
- 53. Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, Buysse JM. 2004. Large-scale identification of genes required for full virulence of Staphylococcus aureus. J. Bacteriol. 186:8478–8489. 10.1128/JB.186.24.8478-8489.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruiz de los Mozos I, Vergara-Irigaray M, Segura V, Villanueva M, Bitarte N, Saramago M, Domingues S, Arraiano CM, Fechter P, Romby P, Valle J, Solano C, Lasa I, Toledo-Arana A. 2013. Base pairing interaction between 5′- and 3′-UTRs controls icaR mRNA translation in Staphylococcus aureus. PLoS Genet. 9:e1004001. 10.1371/journal.pgen.1004001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chien Y, Manna AC, Projan SJ, Cheung AL. 1999. SarA, a global regulator of virulence determinants in Staphylococcus aureus, binds to a conserved motif essential for sar-dependent gene regulation. J. Biol. Chem. 274:37169–37176. 10.1074/jbc.274.52.37169 [DOI] [PubMed] [Google Scholar]
- 56. Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4(1):e00537-12. 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bae T, Glass EM, Schneewind O, Missiakas D. 2008. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 416:103–116. 10.1007/978-1-59745-321-9_7 [DOI] [PubMed] [Google Scholar]
- 58. Ko KS, Lee JY, Song J-H, Baek JY, Oh WS, Chun J, Yoon HS. 2006. Screening of essential genes in Staphylococcus aureus N315 using comparative genomics and allelic replacement mutagenesis. J. Microbiol. Biotechnol. 16:623–632 [Google Scholar]
- 59. Salgado-Pabón W, Schlievert PM. 2014. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat. Rev. Microbiol. 12:585–591. 10.1038/nrmicro3308 [DOI] [PubMed] [Google Scholar]
- 60. Lampe DJ, Churchill ME, Robertson HM. 1996. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 15:5470–5479 [PMC free article] [PubMed] [Google Scholar]
- 61. Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. 2012. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13:578. 10.1186/1471-2164-13-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods. Download
Experimental strategy to determine genes essential for fitness in competitive, infection-relevant contexts. The transposon mutant pool (a) was first cultured in nutrient broth (b) to generate a reproducible inoculum for subsequent experiments. To identify genes contributing to fitness in infection, bacteria were inoculated in duplicate ex vivo into aqueous fluid, vitreous fluid, and whole blood (d). Additionally, genes contributing to persistence and proliferation in an abscess in vivo (e) were identified. Each output was sequenced and compared to nutrient broth outgrowth control (c). Download
Reproducibility of biological replicates. Regression statistics for independent biological replicates. Transposon density is the number of transposon insertions per kilobase, calculated independently for each ORF. dval is the number of transposon insertions found in a given ORF versus the number expected based on the size of the ORF. Download
Analysis of mariner mutant population changes in murine abscess by Tn-seq. (A) Flow chart of experimental strategy. (B) Analyses performed (abscesses generated on hind flank illustrated in yellow). (C) Representative abscesses at 24 h (a) and 48 h (b) prior to excision. Download
Genes contributing to S. aureus fitness across various environments.
Comparison of genes curated as essential after growth in BHI in this study compared to previous study of essentiality in S. aureus.
Genes which are either 4-fold underrepresented or 4-fold overrepresented following growth in infection compared to BHI outgrowth control.
Primers used in this study.