

1. Introduction
For many decades, the design of new nucleoside analogs as potential therapeutic agents focused on both sugar and nucleobase modifications. These nucleoside analogs rely on cellular kinases to undergo stepwise addition of phosphate groups to form the corresponding active nucleoside triphosphate to express their therapeutic effect.1 However, nucleosides triphosphates cannot be considered as viable drug candidates as they usually have poor chemical stability along with high polarity that hinders them from transporting across cell membranes. Within the nucleoside analog phosphate activation process, the first phosphorylation has often been identified as the limiting step, which led medicinal chemists to prepare stable “protected” monophosphate nucleosides capable of delivering nucleoside monophosphates intracellularly. These nucleoside monophosphate prodrugs are designed to efficiently cross the biological barriers (as opposed to nucleoside monophosphates; Figure 1, eq 1) and reach the targeted cells or tissues. Once inside the cell, the biolabile protecting groups are then degraded enzymatically and/or chemically, releasing the free nucleoside analog in the monophosphate form, which can often efficiently express its therapeutical potency by intracellular conversion to the corresponding nucleoside triphosphate (Figure 1, eq 2).
Figure 1.

Mechanism of action of nucleoside monophosphate prodrugs.
Interestingly, the use of such phosph(on)ate prodrugs has not only proved to enhance the activity of parent nucleosides, but also generated potent compounds otherwise inactive in their nucleoside form because of a lack of monophosphorylation. Proof of concept for monophosphate prodrugs has now been clinically validated in the human immunodeficiency virus (HIV), hepatitis B (HBV), and hepatitis C virus (HCV) fields, leading to several potent and selective prodrugs such as the phase II pradefovir,2 the phase III GS-7340 (TAF),3 and the FDA-approved tenofovir disoproxil fumarate (TDF)4 and sofosbuvir (GS/PSI-7977) (Figure 2).5
Figure 2.

Examples of clinical nucleoside prodrugs with anti-HIV, -HBV, or -HCV activities.
Several strategies allowing intracellular delivery of nucleotide analogs were developed over the past 20 years based on the design of many different types of phosphate and phosphonate nucleoside prodrugs (Figure 3). Reviews on nucleoside phosph(on)ate prodrugs generally focus on their enhanced biological activities, potential therapeutic interest, and their physicochemical properties,6,7 but almost completely neglect their sometimes challenging synthetic preparation.8 Herein, we review the most important mono-, di-, and triphosphate and phosphonate prodrug approaches applied to nucleoside analogs (Figure 3) from a chemical point of view, detailing the strengths and limitations of each approach. We will focus on the various synthetic pathways discussing (1) the chemical variation of the biolabile phosph(on)ate masking groups; (2) the reliability of using P(III) and/or P(V) chemistry for both phosphate and phosphonate prodrug synthesis; (3) the influence of the masking group(s) introduction conditions (solvent, temperature, stoichiometry) on the overall outcome for each method; (4) the various protection/deprotection strategies used to impart improved yield and regioselectivity relative to the nature of the nucleobase and the sugar; and (5) the influence of reaction conditions or protective groups on the stereoselectivity (Rp/Sp) observed at the phosphorus center as well as the methods employed to separate both Rp and Sp isomers along with the asymmetric strategies for the synthesis of predominantly single diastereoisomers at the phosphorus center.
Figure 3.

Prodrug approaches detailed in this Review.
2. Nomenclature
Phosphorus is often covalently bonded to common atoms such as C, H, O, N, S, forming various chemical species or functional groups. The different categories of phosphorus functional groups are so extensive that confusion in nomenclature or misused terms is common. As a resource and useful for an in depth understanding of this Review, Table 1 presents an extensive summary of the nomenclature of the principal phosphorus moieties widely used in organic chemistry. Structures and functional group names are classified by the presence of O, C, N, and/or S attached to the phosphorus atom and by its valence (III or V).
Table 1. Functional Group Names of P(III) and P(V) Moieties.

3. Nucleoside Monophosphate Prodrugs
3.1. Nucleoside Phosphates and Phosphonates O-PO(OR)2 and C-PO(OR)2
3.1.1. Carbonyloxymethyl (Including POM, POC)
To date, the only nucleosides phosph(on)ate prodrugs approved by the FDA are the acyclic nucleoside phosphonates adefovir dipivoxil [bis(pivaloyloxymethyl), POM]9 and tenofovir disoproxil fumarate [bis(isopropyloxymethyl carbonate, POC].4 Adefovir dipivoxil was initially developed for HIV,10,11 but studies were stopped due to severe kidney toxicity at the dosage necessary for good antiviral response. In 2002, further investigation of the compound12 for the treatment of HBV infection led to FDA approval of adefovir dipivoxil. The structurally related tenofovir disoproxil fumarate had a more favorable toxicity profile and was approved in 2001 for the treatment of HIV infection. Both POM and POC groups have been shown to increase oral bioavailability13,14 and overall systemic exposure to the parent phosphonic acid compound. More recently, LB80380, a nucleotide bis(POM)-prodrug,15 completed a phase II clinical trial for the treatment of lamivudine resistant HBV infection (Figure 4).16
Figure 4.

Examples of carbonyloxymethyl nucleotide prodrugs approved by the FDA or in clinical trials.
The degradation of POC-prodrugs involves the enzymatic cleavage of the carbonate by an esterase leading to an unstable carboxylate intermediate that undergoes two subsequent chemical degradations to form carbon dioxide, formaldehyde, and the nucleotide POC-monoester. Repetition of this sequence with the second POC group or its direct cleavage by phosphodiesterase frees the nucleoside monophosphate (Figure 5).
Figure 5.

Activation of carbonate-type prodrugs (including POC, R = i-Pr).
In the case of nucleoside prodrugs bearing POM protecting groups, the ester is cleaved to form an unstable hydroxymethyl alcoholate intermediate that undergoes chemical rearrangement to form formaldehyde and the free monophosphate after the second POM degradation (Figure 6).
Figure 6.

Activation of ester-type prodrugs (including POM, R = t-Bu).
The synthetic approaches for carbonyloxymethyl phosphate nucleoside prodrugs are summarized in Figure 7: (A) coupling of a nucleoside monophosphate with a halogeno carbonyloxymethyl derivative (POM-Cl for example), (B) reaction of a bis(carbonyloxymethyl)-phosphorochloridate with a nucleoside under basic conditions, (C) Mitsunobu coupling between a nucleoside and bis(carbonyloxymethyl)-phosphate, and (D) 5′-iodination of a nucleoside followed by bis(POM)-phosphate salt nucleophilic substitution.
Figure 7.

Methods to access carbonyloxymethyl phosphate nucleosides prodrugs.
The more limited methods to access carbonyloxymethyl phosphonates prodrugs are compiled in Figure 8: (A) reaction of the phosphonic acid nucleoside with an halogeno carbonyloxymethyl derivative, and (B) direct conversion of dimethylphosphonate nucleoside using sodium iodide and a halogeno carbonyloxymethyl derivative.
Figure 8.

Methods to access carbonyloxymethyl phosphonates nucleosides prodrugs.
3.1.1.1. Synthesis of Carbonyloxymethyl Phosphates Diesters
In 1984, Farquhar and co-workers were first to report the synthesis of bis(carbonyloxymethyl)phosphate derivatives along with their stability in different buffers, in the presence of liver esterase and in plasma.17,18 They developed two synthetic routes to synthesize the bis(POM)-monophosphate prodrug of 5-FdU 2a,b either by Mitsunobu coupling of (1a,b) with bis(POM)-phosphate 7 or by substitution of a 5-iodo nucleoside 3 with bis(POM)-phosphate silver salt 8 (Scheme 1). The later method was found to be low yielding, and the 3′-acetate could not be removed selectively because of the lack of POM group stability under deprotection conditions. Using this approach, numerous nucleosides of biological interest were transformed in their bis(POM)-monophosphate prodrugs including 5-FdU,17a,18 2′,3′-dideoxyuridine (ddU),19 3′-azido-3′-deoxythymidine (AZT),20 and thymidine.21
Scheme 1. Synthesis of 5-FdU Bis(POM)-monophosphate Prodrug.

Both phosphates 7 and 8 were prepared from common intermediate 6 (Scheme 2), obtained by the reaction between disilver aryl phosphate 5 and iodomethyl pivalate at room temperature. Hydrogenation of 6, precipitation as a cyclohexylammonium salt, and ion exchange on H+-resin provided (7). Subsequent transformation of bis(POM)-phosphate 7 into its sodium salt with Na+-resin and final treatment with an aqueous solution of silver nitrate lead to desired silver salt 8 (Scheme 2).
Scheme 2. Preparation of Reagents 7 and 8.

Rose et al.22 reported the synthesis of α/β 2′-deoxy-4′-thioadenosine bis(POM)-monophosphate prodrug 10 by Mitsunobu coupling between bis(POM)-phosphate 7 and purine nucleosides 9 (Scheme 3).
Scheme 3. Synthesis of 2′-Deoxy-4′-thioadenosine Bis(POM)-monophosphate Prodrug.

Interestingly, applied to the synthesis of 8-bromo-2′-deoxyadenosine bis(POM)-phosphate prodrug, the same method22 led to an unexpected side reaction of elimination/dehydration, yielding exocyclic methylene compound 12 (Scheme 4). To circumvent this elimination problem, nucleoside monophosphate 13 was coupled with commercially available chloromethyl pivalate. According to the authors, the low yield of 8-bromo-2′-deoxyadenosine bis(POM)-prodrug 14 (19%) was due to repeated chromatographic purification.
Scheme 4. Synthesis of 8-Bromo-2′-deoxyadenosine Bis(POM)-phosphate Prodrug.

In 1995, considering the combersome preparation of bis(POM)-phosphate nucleoside prodrugs, Imbach et al. developed a new approach allowing conversion of a nucleoside monophosphate into its corresponding bis(POM)-monophosphate.23 Thus, AZT monophosphate 15 was reacted with iodomethyl pivalate and diisopropyl ethylamine in acetonitrile for 4 days at room temperature to afford AZT bis(POM)-monophosphate prodrug 16 in 22% yield (Scheme 5).
Scheme 5. Conversion of a Monophosphate into Its Corresponding Bis(POM)-monophosphate Nucleoside.

To increase the reactivity of the nucleoside monophosphate during the coupling with POM-I, Kang et al.24 choose to preactivate the phosphate moiety as a tributylstannyl salt by using tributyltin methoxide. As illustrated in Scheme 6, 2′-azido-2′-deoxyuridine monophosphate 17 was first reacted with 2 equiv of tributylstannyl methoxide, then coupled with iodomethyl pivalate in the presence of tetrabutylammonium bromide to deliver bis(POM)-prodrug 18 after purification on reverse phase HPLC. Despite a good overall yield, the use of tin derivatives represents a serious limitation because of the possible presence of toxic tin residues incompatible with further biological evaluations.
Scheme 6. Preactivation of the Phosphate Moiety as a Tributylstannyl Salt.

In 2004, Hwang and Cole developed a new approach using new bis(POM)-phosphorochloridate 21.25 This reagent was synthesized efficiently in five steps from trimethylphosphate by treatment with sodium iodide and chloromethyl pivalate, monodeprotection, and subsequent chlorination with oxalyl chloride. The coupling of AZT with bis(POM)-phosphorochloridate 21 in the presence of triethylamine allowed for the formation of desired AZT bis(POM)-monophosphate prodrug 16 in 47% yield (Scheme 7).
Scheme 7. Use of Bis(POM)-phosphorochloridate.

3.1.1.2. Synthesis of Carbonyloxymethyl Phosphate Monoesters
The POM-phosphate monoesters have also been synthesized. Although these compounds are sometimes evaluated for their biological activities, they are generally prepared as a reference for metabolic degradation studies.
Farquhar et al.18 reported the synthesis of 5-FdU POM-phosphate monoester as a reference during the degradation study of 5-FdU bis(POM)-prodrug. Starting from the dibenzyl phosphate silver salt, the POM-protecting group was introduced by reaction with chloromethyl pivalate. POM-Phosphate 23 was obtained by catalytic hydrogenation, precipitation of cyclohexylammonium salts, and neutralization over acidic resin. The coupling between the dihydrogen POM-phosphate 23 and 5-FdU with DCC in pyridine afforded POM-5-FdU monophosphate monoester prodrug 2b in 53% yield (Scheme 8).
Scheme 8. Synthesis of 5-FdU POM-Phosphate Monoester.

3.1.1.3. Synthesis of 3′-5′-Cyclic Carbonyloxymethyl Phosphates
Tsien et al.26 prepared acetoxymethyl ester prodrugs of N6,O2′-dibutyryl adenosine- and N2,O2′-dibutyryl guanosine-3′,5′-cyclic monophosphate, with the intention of increasing intracellular delivery of second messengers cAMP and cGMP. The coupling of either diisopropylethylammonium or silver salts of adenosine-3′,5′-cyclic monophosphate 24 with acetoxymethyl bromide afforded the acetoxymethyl prodrug as a mixture of two diastereoisomers 25 and 26 (RP/SP) separated by silica gel chromatography. Interestingly, the diastereomeric ratio was found to be dramatically different depending on the method used as the first one afforded a 65:35 mixture in favor of the fast eluting isomer contrary to the 23:77 mixture obtained with the second method (Scheme 9). On the other hand, cGMP prodrug was prepared as a nonseparable mixture of two diastereoisomers (from derivative X = H) using the DIPEA method.
Scheme 9. Synthesis of N2,O2′-Dibutyryl Adenosine-3′,5′-cyclic Monophosphate.

In 2007, Gunic et al.27 reported the synthesis of base modified 2′-C-methyl ribonucleosides cyclic monophosphate prodrugs that exhibited potent anti-HCV activities. 5′-Phosphorylation of nucleosides 27 with POCl3 and P(O)(OMe)3 and subsequent cyclization using DCC in pyridine afforded cyclic monophosphate nucleosides 29 in 30% yield (Scheme 10). Finally, coupling with either chloromethyl pivalate or chloromethyl isopropyl carbonate in the presence of diisopropylethylamine afforded cyclic POM- and POC-prodrugs 30 in low to moderate yields (Scheme 10).
Scheme 10. Synthesis of 2′-C-Methyl Ribonucleosides Cyclic Monophosphates.

3.1.1.4. Carbonyloxymethyl Phosphonates
The first synthesis of bis(carbonyloxymethyl)-nucleoside phosphonate prodrug was reported by Starrett et al.13,28 who prepared the bis(POM)-, bis(isobutyryloxymethyl)-, and bis(propionyloxymethyl)-prodrugs of adefovir (PMEA). At first, the coupling between chloromethyl pivalate or iodomethyl pivalate and various inorganic (Ag+, Li+, K+, Na+, Cs+) or organic salts (Et3NH+, (i-Pr)2N+EtH, n-Bu4N+) of PMEA did not lead to the desired prodrug 31. Finally, bis(POM)-PMEA was obtained in 40% yield from PMEA by using N,N′-dicyclohexylmorpholine carboxamidine (DCMC) as the coupling agent and chloromethyl pivalate. However, the same procedure was not found suitable for 3-hydroxy-2-phosphonomethoxypropyl nucleosides such as HPMP-5-azaC, because the reaction lead to an inseparable mixture of bis(POM)-ester 32 and cyclic POM-monoester phosphonates 33 (Scheme 11). Optimization of the reaction conditions (using other salts in place of DCMC, temperature, and solvents) was not successful.29
Scheme 11. Difference of Reactivity between PMEA versus HPMP-5-azaC Derivatives.

A similar procedure was used by Choi et al.15 for the synthesis of 9-[1-phosphonomethoxy cyclopropyl)methyl]-6-deoxyguanine dipivoxil LB80380. The nucleoside prodrug was obtained in two steps by hydrolysis of the diisopropyl phosphonate diester 34 with trimethylsilyl bromide and coupling of the resulting phosphonic acid 35 with POM-Cl in the presence of triethylamine and 1-methyl-2-pyrrolidinone (Scheme 12).
Scheme 12. Synthesis of LB80380.

The same procedure was used by Tang et al.30 to synthesize several PMEA and PMPA bis(alkyloxymethyl)-carbonate prodrugs. Chloromethyl carbonates 37 were prepared in 60–75% yield from methyl chloroformate, by chlorination with a large excess of sulfuryl chloride in the presence of catalytic AIBN, followed by addition of the corresponding alcohol in pyridine. The coupling of PMEA or PMPA 38 with 4.5 equiv of chloromethyl carbonates, 37, gave crude 39, which were converted into their more stable fumarate salts 40 in 50–70% yield (Scheme 13).
Scheme 13. Synthesis of Several PMEA and PMPA Bis(alkyloxymethyl) Carbonate Prodrugs.

The same procedure was reported by Mackman et al.31 to prepare bis(POC)-5′-phosphonomethoxy prodrugs of potent nucleosides such as d4T, AZT, ddC, or ddT. Phosphonomethoxy-d4T and -ddC derivatives were synthesized by electrophilic addition of dimethyl hydroxymethyl phosphonate to furanoid glycal 41.32,33 After oxidative deselenylation, deprotection of the phosphonate moiety and hydrogenation of the double bond, the resulting phosphonic acid salt 45 was converted to the bis(POC)-prodrug 46 by coupling with chloromethylisopropyl carbonate in the presence of triethylamine (Scheme 14).
Scheme 14. Synthesis of Bis(POC)-prodrug 46.

This method was later used for the synthesis of the bis(POC)-5′-phosphonomethoxy 2′-Fd4A prodrug (GS-9148) as shown in Scheme 15.34,35
Scheme 15. Synthesis of GS9148.

To increase the solubility of highly polar phosphonic acid nucleoside derivative during coupling reactions and also to reduce the formation of side-products, lipophilic protecting groups are often temporarily introduced. Thus, Benzaria et al.36 reported the synthesis of bis(POM)-PMEA 51 by protection of PMEA derivative 48N6-position with a MMTr-group prior to phosphate hydrolysis with TMSBr and subsequent treatment with triethylammonium bicarbonate (Scheme 16). Finally, the reaction of compound 50 with iodomethyl pivalate followed by MMTr-deprotection under acidic conditions allowed for the bis(POM)-PMEA 51 formation in 18% yield over two steps.
Scheme 16. N6-Protection Prior to Bis(POM)-phosphonate Nucleoside Formation.

MMTr-protection was also employed by Chand and co-workers to protect both amine and hydroxyl groups during the synthesis of various C1′-substituted 9-[2-(phosphonomethoxy)ethyl)]adenine37 and 9-[3-(phosphonomethoxy)propyl]adenine38−40 bis(POM)- and bis(POC)-prodrugs derivatives. MMTr-protection of adenosine intermediate 52 and subsequent selective removal of the pivaloyl group with NaOH in MeOH afforded compound 54. The phosphonate moiety was then introduced by coupling with tosylate 55 in the presence of sodium hydride. The protected dialkyl phosphonate 56 was then hydrolyzed with TMSI in the presence of triethylamine to avoid degradation of the MMTr-protecting groups. Finally, the alkylation of 57 with POM-Cl or POC-Cl proceeded efficiently and gave the bis(POM)- and bis(POC)-prodrugs 58 in 69–99% yields, respectively, after deprotection under mild acidic conditions (Scheme 17).
Scheme 17. N6- and Hydroxy Group Protection Prior to Bis(POM)- and Bis(POC)-phosphonates Formation.

In 2011, Agrofoglio and co-workers41 reported the synthesis of 5-substituted uracil butenyl acyclic bis(POM)-phosphonate nucleoside 62 by, first, cross-metathesis reaction between crotylated uracil 60 and dimethyl allylphosphonate 59, followed by direct reaction with chloromethylpivalate and sodium iodide (Scheme 18).
Scheme 18. Synthesis of 5-Substituted Uracil Butenyl Acyclic Bis(POM)-phosphonate Nucleoside 62.

In parallel, the same team developed a more convergent method for the synthesis of 5-substituted uracil butenyl acyclic nucleoside bis(POM)- and bis(POC)-phosphonates 64 and 65 by using a bis(POM)- or bis(POC)-allylphosphonate as cross-metathesis partner.42 Bis(POM)- and bis(POC)-allylphosphonates were generated by reaction of dimethyl allylphosphonate with either POM-Cl and POC-Cl in the presence of sodium iodide (Scheme 19). Interestingly, the authors showed that very low conversion rates were observed when diethylallyl phosphonate was used instead of dimethyl allylphosphonate. The bis(POM)-prodrugs were finally obtained after cross metathesis with crotylated uracil 60 using ruthenium catalyst A at 40 °C. The known instability of carbonates pushed Agrofoglio’s team to find milder reaction conditions; thus, the preparation of bis(POC)-prodrugs was achieved by using IPr indenylidene catalyst B at room temperature (Scheme 19). A similar procedure was used by Montagu et al. for the preparation of 5-substituted analogs.43
Scheme 19. Synthesis of Bis(POM)- and Bis(POC)-allylphosphonates Nucleoside Prodrugs.

Because of the lack of reactivity of ruthenium catalysts in the presence of purines, an alternative strategy was envisaged for the synthesis of butenyl acyclic purine bis(POM)-phosphonate nucleoside 67–73.44 Cross-metathesis between (Z)-2-buten-1,4-diol and bis(POM)-allylphosphonate 63a afforded the desired (E)-bis(POM)-4-hydroxy-but-2-en-1-yl phosphonate reagent 66 in 74% yield (Scheme 20). Mitsunobu coupling between 66 and adenine, 6-chloropurine, or 2-amino-6-chloropurine led to the corresponding bis(POM)-phosphonate nucleosides 67–69. Further acidic hydrolysis with formic acid in water gave hypoxanthine 70 and guanine 71 derivatives in 86% and 85% yields, respectively, while treatment with cyclopropylamine gave 6-cyclopropylamino- 72 and 2-amino-6-cyclopropylamino- 73 derivatives in 82% and 77% yields, respectively.
Scheme 20. Synthesis of Butenyl Acyclic Purine Bis(POM)-phosphonate Nucleoside Prodrugs.

More recently, 5′-methylene phosphonate furanonucleoside bis(POM)-prodrugs have been prepared through a Horner–Wadsworth–Emmons reaction between correctly protected 5′-ketal nucleoside intermediates and a tetra(POM)-bisphosphonate reagent.45 Uridine, N4(Boc)2-cytosine, N6(Boc)2-adenine, 2-N(Boc)2-6-benzyloxy-purine, and 2-N(Boc)2-6-azido-purine 2′-methyl-2′-F-nucleosides 74 underwent oxidation using IBX. Subsequent treatment with deprotonated tetra(POM)-bisphosphonate reagent 75 afforded vinyl phosphonate nucleosides 76. TBDMS deprotection with aqueous formic acid and hydrogenation over palladium hydroxide afforded the desired prodrugs 77 (Scheme 21).
Scheme 21. Synthesis of 5′-Methylene Phosphonate Furanonucleoside Bis(POM)-prodrugs.

3.1.1.5. Carbonyloxymethyl Phosphonate Monoester
Starrett et al.13,28 reported the synthesis of PMEA POM-phosphonate monoester 80. Reaction of diphenyl PMEA 78 with sodium benzoate led to the unexpected formation of benzyl monoester PMEA after spontaneous degradation of the dibenzyl PMEA intermediate. The POM-prodrug 80 was then obtained by coupling the PMEA benzyloxy monoester 79 with chloromethyl pivalate in the presence of triethylamine, and subsequent hydrogenation of the benzyl group with palladium hydroxide on carbon (Scheme 22).
Scheme 22. Synthesis of PMEA POM-Phosphonate Monoester Prodrug.

Tang et al.30 also reported the synthesis of PMEA-carbonyloxymethyl monoester 82 by direct coupling of the phosphonic acid 81 with 1.2 equiv of benzyl or allyl chloromethyl carbonate in the presence of triethylamine (Scheme 23).
Scheme 23. Synthesis of PMEA-Carbonyloxymethyl Monoester Prodrug.

A similar procedure was used by Krecmerova et al.46 for the synthesis of the 2,6-diaminopurine HPMPC (HPMPC-DAP) POM-monoester prodrug 84 by reaction of 83 with POM-Cl in the presence of DCMC (Scheme 24).
Scheme 24. Synthesis of (HPMPC-DAP) POM-Monoester Prodrug 84.

3.1.1.6. Cyclic Carbonyloxymethyl Phosphonate
In 2007, Hóly and co-workers46 reported the synthesis of several cyclic 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine monoester prodrug including POM-derivatives, as an alternative to the bis(POM)-prodrug. However, instability was found in the HPMP series due to the presence of a neighboring hydroxyl group. Following Hostetler’s method,47 cyclic HPMP-5-azaC was obtained in quantitative yield by reacting HPMP-5-azaC with dicyclohexylcarbodiimide and DCMC in dimethylformamide at elevated temperature. cHPMP-5-azaC 85 was converted to its corresponding tributylammonium salt 86 by treatment with a methanolic solution of tetrabutylammonium hydroxide (TBAOH) and finally alkylated with POMCl in dioxane at 100 °C in 52% yield over two steps. Introduction of the POM group induces the formation of a new chiral center at the phosphorus atom with a ratio of 5:2 to 3:2 in favor of the trans-isomer 87b (Scheme 25). Only the cis-isomer 87a was isolated pure on small scale by HPLC purification. The chair conformation was elucidated by inspection of spin–spin coupling constants from 1H NMR spectrum.29 In 2010, Krecmerova et al. took advantage of this later procedure to prepare the 2,6-diaminopurine cyclic POM-monoester prodrugs (cHPMP-DAP). The ratio was found to be 6:1 in favor of the less polar trans-isomer. As before, the diastereoisomers were distinguished by characteristic values of 31P chemical shifts, as well as H–H, H–P, and C–P coupling constants.
Scheme 25. Synthesis of cHPMP-5-azaC POM-Monoester Prodrug.

3.1.1.7. Mixed Prodrugs and Miscellaneous
In 1994, Starrett et al.13 reported the synthesis of PMEA mixed glyoxamide POM-diester as part of their pioneering work on PMEA prodrugs (Scheme 26). Activation of PMEA with thionyl chloride to form the bis(chloro)-intermediate 88 and subsequent reaction with N,N-diethylacetamide generated bis(glyoxamide)-PMEA 89. Selective hydrolysis with sodium hydroxide followed by alkylation with chloromethyl pivalate in the presence of triethylamine gave the desired PMEA mixed glyoxamide POM-diester 91.
Scheme 26. Synthesis of PMEA Mixed Glyoxamide POM-Diester Prodrug.

In 2007, Fu et al.48 reported the synthesis of adefovir bis(l-amino acid)-oxymethyl prodrugs. The desired adefovir prodrugs 92 were obtained by coupling of N-Boc protected l-amino acid chloromethyl esters to PMEA in the presence of DCMC followed by deprotection under acidic conditions (Scheme 27). Interestingly, these compounds were found to be 2 times more potent against HBV and 10 times less toxic than adefovir dipivoxil.
Scheme 27. Synthesis of Adefovir Bis(l-amino acid) Oxymethyl Phosphonate Prodrugs.

3.1.2. S-Acyl-2-thioethyl (SATE) and S-[(2-Hydroxyethyl)sulfidyl]-2-thioethyl (DTE)
In the early 1990s, a French group first reported that mononucleoside phosphotriesters, incorporating a thioethyl chain where the thiol is masked as a thioester (SATE groups, Figure 9), were able to liberate the parent 5′-nucleoside monophosphate inside the cell.49 It has been demonstrated that the decomposition of bis(SATE)-phosphotriester derivatives involves an esterase-dependent activation process leading to an unstable O-2-mercaptoethylphosphotriester. This intermediate decomposes spontaneously via intramolecular nucleophilic displacement into the corresponding phosphodiester with expulsion of ethylene sulfide (Figure 9). Removal of the remaining SATE group follows a similar mechanism giving the desired 5′-O-nucleoside monophosphate.50 The same team also reported a related prodrug containing dithioethanol (DTE) masking groups whose activation to the same mercaptoethyl intermediate is achieved by a reductase (Figure 9). The assumed toxicity concern associated with the ethylene sulfide byproduct has largely limited the advancement of SATE/DTE prodrugs into development, but it is commonly used in the in vitro studies to deliver phosph(on)ates intracellularly.
Figure 9.

Activation of (SATE)- or (DTE)-nucleoside prodrugs.
Nucleosides phosph(on)ate prodrug containing dithioethanol (DTE, Figure 10) can be prepared by (A) coupling of bis(DTE)-phosphate intermediate to the nucleoside, and (B) coupling of a nucleoside phosphonate with 2-substituted (disulfanyl)ethanol derivatives.
Figure 10.

Access to bis(DTE)-phosphotriesters and bis(DTE)-phosphonodiesters.
Bis(SATE)-phosphotriesters nucleosides can be prepared by (A) coupling of a H-phosphonate nucleoside with an hydroxythioester reagent, (B) coupling of a monophosphate nucleoside with an hydroxythioester derivative, and (C) coupling of a N,N-diisopropylphosphoramidite reagent to a nucleoside followed by oxidation of the phosphorus atom (Figure 11).
Figure 11.

Access to bis(SATE)-phosphotriesters and bis(SATE)-phosphonodiesters.
3.1.2.1. Bis(DTE)- and Bis(SATE)-Monophosphate
Historically, the interest for disulfide monophosphate prodrugs began in the early 1990s with the synthesis and the study of dithioethanol (DTE) phosphotriester of AZT23,49 and ddU.51 Dithioethanol is first monoprotected with a MMTr group, then phosphorylated with POCl3 to give bis(MMTr-DTE)-phosphate intermediate 93 in moderate yields. Condensation of either AZT or ddU with compound 93 led to the corresponding bis(DTE)-monosphosphate prodrugs 94a and 94b (Scheme 28).
Scheme 28. Synthesis of Bis(DTE)-monosphosphate Prodrugs.

Direct reaction of nucleosides with bis(SATE)-phosphoramidite is the most commonly used approach to prepare (SATE)-monophosphate prodrugs.
Lannuzel et al.52 described the synthesis of AZT (t-Bu-SATE)-pronucleotide 96 by first preparing AZT-MP 95. The monophosphate derivative was then activated by TPSCl and coupled with the S-pivaloyl-2-thioethanol to give the bis(t-Bu-SATE)-monophosphate prodrug 96 in good yields (Scheme 29).
Scheme 29. Synthesis of Bis(t-Bu-SATE)-Monophosphate Prodrug 96.

Perigaud et al.51 reported the synthesis of bis(SATE)-ddUMP 99 using H-phosphonate chemistry (yields not provided). Dideoxy uridine (ddU) was first converted to the corresponding 5′-hydrogen-phosphonate 97 by reaction with phosphoric acid in the presence of pivaloyl chloride and pyridine (Scheme 30). Compound 97 was then reacted with 2-acetylthioethanol 98 upon pivaloyl chloride activation, to give bis(SATE)-ddUMP 99.
Scheme 30. Synthesis of Bis(MeSATE)-ddUMP Using H-Phosphonate Chemistry.

The most common strategy to prepare (SATE)-phosphate prodrugs involves the coupling of a phosphoramidite intermediate 100 with a nucleoside in the presence of 1H-tetrazole followed by in situ oxidation with tert-butyl hydroperoxide or m-CPBA. This method has been successfully applied to the synthesis of various derivatives of AZT (101),23 adenallene (102),53 9-(2′-β-C-methyl-β-d-ribofuranosyl) substituted purines (103, 104),54,55 pyrrolopyrimidine nucleoside (105),56 and IsoddA (106) (Scheme 31).57
Scheme 31. Traditional (SATE)-Prodrugs Strategies.

However, this method has several limitations related to the nature of the starting materials. For instance, the presence of exocyclic amines on the base can lead to competitive substitution and low solubility of the starting material in commonly used organic solvents.58 Therefore, bases like G or C have been temporarily protected with groups such as MMTr or DMTr (Scheme 32).
Scheme 32. Protection of Base Competitive Sites.

The presence of a 3′-hydroxy group can also lead to the formation of undesired 3′- and 5′,3′-phosphotriester derivatives. Separation of 3′- and 5′-isomers is not always straightforward and can require several steps of difficult chromatographic purification as reported for the synthesis of compound 116 (Scheme 33).59 In other examples, acid labile protective groups such as Boc and TBDMS have been used to circumvent the above-mentioned problem (not shown).60
Scheme 33. Mixtures with Sugar Competitive Sites.

Ribo nucleosides have also been protected by formation of a 2′,3′-isopropylidene group (Scheme 34).22
Scheme 34. 2′,3′-Isopropylidene Group To Mask Competitive Sites.

3.1.2.2. Bis(SATE)- and Bis(DTE)-Phosphonate
2′-C-Methyl adenosine phosphonate prodrug was successfully synthesized by Koh et al.61 as potential anti-HCV inhibitors. The bis(SATE)-prodrug 127 was found to be slightly more potent than its phosphonate parent 126b but also more toxic. Starting from 2′-C-methyl adenosine 123, compound 124 was obtained via a silylation, benzoylation, and desilylation sequence. Oxidation of 124 produced the corresponding 5′-aldehyde, which was subsequently engaged in a Wittig reaction with diphenylphosphoranylidene methylphosphonate to yield the corresponding 5′,6′-vinyl phosphonate (not shown). Catalytic hydrogenation of the double bond followed by transesterification gave the saturated phosphonate ester 125. The 3′-hydroxyl group was protected with a TBDMS group followed by removal of the benzoyl group with ammonia and hydrogenolysis of the benzyl ester to give 3′-protected phophonate 126a. Finally, treatment of 126a with S-(2-hydroxyethyl)-2,2-dimethylpropanethioate followed by desilylation lead to desired bis(t-Bu-SATE)-phosphono nucleoside 127 in good yield (Scheme 35, eq 1). Interestingly, the authors had to go through this long sequence of selective protection/deprotection of the 3′-hydroxyl because direct reaction of S-(2-hydroxyethyl)-2,2-dimethylpropanethioate with phosphonate 126b in the presence of MSNT yielded a 3′,5′-cyclic phosphodiester 128 instead of the desired bis(SATE)-derivative 127 (Scheme 35, eq 2).
Scheme 35. 2′-C-Methyl Adenosine Bis(SATE)-phosphonate Prodrugs.

Benzaria et al.36,49 also prepared and studied bis(SATE)- and bis(DTE)-prodrugs of the antiviral agent PMEA 131 (Scheme 36). Hydroxythioesters precursors were condensed with N-MMtr-protected PMEA derivative 129 in pyridine in the presence of 1-mesitylene-2-sulfonyl-3-nitro-1,2,4-triazole (MSNT) to afford the corresponding phosphonodiesters 130 with monoesters as byproducts. Finally, deprotection under acidic conditions provided the target PMEA prodrugs 131.
Scheme 36. Synthesis of Bis(SATE)- or Bis(DTE)-PMEA Prodrugs.

Li et al.62 prepared the 6′-fluoro-6′-methyl-5′-noradenosine phosphonic acid bis(SATE)-prodrug 133 by reaction of phosphonic acid 132 with S-(2-hydroxyethyl)-2,2-dimethylpropanethioate in the presence of MSNT (Scheme 37).
Scheme 37. Preparation of Bis(SATE)-Prodrug 133.

3.1.2.3. Cyclic Monophosphate Nucleoside Prodrug Bearing SATE Group
Several cyclic monophosphate (cMP) prodrugs of heterobase-modified 2′-C-methyl ribonucleoside were synthesized in 2007 by Gunic et al.63 Coupling of (29) (refer to Scheme 10 for the synthesis) with appropriate hydroxythioester in the presence of MSNT in pyridine gives the corresponding cMP prodrugs 104 and 134 (Scheme 38). Interestingly, (SATE)-cMP prodrugs of nucleosides 29 displayed remarkable improvement in HCV replicon inhibition (7000–11 000-fold) without significant toxicity. Activities of these (SATE)-cMP prodrugs have been shown to be similar to regular 5′-bis(SATE)-MP prodrugs of nucleosides.55
Scheme 38. Synthesis of (SATE)-cMP Prodrugs.

In 2010, Liu et al.64 successfully prepared 3′,5′-cyclic (SATE)-phosphonodiester nucleoside 136 by reacting adenine phosphonic acid 135 with S-(2-hydroxyethyl)-2,2-dimethylpropanethioate in the presence of MSNT (Scheme 39).
Scheme 39. 3′,5′-Cyclic (SATE)-Phosphonodiester Nucleoside Synthesis.

3.1.2.4. Mixed SATE Approach
3.1.2.4.1. Aryl(SATE)-phosphotriester
The main decomposition pathway of these aryl (SATE)-phosphotriesters involves loss of the SATE moiety by action of an esterase, followed by hydrolysis into the corresponding nucleoside monophosphate through phosphodiesterase enzymatic activity (Figure 12).
Figure 12.

Activation of aryl(SATE)-prodrugs.
Aryl(SATE)-phosphotriesters can be prepared by (A) coupling of a N-isopropylphosphoramidite reagent to a nucleoside followed by oxidation of the phosphorus atom, and (B) coupling of an already functionalized phosphorochloridate reagent to a nucleoside (Figure 13).
Figure 13.
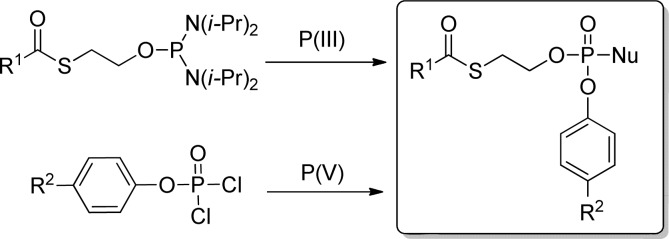
Methods of preparation of aryl(SATE)-nucleoside prodrugs.
Villard et al.65 along with Schlienger et al.66 prepared a series of AZT phenyl(SATE)-phosphotriesters derivatives (Scheme 40). Various thioesters 137 were reacted with phenyldichlorophosphate to give the corresponding SATE derivatives 138, which were directly coupled with AZT in the presence of NMI. Removal of the various protecting groups was carried out using either TFA or aqueous acetic acid to provide the desired prodrugs 139 in high yields.
Scheme 40. AZT Phenyl(SATE)-phosphotriesters Prodrugs.

Perigaud’s team67 reported the synthesis of (SATE)-phosphotriesters bearing modified l-tyrosinyl residues by phosphoramidite P(III) chemistry. Condensation of tyrosinyl precursors with (SATE)-phosphorobis(amidite) reagent 100 led to the corresponding tyrosinyl(SATE)-phosphoramidite intermediates 140. Reaction of (140) with AZT, followed by in situ oxidation with t-BuOOH and treatment of intermediates under acidic conditions (30% HCl in Et2O or 10% TFAA in DCM), afforded the desired prodrugs 141 in good overall yields (Scheme 41).
Scheme 41. Synthesis of (SATE)-Phosphotriesters Bearing Modified l-Tyrosinyl Residues.

3.1.2.4.2. (SATE)-Phosphoramidate Diester68
These (SATE)-phosphoramidate diesters containing either simple aliphatic and aromatic amines or amino acid esters have been shown to deliver 5′-nucleoside monophosphates after simple esterase activation followed by phosphoramidase-catalyzed cleavage of the amino portion (Figure 14).
Figure 14.

Activation pathway of (SATE)-phosphoramidate diester prodrugs.
Perigaud’s research group68 reported the synthesis of AZT phosphoramidate diester bearing one (t-Bu)SATE group and various amino residues using the H-phosphonate chemistry. Thus, key H-phosphonate monoester 142 was first coupled to AZT in the presence of t-BuCOCl to give the corresponding (t-Bu)SATE-AZT H-phosphonate diester 143. Finally, oxidative coupling with various amines afforded the desired AZT (SATE)-phosphoramidate diesters 144,145 (Scheme 42).
Scheme 42. Synthesis of AZT (SATE)-Phosphoramidate Diesters Prodrugs.

Despite the fact that IDX184 development for HCV treatment was stopped in phase IIb in August 2012, the (SATE)-phosphoramidate diester prodrug of 2′-C-methylguanosine remains at present the only example of the successful application of this technology to reach human study. IDX184 was prepared using the H-phosphonate chemistry similar to that described above.69 Thus, the key H-phosphonate monoester precursor was prepared in a few steps from commercially available 2,2-dimethyl-3-hydroxypropanoic acid methyl ester, by protection of the alcohol followed by saponification, leading to compound 146 in 92% yield without purification. Installation of the side chain was performed by peptidic coupling between compound 146 and 2-mercaptoethanol to generate alcohol 147. Finally, treatment of compound 147 with phosphorus acid and pivaloyl chloride, followed by quenching the reaction with triethylammonium bicarbonate (TEAB), generated H-phosphonate monoester precursor 148 in 90% over two steps. 2′-C-Methylguanosine was then reacted with 148 in the presence of pivaloyl chloride to furnish intermediate 149, which was further treated with benzylamine to generate Tr-protected phosphoramidate diester 150 in quantitative yield. Classical deprotection with trifluoroacetic acid led to the isolation of IDX184 in 39% yield (Scheme 43).
Scheme 43. Synthesis of IDX184.

3.1.2.4.3. (SATE)-Glucosyl Phosphorothiolates
This type of mixed (SATE)-phosphodiesters is based on the combination of the iso(SATE)- and the bis(SGTE)-70 prodrugs, two structural modifications previously studied by Perigaud’s group.
The postulated unmasking pathway of the (SATE)-glucosyl phosphorothiolate derivatives may involve an esterase activation leading to the loss of the SATE group and formation of glucosyl phosphorothiolatediesters (Figure 15). These intermediates should then undergo a glucosidase-mediated cleavage of the anomeric bond followed by a rearrangement process similar to the decomposition process proposed for bis(isoSATE)-pronucleotides.71
Figure 15.

Activation of (SATE)-glucosyl phosphorothiolate prodrugs.
The synthesis of such (SATE)-glucosyl phosphorothiolate derivatives involves both P(III) and P(V) intermediates and was developed using AZT as a model system. The (SATE)-H-phosphonate monoester precursor 153 was obtained from S-(2-hydroxyethyl)-2,2-dimethylpropanethioate using salicyl chlorophosphite. Condensation of intermediate 153 with AZT, in the presence of pivaloyl chloride, led to the corresponding H-phosphonate diester, which was in situ oxidized into phosphorothioate 154 using elemental sulfur (diastereoisomeric mixture 1:1). For the glucosyl phosphorothiolate portion, a boron trifluoride etherate-induced glycosylation of a pentaacetyl glucopyranose derivative with 2-bromoethanol gave the corresponding β-glucopyranoside 151. A Finkelstein halogen-exchange reaction with sodium iodide was followed by coupling of the resulting 2-iodoethyl-β-d-glucosides 152 with phosphorothioate diester 154. Phosphorothiolate derivatives 155 were obtained, as a 1:1 diastereomeric mixture (Scheme 44).71
Scheme 44. Synthesis of (SATE)-Glucosyl Phosphorothiolate Derivatives.

3.1.2.4.4. (SATE)-Halogeno Phosphodiesters
Egron et al.72 tried to improve the anti HIV activity of AZT 5′-fluorophosphate by preparing (t-Bu-SATE)-prodrug 156. Starting from H-phosphonate diester 143 (refer to Scheme 44 for preparation), fluorination was achieved using iodine and triethylamine trishydrofluoride. Pure phosphorofluoridate 156 can be obtained as a 1:1 mixture of diastereoisomers using reverse phase column chromatography purification with an isocratic mixture of acetonitrile in water. It is noteworthy that purification of compounds 156 on silica gel column chromatography using MeOH as eluent led to the formation of methylphosphate byproduct 157. However, this approach was not pursued due to the limited chemical stability of the (SATE)-phosphorofluoridate diester, which also provide 158 in buffer media as a side product (Scheme 45).
Scheme 45. Preparation of (t-Bu)SATE Prodrug 156.

3.1.2.4.5. S,S′-Bis(O-acyl-2-oxyethyl) Phosphorodithiolates: Iso(SATE)-pronucleotides
Schlienger et al.70a studied an isomeric form of (SATE)-pronucleotides, mononucleoside S,S′-bis(O-acyl-2-oxyethyl) phosphorodithiolates (iso(SATE)-pronucleotides). The proposed decomposition pathway of the iso(SATE)-pronucleotides involves: (1) an esterase activation leading to intermediate A; (2) nucleophilic attack of the resulting free alcohol on the phosphorus atom, giving rise to five-membered ring intermediate B; and (3) conversion of B into 2-mercaptoethylphosphotriester C followed by spontaneous elimination of episulfide. Removal of the second iso(SATE) functional group is achieved by a similar mechanism or by action of phosphodiesterases, allowing the intracellular delivery of the corresponding nucleoside 5′-monophosphate (Figure 16).
Figure 16.

Decomposition pathway of iso(SATE)-nucleoside prodrugs.
Mononucleoside phosphorodithiolates 161 were obtained in a one-pot procedure involving (pyrrolidino)phosphoramidites 159 and 1H-tetrazole activation, followed by oxidation of 160 with t-BuOOH (Scheme 46).
Scheme 46. One-Pot Procedure Involving (Pyrrolidino)phosphoramidites.

3.1.3. Cyclosaligenyl (cycloSal) Phosphate and Phosphonate Prodrug Approach
cycloSal phosphate and phosphonate prodrugs, originally introduced by Chris Meier and co-workers, are one the most extensively explored types of masked nucleotides.73 This concept is based on the use of salicylic alcohols to mask the phosphate functional group of a nucleoside monophosphate (Nu-MP) and has been successfully applied to the intracellular delivery of a number of antiviral nucleotides (e.g., AZT, d4T, and acyclovir74). Meier’s research group extensively studied this prodrug and demonstrated that the intracellular cleavage of cycloSal pronucleotides is based on an entirely pH-driven chemical hydrolysis mechanism with no enzymatic activation required. Under basic conditions, the aryl ester P–O bond is cleaved first, followed by spontaneous cleavage of the P–O benzyl ester bond (Scheme 47).
Scheme 47. Hydrolysis Pathways of the CycloSal-d4TMP Triesters.

As the cycloSal pronucleotides were designed to release the active drug via a chemical cascade mechanism, the stability and hydrolysis pathways of these pronucleotides have been finely tuned by varying the nature of substituent in the boxed structure (Figure 17). Various diols were obtained by reduction of commercially available or prepared salicylic aldehydes, acids, or esters with NaBH4 or LiAlH4 (Path A). Other variations were achieved using ortho-formylation of substituted phenols followed by reduction (Path B) or mild basic formylation direct hydroxymethylation reactions (Path C). On the other hand, 7-methylated salicyl alcohols were prepared by alkylation of their corresponding aldehydes with methyllithium (Path D).
Figure 17.

Different synthetic methods to access cycloSal-diol precursors.
The coupling of the cycloSal phosphate moiety to the 5′-hydroxyl group of a nucleoside is achieved using either P(III) or P(V) chemistry (Figure 18). However, the strategy using P(III) remains the most common one, due to the usual lack of reactivity of P(V) species. However, the synthesis of cycloSal phosphonates is done exclusively through P(V) chemistry starting directly from nucleoside phosphonates. It is noteworthy that all of these approaches give no diastereoselectivity with respect to the configuration at the phosphorus center. Thus, all compounds are obtained as diastereoisomeric mixtures. Nevertheless, Meier and co-workers filed a patent75 reporting the use of chiral auxiliaries for the synthesis of cycloSal phosphate moieties. Those species can be separated before coupling with the nucleoside, leading to diastereomerically pure cycloSal nucleotide prodrugs.
Figure 18.

Synthesis of cycloSal prodrugs via P(III) or P(V) chemistry.
3.1.3.1. First Generation
3.1.3.1.1. CycloSal Phosphate
In a general manner, cycloSal derivative of nucleosides bearing a pyrimidine base76 such as AZT77 or d4T78 can be easily obtained using two different methods. In the first approach (Scheme 48), diols 162 are reacted with PCl3 to yield the cyclic intermediate saligenylchlorophosphane 163. Target molecules 164 are then obtained in a “one-pot” procedure by coupling nucleosides analogs with (163), followed by in situ oxidation with t-BuOOH.
Scheme 48. P(III) Chemistry To Access cycloSal Phosphate Prodrugs.

An alternative synthetic approach toward such compounds involves the less reactive P(V) chemistry (Scheme 49). The reaction of d4T with phosphorus oxychloride yields phosphodichloridate 165, which is further reacted with salicyl alcohol to give the desired triester 166. However, this last approach leads to yields remarkably lower (37%) than the one obtained with the above P(III) approach.
Scheme 49. P(V) Chemistry To Access cycloSal Phosphate Prodrugs.

Finally, a third approach79 has been used to prepare cycloSal pronucleotides of carbocyclic nucleoside, the phosphorochloridate chemistry (Scheme 50). Alcohol 167 is first reacted with phosphorus oxychloride to give 3-methyl- cyclosaligenylphosphorochloridate 168. Next, reaction of chlorinated intermediate 168 with nucleoside 169 in pyridine gave the phosphate triester 170 in 60% yield. However, this method failed to produce the cycloSal phosphate triester in the case of the 3′-epi isomer of 169, most likely due to steric hindrance or intramolecular cyclization.
Scheme 50. Phosphorochloridate Chemistry To Access CycloSal Phosphate Prodrugs.

In contrast to thymidine nucleosides, cytosine derivatives cause considerable obstacles when reacted with chlorophosphane 172: for instance, cycloSal modifications of 3TC or ddC were achieved in very low yields.80 In this case, the high reactivity of phosphorus(III) chloride is counterproductive leading to a mixture of O- and N,O-di-cycloSal derivatives. To overcome this issue, compound 172 was reacted with diisopropylamine to give the less reactive phosphoramidite 173 (Scheme 51). This compound was then selectively coupled to ddC or 3TC in the presence of pyridinium chloride as an acid catalyst. Finally, oxidation of phosphite intermediate with t-BuOOH afforded the corresponding O-cycloSal derivatives 174 and 175 in 75% and 80% yield, respectively.
Scheme 51. Phosphoramidate Chemistry To Access cycloSal Phosphate Cytosine Prodrugs.

Preparation of cycloSal prodrugs of adenine or guanosine nucleotide derivatives by the same method appears more complicated because of the presence of exocyclic amino groups. However, these amines can be protected with an acid labile group such as a trityl. Common base labile protecting groups have to be avoided due to the potential instability of the target triester derivatives under deprotection conditions. However, for certain substrates such as ddA and d4A,81 the preparation of the corresponding cycloSal derivatives was achieved without any protection because of the known acid-catalyzed cleavage of the glycosydic bond of these particular compounds. In the absence of a protective group on the exocyclic amino group, the regioselective 5′-O-phosphorylation reaction of ddA and d4A was performed at −40 °C to enhance the O- versus N-alkylation (8:1 in favor of the 5′-O-modification, Scheme 52). Using these low temperature conditions, a 1:1.6 instead of 1:1 diastereoisomeric mixture was obtained, the later ratio being usually observed for other nucleosides such as cycloSal-d4TMP.
Scheme 52. Chlorophosphane Chemistry To Access cycloSal Phosphate Adenosine Prodrug Derivatives.

Spáčilova et al. described the synthesis of 6-heteroaryl-7-deazapurine ribonucleosides cycloSal-phosphate pronucleotides 179 as potential adenosine kinase inhibitors.82 Interestingly, they demonstrated the relative stability of the cycloSal prodrug 178 toward Pd-catalyzed transformations: despite the partial decomposition of the cycloSal phosphate under basic conditions, Stille and Suzuki cross-coupling reactions can be performed (Scheme 53). Moreover, like for the protection of exocyclic amines, protection of the sugar moiety was achieved by choosing an acid labile group such as an isopropylidene group that can be easily removed using 90% aqueous trifluoroacetic acid.
Scheme 53. Pd-Catalyzed Reactions with cycloSal Prodrugs.

Meier et al.83 also described the synthesis of cycloSal-BVdUMP triesters 183 from either BVDU 180 or 3′-O-levulinylated BVdU 181 (Scheme 54), using the phosphoramidite/oxidation method previously shown in Scheme 51. Interestingly, after removal of the levulinyl protection under mild condition by treatment with hydrazine hydrate, both methods gave similar overall yields (31–50%).
Scheme 54. Deprotection of a Levulinylate Group on cycloSal-BVdUMP Triesters.

Interestingly, Kortylewicz et al.84 prepared several 5-[125I]iodo-uridine cyclosaligenyl monophosphate prodrugs for cancer imaging and molecular radiotherapy. Nonradioactive iodo analogs 185 were prepared by reaction of nucleosides 184 and phosphochloridates and subsequent oxidation with t-BuOOH. Compounds 185 were then reacted with hexamethylditin to afford the corresponding 5-trimethylstannyl cycloSal-derivatives, which were finally engaged in an electrophilic iodostannylation reaction using Na125I as the source of radioactive iodine to provide the desired radiolabeled prodrugs 187. Separation of each diastereoisomers was achieved by reverse phase HPLC, even though they had close elution profiles (Scheme 55).
Scheme 55. Preparation of Several 5-[125I]Iodo-uridine cycloSal MP Prodrugs.

As mentioned earlier, the chirality of the phosphorus atom leads to the formation of nucleotide prodrugs as mixtures of two diastereoisomers (Rp and Sp) in an almost 1:1 ratio. Moreover, the chromatographic separation of these diastereoisomers, when possible, is often a very difficult task to achieve.
In 2011, Meier and co-workers85−87 reported the first synthetic route to prepare isomerically pure cycloSal-pronucleotides. Their strategies revolved around the use of chiral auxiliaries that were introduced by reaction with phosphorus oxychloride followed by esterification of the resulting dichlorophosphoramidate with salicylic alcohol. At this stage, their strategy required the facile separation of the diastereoisomers by chromatography or recrystallization. Final nucleophilic displacement of the chiral auxiliary by the protected nucleoside generated diastereomerically pure cycloSal-phosphotriesters, provided that this reaction took place with clean inversion of configuration at the phosphorus atom (SNP reaction). Thus, reaction of (S)-4-isopropylthiazolidine-2-thione with POCl3 leads to the formation of intermediate 188 that can be further reacted with 2-(hydroxymethyl)phenol in the presence of DBU to afford a mixture of two diastereoisomers 189a and 189b. At this stage, the two compounds can be separated, and the SP-configuration of (189b) was confirmed by X-ray crystallography. The desired isomer RP-189a is coupled with nucleoside 3′-OAc-dT using t-BuMgCl to give access to the diastereomerically pure monophosphate prodrug 190 (Scheme 56). The authors assigned the stereochemistry of the final products, by assuming that the mechanism of this reaction proceeds with inversion of configuration at the phosphorus atom.
Scheme 56. Synthesis of Diastereomerically Pure Monophosphate Prodrug 190.

Although the reaction conditions worked well for unsubstituted salicylic alcohol, the same sequence was surprisingly not applicable to the synthesis of 3-85 and 5-87 methyl-cycloSal derivatives due to racemization of both the chiral phosphoramidate reagents and the final nucleoside prodrugs. This led the authors to investigate the other chiral auxiliaries 191a–e (Scheme 57).
Scheme 57. Chiral Auxiliaries for 3- and 5-Substituted CycloSal-Derivatives.

Chiral groups 191 were prepared by reaction of amino acid derivatives with dimethylcyano dithioiminocarbonate 192. Ultimately, only 191a and 191e were suitable for the synthesis of 3- and 5-substituted cycloSal phosphotriesters because the diastereoisomers were the only ones that could be separated at the phosphoramidate level. Compounds 191a and 191e were coupled with cycloSal-phosphochloridates generating intermediates 193 as a 1:1 mixture of diastereoisomers.
Diastereoisomers RP-193a and SP-193b were separated by chromatography, and the stereochemistry of SP-193b was confirmed by X-ray crystallography. RP-193a and SP-193b, which were more stable than their SP-and RP-counterparts, were coupled with AZT or d4T to form the expected phosphotriesters 194. After nucleophilic displacement by the nucleoside, both enantiomerically pure (SP)- and (RP)-phosphotriesters 194 could be isolated. Optimization of the reaction conditions for the third step was also investigated. Racemization at the phosphorus atom was suppressed when using [Cu(BEN)](OTf)2 complex in dichloromethane.
It is noteworthy that the authors considered also using chiral thiophosphoramidates,86 but this strategy failed in the last step as the P=S was not electrophilic enough to allow for nucleophilic displacement of the chiral auxiliary by the nucleoside (not shown).
Expending their nucleoside prodrug research program, Meier and co-workers reported the development of bis(cycloSal)-pronucleotides (Figure 19)88 designed to deliver two molecules of active drug for each biomolecule administrated.
Figure 19.

Meier’s bis(cycloSal)-pronucleotides.
Conversion of tetrols 197 into corresponding phosphitylating agents 200 was realized by treatment with PCl3 under basic condition. Careful control of the temperature conditions appeared to be critical to selectively obtain compound 199. Thus, reduction of the reaction temperature from −20 to −40 °C helped decrease the quantity of byproducts resulting from the formation of seven-membered ring 201 (Scheme 58).
Scheme 58. Synthesis of Chlorophosphite 200.

The first attempt of coupling between two molecules of d4T and crude chlorophosphite 200 led to the targeted pronucleotides 203 after tedious chromatography and in poor yield (8%, Scheme 59). The synthesis of these bis(cycloSal) compounds via the phosphoramidite chemistry was also investigated but did not lead to any improvement in yields. As presented before, cycloSal-pronucleotides were always obtained as a mixture of two diastereoisomers (RP/SP configuration). In the case of bis(cycloSal)-d4TMPs, two stereogenic centers are formed in the course of their preparation. Hence, they should be obtained as a mixture of three isomers (RP/RP, RP/SP, and SP/SP configuration) in a ratio approaching 1:2:1 depending on the influence of the nucleoside chirality. In the case of compounds 203, all three diastereoisomers were isolated close to the expected 1:2:1 ratio. However, according to 1H and 31P NMR spectroscopy, compound 203 was obtained as a mixture of three isomers in a ratio of 1:2:2. According to the authors, that stereodifferentiation may be due to steric interactions between the two cycloSal-d4TMP units in 3,3′-bis(cycloSal)-d4TMP 203.
Scheme 59. Synthesis of Bis-cycloSal Pronucleotides.

Another type of bis(cycloSal)-pronucleotides was also developed by Ahmadibeni et al.89 Thus, 3′-fluoro-3′-deoxythymidine (FLT) and 3′-azido-3′-deoxythymidine (AZT) bis(cycloSal)-prodrugs 207 were prepared from tetrol 204 by first formation of bis(chlorophosphite) 205 using PCl3 and then coupling with either AZT or FLT at low temperature. The subsequent oxidation using t-butyl hydroperoxide (TBHP) afforded the desired AZT and FLT bis(cycloSal) derivative as inseparable mixtures of diastereoisomers (Scheme 60).
Scheme 60. Bis-cycloSal Pronucleotides.

3.1.3.1.2. cycloSal Phosphonates
The cycloSal prodrug approach has also been applied to the synthesis of phosphonate nucleosides such as PMEA 48 using P(V) chemistry. First attempts to prepare cycloSal-PMEAs 209, directly from the diethyl ester of PMEA 48, without protection of the exocyclic amino group, led to a complex mixture of reaction products.90 To overcome this problem, the diethyl ester exocyclic amino group was blocked by a monomethoxytrityl protective group (Scheme 61). Treatment of the intermediate with trimethylsilyl bromide then gave the bis(trimethylsilyl) ester that was immediately converted into the corresponding dichloride 208 using PCl5. The dichloro intermediate 208 was then reacted with different salicylic alcohols to give the protected cycloSal-PMEA diesters in low to moderate yields. Finally, the MMTr group was cleaved by treatment with TFA, which led to the target cycloSal-PMEAs 209 in 53–82% yield.
Scheme 61. MMTr Protection/Deprotection To Access cycloSal-PMEAs.

Unexpectedly, cycloSal-PMEA derivatives 209 appeared to be unstable especially in acidic conditions (pH = 2) and led to the design of possibly more stable cycloaminobenzyl-PMEA (i.e., cycloAmb-PMEA) phosphoramidates 211.90 In these molecules, the cycloSal phenolic oxygen atom is replaced by a nitrogen hypothesizing that the less electronegative nitrogen would reduce the electrophilicity of the phosphorus atom and consequently increase the stability of the prodrug. The first attempt to prepare the cycloAmb-PMEAs, using the reaction sequence shown in Scheme 61, led to the isolation of the targeted compounds 211 (Scheme 62) in very poor yield (3–7%). Another approach was then envisaged were PMEA was converted into its corresponding dichloride derivative 210 by treatment with oxalyl chloride. Addition of DMF led to the in situ protection of the nucleobase with a formamidine group. 2-Aminobenzyl alcohols were then condensed to intermediate 210 to provide corresponding cycloAmb-PMEA derivatives 211 in 25–42% yield. Interestingly these cycloAmb-PMEA derivatives 211 appeared dramatically more stable than their cycloSal-PMEA counterparts 209 while still displaying anti-HIV activity.
Scheme 62. Synthesis of cycloAmb-PMEAs Phosphoramidates.

3.1.3.2. “Lock-In” cycloSal-Triesters
Because of the lipophilic character of cycloSal phosphate triesters and their chemically triggered delivery mechanism, a drug concentration equilibrium is generated across the cell membrane. To trap cycloSal triester inside the cells and avoid the formation of this equilibrium, so-called “lock-in” cycloSal pronucleotides were developed.91 These triesters are designed to be enzymatically converted inside the cell into a more polar compound (Figure 20).
Figure 20.

Mechanism of action for “lock-in” cycloSal pronucleotides.
Elaborated acyloxy systems, such as the acetoxymethyl (AM), isopropyloxycarbonyloxymethyl (POC), pivaloyloxymethyl (POM),92 and amino acid,93 were used to release the corresponding carboxylates. Starting from compound 212, obtained using the standard chlorophosphite procedure, deprotection with TFA led to cycloSal-d4TMP acid 213. POC and POM groups can be introduced by reaction of (213) with the corresponding chloromethyl alkyl reagent to give compounds 214. On the other hand, a peptidic coupling between (213) and various amino acids leads to the corresponding amide-containing cycloSal derivatives 215 (Scheme 63).
Scheme 63. Elaborated Acyloxy Systems.

Meier et al. developed another type of “lock-in” cycloSal-pronucleotide that bears a (carboxy)esterase-cleavable geminal dicarboxylate91,94 or an acetoxyvinyl95 group attached to the aromatic ring of the saligenyl unit. Those new “lock-in” cycloSal-pronucleotides are enzymatically transformed into a more polar aldehyde or ketone inside cells (Figure 21).
Figure 21.

“Lock-in” cycloSal-pronucleotides bearing geminal dicarboxylate or acetoxyvinyl groups.
The synthesis of these compounds starts with the conversion of 4-formylsalicylic alcohols 218 into cycloSal triesters 220 using a standard P(III)-chemistry route. Next, triesters 220 are reacted with acetic anhydride and zirconium(IV) chloride to give the corresponding final prodrugs 221 in 23–45% yield. Interestingly, for some compounds, a separation of the two diastereoisomers (Rp or Sp) was achieved. The Sp form of the cycloSal triesters demonstrated improved antiviral activity as compared to the Rp form (Scheme 64).
Scheme 64. Synthesis of “Lock-In” cycloSal-Pronucleotides Bearing Geminal Dicarboxylate Groups.

3.1.4. Cyclic 1-Aryl-1,3-propanyl Ester HepDirect
HepDirect prodrugs are aryl substituted cyclic 1,3-propanyl esters developed in the early 2000s by Metabasis Therapeutics, Inc. as a liver-directed prodrug combining high plasma and tissue stabilities. So far, three drugs including MB07811(96) and two nucleosides pradefovir2 and MB07133(97) have been advanced to human clinical trials (Figure 22). Pradefovir is a 3-chlorophenyl HepDirect prodrug of Adefovir in development for hepatitis B infection therapy, while MB07133, a 4-pyridyl HepDirect prodrug of cytarabine, has been developed for hepatocellular carcinoma treatment. MB07811 was considered as a candidate for the treatment of hyperlipidemia.
Figure 22.

HepDirect prodrugs in clinical trial.
These cyclic 1,3-propanyl esters were designed to undergo oxidative cleavage catalyzed by the cytochrome P450 (CYP) enzyme 3A, expressed predominantly in the liver. The hemiketal intermediate can undergo ring opening to form a negatively charged phosph(on)ate, which subsequently delivers the free phosph(on)ate nucleoside after spontaneous β-elimination. The aryl vinyl ketone released during the process of the reaction is then rapidly detoxified by glutathione S-transferase, an enzyme present in high concentration in liver cells.
Interestingly, it was shown that the cleavage of the prodrug portion depends on the stereochemistry at the benzylic position. Indeed, only the phosphates with a cis-relationship between the aryl group and the nucleoside portion (and not the trans) were found to be activated by CYP3A. In addition, modifications at the phenyl moiety revealed the importance of an electron-withdrawing group for sufficient chemical stability (Figure 23).2b,98
Figure 23.

Mechanism of activation for HepDirect nucleoside prodrugs.
HepDirect phosphate prodrugs can be prepared by coupling a nucleoside with a phosphorylating agent derived from a 1-arylpropane-1,3-diol using either P(III) (diisopropylphospharamidite reagent) or P(V) (nitrophenylphosphate) chemistry. On the other hand, synthesis of phosphonates is achieved by direct coupling of 1-arylpropane-1,3-diol with a phosphonate nucleoside (Figure 24).
Figure 24.

Methods to access HepDirect phosphate or phosphonate nucleoside prodrugs.
Because HepDirect prodrugs have two chiral centers (the benzylic position and the phosphorus atom), nonselective HepDirect prodrug formation results in the formation of four diastereoisomers. However, starting from an enantiomerically pure diol results in the formation of only two diastereoisomers identified as cis and trans that differ only in the configuration of the newly formed phosphorus chiral center (Figure 25).
Figure 25.
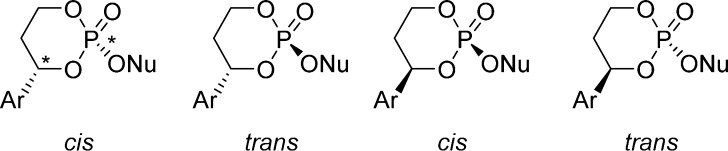
Chirality in HepDirect prodrugs.
3.1.4.1. Synthesis of Aryl-Substituted Cyclic 1,3-Propanyl Esters
Enantiomerically pure (R)- and (S)-1-aryl-propane-1,3-diols 223 were obtained through chromatographic separation of diastereomeric (−)-menthone ketals (Scheme 65). Alternatively, they can be synthesized by asymmetric reduction of the aryl ketoacid 224 with (−)- or (+)-B-chlorodiisopinocampheylborane (DIP-Cl) followed by reduction of the resulting β-hydroxy acid with LiAlH4 or BH3.Et2O with ee’s > 96%.2d,98
Scheme 65. Preparation of Enantiomerically Pure (R)- and (S)-1-Aryl-propane-1,3-diols Using (−)-Menthone.

In certain cases such as 4-pyridyl derivatives, the separation of diastereoisomers as menthone ketals is difficult, and thus other chiral moieties have been employed. Esterification of racemic β-hydroxy ester 225 with N,N-dimethyl phenylalanine led to an easy separation of both diastereoisomers 226 in high optical purities and gave the desired diol S-226b after removal of the phenylalanate portion (Scheme 66).97
Scheme 66. Preparation of Enantiomerically Pure (R)- and (S)-1-Aryl-propane-1,3-diols Using N,N-Dimethyl-phenylalanine.

Enantiomeric enriched (S)-1-(4-pyridyl)-propane-1,3-diol was also obtained by lipase-mediated resolution in the presence of porcine pancreatic lipase (PPL) and vinyl acetate in 35–40% conversion and >95% ee. Final hydrolysis of the acetate groups led to compound S-226b.99
3.1.4.2. HepDirect Phosphate Prodrugs
The first method developed by Erion et al.98 used P(III) chemistry and the reaction of a phosphoramidite and a free nucleoside followed by the oxidation of the phosphate intermediate.
Phosphoramidite 228 is synthesized by reaction of diol S-223 and commercially available 1,1-dichloro-N,N-diisopropylphosphinamine 227 (Scheme 67). Compound 228 was stable and was purified by column chromatography on silica gel. The desired HepDirect prodrug of Lamivudine 229 was obtained as a mixture of cis- and trans-phosphate cyclic diesters after coupling of phosphoramidite 228 with 3TC followed by oxidation with t-BuOOH.
Scheme 67. Synthesis of the HepDirect Prodrug of Lamivudine.

Reddy et al. used the same phosphite approach to prepare 4-pyridyl HepDirect prodrug of ara-A.99 The phosphorylation step was found to be almost instantaneous at 0 °C, giving a mixture of cis and trans isomers after oxidation. However, it was found that the thermal epimerization of the cis–trans mixture (60 °C, 3 h) enables the selective formation of the thermodynamically more stable trans-phosphoramidite. Finally, the stereospecific oxidation of P(III) phosphite 231 into P(V) phosphate derivative resulted in the exclusive formation of trans-HepDirect phosphate prodrug 232 (Scheme 68).
Scheme 68. Formation of trans-HepDirect Phosphate Prodrug 232.

The stereochemistry of the trans isomer 232 was established using NOE studies, 31P NMR, and comparison with similar prodrugs previously reported in the literature.
This coupling reaction was also studied to develop a high throughput synthesis of HepDirect prodrugs.100 DMSO can be also used as a cosolvent when nucleosides are not totally soluble in DMF (i.e., G nucleosides). The reaction failed to proceed in low polarity solvents because of the poor solubility of unprotected nucleosides. Optimization of the stoichiometry of phosphoramidite relative to coupling agent shows that the best yield (31% ± 14%) can be obtained when 6 equiv of both reagents were used. These conditions were applied to 148 different nucleosides and show an excessive production of doubly phosphorylated products. Moreover, the desired monophosphorylated derivative was only obtained for 52% of the substrates. The use of 2 equiv generally resulted in a decreased yield (11% ± 9%), but led to a better rate of success with 80% of cases giving the desired phosphorylated products. The stoichiometry 1:2:2 (nucleoside:phosphoramidate:coupling agent) is the one generally used for creating nucleoside libraries. For purification, the most efficient method was determined to be preparative reverse-phase HPLC with mass-based fraction collection after filtration of the crude reaction mixture. The process was chosen for its automation capabilities and ease of HepDirect prodrug preparation. Normal-phase silica gel cartridge-based purification can also be used but was less efficient because several sample preparation steps were needed prior to chromatography. The HPLC purity of these compounds (obtained with the stoichiometry 1:2:2) was acceptable (90% ± 7%), and the cis–trans ratio was slightly in favor of the cis-compound.
To obtain the desired cis-isomer prodrugs in a completely selective manner, Erion et al.98 developed a chiral p-nitrophenylphosphate reagent that can react through a SN2-type reaction with a nucleoside.
The p-nitrophenylphosphate trans-isomer can be prepared by reaction of p-nitrophenyl phosphorodichloridate and 1,3-propanediols to give 234 as a 40:60 cis:trans mixture of diastereoisomers (Scheme 69). Interestingly, stirring the reaction mixture overnight in the presence of an excess of p-nitrophenol in Et3N shifts the equilibrium toward the more thermodynamically favored trans-isomer with less than 3% of cis compound remaining. In the same manner, the reaction of diisopropylphosphoramidites 233 with p-nitrophenol can be driven to the exclusive formation of the single trans-isomer 235 when stirred at room temperature for 8 h. Subsequent oxidation with t-BuOOH gives access to the desired p-nitrophenylphosphate trans-isomer 236b.99 The stereochemistry of the final compounds was determined by NOE experiments and comparison of benzylic methane proton chemical shift with literature examples.
Scheme 69. Preparation of Enantiomerically Pure trans p-Nitrophenylphosphates.

Unexpectedly, the coupling of p-nitrophenylphosphate reagents 236a,b with a free nucleoside in the presence of bases such as LiH, KOt-Bu, or KNH2 led to the formation of a cis–trans mixture and/or extensive hydrolysis of the phosphate ester. Surprisingly, the use of sodium hydride selectively generated the trans-isomer nucleoside prodrug (in low yield) through an unknown reaction mechanism. Finally, it was found that the use of t-BuMgCl on 2′,3′-protected nucleosides resulted in the exclusive formation of cis-isomers as illustrated in Scheme 70 with cytarabine (52%)99 and 2′-Me-A (35%).101 Determination of the stereochemistry of the final product was established by comparison of NMR data with literature examples. Isopropylidene and TBS protective groups were finally removed after phosphorylation under acidic condition or by treatment with a source of fluorine (TEAF, TBAF). It is noteworthy that substrates, bearing leaving groups such as chloro, 4-chlorophenoxy, and 2,4-dichlorophenoxy groups in place of the nitrophenoxy group, were also tested, but were found to epimerize during coupling with the nucleoside.
Scheme 70. Formation of cis-Isomers.

Bookser et al.102 prepared 3′-amino-3′-deoxyguanosine monophosphate HepDirect prodrug 242 using temporary protection of purine 2-NH2 and sugar 3′-hydroxyl with N,N-dibenzylformamidine and TBS groups, respectively. These protections served two purposes: first, they render the extremely polar guanosine more manageable in term of solubility and purification, and they also prevent side reactions. Thus, protected compound 240 was reacted with p-nitrophenylphosphate reagent 236a in the presence of t-BuMgCl to generate corresponding HepDirect intermediate 241 in 93% yield. Finally, treatment with triethylammonium formate (TEAF) then TFA allowed for the removal of both the formamidine and the TBS groups. The 2′-N3 group was subsequently reduced under classical Staudinger reaction conditions to give desired 3′-amino-3′-deoxyguanosine monophosphate HepDirect prodrug 242 in quantitative yield (Scheme 71). Determination of the stereochemistry of the final product was established by comparison of NMR data with literature examples.
Scheme 71. Synthesis of 3′-Amino-3′-deoxyguanosine Monophosphate HepDirect Prodrug.

Boyer et al.97 also showed that in the case of cytosine nucleosides, such as ara-C, N4-protection was necessary to avoid N4-phosphorylation. Starting from dimethylformamidine derivative 243, coupling with p-nitrophenylphosphate reagent 236b in the presence of t-BuMgCl followed by deprotection under acidic conditions allowed for the preparation of 4-pyridyl ara-C HepDirect prodrug 244 in gram quantities (Scheme 72). Unambiguous structural assignement was made by single-crystal X-ray structure determination of the final product and confirmed the relative stereochemistry between the aryl ring and the nucleoside as cis.
Scheme 72. Synthesis of ara-C-HepDirect Prodrug 244.

3.1.4.3. HepDirect Phosphonate Prodrugs
HepDirect phosphonate prodrugs can be readily prepared from a phosphonic acid nucleotide. In fact, adefovir HepDirect prodrug was initially prepared by peptidic coupling conditions of (S)-1-(3-chlorophenyl)-propane-1,3-diol as a mixture of racemic cis- and trans-isomers (ratio from 55:45 to 60:40, favoring the cis isomer) separable by chromatography and fractional crystallization (Scheme 73). Stereochemistry of the cis versus the trans isomers was determined by 1H and 31P NMR experiments as well as comparison with known similar examples from the literature.
Scheme 73. Adefovir HepDirect Phosphonate Prodrugs via Peptidic Coupling Conditions.

To favor the formation of the cis-isomers, alternative coupling procedures and conditions were evaluated. Boyer and collaborators2d,98 found that nucleophilic substitution at low temperature of an activated bis-chlorophosphonate 210 led to the formation of 246 in a 75:25 cis:trans ratio (Scheme 74). Finally, the cis-isomer 247 was obtained after deprotection of the imine group with acetic acid and purification by chromatography in de >95%. The stereochemistry was assigned on the basis of 1H and 31P NMR and comparison with the literature, but was ultimately confirmed by X-ray crystallography.
Scheme 74. Adefovir HepDirect Phosphonate Prodrugs via a Bis-chlorophosphonate.

3.1.5. 3′,5′-Cyclic Phosphate Ester Prodrugs
3′,5′-Cyclic phosphate ester prodrugs (Figure 26) are part of an interesting prodrug concept that led to the discovery of PSI-352938, a compound that demonstrated anti-HCV efficacy in vitro and in human phase 1 trials. The activation of these derivatives to the monophosphate involves, first, an enzymatic P–O-dealkylation by CYP3A4 and then cleavage of the 3′-phosphorus–oxygen bond by phosphodiesterases.103
Figure 26.

Mechanism of activation for 3′,5′-cyclic phosphate nucleoside prodrugs.
PSI-352938(104) and related analogs were prepared by reacting 6-substituted purine nucleoside 248a with tetra-isopropyl phosphorodiamidite in the presence of 4,5-dicyanoimidazole (DCI) and then oxidation to the corresponding cis- and trans-cyclic phosphate 250 using either I2 or t-BuOOH (Scheme 75). Interestingly, the authors found that by heating the mixture of cis and trans phosphite isomers 249 at 50 °C for several hours, the thermodynamically more stable intermediate cis-249 was favored (>95%). It is noteworthy that cis- and trans-phosphate isomers cis-249 and trans-249 can be isolated by simple column chromatography and that the structure and stereochemistry of cis-249 was elucidated using X-ray crystallographic analysis.
Scheme 75. Synthesis of PSI-3529386 Using P(III) Chemistry.

Yields not provided.
An alternative approach using P(V) chemistry was developed to stereoselectively prepare PSI-352938 on multigram scale (Scheme 76). Thus, after optimization of the reaction conditions, the desired cis-cyclic phosphate PSI-352938 was obtained as the major isomer by reacting nucleoside 248b with isopropyldichlorophosphate in the presence of NMI and Et3N.105 The target compound PSI-352938 was obtained with a purity above 99.5% after either column chromatography or recrystallization.
Scheme 76. Stereoselective Synthesis of PSI-352938.

3.2. Alkoxyalkyl Monoester (HDP, ODE)
Alkoxyalkyl monoesters prodrugs, including the hexadecyloxypropyl (HDP) and octadecyloxyethyl (ODE), are ether lipid phospho-conjugates (LPC) developed by Hostetler and co-workers in the mid 1990s.106 This strategy led to the discovery of CMX-001, a HDP prodrug of cidofovir currently in phase II clinical trials for CMV and adenovirus infections, and to CMX-157, a HDP prodrug of adefovir, currently in clinical development for treatment of HIV infection. Using a similar approach, fozivudine tidoxil, a thioether lipid prodrug of AZT, reached phase II clinical trials for the treatment of HIV infection (Figure 27).
Figure 27.

Alkoxyalkyl monoester prodrugs in clinical trial.
The concept of these prodrugs is based on the mimicking of lysophosphatidylcholine (LPC), a naturally occurring phospholipid. By replacing the choline moiety by a drug, the prodrug is supposed to use the LPC natural uptake pathway in the small intestine to reach targeted tissues and achieve high oral availability. Once delivered into the desired tissue, specific intracellular enzymes such as phospholipase C cleave the lipid carrier to free the nucleoside monophosphate (Figure 28).
Figure 28.
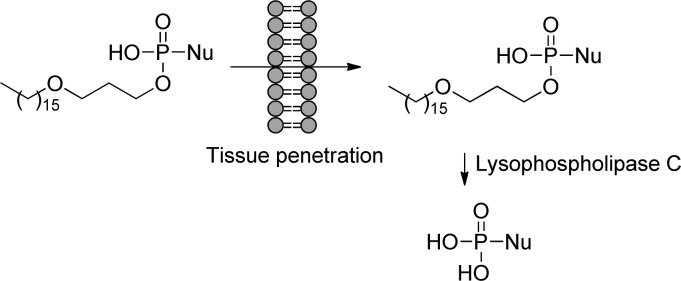
Metabolism drives the pathway of HDP/ODE prodrugs.
To build a more robust prodrug and prevent undesired metabolic reactions, the initial LCP structure was modified over the years. Thus, the acyl linkage at the sn-1 position of the glycerol backbone was changed to an ether linkage to prevent cleavage by lysolecithinase, and the hydroxyl group at the sn-2 position was either reduced or substituted to prevent a second acylation by acyl transferases. Overall, alkoxyalkyl monoesters prodrugs, such as HDP, are the result of a series of chain length, substitutions, and saturation optimizations (Figure 29).
Figure 29.

Modifications of LCP structures.
The synthesis of nucleosides alkoxyalkyl phosphate prodrugs, summarized in Figure 30, involves (A) Mitsunobu coupling between a nucleoside and an alkoxyalkyl phosphate moiety, (B) phosphorylation of a nucleoside and subsequent introduction of the alkoxyalkyl alcohol, (C) coupling between a nucleoside and an alkoxyalkyl phosphoramidite moiety followed by oxidation of the phosphorus atom, and (D) phosphite condensation and subsequent oxidation.
Figure 30.

Methods to synthesize nucleosides alkoxyalkyl phosphate prodrugs, PG = 2-chlorophenyl cyanoethyl, AA = alkoxy alkyl.
On the other hand, the synthesis of alkoxyalkyl phosphonate prodrugs, shown in Figure 31, has been achieved by (A) monosaponification of bis(alkoxyalkyl)-nucleoside phosphonate, (B) direct coupling of a nucleoside phosphonic acid with an alkoxyalkyl alcohol under Mitsunobu or DCC coupling conditions (strategy employed in most cases), (C) substitution of monochloro activated nucleoside phosphonates, (D) direct alkylation of nucleoside phosphonic acid with alkoxyalkyl halides, and (E) direct introduction of the phosphonate moiety bearing the alkoxyalkyl chain in a single step by substitution with a phosphonoalkoxyalkyl oxymethylmethyl tosylate.
Figure 31.

Methods to synthesize nucleosides alkoxyalkyl phosphonate prodrugs, AA = alkoxy alkyl.
3.2.1. Alkoxyalkyl Phosphate Monoester Prodrugs
The first synthesis of nucleoside alkoxyalkyl phosphate monoester was reported by Piantadosi in 1991 who prepared ether lipid conjugates monophosphate prodrugs of AZT and ddI.107
Alkoxyalkyl monophosphates 251 were prepared by three possible methods (Scheme 77): (1) by reacting alkoxyalkyl alcohols with diphenyl chlorophosphate followed by catalytic hydrogenation; (2) by reacting alkoxyalkyl alcohols with phosphorus oxychloride followed by hydrolysis of the chlorinated intermediate; or (3) by Arbuzov rearrangement (reaction of an alkoxyalkyl bromide derivative and trimethylphosphite) and subsequent removal of the methoxy groups using trimethylsilyl bromide.
Scheme 77. Syntheses of Alkoxyalkyl Phosphate Derivatives.

The DCC-mediated coupling of the alkoxyalkyl phosphate derivatives 251a–c with AZT afforded the corresponding alkoxyalkyl phosphate prodrugs 252 in 22–28% yields (Scheme 78).
Scheme 78. AZT Alkoxyalkyl Phosphate Prodrug, AA = Alkoxy Alkyl.

Mavromoustakos et al.108 demonstrated that AZT alkoxyalkyl monophosphate prodrugs can be prepared in a more efficient manner by simply using temporarily protected phosphate derivative 258 (Scheme 79). Thus, starting from protected glycerol derivative 255, reaction with hexadecyl bromide followed by acidic removal of the trityl group provided intermediate 256. Compound 256 was then reacted with o-chlorophenyl phosphodi-1,2,4-triazolide 254 and treated with triethylamine and water to afford the desired alkoxyalkyl triethylammonium phosphate salt 257. Finally, the coupling of AZT with compound 257 in the presence of MSNT was followed by deprotection with TBAF, which allowed for the preparation of AZT prodrug 252 in 68% yield.
Scheme 79. AZT Alkoxyalkyl Monophosphate Prodrugs.

In 1997, Hostetler et al.109 reported a new chemical approach for the synthesis of octadecyl glycerol (ODG) and HDP phosphate prodrugs involving the formation of the nucleoside monophosphate before introduction of the alkyloxyalkyl chain. Thus, the ODG-acyclovir phosphate prodrug 261 (Scheme 80) was prepared in three steps from the N2-acetyl protected acyclovir 259. The phosphate moiety was first introduced on the protected nucleoside 260 by a DCC-mediated coupling with cyanoethyl phosphate. Resulting nucleoside cyanoethyl phosphate 260 was subsequently coupled with 1-octadecyl-glycerol in the presence of MSNT and NMI in a low 17% yield. Cleavage of the cyanoethyl and N2-acetyl protective groups with ammonia afforded the desired prodrug 261 in 92% yield.
Scheme 80. Synthesis of ODG-Acyclovir Monophosphate Prodrug, MSNT = 1-Mesitylenesulfonyl-3-nitro-1,2,4-triazole.

In a similar manner, ODG-AZT was obtained by direct DCC-mediated coupling of 1-octadecyl-glycerol with AZT monophosphate in 25% yield (Scheme 81).
Scheme 81. Synthesis of ODG-AZT Monophosphate Prodrug.

Beadle and co-workers110 reported the synthesis of HDP-acyclovir phosphate prodrug 265 by coupling of 2-chlorophenyl phosphodi-1,2,4-triazolide 254 with N2-MMTr-protected acyclovir in the presence of HDPOH and NMI (Scheme 82). The subsequent removal of the MMTr group with acetic acid was followed by deprotection of the phenol group under basic conditions to afford the desired acyclovir HDP phosphate prodrug 265 in 78% yield.
Scheme 82. Synthesis of HDP-Acyclovir Phosphate Prodrug.

A similar procedure was used by Liang et al. in 2006 for the preparation of HDP- and ODE-(−)-β-d-(2R,4R)-dioxolane-thymine (DOT) monophosphate prodrug in 60% yield.111 The 2-chlorophenyl deprotection was conducted with a 0.5 N NaOH solution in THF to afford the desired prodrug in 93% yield (Scheme 83).
Scheme 83. Preparation of HDP and ODE Dioxolane Prodrugs.

Ludwig et al.112 prepared an alkoxyalkyl phosphate monoester prodrug of 5-fluoro-2′-deoxyuridine 272 using P(III) chemistry (Scheme 84). The alkoxyalkyl hydrogen phosphonate reagent was prepared by reacting 1-O-octadecyl-2-O-acetyl-glycerol with salicylchlorophosphite in the presence of pyridine followed by hydrolysis. Reaction of phosphite reagent 270 with 3′-acetyl-5-fluorodeoxyuridine in the presence of pivaloyl chloride formed the nucleoside hydrogen phosphonate intermediate 271. Oxidation of P(III) to P(V) with iodine in water and removal of the acetate group using ammonia in methanol afforded the desired alkoxyalkyl phosphate prodrug 272 in 82% yield.
Scheme 84. Synthesis of Alkoxyalkyl Phosphate Monoester Prodrug of 5-Fluoro-2′-deoxyuridine.

P(III) chemistry has also been used by Sigmund et al.113 for the synthesis of AZT and 3′-deoxyadenosine phospholipid conjugates. 2-(4-Nitrophenyl)ethoxy-protected (NPE) alkoxyalkyl phosphoramidites 274 were first obtained by reacting alkoxyalkyl alcohols 273 and diisopropy1amino[2-(4-nitrophenyl)ethoxy] chloro phosphate. 1H-Tetrazole-mediated coupling of NPE-protected 3′-deoxyadenosine with phosphoramidite 275 followed by phosphorus oxidation with iodine afforded the desired protected prodrugs 276 in excellent yields. The subsequent deprotection by treatment with DBU in pyridine afforded the desired prodrug 277 obtained in 71% yield (Scheme 85).
Scheme 85. Synthesis of 3′-Deoxyadenosine Phospholipid Conjugates, NPE = 2-(4-Nitrophenyl)ethoxycarbonyl.

3.2.2. Alkoxyalkyl Phosphonate Monoester Prodrugs106a
In 2002, Hostetler et al. reported the first synthesis of HDP and ODE prodrugs of cyclic and noncyclic cidofovir. By increasing the oral bioavailability of the parent molecule cidofovir,114 its corresponding prodrugs exhibit increased in vitro antiviral activity against poxviruses,115 CMV, and other herpes viruses.116 To extend the alkoxyalkyl prodrug technology to other HPMP and PME acyclic nucleoside, several efficient syntheses were developed.
Alkoxyalkyl cidofovir HDP prodrug was initially obtained by intramolecular cyclization of HPMPC using DCC and DCMC. The formed cHPMPC salt was then alkylated with alkoxyalkyl bromide in DMF at 80 °C, which led to HDP cyclic cidofovir prodrug 278 (HDP-cHPMPC) in 33% yield. The subsequent saponification allowed for the ring opening and generation of HDP cidofovir prodrug 279 in 58% yield (Scheme 86).115
Scheme 86. Synthesis of Alkoxyalkyl Cidofovir HDP Prodrug.

A similar strategy was later used in an attempt to synthesize 5-aza-HPMPC117 alkoxyalkyl prodrug 281. 5-Aza-cHPMPC cyclic phosphonate 86 was synthesized as shown in Scheme 25. This latter compound was reacted with hexadecyloxyethyl bromide affording the alkoxyalkyl prodrug in 53% yield as a 3:2 trans/cis ratio at the newly formed phosphorus chiral center. Surprisingly, Mitsunobu coupling conditions between phosphonic acid and alkoxyalkyl alcohol afforded the corresponding prodrug 280 in only 6.5% yield with recovery of the starting material. However, the authors were unable to obtain the desired HPMP-5-aza-C alkoxyalkyl prodrug 281 due to 5-aza-cytosine instability29 under basic conditions (Scheme 87).
Scheme 87. Synthesis of 5-Aza-HPMPC.

This strategy was also used by Krecmerova et al.46 for the synthesis of the alkoxyalkyl prodrugs of 2,6-diaminopurine HPMP analog (HPMPDAP). Pure trans-cHPMPDAP 283 was first obtained by reacting HPMPDAP with DCC and DCMC. Conversion of phosphonic acid 283 into its corresponding tetrabutylammonium salt and subsequent alkylation with hexadecyloxypropyl bromide afforded the corresponding HDP prodrug 284 in 46% yield as a mixture of trans and cis isomers (1.6:1), partially separable by chromatography. The stereochemistry of the cyclic phosphonate 283 and phosphonate esters cis-284 and trans-284 was assigned on the basis of 1H and 31P NMR and comparison with the literature. Final saponification of the mixture of diasteroisomers 284 afforded ring opening and formation of the HDP-PMPDAP prodrug 285 in 54% yield (Scheme 88).
Scheme 88. Synthesis of HPMPDAP Alkoxyalkyl Prodrugs.

An alternative method for the synthesis of alkoxyalkyl cidofovir prodrug analogs was developed by Wan et al. and involves a Mitsunobu-type coupling between cHPMPC 286 and oleyloxypropyl alcohol in the presence of triphenylphosphine and diisopropyl azadicarboxylate (DIAD). Ring opening of 287 under basic conditions and subsequent neutralization with acetic acid afforded the desired OLE-HPMPC prodrug 288 in 42% yield (Scheme 89).118 This method was later used for the synthesis of glycero prodrug derivatives such as 1-O-octadecyl-2-O-benzyl-sn-glycero-3-cidofovir. This prodrug was shown to target the lungs more specifically.119
Scheme 89. Synthesis of OLE-HPMPC Prodrug.

Valiaeva et al. reported the preparation of HDP-PMEG prodrug 292.120 2-Amino-6-chloropurine phosphonic acid 290 was synthesized121 by, first, alkylation of diisopropyl 2-chloroethoxymethylphosphonate with 2-amino-6-chloropurine in the presence of DBU, followed by phosphonate deprotection with TMSBr. Interestingly, introduction of the alkoxyalkyl chain was carried out at this stage of the synthesis on the 2-amino-6-chloropurine nucleoside phosphonic acid, presumably to avoid competitive alkylation at the O6-position of guanosine and/or increase the solubility of the nucleoside. The coupling reaction of phosphonic acid 290 with HDPOH in the presence of DCC afforded the corresponding 2-amino-6-chloropurine acyclic nucleoside phosphonate prodrug 291 in 47% yield. Subsequent acidic hydrolysis with a 1 M HCl solution and basic neutralization converted nucleotide 292 into the desired guanosine derivative in 75% yield (Scheme 90).
Scheme 90. Preparation of HDP-PMEG Prodrug.

This procedure was also used for the synthesis of phosphonopropoxymethyl guanine and 2,6-diaminopurine alkoxyalkyl prodrugs, phosphonate isoster of acyclovir phosphate.122 Alkylation of diethyl-3-chloromethoxypropylphosphonate with 2-amino-6-chloropurine and subsequent deprotection of the phosphonate moiety with TMSBr afforded 2-amino-6-chloropurine nucleoside phosphonic acid 294. The phosphonic acid was converted into its ODE prodrug 295 as a dimethylamine pyridinium salt, by DCC coupling in the presence of DMAP. Subsequent basic hydrolysis with 1 M aqueous sodium hydroxide and neutralization with acetic acid afforded the ODE-guanosine nucleoside phosphonate prodrug 296 (Scheme 91).
Scheme 91. Synthesis of Phosphonopropoxymethyl Guanine Alkoxyalkyl Prodrugs.

Choo et al. reported the preparation of the alkoxyalkyl cis-5-phosphono-pent-2-en-1-yl nucleoside prodrug 299.123 Uracil diethyl phosphonate derivative 297 was first synthesized by Mitsunobu coupling between N3-benzoylated uracil and (Z)-diethyl (5-hydroxypent-3-en-1-yl)phosphonate followed by debenzoylation with ammonia in methanol. Triisopropylphenylsulfonylation and subsequent aminolysis converted the uracil diethylphosphonate 297 into its corresponding cytosine derivative 298 in 75% yield. Deprotection of the phosphonate moiety with TMSBr afforded phosphonic acid, which was subsequently coupled to HDPOH in the presence of DCC and DMAP (Scheme 92).
Scheme 92. Preparation of Alkoxyalkyl cis-5-Phosphono-pent-2-en-1-yl Nucleoside Prodrug 299.

Beadle et al.124 developed an attractive alternative and more convergent strategy for the synthesis of related HPMP adenine prodrug 303 based on the introduction of a phosphonate moiety already bearing the alkoxyalkyl chain. The key alkoxyalkyl tosylate 301 was synthesized from the diethyl derivative 300 by, first, deprotection with TMSBr, chlorination of the resulting phosphonic acid, selective substitution with alkoxyalkyl alcohol, and, finally, hydrolysis with a saturated NaHCO3 solution. The HPMP-adenine analog 303 was readily obtained by alkylation of (S)-9-(3-trityloxy-2-hydroxypropyl)-N6-trityl-adenine 302 with alkoxyalkyl toluenesulfonyloxy methylphosphonate 301 followed by deprotection under acidic condition (Scheme 93).
Scheme 93. Synthesis of HPMP-Adenine Prodrug 303.

Alternative preparations of the related HDP-tosylate 309, through a bis(HDP) P(III) derivative, have been reported by Vrbkova et al. (Scheme 94).125 HDPOH was first treated by PCl3 in pyridine to afford bis(HDP)-phosphite 304. Standard hydroxymethylation with paraformaldehyde and triethylamine was followed by tosylation, and afforded bis(HDP)-tosylate 308 in 25% yield. A more efficient synthetic pathway to bis(HDP)-tosylate 308 involves bis-activation of phosphonic acid 307 with oxalyl chloride and subsequent substitutions with HDPOH. Following a known procedure,126 monodeprotection by treatment with excess LiN3 in DMF at high temperature led to HDP hydroxymethyl tosylate 309 in high yield (Scheme 94).
Scheme 94. Preparations of HDP-Tosylate 309.

Bis(phosphonomethoxy)-acyclic nucleoside 311 was prepared in a 60% yield by reacting 310 with 2 equiv of HDP tosylate 301 in DMF. Monosubstituted compound 312 was obtained as a side product in 25% yield (Scheme 95).
Scheme 95. Preparation of Bis(phosphonomethoxy)-acyclic Nucleoside 311.

Similarly, the 5-fluorocytosine HPMP derivative 316 was obtained in 69% yield in three steps: (1) alkylation of the free alcohol of 313 with HDP tosylate 301, (2) Bz-deprotection by aminolysis, and (3) trityl removal in acidic conditions. The direct deprotection of 315 with 80% acetic acid afforded the 5-fluorouracil derivative in 54% yield (Scheme 96).120
Scheme 96. Synthesis of 5-Fluorocytosine HPMP Derivatives 315 and 316.

Interestingly, the 2,6-diaminopurine HPMP derivative could be obtained following the same procedure, but without nucleobase protection (Scheme 97).120
Scheme 97. Synthesis of 2,6-Diaminopurine HPMP Derivative Requires Only Hydroxyl Protective Group.

Using the same key intermediate 301, HDP-PMPDAP (2,6-diaminopurine) alkoxyalkyl prodrugs 321a and its 2-amino-6-cyclopropyl analog 321b were synthesized by Valiaeva et al.120 Thus, purines were reacted with 1,3-dioxolan-2-one, and subsequent alkylation with alkoxyalkyl tosylate 301 afforded the desired alkoxyalkyl prodrugs 321 (Scheme 98).
Scheme 98. Synthesis of HDP-PMPDAP Alkoxyalkyl Prodrugs and Its 2-Amino-6-cyclopropyl Analog.

In a similar manner, HDP-PMEDAP, an open ring analog based on the 2,4,6-triaminopyrimidine, was obtained in 19% yield by reaction of tetrahydropyranyloxyethylamine with 2,4-diamino-6-chloropyrimidine 322 followed by acidic hydrolysis. Subsequent alkylation of compound 323 with HDP tosylate 309 afforded HDP-PMEDAP 324 in 15% yield (Scheme 99).120
Scheme 99. Synthesis of HDP-PMEDAP Prodrug.

Valiaeva et al.127 prepared ODE-(S)-MPMP guanosine compound 327, which was found to be active against HCV. Compound 327 was easily synthesized in two steps from the O-benzylated guanosine derivative 325 by first coupling with alkoxyalkyl tosylate 301 in the presence of t-BuONa, followed by removal of the benzyl group under acidic conditions (Scheme 100).
Scheme 100. Preparation of ODE-(S)-MPMP Guanosine Compound 327.

Finally, an alternative approach allowing direct conversion of the free PME-C, -G, and -A nucleosides into their corresponding alkoxyalkyl prodrugs was developed by Vrbkova et al.125 Thus, reaction of PMEG with oxalyl chloride in DMF allowed for the one-pot chlorination of the phosphorus atom and formation of a formamidine functional group at the C-2 position. Intermediate 328 was reacted with HDPOH in pyridine to form a bis(HDP)-substituted compound. Subsequent deprotection in 80% acetic acid and removal of one of the alkoxyalkyl chain by treatment with an excess of LiN3 afforded HDP prodrug 330 in high yield (Scheme 101).
Scheme 101. Synthesis of HDP-PMEG Prodrug.

Tichy et al.128 also used a selective hydrolysis of bis(HDP)-monophosphate derivatives to prepare HDP-(S)-HPMP and HDP-2-(2-phosphonoethoxy)ethyl (PEE) prodrugs 333 and 336. Thus, starting from bis(isopropyl)-phosphonoethoxyethyl chloride, treatment with TMSBr, chlorination of the resulting phosphonic acid, and coupling with HDPO gave the desired bis(HDP)-chloro derivative 331. 2-Amino-6-chloropurine was then introduced under basic condition, and the resulting intermediate was hydrolyzed with AcOH to give the bis(HDP)-guanine prodrug 332. Finally, treatment with LiN3 in DMF afforded the desired HDP-PEE prodrug 333 (Scheme 102).
Scheme 102. Preparation of HDP-PEE by Hydrolysis of Bis(HDP)-MP Derivatives.

The related HDP-(S)-HPMP prodrug 336 was prepared in six steps from bis(isopropyl)-phosphonoethoxyethyltosylate 306. Thus, 306 was reacted with TMSBr to give the corresponding phosphonic acid, which was then chlorinated and reacted with HDPOH. The resulting bis(HDP)-phosphonate 308 was then reacted with 334 in the presence of NaH to give the bis(HDP)-(S)-HPMPG 335 after deprotection under acidic conditions. Finally, selective hydrolysis was achieved by treatment with NaOH in a mixture of dioxane and water to provide the desired HDP-HPMPG prodrug 336. Interestingly, the authors also prepared the cyclic monoester 337 by treatment of 336 with PyBOP in the presence of i-Pr2EtN (Scheme 103).
Scheme 103. Hydrolysis of Bis(HDP)-MP Derivatives To Prepare HDP-(S)-HPMPG.

3.2.3. Alkoxyalkyl Phosphoramidates
Liang et al.111 reported the synthesis of alkoxyalkyl phosphoramidate DOT prodrug 339. This mixed prodrug was generated by reaction of diphenyl phosphite with DOT, followed by addition of alkoxyalkyl alcohol. The amino acid portion of the prodrug was finally introduced by reaction of phosphite intermediate 338 with alanine (Scheme 104).
Scheme 104. Synthesis of Alkoxyalkyl Phosphoramidate DOT Prodrug 339.

3.3. Phosphoramidates and Phosphonamidates O-PO(OR)(NR2) and C-PO(OR)(NR2)
3.3.1. Aryloxy Amino Acid Amidate ProTide
Aryloxyphosphoramidate prodrugs, also called “ProTides”, contain a phosphorus atom bearing an amino acid alkyl ester and an aryloxy group. Pioneered in the early 1990s by McGuigan and co-workers, this prodrug approach was the result of several years of SAR studies during which several types of masked phosphate moieties were evaluated including bis(alkyloxy)- and haloalkyloxyphosphates,129 bis(aryloxyphosphate),130 cyclic131 and noncyclic alkyloxyphosphoramidates,129i,132 and phosphorodiamidates.133
Because of their ability to increase or even reveal activity of nucleosides, but also because they are relatively easy to prepare, this type of prodrug was used in drug discovery settings by medicinal chemists for the biological evaluation of new nucleosides/tides candidates in vitro.134 The proof-of-principle in humans demonstrated with sofosbuvir (PSI-7977, originally discovered by Pharmasset, Inc., and approved for the treatment of HCV) paved the way for the development of several other aryloxyphosphoramidate prodrugs that have now advanced to clinical trials for HIV treatment (GS-7340 to phase III,135GS-9131 to phase II,136 stampidine, to phase I137), cancer (thymectacin, in phase I/II for the treatment of colon cancer138), and HCV treatment (INX-08189139 to phase II, PSI-353661(140) to phase I) (Figure 32).
Figure 32.

ProTides nucleoside in clinical trials or FDA-approved.
The mode of action of these aryloxyphosphoramidates, leading to the intracellular delivery of active nucleoside monophosphates, has been studied in detail over the years.141 After crossing the cell membrane, the monophosphate deprotection is initiated by an esterase or cathepsin A producing carboxylate A.142 A spontaneous intramolecular cyclization to a five-member ring occurs, releasing a molecule of phenol. Cyclic intermediate B undergoes chemical opening in the presence of water leading to phosphoramidate diester C. Finally, cleavage of C by intracellular phosphoramidase or histidine triad nucleotide-binding protein 1 (HINT-1)143 frees the nucleoside monophosphate (Figure 33).141a,144
Figure 33.

Mode of action of aryloxyphosphoramidates/phosphonamidates.
Aryloxyphosphoramidate nucleoside prodrugs are generally prepared by three different methods highlighted in Figure 34: (A) coupling of the nucleoside with a phosphorochloridate reagent, (B) coupling of a nucleoside with a diarylphosphite and subsequent oxidative amination, and (C) coupling of an amino acid to a nucleoside aryl phosphate.
Figure 34.

Methods to access phosphoramidates nucleoside prodrugs.
It is noteworthy that these different synthetic approaches generally lead to approximate 1:1 mixtures of diastereoisomers at the phosphorus center often inseparable by flash chromatography. The discovery that Sp and Rp isomers had different in vitro biological properties lead to the development of a diastereoselective approach using enantiomerically pure aryloxy phosphoramidate reagents (Method A′, Figure 34).
On the other hand, the aryloxyphosphonamidate nucleoside prodrugs are obtained from the nucleoside phosphonic acid as shown in Figure 35: (A) bis-chlorination and subsequent substitutions with phenols and amino acids, (B) DCC coupling with phenols (1 equiv) followed by chlorination of the nucleoside aryl phosphate and subsequent substitution with an amino acid, and (C) selective saponification leading to a nucleoside monoaryl phosphate and subsequent coupling with an amino acid.
Figure 35.

Methods to access phosphonamidates nucleoside prodrugs (AA = amino acid).
3.3.1.1. Aryloxy Amino Acid Phosphoramidate
3.3.1.1.1. Phosphorochloridate Coupling
Aryloxyphosphoramidate nucleoside prodrugs are generally prepared by coupling of nucleosides with phosphorochloridate by either activation of the imidazolium intermediate with NMI145 or by 5′-deprotonation of the nucleoside with t-BuMgCl146 and subsequent substitution with the chlorophosphoramidate (Figure 36).147
Figure 36.

Mechanism to generate phosphoramidates nucleoside prodrugs.
Over the past 20 years, substitution of the phosphorochloridate reagent has been explored by modifying (1) the nature of the aryloxy portion (substituted phenols or napthols), (2) the amino acid, and (3) the amino acid ester. Key phosphorochloridate reagents are generally prepared by reaction of phosphorus oxychloride with an aryl alcohol in the presence of triethylamine followed by addition of the appropriate amino acid alkyl ester.148
Phosphorochloridates are generally obtained as a 1:1 mixture of Rp and Sp diastereoisomers. They are often used crude after a simple extraction or filtration, but cleaner reaction and higher yields are observed when purified by a flash chromatography. From all of the natural amino acids, l-alanine is the most commonly used, while the nature of the aryl group and carboxyl ester portion is dependent on the nucleoside and/or its application. In a general manner, the replacement of the natural amino acid with, for instance, d-amino acids149 or dialkyl glycine150 led to significant loss of activity. The only counter-example is the dimethyl glycine that showed potency almost similar to that of l-alanine.151 In the same way, any attempts to replace the amino acid moiety by simple amines152 or to incorporate methylene linker between the nitrogen and the ester group of the amino acids153 led to almost total loss of in vitro activity.
Since the first use of the NMI-mediated coupling for the synthesis AZT aryloxyphosphoramidate prodrug by McGuigan et al. in 1992,130a numerous nucleoside prodrugs have been successfully prepared using this approach including AZT,154,155 d4T,141b,150,151,156−159 ddU,160 BVdU,161 DOT,111,162 5-trimethylsilyl-arabinofuranosyl uracil,163 spiropentane nucleoside,164 8-aza-isoddA,165 9-deaza-guanosine,166 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine,167 AraU,168 carbocyclic 2′-methyl-2′-fluoro uridine,169 and 2′-C-methyl-6-hydrazinopurine ribonucleoside analogs (Figure 37).55
Figure 37.

NMI method.
On the other hand, the method employing t-BuMgCl as a reagent has been successfully employed to prepare d4U and ddU,170 ddA and d4A,171d- and l-carbocyclic d4A and ddA,172l-2′-deoxythreofuranosyl 3′-aryloxyphosphoramidate prodrugs,173 3TC,174l-carbocyclic 2′,3′-dideoxy-2′,3′-didehydro-7-deazaadenosine,175 2′,5′-dideoxyadenosine,176 2′,3′-dideoxy-3′-fluoroadenosine,177 2′-fluoro-6′-methylene-carbocyclic adenosine,178 4′-azidouridine,179 cytidine180 and adenosine,181 and 2′-methyl-4′-azidouridine and -cytidine182 prodrugs (Figure 38).
Figure 38.

t-BuMgCl method.
These two approaches are substrate-dependent, and therefore it is very difficult to predict a priori which one to use for the best outcome. In some cases, they both provide similar yields like in the case of 2′-C-methyl-2-amino-6-substituted-purine ribonucleoside analogs183 and 5-FdU (Scheme 105).184
Scheme 105. Comparable Efficiency between the NMI and t-BuMgCl Methodologies.

In most cases, both approaches provide the expected product prodrug, albeit one in better yields than the other as observed by Kumar et al.185 during the synthesis of 6-thio-7-deaza-2′-deoxyguanosine phosphoramidate 364 (Scheme 106, eq 1). Finally, one approach will sometimes afford the expected prodrug, while the other one will be completely ineffective. For instance, McGuigan et al.186 were unable to synthesize abacavir and carbovir prudrugs using the NMI-mediated coupling. However, when 365a and 365b were treated with 1.1 equiv of t-BuMgCl, before adding, respectively, 2.2 and 3 equiv of phosphorochloridate, abacavir was phosphorylated in 43% yield after 36 h, while carbovir was converted to the corresponding prodrug 366b in 23% yield after 1 week (Scheme 106, eq 2). On the contrary, Yoo et al.187 had to use the NMI-approach to prepare 2′-deoxyzebularine because the treatment of (367) with t-BuMgCl and subsequent addition of phosphorochloridate never provided the expected compound 368 (Scheme 106, eq 3).
Scheme 106. NMI versus t-BuMgCl Methodology.

These methodologies present some limitations mainly in the formation of byproducts that requires, in some cases, the protection of the nucleobase and/or the sugar moiety.
3.3.1.1.1.1. Byproducts
Reaction efficiency depends essentially on the presence of the following.
(1) Other free hydroxyls groups on the sugar backbone of the nucleoside: For instance, the phosphorylation of unprotected nucleosides bearing competitive hydroxyl group(s) can lead to a mixture of 5′-mono and 3′,5′-bisphosphorylated products (often separable by chromatography). However, the same reaction can also produce, in certain cases, a 3′-monophosphorylated regioisomer hardly separable from the 5′-phosphorylated product.5,184b,188
(2) The nature of the nucleobase: Uridine and adenine nucleoside analogs can, in general, be phosphorylated with both methods. That is, no side products resulting from nucleobase phosphorylation are typically observed for the uridine derivatives, while minor N6-phosphorylation of adenine can be observed, but are easily removed during chromatography.189,190 Cytosine nucleoside prodrugs can be obtained by NMI-mediated coupling;191 however, the high nucleophilicity of the amine can lead to partial N4-phosphorylation, and therefore the anionic method (t-BuMgCl) is generally preferred.174,180 Similarly, conversion of guanosine, 2,6-diaminopurine, and hypoxanthine nucleoside analogs to their corresponding phosphoramidate prodrugs can often be problematic. Indeed, competitive O6-phosphorylation can occur, and their low solubility often limits the efficiency of the reaction. However, both NMI and t-BuMgCl methods can be used; nevertheless, the NMI-mediated phosphorylation can fail when solubility of the substrate is very low.186
Thus, reaction of allene derivative 369 with 2.15 equiv of phosphorochloridate in the presence of 4.15 equiv NMI led to the formation of allenic phosphoramidate 370 along with a side product presumably identified as the O6-phosphorylated compound. Interestingly, this bis-phosphorylated side product was not isolated, because treatment with silica gel and methanol led to its disappearance. A similar byproduct was observed during the synthesis of butenol nucleoside prodrug 372, in which the reaction mixture was treated with 80% AcOH to hydrolyze the O6-phosphoramidate prior to chromatography (Scheme 107).
Scheme 107. Competitive O6-Phosphorylation, Separation of Mixtures.

Qiu et al. observed the same phenomena during the synthesis of E- and Z-methylenecyclopropane acyclic purine nucleoside aryloxyphosphoramidate prodrugs.190,192 The phosphorylation reaction was performed by treating nucleoside 373(193) with 5 equiv of phosphorochloridate and 10 equiv of NMI with added pyridine as a solubility enhancing cosolvent. As a result, the bis-phosphorylated derivate 374 was formed as the major product. This compound could then be converted into the desired prodrug 375 in 80% yield after acidic hydrolysis (Scheme 108).
Scheme 108. Competitive O6-Phosphorylation, Hydrolysis to Desired Prodrug 375.

3.3.1.1.1.2. Protection of Competitive Sites on the Nucleobase
In addition to the use of polar cosolvents such as pyridine or DMF that can sometimes be beneficial, temporary protection of the nucleobase or the sugar moiety can alternatively be used to increase the solubility of the nucleoside and mask other competitive hydroxy and amino groups.
Thus, Ambrose et al.194 prepared cytosine methylenecyclopropane acyclic nucleoside 379 by first N4-protection of compound 376 with a benzoyl group, followed by reaction with phosphorochloridate 340 via the NMI-mediated coupling. In this case, the benzoyl group prevents N4-phosphorylation reaction but also allows for the separation of Z and E nucleoside isomers 377a and 377b. Selective benzoyl group deprotection was carried out by treatment with hydrazine in a 4:1 mixture of pyridine/AcOH to give the desired prodrug 379 in 29% yield (Scheme 109).
Scheme 109. Competitive N4-Phosphorylation, Benzoyl Protection.

Because direct phosphorylation of acyclovir was low-yielding with the t-BuMgCl method (11%) and ultimately failing with the NMI-mediated coupling, McGuigan et al. reported the preparation of acyclovir aryloxyphosphoramidate prodrug 381 from N2- dimethylformamidine protected nucleoside 382 (Scheme 110).195 More soluble than the free nucleoside, the DMF-protected acyclovir was efficiently phosphorylated with the NMI method in 51% yield. Deprotection in refluxing propanol afforded the desired acyclovir prodrug in 90% yield. Additional aryloxyphosphoramidate N2-dimethylformamidine protected analogs were later prepared with the t-BuMgCl method in yields ranging from 31% to 93%.196 However, the deprotection step usually led to modest yields (2–25%). These low yields were partially due to the additional HPLC reverse phase purification step after the classical flash chromatography.
Scheme 110. N2-Protection as a Formamidine Group.

Formamidine protections can also be used to temporarly protect the cytidine N4-exocyclic amine. Thus, Nilsson et al.197 synthesized 4′-azido-2′-deoxy-2′-C-methylcytidine prodrug 386 by first reaction of (383) with dimethylformamide dimethylacetal, followed by reaction with a chlorophosphoramidate in the presence of NMI and final deprotection of the amino group under acidic conditions (Scheme 111).
Scheme 111. Competitive N4-Phosphorylation, Formamidine Protection.

3.3.1.1.1.3. Protection of Competitive Sites on the Sugar Moiety
Along with the nucleobase protection, temporary groups can also be introduced on the sugar moiety to increase the solubility of the starting material and avoid competitive phosphorylation.
For instance, while the direct phosphorylation of 4′-azido adenosine181 afforded the desired prodrug 390 in very low yield (6%), the 2′,3′-diol protection with a cyclopentylidene moiety allowed the coupling of (388) with chlorophosphoramidates in yields ranging from 71% to 92%. Mild acidic deprotection using 80% formic acid in water for 4 h afforded free prodrugs 390 in 47–55% yield (Scheme 112).
Scheme 112. Competitive OH Groups, Protection with a Cyclopentylidene Moiety.

McGuigan et al.198 protected nucleoside 391 with an isopropylidene group using a catalytic amount of perchloric acid in acetone. Phosphorylation of 392 with 2 equiv of t-BuMgCl and various phosphorochloridates afforded the corresponding protected aryloxyphosphoramidate nucleosides in 30–88% yield. The following deprotection was carried out in acidic conditions to afford the desired prodrugs 393 in moderate to good yields (Scheme 113). Similar alkylidene protection strategies were also employed for the preparation of several nucleoside analogs such as 5-substituted uridine,199 4′-azidocytidine180 and inosine,200 2′-C-Me-cytidine201 and adenosine,198 or ribavirin,202 using the t-BuMgCl method for the phosphorylation reaction (not shown).
Scheme 113. Competitive OH Groups, Protection with an Isopropylidene Moiety.

Alternative protecting groups such as benzyloxycarbonyl (Cbz) have also proven to be compatible with the synthesis of phosphoramidate prodrugs. Thus, Cho et al.203 prepared Cbz-protected A, U, G, and C derivatives 397 using a high-yielding three-step process: first 5′-hydroxy TBDMS-protection, followed by 2′,3′-hydroxyl groups Cbz-protection (along with the N4-position in the case of cytosine derivative), and final TBDMS-removal using Et3N·HF. The NMI-mediated coupling of 395 with phosphorochloridate 340 afforded corresponding derivatives in yields ranging from 94% to 98%. Finally, catalytic hydrogenolysis of (396) delivered the desired A, U, G, and C prodrugs in almost quantitative yields (Scheme 114). It is noteworthy that catalytic hydrogen transfer with cyclohexa-1,4-diene and palladium on charcoal was preferred for uridine derivatives to avoid partial reduction of the C(5)–C(6) double bond. Despite a long sequence, the excellent overall yield represents a real improvement to the direct phosphorylation of unprotected nucleoside (86% to 10% from cytidine, respectively). This method was eventually applied to the synthesis of aryloxyphosphoramidate prodrugs of both 2′-deoxy-2′-α-fluoro-2′-β-C-methyl uridine and cytidine in 87% and 86% yields.203
Scheme 114. Competitive OH Groups, Cbz-Protection.

More recently, Cho et al.204 showed that N6-carbamoyl adenosines nucleosides 399 can also be efficiently coupled with chlorophosphoramidate 340 using t-BuMgCl (Scheme 115).
Scheme 115. NMI, Used as Coupling Agent for the Cbz-Protection of the Purine Nucleoside.

A temporary levulinate protecting group has also been reported by Shen et al. for the synthesis of the vidarabine aryloxyphosphoramidate prodrug 404 (Scheme 116).205 Vidarabine was sequentially silylated at the 5′-position and acylated at the 2′- and 3′-positions with levulinic anhydride. Selective desilylation using TBAF in acetic acid afforded the correctly protected nucleoside 403 in 87% yield. Interestingly, acetic acid was critical in this reaction to prevent the levulinyl group from shifting from the 3′- to the 5′-position. Desired prodrugs were finally obtained by NMI-mediated phosphorylation and subsequent deprotection with hydrazine hydrate in a pyridine/acetic acid buffer (Scheme 116).
Scheme 116. Competitive OH Groups, Lev-Protection.

Shen et al.206 also used a similar sequence involving temporary protection of compound 405 with levulinates to prepare the triciribine prodrug 407 (Scheme 117).
Scheme 117. Competitive OH Groups, Lev-Protection.

Interestingly, Di Francesco et al.207 found the direct formation of 7-substituted phosphoramidate prodrug 410 from the corresponding parent nucleoside to be problematic and decided to use tetrahydropyranyl (THP) groups to both protect the secondary hydroxyl group and the pyrazole moiety. Thus, key intermediate 409 was obtained in four steps from 408 by 5′-silylation followed by protection of the 3′-hydroxyl, selective desilylation using TBAF, and Suzuki coupling with THP-protected pyrazole boronic acid. Finally, reaction of 408 with chlorophosphoramidate 340 in the presence of t-BuMgCl followed by removal of the two THP groups in AcOH afforded the desired 7-substituted 7-deaza-adenine nucleoside prodrug 409 (Scheme 118).
Scheme 118. Synthesis of 7-Substituted 7-Deaza-adenine Nucleoside Prodrug.

In the presence of competitive 5′- and 3′-hydroxyl groups, it is worth mentioning that reaction conditions can sometimes be optimized to minimize the formation of undesired species without the use of protective groups (Scheme 119). Lehsten et al.208 reported reaction conditions for the large-scale synthesis of NB1011, the phosphoramidate of (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU). They found that the temperature and the rate of addition of the electrophilic phosphoramidating species were critical factors to selectively phosphorylate the 5′- over the 3′-hydroxyl groups. The optimized conditions used dichloromethane as the solvent for the entire process. A ratio of 1.4:1 for the amino acid HCl salt to PhOP(=O)Cl2 allowed for the optimum formation of B. Maintaining the temperature between −10 and 0 °C, a solution of NMI in dichloromethane was then added dropwise. The reactive mixture is transferred slowly into a mixture of BVDU in dichloromethane at −5 °C. The nucleoside, poorly soluble in DCM, slowly dissolves in the presence of the excess NMI, and this allows further control of the reaction providing ∼1 kg of NB1011 of high purity (>99% by HPLC) in 53% yield after silica gel chromatography.
Scheme 119. Optimized Reaction Conditions for the Synthesis of NB1011, without the Use of Protective Groups.

3.3.1.1.2. Phosphite Approach
An alternative approach to the synthesis of aryloxyphosphoramidate nucleoside prodrugs involves the reaction of a nucleoside with a diaryl phosphite and subsequent amination under Atherton–Todd conditions with amino acids.
This method was developed by Li et al. using d4T and AZT as models.209 The key diaryl phosphite was prepared in a two-step procedure involving reaction of phenol with phosphorus trichloride and subsequent reaction with phosphorous acid (Scheme 120).210 The aryloxyphosphoramidate nucleoside prodrugs are then formed in a one-pot two-step procedure. First, the addition of the nucleoside to a mixture of 1.5 equiv of diarylphosphite and catalytic amounts of triethylamine in THF at low temperature quickly yielded the nucleoside aryl phosphite.211
Scheme 120. Preparation of Key Diaryl Phosphite Species.

The Atherton–Todd reaction was subsequently carried out by addition of amino acid methyl ester, triethylamine, and hexachloroethane. AZT and d4T prodrugs 413 and 414 were obtained in 60–72% yield over two steps (Scheme 121, eq 1).209 Despite this encouraging result, this reaction appeared limited and was reported later to lead to a complex mixture of products due to the excess of diaryl phosphite used to avoid nucleoside dimerization. Ora et al. used the same approach to prepare thymidine aryloxyphosphoramidate prodrug 415.212 Thymidine was reacted with diphenyl phosphite in pyridine and subsequently with alanine methyl ester (yields not reported, Scheme 121, eq 2).
Scheme 121. Phosphoramidates via Diaryl Phosphites.

Like for the phosphorochloridate coupling, the phosphite approach presents some limitations mainly with the formation of byproducts, which necessitates the protection of the nucleobase and/or the sugar moiety.
3.3.1.1.2.1. Byproducts
One drawback of this phosphite approach is the potential formation of complex products mixtures including dinucleotides and diamino acids species. To overcome these problems, a different synthetic pathway to the key nucleoside aryl phosphite intermediate was reported by Jiang et al. based on P(III) substitution.213 Using d4T and AZT as models, this three-component Arbuzov reaction214 is initiated by reacting the nucleoside with phosphorodichloridate (1 equiv), t-BuOH, and triethylamine to yield the corresponding nucleoside aryl phosphate 416 (Scheme 122). These conditions afforded the nucleoside aryl phosphite cleanly and in high yield (86% for the p-methoxy phenol derivative). The addition of 1 equiv of the amino acid, NCS, and 4 equiv of triethylamine to the solution containing the intermediate aryl nucleoside 5′-phosphite afforded almost quantitatively the desired aryloxyphosphoramidate prodrugs 417 in overall yields that ranged from 63% to 79% over two steps.
Scheme 122. Three-Component Arbuzov Reaction.

3.3.1.1.2.2. Protection of Competitive Sites on the Nucleobase and/or the Sugar Moiety
The overall yield of the sequence can usually be improved by masking competitive nucleophilic sites, which also increases the substrate solubility in commonly utilized solvents.
For example, Leisvuori et al.215 prepared 2′-OMe cytidine aryloxyphosphoramidate prodrug 422 by first 5′-hydroxyl silylation of 418, tritylation of the N4-position, and protection of the 3′-hydroxyl with levulinic acid and DCC in dioxane. Selective 5′-desilylation carried out with TBAF in a mixture of THF and acetic acid afforded the appropriately protected nucleoside 420. This latter compound was reacted with 1.2 equiv of diphenyl phosphite in pyridine followed by 1.5 equiv of alanine methyl ester in the presence of carbon tetrachloride and triethylamine to afford the protected nucleoside prodrug 421 in 70% yield. Treatment of 421 with hydrazine, acetic acid, and pyridine cleaved the levulinoyl group, while the MMTr group was removed by 80% aqueous AcOH at 65 °C, affording the desired prodrug 422 in 50% yield (Scheme 123).
Scheme 123. Protection of Competitive Sites – Use of MMTr for the Nucleobase and Levulinate for the Sugar.

In the same vein, Leisvuori and co-workers215 used levulinate groups to prepare ribavirin phosphoramidate prodrug 426. 2′,3′-Bis-levulinoylated ribavirin 423 was reacted with 1.5 equiv of diphenyl phosphite in pyridine to allow formation of the nucleoside phosphite phenyl ester and subsequently to alanine methyl ester in the presence of carbon tetrachloride and triethylamine to form the protected nucleoside prodrug 425 in 67% yield. Deprotection of the levulinoyl groups afforded the desired ribavirin aryloxyphosphoramidate prodrug 426 in 60% yield (Scheme 124).
Scheme 124. Protection of Competitive Sites – Levulinate Sugar Potection.

In the case of a ribonucleoside, protection of the 2′,3′-diol with an isopropylidene group can also be envisaged. Thus, Donghi et al. used this approach for the synthesis of 2′-C-Me-cytidine aryloxyphosphoramidate prodrugs bearing β-amino alcohols (Scheme 125).216 2′-C-Me-cytidine was first protected with an isopropylidene group after treatment with 2,2-dimethoxypropane and p-TsOH in acetone in 80% yield. The protected nucleoside was reacted with diphenyl phosphite in pyridine followed by amino alcohol to give 429 in 40% yield over two steps. The final deprotection with TFA in water afforded the desired prodrugs 430 as a mixture of phosphorus diastereoisomers that were later separated by HPLC or supercritical fluid chromatography (SFC).
Scheme 125. Protection of Competitive Sites – Isopropylidene Group.

3.3.1.1.3. Miscellaneous Approaches
Another approach involving P(V) chemistry was developed by Nillroth et al. for the synthesis of FLT-prodrugs.217 FLT was first reacted with 2 equiv of o-chlorophenyl phosphorodichloridate and excess 1,2,4-triazole in the presence of triethylamine to form the nucleoside aryloxy triazolide phosphoramidate intermediate 431. The subsequent addition of glycine methyl ester hydrochloride and triethylamine afforded the FLT-aryloxyphosphoramidate prodrug 432 in 80% yield (Scheme 126).
Scheme 126. Other Approach Using P(V) Chemistry.

In the same study,217 the use of a cyclic phosphorodiamidate reagent for the synthesis of a FLT o-(methynesulfonamino)phenyl methoxy glycine prodrug analog was also reported (Scheme 127). Cyclic phosphorochloridate 433 was reacted with glycine methyl ester in the presence of triethylamine to form phosphorodiamidate reagent 434. The crude compound was then directly reacted with FLT, affording the corresponding FLT-prodrug 435 in 86% yield.
Scheme 127. Use of Cyclic Phosphorochloridate.

Because the direct coupling of dipropylglycine phosphorochloridate with d4T afforded the desired phosphoramidate prodrug 437 in only 2% yield, McGuigan et al. designed an alternative approach for its synthesis.150 However, the coupling of d4T 5′-monophenyl phosphate 436(218) with 2 equiv of dipropyl glycine methyl ester and 2.5 equiv of MSNT in pyridine afforded the desired prodrug 437 in a low 7% yield (Scheme 128).
Scheme 128. Synthesis of D4T Phosphoramidate Prodrug 437.

3.3.1.1.4. Asymmetric Synthesis
It has been proven over time that Sp and Rp diastereoisomers can display different biological profiles, and it is not uncommon to see 10-fold or more difference in terms of in vitro potency between two phosphorus diastereomers.5,219 The separation of phosphorus diastereomeric mixtures can be realized, in some cases, by HPLC, selective crystallization, or flash chromatography on silica gel. However, chemists have more recently developed diastereoselective approaches based on a phosphorus SN2-type mechanism with chiral phosphor(odi)amidate reagents.
Thus, Román et al.219,220 designed a phosphorodiamidate reagent bearing a (S)-4-isopropylthiazolidine-2-thione as chiral auxiliary (Scheme 129). This chiral auxiliary allows the separation of the Sp and Rp diastereoisomers and acts as a leaving group during a SN2 reaction with a nucleoside. (S)-4-Isopropylthiazolidine-2-thione was reacted with phosphoryl chloride in the presence of triethylamine to give dichlorophosphate 188. The diastereoselective introduction of the aryl moiety was then carried out with either DBU or Et3N in acetone at −91 °C. The phosphorochloridates 438 were obtained in 59–93% yields and diastereomeric excess (de) between 28% and 87% depending on the nature of the aromatic moiety. It is noteworthy that the use of substoichiometric amounts of phenol was required to avoid formation of diaryl phosphoramidate byproducts that are hardly separable from the desired product. The introduction of the amino acid was carried out by reacting the diastereomerically enriched mixture of phosphorochloridates with a single equivalent of l-alanine methyl or benzyl ester hydrochloride and 3 equiv of triethylamine in dichloromethane. Interestingly, the diastereomeric ratios were found generally lower (∼15–85%) than those for the starting phosphorochloridate, pointing out a possible isomerization. However, these phosphorodiamidate reagents 439 can be separated via flash chromatography to deliver pure diastereoisomers (de > 95%). Crystals of the major diasteroisomer were obtained, and its structure and (R)-configuration were confirmed by X-ray crystallography. Finally, the coupling of d4T with 1 equiv of diastereomerically pure phosphorodiamidate 439 and 3 equiv of t-BuMgCl in a mixture of THF and pyridine (1:1) for 5 days at room temperature afforded the desired prodrugs 440 as single diastereoisomers in 11–50% yields (85–95% de). The stereochemistry of the (SP)-diasteromer was assigned by comparison with analytical data from the literature.
Scheme 129. Use of a Chiral Auxiliary.

Ross et al.221 also prepared several chiral phosphoramidate reagent bearing substituted phenols that would act as leaving groups during the phosphorylation step. p-Nitrophenyl phosphoramidate reagent 441 was prepared from the commercially available p-nitrophenyl dichlorophosphate by reaction with phenol and amino acid hydrochloride (Scheme 130). At this stage, two successive crystallizations in diisopropyl ether afforded the pure Sp-reagent in 22% yield (96% de). The stereochemistry of the phosphorus center was assigned by X-ray crystallographic analysis. On the other hand, pure Rp-isomer was obtained (de > 99.9%) by purification of the enriched mixture by supercritical fluid chromatography using a chiral stationary phase. The synthesis of PSI-7977 was then conducted with the Sp isomer 441 and t-BuMgCl, affording the desired prodrug 442 in 40% yield (99.7% de after two recrystallizations from dichloromethane).
Scheme 130. Crystallization of Phosphoramidate Reagent.

In the same paper, the authors also investigated the influence of other electronegatively substituted phenol moieties (nitro groups and halogens). 2,4-Dinitrophenol and pentafluorophenol phosphoramidates were found to be the most reactive reagents. The 2,4-dinitrophenol phosphoramidate had low selectivity between 3′- and 5′-hydroxy groups, leading to a higher proportion of 3′,5′-bis(phosphorylated)-nucleoside, whereas the pentafluorophenyl reagent was more selective and therefore was selected in further studies.
Compound 443 was prepared by reaction of phenyl dichlorophosphate with l-alanine isopropyl ester hydrochloride followed by pentafluorophenol addition (Scheme 131). After filtration of the salts, the crude solid was triturated in a mixture of 20% ethyl acetate in hexanes solubilizing only the desired Sp isomer (de >98%). The stereochemistry of (SP)-443 was determined by X-ray crystallography. Coupling conditions of (443) with 2′-Me,2′F-nucleoside were optimized, and it was found that low temperature of reaction (−5 °C) and slow addition of reagent lowered the formation of both 3′-phosphorylated and 3′,5′-bisphosphorylated side products while maximizing reaction conversion. Finally, the multigram scale synthesis of PSI-7977 was carried out by treating 2′-C-Me-2′-F-nucleoside with 2.1 equiv of t-BuMgCl in THF at −5 °C followed by the addition of 1.2 equiv of pentafluorophenol phosphoramidate 443. After being stirred for 18 h at 5 °C and two successive crystallizations, PSI-7977 was obtained in an excellent 68% yield (de > 99.7%).
Scheme 131. Synthesis of PSI-7977 (Sofosbuvir).

3.3.1.1.5. Post Modifications of Phosphoramidate Nucleoside Prodrugs
Interestingly, aryloxyphosphoramidate nucleoside prodrugs have proven to be stable enough to undergo further modifications. Thus, Velázquez et al. prepared AZT, d4T, and thymidine heterodimers with TSAO-T as potential inhibitors of HIV-1 reverse transcriptase.222 Thymidine aryloxyphosphoramidate prodrug formation was performed using 2 equiv of phosphorochloridate 340 and 6 equiv of NMI (Scheme 132). The corresponding nucleoside phosphoramidate underwent N3-alkylation with 1,3-dibromopropane, and 444 was then coupled to TSAO-T using potassium carbonate. Desired heterodimer 446 was obtained in 81% yield.
Scheme 132. Post Modifications – Alkylation Reactions.

As seen previously for the removal of Cbz groups, hydrogenation conditions are compatible with the aryloxyphosphoramidate moiety. Thus, reduction of l-Cd4A prodrug 447 gave the corresponding l-ddA phosphoramidate 449 in 49% yield (Scheme 133).172
Scheme 133. Hydrogenation of l-Cd4A to l-ddA phosphoramidite.

Similarly, Hatton et al. reported the reduction of the 4′-C-3′-O-propylene-linked bicyclic pyrimidine nucleoside (Scheme 134).223 While the cytosine analog 450a was hydrogenated with hydrogen over Pd/C, the uracil derivative 450b was hydrogenated by transfer hydrogenation over Pd/C to minimize the simultaneous uracil base hydrogenation.
Scheme 134. Post Modifications – Hydrogenation Reactions.

Postmodification of phosphoramidate nucleoside prodrugs by palladium-catalyzed reactions has also been reported by Perlikova et al.224 (Scheme 135). 6-Chloro-7-deazapurine ribonucleoside was first protected with an isopropylidene group before reaction with t-BuMgCl and phosphorochloridates. Phenyl, furyl, thienyl, and dibenzofuryl groups were then introduced at the 6-position using Suzuki–Miyaura or Stille cross-coupling reactions. Finally, isopropylidene deprotection with 90% TFA at room temperature led to the desired aryloxyphosphoramidate prodrugs 454 in yields ranging from 40% to 87%. It is noteworthy that partial hydrolysis of aryloxyphosphoramidate ester group was observed during the deprotection reaction.
Scheme 135. Post Modifications – Palladium-Catalyzed Cross-Coupling Reactions.

3.3.1.2. Aryloxy Amino Acid Phosphonamidate
Despite its long running success with regular phosphate nucleosides, ProTide technology has not been widely exploited with phosphonate nucleoside until recently.
One method developed by Ballatore et al. involves the bis-chlorination of the phosphonic acid nucleoside (PMPA) with thionyl chloride and subsequent substitutions with phenol and l-alanine methyl ester in the presence of triethylamine.225 Interestingly, the nucleoside was reacted again with thionyl chloride between the two substitutions, presumably to reactivate the potential hydrolyzed product. In these conditions, PMPA-aryloxyphosphonamidate prodrug 456 was only obtained in 5% yield (Scheme 136).
Scheme 136. Synthesis of PMPA-Aryloxy Phosphonamidate Prodrug.

A more efficient method was developed by Chapman et al. for the kilogram scale synthesis of GS-7171, an isopropyl ester aryloxyphosphoramidate prodrug of PMPA (Scheme 137).226 This method first involves a DCC-mediated coupling between PMPA and phenol in NMP at high temperature. The activation of the remaining hydroxyl with thionyl chloride and the subsequent substitution with the amino acid isopropyl ester at −78 °C afforded GS-7171 in 47% yield. At this stage, Rp and Sp isomers were separated using simulated moving bed chromatography to give GS-7340 (de > 98%).
Scheme 137. Syntheses of Both GS-7171 and GS-7340.

Mackman et al. used a third strategy for the synthesis of the phosphonomethyloxy cyclic nucleoside GS-9131 (Scheme 138).35 The conversion of nucleoside 458 into phosphonate intermediate 461 was accomplished by oxidation of the 5′-hydroxyl using Jones’ reagent, glycal formation under Mitsunobu conditions, and treatment with IBr and diphenyl hydroxymethyl phosphate. Oxidation of the iodine with NaOCl and treatment with aqueous ammonia afforded the nucleoside phenyl phosphonate monoester 462 in 18% yield. Coupling of this compound with l-alanine ethyl ester hydrochloride and PyBOP afforded the desired prodrug GS-9131 in 19% yield.
Scheme 138. Synthesis of GS-9131.

3.3.2. 3′,5′-Cyclic Phosphoramidate
3′,5′-Cyclic phosphoramidates have been recently designed as an alternative to McGuigan Protides to mainly eliminate the potential toxicity associated with the realease of phenol moieties. Thus, Gardelli et al.227 prepared 2′-C-methylcytidine-3′-5′-cyclic phosphoramidate 464 by reacting 2′-C-Me-C with a chlorophosphoramidate reagent bearing a 4-chlorophenol, in the presence of t-BuMgCl. At this stage, both fast eluting (464a F.E.) and slow eluting (464b S.E.) isomers were separated by RP-HPLC and reacted with t-BuOK to form the desired cyclic prodrugs. Interestingly, isomer 464a F.E. was found to give the desired cyclic prodrug 465a (SP) in 67% yield, while the other isomer 464b S.E. yields the corresponding cyclic compound 465b F.E. (RP) in only 35% along with monophosphate 466 (Scheme 139). The absolute stereochemistry of the phosphorus center on both cyclic compounds (SP)-465a and (RP)-465b was assigned by NOE experiments.
Scheme 139. 2′-C-Me-Cytidine-3′-5′-cyclic Phosphoramidate.

Jain et al.228 developed a one-step method for the synthesis of 3′,5′-cyclic phosphoramidate prodrug using novel phosphoramidate reagent 468 that was prepared in two steps from 4-nitrophenol by first reaction with POCl3 to give chloro intermediate 467, and then reaction with alanine methyl ester in the presence of Et3N. Reaction of 5-fluoro-2′-deoxyuridine (FdUrd) with intermediate 468 in the presence of DBU afforded cyclic prodrug 469 as a 5:1 mixture of diastereoisomers as determined by 31P NMR (Scheme 140).
Scheme 140. 2′-C-Me-FdU 3′-5′-Cyclic Phosphoramidate.

3.3.3. Amino Acid Amidate Monoester
Amino acid phosphoramidate nucleoside monoester prodrugs were pioneered in the 1990s by Wagner and co-workers.229 This prodrug was designed as a modification of the aryloxyphosphoramidate strategy detailed previously. The intention was to explore whether the lipophilic aryl group was indispensable or not while increasing the water solubility of the prodrug and losing the chirality of its phosphorus center. It was designed in such a way that the amino acid phosphoramidate mono ester biodegradation involves the direct cleavage of the amino acid group by the action of a phosphoramidase (Figure 39).144b,144c,230,231
Figure 39.
Decomposition pathway for amino acid amidate monoester nucleosides prodrugs.
Interestingly, amino acid phosphoramidate nucleoside monoesters regained some interest in recent years with the discovery, by Herdewijn et al., of their ability to act as a triphosphate mimic and thus to be substrates of reverse transcriptases (including HIV-1). It was demonstrated that amino acid phosphoramidate monoester nucleosides with specific amino acids such as l-aspartic acid and particularly l-histidine, in there acid form, are fulfilling the requirements of structural and electronic properties that allow proper alignment of α-phosphorus atom in the polymerase active site, mimicking a nucleoside triphosphate (Figure 40).232,233 The success of this recent approach led to its extension to modified or unnatural amino acids derivatives.234
Figure 40.
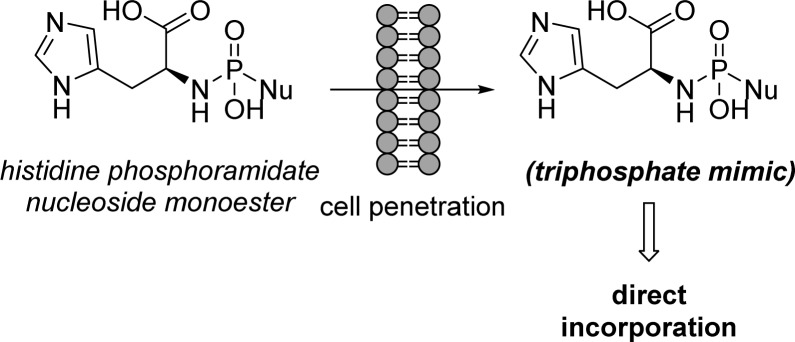
Histidine phosphoramidate nucleoside monoester acting as a triphosphate mimic.
The synthesis of amino acid phosphoramidate mono ester nucleosides prodrugs can be achieved by different synthetic pathways shown in Figure 41: (A) formation of a hydroxyl cyanoethyl nucleoside phosphite, oxidation to the monophosphate, and subsequent coupling/deprotection, (B) formation of a methyl cyanoethyl nucleoside phosphite and subsequent oxidative amination/deprotection, (C) formation of a protected H-phosphonate, and subsequent oxidative amination/deprotection, (D) hydrolysis of phosphorothioates, (E) hydrolysis of phosphoramidates, (F) transformation of the nucleoside di- or triphosphate by transient persilylation and substitution of a mono or diphosphate unit, and (G) direct coupling of an amino acid with a nucleoside monophosphate.
Figure 41.

Amino acid phosphoramidate mono ester nucleoside formation.
Their phosphonate counterparts can be prepared by coupling one amino acid to phosphonate nucleoside (Figure 42).
Figure 42.

Amino acid phosphonate mono ester nucleoside formation.
3.3.3.1. Amino Acid Phosphoramidate Monoester
In 1994, the first synthesis of amino acid nucleoside phosphoramidate monoester was reported by Wagner and co-workers235 to synthesize new AZT, FLT, and d4T prodrugs. AZT was first reacted with 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite and subsequently treated with 1H-tetrazole and methanol to yield the methyl cyanoethyl nucleoside phosphite intermediate 470. Subsequent reaction with phenyl alanine methyl ester in the presence of iodine and final hydrolysis afforded nucleoside phosphoramidate 471 in 38% yield. Removal of the cyanoethyl group with ammonia in methanol and purification on acidic resin afforded the desired prodrug 472 in 88% yield (Scheme 141).
Scheme 141. First Synthesis of Amino Acid Nucleoside Phosphoramidate Monoester.

In 1996, a more commonly used method was reported by Abraham et al.230a for the synthesis of amino acid phosphoramidate mono ester prodrugs of Ara-C and 5-FdU and involves the direct coupling of the amino acid to the nucleoside monophosphate. Thus, Ara-C was selectively phosphorylated at the 5′-position with phosphorus oxychloride in triethyl phosphate. After hydrolysis, a DCC-mediated coupling with phenylalanine and tryptophan methyl esters afforded the desired amino acid Ara-C phosphoramidates 474a and 474b (Scheme 142).
Scheme 142. Ara-C Monoester Phosphoramidates.

In 1997, the same approach was used by Abraham et al. for the preparation of amino acid phosphoramidate monoester of acyclovir (Scheme 143).236 After phosphorylation of acyclovir, a DCC-mediated coupling with various amino esters afforded the desired prodrugs 475 in 28–89% yield. It is noteworthy that the coupling of ACV with cyanoethyl phosphate afforded a mixture of products and was abandoned.
Scheme 143. Amino Acid Phosphoramidate Monoester of Acyclovir.

EDC has also been used as a coupling agent with 2′-deoxyadenosine to perform the reaction at room temperature, and compound 476 was obtained in an improved 63% yield (Scheme 144).230,234d
Scheme 144. EDC for Milder Coupling Conditions.

Interestingly, Abraham et al.237 observed a lack of selectivity between 5-FdU’s 5′- and 3′-hydroxy groups during phosphorylation, and thus had to use a series of temporary protecting groups. First, an MMtr group was introduced at the 5′-position of 5-FdU before acetylation of the 3′-hydroxy group. Selective 5′-deprotection and DCC-mediated coupling with cyanoethyl phosphate afforded protected 5-FdU intermediate 477. Deprotection under basic conditions followed by coupling with phenylalanine or tryptophan methyl esters afforded the desired 5-FdU prodrugs 479 (Scheme 145).
Scheme 145. 5-FdU Prodrugs.

Adelfinskaya et al. prepared different amino acid 2′-deoxy adenosine phosphoramidate mono esters (Scheme 146).232 DCC-mediated coupling between 2′-deoxy adenosine monophosphate 480 and various protected amino acids afforded the amino acid methyl ester nucleoside phosphoramidates 481 in 39–94% yield. The subsequent saponification was carried out with 0.4 M NaOH to afford the amino acid nucleoside phosphoramidates 482 in 22–94% yield. Alternative deprotection with potassium carbonate in 2:1 MeOH:water could also be used.234a A similar procedure was used for the synthesis of 1-deaza-, 3-deaza-, and 7-deaza- adenine analogs (not shown).238
Scheme 146. Preparation of Amino Acid 2′-Deoxy Adenosine Phosphoramidate Mono Esters.

Nucleoside phosphoramidate monoesters bearing amino acid in their acidic form, such as 484 and 486, are also intermediates of aryloxyphosphoramidate bioactivation and were synthesized as part of the activation studies (Scheme 147). These compounds were prepared by simple saponification of their corresponding aryloxyphosphoramidates 483 and 485, in aqueous triethylamine.184b,201
Scheme 147. Synthesis of Phosphoramidate Mono Acids 484 and 486.

Alternatively, the amino acid nucleoside phosphoramidate monoesters can be generated using H-phosphonate intermediates.239 AZT triethylammonium H-phosphonate was generated in 69% yield from AZT by first treatment with diphenyl phosphite in pyridine and subsequent hydrolysis in aqueous triethylamine. Transient silylation of 487 followed by oxidation with iodine, substitution with amino acids methyl ester, and basic hydrolysis afforded the desired amino acid nucleoside phosphoramidate 488 in 31–70% yield. Treatment with a methylamine solution in methanol gave the corresponding methyl amide analogs 489 in good to quantitative yields (Scheme 148).
Scheme 148. Amino Acid Nucleoside Phosphoramidate Monoesters, Generated Using H-Phosphonate Intermediates.

Other alkyl amines derivatives were directly prepared from the H-phosphonate nucleoside as previously described for the methyl ester analogs in 28–56% yield (Scheme 149).
Scheme 149. Preparation of Alkyl Amine Derivatives from H-Phosphonate Nucleosides.

The same method was used by Chang et al. for the preparation of amino acid 2′,3′-dideoxyadenosine (ddA) phosphoramidate monoester without requiring nucleoside protection.240 The oxidative amination of the nucleoside H-phosphonate with different amino acids afforded the desired ddA prodrugs in yields ranging from 28% to 51% (Scheme 150).
Scheme 150. Preparation of Amino Acid ddA Phosphoramidate Monoester Prodrugs.

One major drawback of such strategies is the need to use reverse phase or ion-exchange chromatography for the purification of these very polar nucleoside phosphate monoesters. To overcome this limitation, Zhu et al.241 used a fluorenylmethyl protecting group (Scheme 151). A slight excess of diphenyl phosphite was reacted successively with FmOH and with adenosine in pyridine to afford nucleoside H-phosphonate intermediate 493. The Atherton–Todd amination with amino acids hydrochloride, carbon tetrachloride, and triethylamine afforded the protected nucleoside phosphoramidates 494 in 73–82% yield. Deprotection with piperidine in dichloromethane led to the desired amino acid adenosine phosphoramidate mono esters 495 in 65–75% yields. Ora et al. used a similar strategy to prepare l-alanine thymidine phosphoramidate monoester (not shown).212
Scheme 151. Fluorenylmethyl Protecting Group To Ease the Purification of Highly Polar Nucleoside Phosphate Monoesters.

A benzyl group was also used as a temporary protecting group by Gardelli et al. for the preparation of amino acids 2′-C-Me-C phosphoramidate prodrugs.242 Diphenyl phosphite was successively treated with the 2′-C-Me-C nucleoside and benzyl alcohol to form nucleoside H-phosphonate 496 (Scheme 152). Oxidative amination with l-alanine 2-propenylpentyl ester under Atherton–Todd conditions afforded nucleosides 497. Subsequent acidic deprotection with 80% TFA in water and hydrogenolysis yielded the desired prodrugs 498 (yields not provided).
Scheme 152. Benzyl Protecting Group To Ease the Purification of Highly Polar Nucleoside Phosphate Monoesters.

Phosphite intermediates can also be generated through phosphoramidites as reported by Whalen et al. for the preparation of cytidine phosphoramidates monoester.243 Tribenzoyl cytidine phosphoramidite 501 was prepared by a tetrazole-mediated coupling of tribenzoyl cytidine with allyl phosphoramidite 500. Hydrolysis and oxidative amination under Atherton–Todd conditions afforded the protected nucleoside phosphoramidates 502 in 30–78% yield. Removal of the allyl group with Pd(PPh3)4 and subsequent treatment with NaOMe and NaOH afforded the desired phosphoramidates 503 in 80% yield for all examples (Scheme 153).
Scheme 153. Use of Phosphoramidites To Generate Phosphoramidate Prodrugs.

Fu et al. reported the synthesis of amino acid nucleoside phosphoramidate monoester from nucleosides di- and triphosphates (Scheme 154).244 Treatment of ADP with TMSCl and various amino acid methyl ester in pyridine and subsequent hydrolysis with 2 M ammonium hydroxide afforded the desired nucleoside monophosphate prodrugs in roughly 50% yield.
Scheme 154. Synthesis of Amino Acid Nucleoside Phosphoramidate Monoester from Nucleosides Di- and Triphosphates.

Using a similar approach, Zhu et al. prepared various amino acid nucleoside phosphoramidate monoesters from thymidine, uridine, adenosine, and guanosine triphosphates (Scheme 155).245
Scheme 155. Amino Acid Nucleoside Phosphoramidate Monoesters from T, U, A, and G Triphosphates.

Amino acid nucleoside phosphoramidate monoesters can also be obtained by hydrolysis of phosphorothioamidate derivatives (Scheme 156).216,246 2-Chloro-3-methyl-1,3,2-thiazaphospholidin-4-one 2-oxide was first reacted with glycine methyl ester hydrochloride in the presence of triethylamine to provide intermediate 506. Subsequent reaction with FLT and hydrolysis with 10% Et3N in dichloromethane over silica gel at 40 °C afforded the amino acid nucleoside phosphoramidate monoesters 508.
Scheme 156. Hydrolysis of Phosphorothioamidates.

De Napoli et al. used thymidine as a model for the synthesis of nucleoside phosphoramidates monoester libraries on solid phase.247 Tentagel HL resin was first linked to the thymidine phosphoramidite 509 via a 3-chloro-4-hydroxyphenylacetic linkage by a tetrazole-mediated coupling. After oxidation with iodine in pyridine and water, the 3′-trityloxy group was replaced by an acetoxy group, and the cyanoethyl group was cleaved in the presence of triethylamine in pyridine. With key intermediate 511 in hand, introduction of various amino acids was carried out successfully after tosylation of the phosphate ester. For optimal results, the coupling was repeated three times before the desired nucleoside phosphoramidate was detached from the solid support with concentrated aqueous ammonia (Scheme 157).
Scheme 157. Synthesis of Nucleoside Phosphoramidates Monoester Libraries on Solid Phase.

3.3.3.2. Amino Acid Phosphonamidate Monoester
The synthesis of amino acid nucleoside phosphonamidate monoester, such as 513, is much less represented in the literature when compared to their phosphoramidate counterparts. McKenna et al.248 reported the coupling of cidofovir with valine methyl and ethyl esters in the presence of EDC in water in 35–40% (Scheme 158).
Scheme 158. Synthesis of Cidofovir Phosphonamidate Monoester Prodrug.

A similar procedure was used by Adelfinskaya et al. for the synthesis of an aspartic acid adefovir derivative.232b DCC-mediated coupling of aspartic acid and adefovir and subsequent saponification with sodium hydroxide in methanol and water afforded the desired adefovir prodrug 514 in 58% yield (Scheme 159).
Scheme 159. Phosphonamidate of Adefovir.

3.3.4. Borch’s Methylaryl Haloalkylamidates
As part of their research program on nucleosides, the Borch team developed a neutral methyl aryl haloalkyl phosphoramidate prodrug capable of passing through the cell membrane. These prodrugs are designed to undergo intracellular activation to generate unstable phosphoramidate anion intermediate B, which in turn undergoes spontaneous cyclization and P–N bond cleavage by water to liberate the nucleoside monophosphate (Figure 43). It is noteworthy that their first prodrug approach involved a haloethyl (instead of halobutyl) nucleoside phosphoramidate.249 However, further mechanistic studies revealed that after cyclization of the haloethyl phosphoramidate anion, nonselective nucleophilic attack of water at the carbon and phosphorus centers of the pyrolidinium ion intermediate was observed, delivering the NuMP, but also an undesired hydroxyethyl byproduct.250
Figure 43.

Decomposition pathway of Borch’s methylaryl haloalkylamidates prodrugs.
The coupling of the methyl aryl haloalkyl phosphoramidate moiety to the nucleosidic part was achieved either using either P(III) or P(V) chemistry (Figure 44). No chiral synthesis has been developed so far, and final methylaryl haloalkyl phosphoramidate prodrugs are always obtained as a mixture of diastereoisomers at the phosphorus center.
Figure 44.

P(III) versus P(V) chemistry.
This prodrug approach has been successfully applied to the intracellular delivery of anticancer nucleotide 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP). This compound was synthesized using P(III) chemistry as shown in Scheme 160.251 Phosphorus trichloride is reacted with the corresponding alcohol in the presence of diisopropylethylamine followed by reaction with N-methyl-N-(4-chlorobutyl)amine hydrochloride to generate chlorophosphoramidite 515. This intermediate is directly reacted with 5FdU in situ and then oxidized with tert-butyl hydroperoxide to yield 5-nitrofurfuryl N-methyl-N-(4-chlorobutyl) phosphoramidate 517 in 34% yield.
Scheme 160. FdU Borch’s Phosphoramidate.

Wu et al.252 investigated the influence of hydrophilic modification of 5-FdU phosphoramidates by replacing the N-methyl group with an N-dihydroxypropyl chain. Selective phosphorylation on the 5′-hydroxyl group of 5-FdU using phosphoramidite 518, generated in situ by reaction of N-allyl-4-chlorobutan-1-amine hydrochloride with POCl3, provided compound 520. The -OBt moiety was then displaced by either benzyl alcohol or 5-nitrofurfuryl alcohol in the presence of DMAP to furnish the corresponding methylaryl haloalkyl phosphoramidates derivatives. Dihydroxylation with OsO4/NMO afforded the final N-2,3-dihydroxypropyl-N-(4-chlorobutyl) phosphoramidates 521 (Scheme 161).
Scheme 161. N-Dihydroxypropyl Phosphoramidates.

Interestingly, attempts to synthesize directly the methylaryl haloalkyl phosphoramidate of cytosine derivatives, such as cytarabine253 or gemcitabine,254 using either P(III) or P(V) approaches, were unsuccessful presumably because of possible side reactions and very low solubility of the nucleosides. To circumvent this problem, the cytosine amino group had to be protected with an allyloxycarbonyl group that was removed, after the phosphorylation step, by treatment with Pd(PPh3)4 and p-toluenesulfinate (Scheme 162).
Scheme 162. Allyloxycarbonyl Group as Transient Protective Group.

3.4. Phosphorodiamidates and Phosphonodiamidates O-PO(NR2)2 and C-PO(NR2)2
Phosphorodiamidate prodrugs have rarely been used for the last 20 years, probably due to the success of aryloxyphosphoramidates. It has only been recently that this prodrug approach was reinvestigated because, unlike aryloxyphosphoramidates, it bears an achiral phosphorus center and releases only natural amino acids upon metabolism.
A putative mechanism of unmasking to the monophosphate was proposed by McGuigan et al.255 in which carboxypeptidase cleaves the ester function of the amino acid, inducing spontaneous cyclization of the carboxylate of the free amino acid onto the phosph(on)ate moiety. After a spontaneous hydrolysis, the nucleoside phosphoramidate monoester is cleaved into the free nucleoside monophosph(on)ate by action of phosphoramidase (Figure 45).
Figure 45.

Mechanism of action of O- and C-phosphorodiamidate nucleoside prodrugs.
Bis(amino acid) nucleoside phosphorodiamidates can be prepared as shown in Figure 46: (A) phosphorylation of the nucleoside with phosphorus oxychloride and subsequent bis-substitution with amino acids, and (B) chlorination of a nucleoside monophosphate and coupling with the amino acids.
Figure 46.
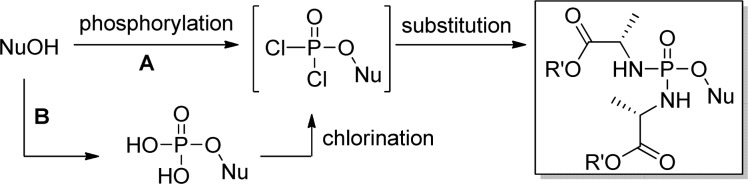
Methods to access O-phosphorodiamidate nucleoside prodrugs.
On the other hand, the synthesis of the bis(amino acid) nucleoside C-phosphorodiamidate involves three different methods highlighted in Figure 47: (A) direct coupling of the phosphonic acid nucleoside with amino acids, (B) TMSBr-deprotection of the phosphonate alkyl ester nucleoside and subsequent coupling with amino acids, and (C) chlorination of a nucleoside phosphonate and coupling with amino acids.
Figure 47.
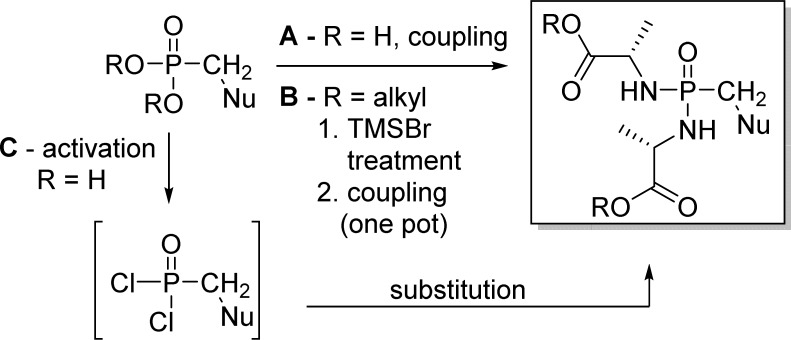
Methods to access C-phosphorodiamidate nucleoside prodrugs.
3.4.1. Bis(amino acid) O-Phosphorodiamidates
The first syntheses of bis(amino acid) O-phosphorodiamidate nucleosides were described in 1991 by McGuigan and co-workers who used AZT256 and FdU257 as substrates. AZT was reacted with phosphorus oxychloride in triethylphosphate to generate AZT monophorodichloridate 525. Subsequent substitution with excess amino acids in the presence of triethylamine afforded the corresponding AZT phosphorodiamidate prodrugs 526 in 21–44% yield (Scheme 163).
Scheme 163. AZT O-Phosphorodiamidates Synthesis.

McGuigan et al. also reported the preparation of bis(amino acid)-2′-methyl-6-methoxyguanosine O-phosphorodiamidate prodrug as part of an extensive SAR study (Scheme 164).255 Compounds were prepared either using the conditions described above or by slight modification of the procedure. Nucleoside was first phosphorylated with POCl3 at −78 °C in THF. Subsequent displacement with amino acids was carried out in the presence of diisopropylethylamine.
Scheme 164. 2’-Methyl-6-alkoxyguanosine Phosphorodiamidate Prodrug.

AA = amino acid.
Nonsymetrical O-phosphorodiamidates have been obtained in yields ranging from 4% to 17%255 following a similar protocol with successive addition of two different amino acids (Scheme 165).
Scheme 165. Nonsymetrical O-Phosphorodiamidates.

Key phosphorodichloridate intermediates can be alternatively generated from a nucleoside monophosphate as reported by Korboukh et al.258 Nucleoside monophosphate 532 was obtained in three steps through phosphoramidite coupling, subsequent oxidation, and cleavage of the tert-butyl groups under acidic conditions. Compound 532 was then reacted with 3 equiv of oxalyl chloride and a catalytic amount of DMF to form phosphorodichloridate intermediate 533. Subsequent reaction with 2.5 equiv of ethyl glycine hydrochloride in the presence of DIPEA, and isopropylidene deprotection with para-toluene sulfonic acid in methanol at 60 °C afforded desired prodrug 534 in 7% yield (Scheme 166).
Scheme 166. O-Phosphorodiamidates Nucleoside Prodrugs from Phosphate Nucleosides.

3.4.2. Bis(amino acid) C-Phosphorodiamidates
The bis(amino acid) nucleoside phosphorodiamidates are generally obtained from phosphonic acid intermediates after activation as phosphorodichloridates. Thus, Serafinowska et al. reported the synthesis of acyclophosphonate prodrug 538(259) in 15% yield by treatment of ethyl phosphonate derivative 535 with TMSBr, reaction of the corresponding silyl ester 536 with PCl5, and reaction with alanine methyl ester hydrochloride in the presence of triethylamine and NMI (Scheme 167).
Scheme 167. Synthesis of [(Phosphonomethoxy)ethoxy]adenine Prodrug 538.

Formation of such bis(amino acid) nucleoside phosphorodiamidates can also sometimes require temporary protection of the nucleobase. Thus, Dang et al. (Scheme 168, eq 1)260 treated PMEA with oxalyl chloride in the presence of DMF, allowing simultaneous chlorination of the phosphonic acid and protection of the N6-position. Subsequent reaction with 2-methylalanine ethyl ester and triethylamine gave the protected diamidates prodrugs 541. Finally, hydrolysis of the formidamide protection with acetic acid in isopropanol afforded the desired PMEA prodrug 542 in 16% yield. Interestingly, reactions carried out with the glycine methyl ester without nucleobase protection failed to produce the corresponding prodrug.
Scheme 168. Synthesis of PMEA Bis(amino acid) Nucleoside Phosphorodiamidates with Protective Groups.

Bis(amino acid) PMEA prodrugs such as 543(260) have also been prepared by direct coupling of PMEA with glycine ethyl ester in pyridine and treatment with a premixed solution of triphenylphosphine and 2,2′-dipyridyl disulfide (Scheme 168, eq 2). Interestingly, the reaction was reported to fail when using 2-methylalanine ethyl ester presumably because of the steric hindrance at the nitrogen.
A similar procedure was used by Mackman et al.35 for the synthesis of GS-9148 bis(amino acid) prodrug derivative (Scheme 169). Diethyl nucleoside phosphonate 544 was first treated with ammonium hydroxide generating 6-aminopurine nucleobase while deprotecting one of the phosphonate esters. Subsequent treatment with TMSBr afforded phosphonic acid 545. The coupling of different amino acids in the presence of 2,2′-dithiopyridine, triphenyl phosphine, and triethylamine in pyridine afforded the desired prodrugs 546 in 11–73% yield.
Scheme 169. GS-9148 Bis(amino acid) Prodrug.

Jansa et al.261 reported the synthesis of bis(amino acid) nucleoside phosphonamidate prodrugs directly from the bis(alkyl) nucleoside phosphonate 549 by coupling of transient silyl ester phosphonate intermediate 548 with amino acids (Scheme 170). This procedure prevents tedious isolation of nucleoside phosphonic acids.
Scheme 170. Nucleoside Phosphonamidate Prodrugs Directly from the Bis(alkyl) Nucleoside Phosphonates.

4. Nucleoside Di- and Triphosphate Prodrugs
For two decades, numerous prodrug strategies have been developed to deliver nucleoside monophosphates into the cells. The monophosphate’s delivery allows bypassing the first phosphorylation, which is often the rate-limiting step to NTP formation. However, di- and triphosphate prodrugs have rarely been studied. This lack of research can be explained by the generally efficient second and third phosphorylations (for most nucleosides) and the inherent instability of the phosphate anhydride bond. This bond is only kinetically stable because of the negative charge resonance that avoids the nucleophilic attack at phosphorus moiety.
The di- and triphosphate prodrug strategy has been mainly applied to AZT. AZT is a highly potent anti-HIV drug that was the first FDA-approved nucleoside analog for treatment of HIV infection. AZT is efficiently converted to the monophosphate, but only slowly to the diphosphate,262 resulting in intracellular accumulation of AZT-MP, which is responsible for some of its side effects.263 In an ideal situation, the delivery of AZT-DP or AZT-TP would retain the antiviral activity of the parent compound, but avoid the toxicity associated with the AZT-MP. This approach was also widely applied to ara-C to increase its bioavailability and to avoid base deamination.
The main strategy for the synthesis of di- and triphosphate prodrugs developed in the early 1980s involves the introduction of an alkyl or acyl lipophilic chain to the last phosphate unit (β-phosphorus for diphosphate and γ- for triphosphate). The synthesis of these lipophilic di- and triphosphate prodrugs commonly involves the coupling of a lipophilic chain bearing a phosphate or pyrophosphate moiety to a NDP or NTP. The lipophilic chain itself can be also directly coupled to a NDP or NTP. Recently, a new strategy was developed as acyloxybenzyl β-diester diphosphate using P(III) chemistry with the coupling of a phosphoramidite and a NuMP (Figure 48).
Figure 48.

BLG = biolabile group.
4.1. Nucleoside Di- and Triphosphate Glycerides
Several nucleoside di- and triphosphate prodrugs bearing acyl and alkyl glyceride moieties have been reported. The rational design of these prodrugs was based on naturally occurring phospholipid cytidine diphosphate diglyceride, which is a natural intermediate in the biosynthesis of anionic glycerophospholipid in mammalian cells. These prodrugs were mainly developed to reach HIV reservoirs such as macrophages and related cells involved in phagocytosis and antigen presentation. However, administration of antiviral nucleosides such as AZT, ddC, or 3′-deoxythymidine as nucleoside diphosphate diglycerides was found to deliver monophosphorylated anti-HIV agents intracellularly, due to the cleavage of the pyrophosphate unit between the α- and β-phosphorus (Figure 49).264
Figure 49.

Mechanism of action for nucleoside diphosphate glycerides.
Phosphatidic acid was commonly used for the synthesis of nucleosides di- and triphosphate diglycerides. Historically, the direct coupling of nonactivated phosphatidic acid and nucleoside monophosphate was first reported on natural nucleosides but provided only low yields.265 Therefore, the most common strategy involves the coupling of phosphatidic acid to a nucleoside 5′-monophosphate activated as a morpholidate (Scheme 171).266,267 The activation of the phosphatidic acid rather than the nucleoside monophosphate was reported later to give better yields and to facilitate the purification step.268 NTPs diglycerides were less described and generally synthesized by coupling of phosphatidic acid and an activated NDP in low yield: for instance, AZT-TP distearoylglycerol was prepared by condensation of AZT diphosphate with distearoylphosphatidic acid morpholidate. It was shown to deliver a mixture of AZT and AZT-MP to the cells.269 Numerous saturated and unsaturated lipophilic chains linked to the glycerol moiety have been reported such as myristyl, palmityl, stearyl, or oleyl.
Scheme 171. AZT DP-Prodrug.

The coupling of a morpholidate activated phosphatidic acid was also used for the preparation of a myristoyl glyceride DP derivative of acyclovir.268b This compound (not shown) was found to be active on ACV resistant herpes TK–, indicating an efficient delivery of ACV-MP.270
This prodrug approach was also applied to vidarabine (ara-A) and cytarabine (ara-C), which are known for their antiviral and anticancer activity, respectively (not shown). These compounds bearing free hydroxyl groups in 2′- and 3′-position did not require any protection to perform the synthesis of their diphosphate prodrugs. While NDP-prodrugs were found less active that parent ara-C in in vitro antiproliferative studies, they were actually much more potent in mice.271 In the form of a diphosphate prodrug, ara-C was found to be protected from the cytosine deamination, which leads to the biologically ineffective ara-U.272
Some studies also reported the synthesis of oxyalkyl and thioalkyl ether glyceride of anti-HIV273 and anticancer274 agents. The synthetic strategy remains the same with the coupling of the glycerophospholipid part to a NMP activated as morpholidate (Scheme 172). The thioalkyl and oxyalkyl ether glycerophospholipid were previously synthesized by successive alkylation and acylation. After removal of the protective trityl group, the alcohol is treated with POCl3 followed by hydrolysis.
Scheme 172. Synthesis of Oxyalkyl and Thioalkyl Ether Glycerides Ara-C-DP Prodrugs.

4.2. Lipids and Steroids Nucleoside Di- and Triphosphates or Phosphonophosphates
4.2.1. Acyl Phosphates
The literature reports several examples of lipophilic acyl chains linked to the NDP or NTP. After cell penetration, the acylphosphate is expected to be cleaved by a hydrolase to give the corresponding NDP or NTP (Figure 50). Interestingly, the preferential cleavage of the mixed carboxylic phosphoric anhydride part (C–O–P) over the phosphoric anhydride (P–O–P) was observed in buffer and in culture media. Thus, the instability of this prodrug in cell culture media did not allow for an efficient transmembrane diffusion resulting in poor cellular uptake.275
Figure 50.

Expected and observed mechanisms of acyl phosphate nucleoside prodrugs.
The syntheses of octanoyl, lauroyl, myristoyl, and palmitoyl acyl nucleoside diphosphates of AZT (not shown) and d4T (560) were first reported by coupling an acyl pyrophosphate unit to a nucleoside with DCC. It is noteworthy that better yields were observed when the tetrabutylammonium counterions of the acyl pyrophosphate were exchanged for tributylammonium. Acyl nucleoside triphosphates (561) on the other hand were obtained by coupling of a phosphoro morpholidate nucleoside and an acyl pyrophosphate (Scheme 173).276
Scheme 173. Synthesis of d4T Octanoyl, Lauroyl, Myristoyl, and Palmitoyl Acyl Nucleoside Diphosphates and Triphosphates.

An alternative procedure for direct DCC-coupling of an acyl chain to the NDP or NTP was developed by Kreimeyer et al.277 This method appeared to be efficient for the formation of 2′,3′-deoxynucleosides prodrugs, but low yields were observed with ribofuranosyl purine nucleotides due to additional potential acylation sites. To circumvent this problem, the authors used an ethyl chloroformate activated form of myristoic acid that selectively reacted with ADP or ATP in good yields (Scheme 174).
Scheme 174. ADP or ATP Prodrugs.

Kreimeyer et al.278 used a similar approach to prepare a cholesterol carbonate prodrug of adenosine triphosphate 563 and showed that this compound was effectively transported across the membrane bilayer of liposomes (Figure 51).
Figure 51.

Cholesterol carbonate prodrug of ATP 563.
4.2.2. Ether Phosphates
Steroids and lipids ether diphosphates nucleosides were also developed by Hong et al. Because of the ether linkage between the lipid chain and the phosphorus moiety, hydrolysis by hydrolase is impossible, but the intracellular cleavage of the pyrophosphate unit allows for the delivery of nucleoside monophosphate (Figure 52).279
Figure 52.

Mechanism of action of ether phosphates.
Steroids diphosphate derivatives of ara-C were prepared by coupling between ara-CMP morpholidate and various phosphocorticosteroids (Scheme 175). These phosphocorticosteroids were synthesized by either condensation of the steroid 564 with 2-cyanoethylphosphate in the presence of DCC followed by deprotection of the cyanoethyl group or by treatment of 21-iodocorticosteroid 565 with phosphoric acid. This second method was generally preferred because of easy purification of product 566 by simple crystallization. It is noteworthy that these corticosteroid diphosphate prodrugs of ara-C, 567, showed similar activities in vitro as compared to their corresponding monophosphate prodrugs, but were found to be generally less active in vivo. These differences were attributed to the high hydrolysis rate of the phosphoric anhydride bond of the diphosphate prodrugs.
Scheme 175. Preparation of Ara-C Steroids Diphosphate Derivatives.

4.2.3. Phosphonophosphates Derivatives
Alkyldiphosphate and alkylphosphonophosphate derivatives of naturally occurring nucleosides such as cytidine, deoxycytidine, thymidine, and adenosine have been reported to exhibit antiproliferative activities that were attributed to the phospholipidic chain.280,281 Thus, based on this work, alkylphosphono phosphate ara-C derivatives, (compound 568 is shown in Scheme 176 as a representative example) were prepared as a prodrug that would increase the bioavailability of the nucleoside while avoiding the deamination of the cytosine occurring at the nucleoside level and deliver ara-CTP and a phospholipidic chain, two cytotoxic principles.282
Scheme 176. Preparation of ara-C Alkylphosphono Phosphate Derivatives.

Ruiz et al.283 reported the synthesis of PMEA and HPMPC phosphonophosphate HDP and ODE prodrugs (Scheme 177). The phosphate bearing the lipophilic group was obtained by reaction of phosphorus oxychloride and HDP–OH or ODE–OH. Alkoxyalkylphosphates were then activated as phosphomorpholidates using DCC, followed by reaction with DMTr-protected HPMPC in the presence of pyridine. Following DMT deprotection with TFA, phosphonophosphates HDP and ODE prodrugs were afforded in 40% and 20% yield, respectively. Unfortunately, these compounds were found to exert less antiviral activity than their HDP and ODE phosphonate prodrugs.
Scheme 177. Synthesis of PMEA and HPMPC Phosphonophosphate HDP and ODE Prodrugs.

4.3. para-Methoxybenzyl Diphosphate Diester
More recently, Meier’s group proposed to use various biolabile protecting groups to synthesize diphosphate prodrugs to efficiently deliver diphosphate nucleosides.284 The first attempt was realized using a cycloSal (see section 3.1.3) protecting group to mask both hydroxyl groups of the β-phosphate moiety. However, after preparation of several aryl substituted cycloSal diphosphate (synthesis not reported), they observed the predominant release of NMP, by hydrolysis of the phosphorus anhydride bond. To circumvent the hydrolysis of the pyrophosphate unit, the use of a para-acyloxybenzyl (see section 3.1.3) protecting group was investigated (Figure 53). Unlike the cycloSal, deprotection of para-acyloxybenzyl is initiated by enzymatic or chemical cleavage of the ester group, and not by nucleophilic attack at the phosphorus moiety.
Figure 53.

Use of a para-acyloxybenzyl group to synthesize diphosphate prodrugs.
AZT (not shown) and d4T (574) DP prodrugs were synthesized using P(III) chemistry via a dicyanoimidazole-mediated coupling of bis-para-acyloxybenzylphosphoramidite, 573, and bis(tetra-n-butylammonium) nucleoside monophosphates, followed by a subsequent oxidation with tert-butyl hydroperoxide. Phosphoramidites were previously synthesized by reaction of para-acyloxybenzylalcohol and diisopropyl phosphoramidous dichloride (Scheme 178). Interestingly, these compounds proved to possess a high chemical stability in buffer but also to undergo fast and highly selective enzymatic cleavage in cell extract to deliver NDPs. The retained antiviral activities of d4T diphosphate prodrugs (no marked toxicity) proved their ability to penetrate the cells and release biologically active metabolites intracellularly.
Scheme 178. AZT and d4T Bis(para-scyloxybenzyl)-phosphoramidites DP Prodrugs.

5. Conclusion
Despite that the concept of phosph(on)ates prodrugs was originally developed in the 1990s and led to the FDA approval of potent antiviral such as TDF in 2001, it is only very recently that the synthesis of phosph(on)ate prodrugs became systematic in the nucleoside field. Indeed, the large number of examples in the literature of phosph(on)ate prodrugs increasing the activity of a nucleoside or even better, revealing the activity of a inactive parent nucleoside, has led the nucleoside community to consider prodrug evaluation as indispensable. As presented in this Review, multiple synthetic methodologies were developed to prepare a large variety of phosph(on)ates prodrugs. However, several challenges remain, including the development of efficient methods for the preparation of chiral phosph(on)ates prodrugs because one diastereomer may possess overall biological properties superior to those of the other. Improvements in the targeting of prodrugs to particular organs and cellular compartment as well as the development of nanoparticles containing nucleoside prodrugs are needed. Finally, the ultimate goal remains the development of efficient triphosphate prodrugs that would completely overcome the phosphorylation issues by delivering the active compound directly to the target polymerase. The application of prodrug technology has had a large impact on the development of nucleoside and nucleotide antiviral therapies, and provides great hope for persons suffering from deadly viruses such as HIV, HBV, and HCV. Finally, it is likely that the lessons learned from these viruses with novel nucleoside prodrugs will be applied to new emerging viruses such as Noro, Hendra, Dengue, and Chikungunya viruses.
Acknowledgments
This work was supported in part by NIH grants 2P30-AI-050409 and the Department of Veterans Affairs.
Biographies

Ugo Pradere was born in Poitiers (France) in 1983 where he studied chemistry until his master degree. In 2006, he joined the University of Orleans (France) and obtained his Ph.D. in 2009 under the direction of Professor Luigi A. Agrofoglio working on the metallo-catalyzed synthesis of nucleoside analogs and the development of a new convergent synthetic pathway for the preparation of phosphonate prodrugs. In 2010, he joined Dr. Raymond F. Schinazi at Emory University (Atlanta, GA) as a Postdoctoral Fellow where he focused on the synthesis of nucleoside phosphate and phosphonate prodrugs targeting HCV inhibition with an emphasis on the conversion of furanonucleoside analogs into their corresponding phosphonate prodrug derivatives. In 2011, he joined Professor Jonathan Hall at the ETH of Zurich for a second postdoctoral fellow and is currently working on the synthesis of multiple labeled long modified oligoribonucleotides (RNA) and their use in biological assays. He is now a senior scientist in Hall’s group.

Ethel C. Garnier-Amblard received a M.Sc. degree in Chemistry from the University of Orleans (France) in 2000. She then began graduate studies at the same university in collaboration with the Commissariat a l’Energie Atomique (CEA, Le Ripault), where she received a Ph.D. in organic chemistry under the direction of Professor G. Guillaumet in 2004. She conducted postdoctoral studies first with Professor Lanny S. Liebeskind (2005), working on organometallic chemistry and catalysis, and then with Professor Dennis C. Liotta (2007) at Emory University (Atlanta, GA), working on medicinal projects involving sphingolipid analogs for oncology applications. Two years later, she was appointed Faculty at the Emory University - Department of Pharmacology, where she worked in the field of heterocyclic and nucleosidic chemistry. In 2013, she joined RFS Pharma, LLC (a biopharmaceutical company focused on developing novel, differentiated therapeutics for the treatment of hepatitis viruses) where she is currently a Senior Scientist. Dr. Garnier-Amblard’s research interests concern the discovery of new drugs for the treatment of HIV-1 and hepatitis infections as well as emerging viruses. Her research interests include the development of practical new methodologies, the synthesis of active pharmaceutical ingredients, and asymmetric synthesis in general.

Steve Coats obtained his doctorate in organic chemistry under the direction of Albert Padwa at Emory University in Atlanta, Georgia. He completed a postdoctoral fellowship with Harry Wasserman at Yale University in New Haven, Connecticut. He then spent 3 years at Helios Pharmaceuticals in Louisville, Kentucky and 7 years at Johnson & Johnson Pharmaceutical Research and Development in Philadelphia, Pennsylvania. In 2006 he moved to RFS Pharma in Atlanta, Georgia, where he is currently Senior Director of Chemistry. His research interests include medicinal chemistry, heterocycles, nucleosides, nucleotides, and prodrugs.

Franck Amblard was born in Châteauroux, France. He studied chemistry at the University of Orléans (France), where he received his Ph.D. in 2004 under the guidance of Professor Luigi A. Agrofoglio working on the synthesis of new nucleosides analogs using metathesis and palladium-catalyzed reactions. In 2005, he moved to the U.S. to join Professor Raymond F. Schinazi’s research group at Emory University (Atlanta, GA) and worked, as a postdoctoral fellow, on new nucleosides and nucleotides prodrugs. He is now Assistant Professor at the Department of Pediatrics, Emory University School of Medicine. His main research interests include the study of nucleosides analogs and small molecules as potential antiviral agents as well as the isolation and characterization of natural compounds from traditional medicines.

Dr. Raymond F. Schinazi is the Frances Winship Walters Professor of Pediatrics and Director of the Laboratory of Biochemical Pharmacology at Emory University. He serves as Senior Research Career Scientist at the Atlanta Department of Veterans Affairs, and Director of the Scientific Working Group on Viral Eradication within the NIH-sponsored Emory University Center for AIDS Research. Dr. Schinazi is a world leader in the area of nucleoside chemistry having published more than 500 papers and been issued 100 U.S. patents. He is the founder of several biotechnology companies focusing on antiviral drug discovery and development, including Pharmasset, Inc., Triangle Pharmaceuticals, Idenix Pharmaceuticals, and RFS Pharma, LLC. He is best known for his innovative and pioneering work on stavudine, lamivudine, emtricitabine, telbivudine, and sofosbuvir, all of which have been approved by the United States Food and Drug Administration. More than 94% of HIV-infected individuals take at least one of the drugs he invented.
Dr. Schinazi is the founder and chairman of RFS Pharma, LLC. He was a Founder of Idenix and Pharmasset, Inc., now acquired by Merck and Gilead Sciences, respectively.
Funding Statement
National Institutes of Health, United States
References
- Van Rompay A. R.; Johansson M.; Karlsson A. Pharmacol. Ther. 2000, 87, 189. [DOI] [PubMed] [Google Scholar]
- a Lin C.-C.; Yeh L.-T.; Vitarella D.; Hong Z.; Erion M. D. Antiviral Chem. Chemother. 2004, 15, 307. [DOI] [PubMed] [Google Scholar]; b Erion M. D.; van Poelje P. D.; MacKenna D. A.; Colby T. J.; Montag A. C.; Fujitaki J. M.; Linemeyer D. L.; Bullough D. A. J. Pharmacol. Exp. Ther. 2005, 312, 554. [DOI] [PubMed] [Google Scholar]; c Erion M. D.; Bullough D. A.; Lin C.-C.; Hong Z. Curr. Opin. Invest. Drugs 2006, 7, 109. [PubMed] [Google Scholar]; d Reddy K. R.; Matelich M. C.; Ugarkar B. G.; Gomez-Galeno J. E.; DaRe J.; Ollis K.; Sun Z.; Craigo W.; Colby T. J.; Fujitaki J. M.; Boyer S. B.; van Poelje P. D.; Erion M. D. J. Med. Chem. 2008, 51, 666. [DOI] [PubMed] [Google Scholar]
- Eisenberg E. J.; He G. X.; Lee W. A. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1091. [DOI] [PubMed] [Google Scholar]
- a Naesens L.; Bischofberger N.; Augustijns P.; Annaert P.; Van den Mooter G.; Arimilli M. N.; Kim C. U.; De Clercq E. Antimicrob. Agents Chemother. 1998, 42, 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Robbins B. L.; Srinivas R. V.; Kim C.; Bischofberger N.; Fridland A. Antimicrob. Agents Chemother. 1998, 42, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schooley R. T.; Ruane P.; Myers R. A.; Beall G.; Lampiris H.; Berger D.; Chen S.-S.; Miller M. D.; Isaacson E.; Cheng A. K. AIDS 2002, 16, 1257. [DOI] [PubMed] [Google Scholar]; d De Clercq E. Clin. Microbiol. Rev. 2003, 16, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H. R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Steuer H. M.; Niu C.; Otto M. J.; Furman P. A. J. Med. Chem. 2010, 53, 7202. [DOI] [PubMed] [Google Scholar]
- For recent reviews on nucleoside prodrugs, see:; a Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. J. Med. Chem. 2012, 55, 2481. [DOI] [PubMed] [Google Scholar]; b Madela K.; McGuigan C. Future Med. Chem. 2012, 4, 625. [DOI] [PubMed] [Google Scholar]; c Bobeck D. R.; Schinazi R. F.; Coats S. J. Antiviral Ther. 2010, 15, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hurwitz S. J.; Schinazi R. F. Curr. Opin. HIV AIDS 2013, 8, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sofia M. J. Adv. Pharmacol. 2013, 67, 39. [DOI] [PubMed] [Google Scholar]; f Ray A. S.; Hostetler K. Y. Antiviral Res. 2011, 92, 277. [DOI] [PubMed] [Google Scholar]; g Chhikara B. S.; Parang K. Expert Opin. Drug Delivery 2010, 7, 1399. [DOI] [PubMed] [Google Scholar]; h Zhang Y.; Gao Y.; Wen X.; Ma H. Asian J. Pharm. Sci. 2014, 9, 65. [Google Scholar]
- For a recent review dealing specifically with nucleoside phosphonate prodrugs, see:Pertusati F.; Serpi M.; McGuigan C. Antiviral Chem. Chemother. 2012, 22, 181. [DOI] [PubMed] [Google Scholar]
- To the best of our knowledge, there is no extensive review covering the chemical synthesis of nucleoside/nucleotide monodi and tri phosph(on)ate prodrugs. The following reviews cover some specific chemistry:; a Schultz C. Bioorg. Med. Chem. 2003, 11, 885. [DOI] [PubMed] [Google Scholar]; b Mackman R. L.; Cihlar T. Annu. Rep. Med. Chem. 2004, 39, 305. [Google Scholar]; c Jochum A.; Schlienger N.; Egron D.; Peyrottes S.; Périgaud C. J. Organomet. Chem. 2005, 690, 2614. [Google Scholar]; d Meier C. Eur. J. Org. Chem. 2006, 5, 1081. [Google Scholar]; e Li P.; Sergueeva Z. A.; Dobrikov M.; Shaw B. R. Chem. Rev. 2007, 107, 4746. [DOI] [PubMed] [Google Scholar]; f Krecmerova M.; Holy A. PharmaChem 2009, 6. [Google Scholar]; g Peyrottes S.; Perigaud C.. Mononucleotide Prodrug Synthetic Strategies. In Chemical Synthesis of Nucleoside Analogues; Merino P., Ed.; John Wiley and Sons: New York, 2013; Chapter 6, p 229. [Google Scholar]
- a Naesens L.; Neyts J.; Balzarini J.; Bischofberger N.; De Clercq E. Nucleosides Nucleotides 1995, 14, 767. [Google Scholar]; b Noble S.; Goa K. L. Drugs 1999, 58, 479. [DOI] [PubMed] [Google Scholar]
- GS840, a PMEA prodrug, enters trials for HIV-infection.Antiviral Agents Bull. 1994, 7, 232. [Google Scholar]
- Srinival R. V.; Robbins B. L.; Connelly M. C.; Gong Y.-F.; Bischofberger N.; Fridland A. Antimicrob. Agents Chemother. 1993, 37, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hadziyannis S. J.; Tassopoulos N. C.; Heathcote E. J.; Chang T.-T.; Kitis G.; Rizzetto M.; Marcellin P.; Lim S. G.; Goodman Z.; Wulfsohn M. S.; Xiong S.; Fry J.; Brosgart C. L. N. Engl. J. Med. 2003, 348, 800. [DOI] [PubMed] [Google Scholar]; b Marcellin P.; Chang T.-T.; Lim S. G.; Tong M. J.; Sievert W.; Shiffman M. L.; Jeffers L.; Goodman Z.; Wulfsohn M. S.; Xiong S.; Fry J.; Brosgart C. L. N. Engl. J. Med. 2003, 348, 808. [DOI] [PubMed] [Google Scholar]
- Starrett J. E. Jr.; Tortolani D. R.; Russell J.; Hitchcock M. J. M.; Whiterock V.; Martin J. C.; Mansuri M. M. J. Med. Chem. 1994, 37, 1857. [DOI] [PubMed] [Google Scholar]
- Arimilli M. N.; Kim C. U.; Dougherty J.; Mulato A.; Oliyai R.; Shaw J. P.; Cundy K. C.; Bischofberger N. Antiviral Chem. Chemother. 1997, 8, 557. [Google Scholar]
- Choi J. R.; Cho D. G.; Roh K. Y.; Hwang J. T.; Ahn S.; Jang H. S.; Cho W. Y.; Kim K. W.; Cho Y. G.; Kim J.; Kim Y. Z. J. Med. Chem. 2004, 47, 2864. [DOI] [PubMed] [Google Scholar]
- Yuen M. F.; Han K. H.; Um S. H.; Yoon S. K.; Kim H. R.; Kim J.; Kim C. R.; Lai C. L. Hepatology 2010, 51, 767. [DOI] [PubMed] [Google Scholar]
- a Freed J. J.; Farquhar D.; Hampton A. Biochem. Pharmacol. 1989, 38, 3193. [DOI] [PubMed] [Google Scholar]; b Farquhar D.; Srivastva D. N.; Kuttesch N. J.; Saunders P. P. J. Pharmacol. Sci. 1983, 72, 324. [DOI] [PubMed] [Google Scholar]
- Farquhar D.; Khan S.; Srivastva D. N.; Saunders P. P. J. Med. Chem. 1994, 37, 3902. [DOI] [PubMed] [Google Scholar]
- Sastry J. K.; Nehete P. N.; Khan S.; Nowak B. J.; Plunkett W.; Arlinghaus R. B.; Farquhar D. Mol. Pharmacol. 1992, 41, 441. [PubMed] [Google Scholar]
- Pompon A.; Lefebvre I.; Imbach J. L.; Kahn S.; Farquhar D. Antiviral Chem. Chemother. 1994, 5, 91. [Google Scholar]
- a Khan S.; Nowak B.; Plunkett W.; Farquhar D. Proc. Am. Assoc. Cancer Res. 1990, 31, 425. [Google Scholar]; b Khan S. R.; Nowak B.; Plunkett W.; Farquhar D. Biochem. Pharmacol. 2005, 69, 1307. [DOI] [PubMed] [Google Scholar]
- Rose J. D.; Parker W. B.; Secrist J. A. III. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 809. [DOI] [PubMed] [Google Scholar]
- Lefebvre I.; Périgaud C.; Pompon A.; Aubertin A.-M.; Girardet J.-L.; Kirn A.; Gosselin G.; Imbach J.-L. J. Med. Chem. 1995, 38, 3941. [DOI] [PubMed] [Google Scholar]
- a Kang S. H.; Sinhabadu A. K.; Cory J. G.; Mitchell B. S.; Thakker D. R.; Cho M. J. Pharm. Res. 1997, 14, 706. [DOI] [PubMed] [Google Scholar]; b Kang S. H.; Sinhabadu A. K.; Cho M. J. Nucleosides Nucleotides 1998, 17, 1089. [DOI] [PubMed] [Google Scholar]
- Hwang Y.; Cole P. A. Org. Lett. 2004, 6, 1555. [DOI] [PubMed] [Google Scholar]
- Schultz C.; Vajanaphaniche M.; Harootunian A. T.; Sammak P. J.; Barrette K. E.; Tsien R. Y. J. Biol. Chem. 1993, 268, 6316. [PubMed] [Google Scholar]
- Gunic E.; Girardet J.-L.; Ramasamy K.; Stoisavljevic-Petkov V.; Chow S.; Yeh L.-T.; Hamatake R. K.; Raney A.; Hong Z. Bioorg. Med. Chem. Lett. 2007, 17, 2452. [DOI] [PubMed] [Google Scholar]
- Starrett J. E. Jr.; Tortolani D. R.; Hitchcock M. J. M.; Martin J. C.; Mansuri M. M. Antiviral Res. 1992, 19, 267. [DOI] [PubMed] [Google Scholar]
- Krecmerova M.; Holy A.; Pohl R.; Masojidkova M.; Andrei G.; Naesens L.; Neyts J.; Balzarini J.; De Clercq E.; Snoeck R. J. Med. Chem. 2007, 50, 5765. [DOI] [PubMed] [Google Scholar]
- Tang Y.-B.; Peng Z.-G.; Liu Z.-Y.; Li Y.-P.; Jiang J.-D.; Li Z.-R. Bioorg. Med. Chem. Lett. 2007, 17, 6350. [DOI] [PubMed] [Google Scholar]
- a Mackman R. L.; Zhang L.; Prasad V.; Boojamra C. G.; Douglas J.; Grant D.; Hui H.; Kim C. U.; Laflamme G.; Parrish J.; Stoycheva A. D.; Swaminathan S.; Wang K.; Cihlar T. Bioorg. Med. Chem. 2007, 15, 5519. [DOI] [PubMed] [Google Scholar]; b Mackman R. L.; Zhang L.; Prasad V.; Boojamra C. G.; Chen J.; Douglas J.; Grant D.; Laflamme G.; Hui H.; Kim C. U.; Parrish J.; Stoycheva A. D.; Swaminathan S.; Wang K. Y.; Cihlar T. Nucleosides Nucleotides 2007, 26, 573. [DOI] [PubMed] [Google Scholar]
- Kim C. U.; Luh B. Y.; Martin J. C. J. Org. Chem. 1991, 56, 2642. [Google Scholar]
- Mackman R. L.; Boojamra C. G.; Prasad V.; Zhang L.; Lin K.-Y.; Petrakovsky O.; Babusis D.; Chen J.; Douglas J.; Grant D.; Hui H. C.; Kim C. U.; Markevitch D. Y.; Vela J.; Ray A.; Cihlar T. Bioorg. Med. Chem. Lett. 2007, 17, 6785. [DOI] [PubMed] [Google Scholar]
- Boojamra C. G.; Mackman R. L.; Markevitch D. Y.; Prasad V.; Ray A. S.; Douglas J.; Grant D.; Kim C. U.; Cihlar T. Bioorg. Med. Chem. Lett. 2008, 18, 1120. [DOI] [PubMed] [Google Scholar]
- Mackman R. L.; Ray A. S.; Hui H. C.; Zhang L.; Birkus G.; Boojamra C. G.; Desai M. C.; Douglas J. L.; Gao Y.; Grant D.; Laflamme G.; Lin K.-Y.; Markevitch D. Y.; Mishra R.; McDermott M.; Pakdaman R.; Petrakovsky O. V.; Vela J. E.; Cihlar T. Bioorg. Med. Chem. 2010, 18, 3606. [DOI] [PubMed] [Google Scholar]
- Benzaria S.; Pélicano H.; Johnson R.; Maury G.; Imbach J.-L.; Aubertin A.-M.; Obert G.; Gosselin G. J. Med. Chem. 1996, 39, 4958. [DOI] [PubMed] [Google Scholar]
- Wu M.; El-Kattan Y.; Lin T. H.; Ghosh A.; Kumar V. S.; Kotian P. L.; Cheng X.; Bantia S.; Babu Y. S.; Chand P. Nucleosides Nucleotides 2005, 24, 1569. [DOI] [PubMed] [Google Scholar]
- Wu M.; El-Kattan Y.; Lin T. H.; Ghosh A.; Vadlakonda S.; Kotian P. L.; Babu Y. S.; Chand P. Nucleosides Nucleotides 2005, 24, 1543. [DOI] [PubMed] [Google Scholar]
- El-Kattan Y.; Lin T. H.; Wu M.; Kumar V. S.; Kotian P. L.; Ghosh A.; Cheng X.; Bantia S.; Babu Y. S.; Chand P. Nucleosides Nucleotides 2005, 24, 1597. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; El-Kattan Y.; Wu M.; Lin T. H.; Vadlakonda S.; Kotian P. L.; Babu Y. S.; Chand P. Nucleosides Nucleotides 2005, 24, 1587. [DOI] [PubMed] [Google Scholar]
- Topalis D.; Pradere U.; Roy V.; Caillat C.; Azzouzi A.; Broggi J.; Snoeck R.; Andrei G.; Lin J.; Eriksson S.; Alexandre J. A. C.; El-Amri C.; Deville-Bonne D.; Meyer P.; Balzarini J.; Agrofoglio L. A. J. Med. Chem. 2011, 54, 222. [DOI] [PubMed] [Google Scholar]
- Pradere U.; Clavier C.; Roy V.; Nolan S. P.; Agrofoglio L. A. Eur. J. Org. Chem. 2011, 36, 7324. [Google Scholar]
- Montagu A.; Pradere U.; Roy V.; Nolan S. P.; Agrofoglio L. A. Tetrahedron 2011, 67, 5319. [Google Scholar]
- Pradere U.; Roy V.; Montagu A.; Sari O.; Hamada M.; Balzarini J.; Snoeck R.; Andrei G.; Agrofoglio L. A. Eur. J. Med. Chem. 2012, 57, 126. [DOI] [PubMed] [Google Scholar]
- Pradere U.; Amblard F.; Coats S. J.; Schinazi R. F. Org. Lett. 2012, 14, 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krecmerova M.; Holy A.; Andrei G.; Pomeisl K.; Tichy T.; Brehova P.; Masojidkova M.; Dracinsky M.; Pohl R.; Laflamme G.; Naesens L.; Hui H.; Cihlar T.; Neyts J.; De Clercq E.; Balzarini J.; Snoeck R. J. Med. Chem. 2010, 53, 6825. [DOI] [PubMed] [Google Scholar]
- Kern E. R.; Hartline C.; Harden E.; Keith K.; Rodriguez N.; Beadle J. R.; Hostetler K. Y. Antimicrob. Agents Chemother. 2002, 46, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X.; Jiang S.; Li C.; Xin J.; Yang Y.; Ji R. Bioorg. Med. Chem. Lett. 2007, 17, 465. [DOI] [PubMed] [Google Scholar]
- Puech F.; Gosselin G.; Lefebvre I.; Pompon A.; Aubertin A.-M.; Kirn A.; Imbach J.-L. Antiviral Res. 1993, 22, 155. [DOI] [PubMed] [Google Scholar]
- For detailed reviews on in vitro and in vivo results obtained with pronucleotides bearing SATE groups see:; a Peyrottes S.; Egron D.; Lefebvre I.; Gosselin G.; Imbach J.-L.; Perigaud C. Mini-Rev. Med. Chem. 2004, 4, 395. [DOI] [PubMed] [Google Scholar]; b Jochum A.; Schlienger N.; Egron D.; Peyrottes S.; Perigaud C. J. Organomet. Chem. 2005, 690, 2614. [Google Scholar]
- Perigaud C.; Gosselin G.; Lefebvre I.; Girardet J.-L.; Benzaria S.; Barber I.; Imbach J.-L. Bioorg. Med. Chem. Lett. 1993, 3, 2521. [Google Scholar]
- Lannuzel M.; Egron D.; Imbach J.-L.; Gosselin G.; Perigaud C. Nucleosides Nucleotides 1999, 18, 1001. [DOI] [PubMed] [Google Scholar]
- Egron D.; Perigaud C.; Gosselin G.; Aubertin A.-M.; Gatanaga H.; Mitsuya H.; Zemlicka J.; Imbach J.-L. Bioorg. Med. Chem. Lett. 2002, 12, 265. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Girardet J.-L.; Hong Z.; Lai V. C. H.; An H.; Koh Y.-H.; Shaw S. Z.; Zhong W. Bioorg. Med. Chem. Lett. 2005, 15, 709. [DOI] [PubMed] [Google Scholar]
- Gunic E.; Chow S.; Rong F.; Ramasamy K.; Raney A.; Li D. Y.; Huang J.; Hamatake R. K.; Hong Z.; Girardet J.-L. Bioorg. Med. Chem. Lett. 2007, 17, 2456. [DOI] [PubMed] [Google Scholar]
- Koh Y.-H.; Shim J. H.; Girardet J.-L.; Hong Z. Bioorg. Med. Chem. Lett. 2007, 17, 5261. [DOI] [PubMed] [Google Scholar]
- Valette G.; Pompon A.; Girardet J.-L.; Cappellacci L.; Franchetti P.; Grifantini M.; La Colla P.; Loi A. G.; Perigaud C.; Gosselin G.; Imbach J.-L. J. Med. Chem. 1996, 39, 1981. [DOI] [PubMed] [Google Scholar]
- Perigaud C.; Gosselin G.; Girardet J.-L.; Korba B. E.; Imbach J.-L. Antiviral Res. 1999, 40, 167. [DOI] [PubMed] [Google Scholar]
- Prakash T. P.; Prhavc M.; Eldrup A. B.; Cook P. D.; Caroll S. S.; Olsen D. B.; Stahlhut M. W.; Tomassini J. E.; MacCoss M.; Galloway S. M.; Hilliard C.; Bhat B. J. Med. Chem. 2005, 48, 1199. [DOI] [PubMed] [Google Scholar]
- Bazzanini R.; Gouy M.-H.; Peyrottes S.; Gosselin G.; Perigaud C. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 1635. [DOI] [PubMed] [Google Scholar]
- Koh Y.-H.; Shim J. H.; Wu J. Z.; Zhong W.; Hong Z.; Girardet J.-L. J. Med. Chem. 2005, 48, 2867. [DOI] [PubMed] [Google Scholar]
- Li H.; Yoo J. C.; Baik Y. C.; Lee W.; Homg J. H. Bull. Korean Chem. Soc. 2010, 31, 2514. [Google Scholar]
- Gunic E.; Girardet J.-L.; Ramasamy K.; Stoisavljevic-Petkiov V.; Chow S.; Yeh L.-T.; Hamatake R. K.; Raney A.; Hong Z. Bioorg. Med. Chem. Lett. 2007, 17, 2452. [DOI] [PubMed] [Google Scholar]
- Liu L. J.; Seo R. S.; Yoo S. W.; Choi J.; Hong H. J. Bull. Korean Chem. Soc. 2010, 31, 915. [Google Scholar]
- Villard A.-L.; Coussot G.; Lefebvre I.; Augustijns P.; Aubertin A.-M.; Gosselin G.; Peyrottes S.; Perigaud C. Bioorg. Med. Chem. Lett. 2008, 16, 7321. [DOI] [PubMed] [Google Scholar]
- Schlienger N.; Beltran T.; Perigaud C.; Lefebvre I.; Pompon A.; Aubertin A.-M.; Gosselin G.; Imbach J.-L. Bioorg. Med. Chem. Lett. 1998, 8, 3003. [DOI] [PubMed] [Google Scholar]
- a Schlienger N.; Peyrottes S.; Kassem T.; Imbach J.-L.; Gosselin G.; Aubertin A.-M.; Perigaud C. J. Med. Chem. 2000, 43, 4570. [DOI] [PubMed] [Google Scholar]; b Peyrottes S.; Coussot G.; Lefebvre I.; Imbach J.-L.; Gosselin G.; Aubertin A.-M.; Perigaud C. J. Med. Chem. 2003, 46, 782. [DOI] [PubMed] [Google Scholar]
- a Beltran T.; Egron D.; Pompon A.; Lefebvre I.; Perigaud C.; Gosselin G.; Aubertin A.-M.; Imbach J.-L. Bioorg. Med. Chem. Lett. 2001, 11, 1775. [DOI] [PubMed] [Google Scholar]; b Beltran T.; Egron D.; Lefebvre I.; Perigaud C.; Pompon A.; Gosselin G.; Aubertin A.-M.; Imbach J.-L. Nucleosides Nucleotides 1999, 18, 973. [DOI] [PubMed] [Google Scholar]; c Egron D.; Perigaud C.; Gosselin G.; Aubertin A.-M.; Imbach J.-L. Nucleosides Nucleotides 1999, 18, 981. [DOI] [PubMed] [Google Scholar]; d Egron D.; Perigaud C.; Gosselin G.; Aubertin A.-M.; Imbach J.-L. Nucleosides Nucleotides 2001, 20, 751. [DOI] [PubMed] [Google Scholar]
- Sommadossi J.-P.; Gosselin G.; Pierra C.; Perigaud C.; Peyrottes S.. Compounds and pharmaceutical compositions for the treatment of viral infections. PCT Int. Appl. WO 2008082601, 2008.
- a Schlienger N.; Perigaud C.; Gosselin G.; Imbach J.-L. Nucleosides Nucleotides 1997, 16, 1325. [DOI] [PubMed] [Google Scholar]; b Schlienger N.; Perigaud C.; Aubertin A.-M.; Thumann C.; Gosselin G.; Imbach J.-L. Helv. Chim. Acta 1999, 82, 2044. [Google Scholar]
- Jochum A.; Schlienger N.; Gosselin G.; Imbach J.-L.; Aubertin A.-M.; Perigaud C. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 899. [DOI] [PubMed] [Google Scholar]
- Egron D.; Arzumanov A. A.; Dyatkina N. B.; Aubertin A.-M.; Imbach J.-L.; Gosselin G.; Krayesvsky A.; Perigaud C. Bioorg. Chem. 2001, 29, 333. [DOI] [PubMed] [Google Scholar]
- For reviews on the cycloSal-prodrug approach, see:; a Meier C.; Balzarini J. Antiviral Res. 2006, 71, 282–292. [DOI] [PubMed] [Google Scholar]; b Meier C. Eur. J. Org. Chem. 2006, 1081. [Google Scholar]
- Meier C.; Habel L.; Haller-Meier F.; Lomp A.; Herderich M.; Klöcking R.; Meerbach A.; Wutzler P. Antiviral Chem. Chemother. 1998, 9, 389. [DOI] [PubMed] [Google Scholar]
- Meier C.; Thomann J. O. Pat. Appl. US 20110028705.
- Bryld T.; Sørensen M. H.; Nielsen P.; Koch T.; Nielsen C.; Wengel J. J. Chem. Soc., Perkin Trans. 1 2002, 1655. [Google Scholar]
- Meier C.; De Clercq E.; Balzarini J. Eur. J. Org. Chem. 1998, 837. [Google Scholar]
- a Meier C.; Lorey M.; De Clercq E.; Balzarini J. J. Med. Chem. 1998, 41, 1417. [DOI] [PubMed] [Google Scholar]; b Ducho C.; Wendicke S.; Görbig U.; Balzarini J.; Meier C. Eur. J. Org. Chem. 2003, 4786. [Google Scholar]; c Meier C.; Lorey M.; De Clercq E.; Balzarini J. Bioorg. Med. Chem. Lett. 1997, 7, 99. [Google Scholar]
- Ludek O. R.; Krämer T.; Balzarini J.; Meier C. Synthesis 2006, 1313. [Google Scholar]
- Mugnier F.; Meier C. Nucleosides Nucleotides 1999, 18, 941. [DOI] [PubMed] [Google Scholar]
- a Meier C.; Knispel T.; De Clercq E.; Balzarini J. J. Med. Chem. 1999, 42, 1604–1614. [DOI] [PubMed] [Google Scholar]; b Knispel T.; Meier C. Nucleosides Nucleotides 1999, 18, 945. [DOI] [PubMed] [Google Scholar]
- Spáčilova P.; Nauš P.; Pohl R.; Votruba I.; Snášel J.; Zábranská H.; Pichová I.; Ameral R.; Birkuš G.; Cihlář T.; Hocek M. ChemMedChem 2010, 5, 1386. [DOI] [PubMed] [Google Scholar]
- Meier C.; Lomp A.; Meerbach A.; Wutzler P. J. Med. Chem. 2002, 45, 5157. [DOI] [PubMed] [Google Scholar]
- Kortylewicz Z. P.; Kimura Y.; Inoue K.; Mack E.; Baranowska-Kortylewicz J. J. Med. Chem. 2012, 55, 2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales E. H. R.; Balzarini J.; Meier C. Chem.—Eur. J. 2011, 17, 1649. [DOI] [PubMed] [Google Scholar]
- Morales E. H. R.; Roman C. A.; Thomann J. O.; Meier C. Eur. J. Org. Chem. 2011, 4397. [Google Scholar]
- Morales E. H. R.; Balzarini J.; Meier C. J. Med. Chem. 2012, 55, 7245. [DOI] [PubMed] [Google Scholar]
- a Ducho C.; Görbig U.; Jessel S.; Gisch N.; Balzarini J.; Meier C. J. Med. Chem. 2007, 50, 1335. [DOI] [PubMed] [Google Scholar]; b Gisch N.; Balzarini J.; Meier C. J. Med. Chem. 2009, 52, 3464. [DOI] [PubMed] [Google Scholar]
- Ahmadibeni Y.; Tiwari R.; Swepson C.; Pandhare D. C.; Doncel G. F.; Parang K. Tetrahedron Lett. 2011, 52, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Meier C.; Görbig U.; Müller C.; Balzarini J. J. Med. Chem. 2005, 48, 8079–8086. [DOI] [PubMed] [Google Scholar]; b Görbig U.; Balzarini J.; Meier C. Nucleosides Nucleotides 2007, 26, 831. [DOI] [PubMed] [Google Scholar]
- Gisch N.; Balzarini J.; Meier C. J. Med. Chem. 2008, 51, 6752. [DOI] [PubMed] [Google Scholar]
- Meier C.; Ducho C.; Jessen H.; Vukadinovic-Tenter D.; Balzarini J. Eur. J. Org. Chem. 2006, 197. [Google Scholar]
- Jessen H.; Meier C.; Balzarini J. J. Med. Chem. 2008, 51, 6592. [DOI] [PubMed] [Google Scholar]
- Gisch N.; Balzarini J.; Meier C. J. Med. Chem. 2007, 50, 1658. [DOI] [PubMed] [Google Scholar]
- Gisch N.; Pertenbreiter F.; Balzarini J.; Meier C. J. Med. Chem. 2008, 51, 8115. [DOI] [PubMed] [Google Scholar]
- For more information concerning MB07811, see:; a Erion M. D.; Cable E. E.; Ito B. R.; Jiang H.; Fujitaki J. M.; Finn P. D.; Zhang B. H.; Hou J.; Boyer S. H.; van Poelje P. D.; Linemeyer D. L. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 15490. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fujitaki J. M.; Cable E. E.; Ito B. R.; Zhang B.-H.; Hou J.; Yang C.; Bullough D. A.; Ferrero J. L.; van Poelje P. D.; Linemeyer D. L.; Erion M. D. Drug Metab. Dispos. 2008, 36, 2393. [DOI] [PubMed] [Google Scholar]
- Boyer S. H.; Sun Z.; Jiang H.; Esterbrook J.; Gomez-aleno J. E.; Craigo W.; Reddy K. R.; Ugarkar B. G.; MacKenna D. A.; Erion M. D. J. Med. Chem. 2006, 49, 7711. [DOI] [PubMed] [Google Scholar]
- Erion M. D.; Reddy K. R.; Boyer S. B.; Matelich M. C.; Gomez-Galeno J.; Lemus R. H.; Ugarkar B. G.; Colby T. J.; Schanzer J.; van Poelje P. D. J. Am. Chem. Soc. 2004, 126, 5154. [DOI] [PubMed] [Google Scholar]
- Reddy K. R.; Boyer S. H.; Erion M. D. Tetrahedron Lett. 2005, 46, 4321. [Google Scholar]
- Bookser B. C.; Raffaele N. B. J. Comb. Chem. 2008, 10, 567. [DOI] [PubMed] [Google Scholar]
- Hecker S. J.; Reddy K. R.; van Poelje P. D.; Sun Z.; Huang W.; Varkhedkar V.; Reddy M. V.; Fujitaki J. M.; Olsen D. B.; Koeplinger K. A.; Boyer S. H.; Linemeyer D. L.; MacCoss M.; Erion M. D. J. Med. Chem. 2007, 50, 3891. [DOI] [PubMed] [Google Scholar]
- Bookser B. C.; Raffaele N. B.; Reddy R. K.; Fan K.; Huang W.; Erion M. D. Nucleosides, Nucleotides Nucleic Acids 2009, 28, 969. [DOI] [PubMed] [Google Scholar]
- Du J.; Bao D.; Chun B.-K.; Jiang Y.; Reddy P. G.; Zhang H.-R.; Ross B. S.; Bansal S.; Bao H.; Espiritu C. L.; Lam A. M.; Murakami E.; Niu C.; Micholochick Steuer H.; Furman P. A.; Otto M. J.; Sofia M. J. Bioorg. Med. Chem. Lett. 2012, 22, 5924. [DOI] [PubMed] [Google Scholar]
- Reddy P. G.; Bao D.; Chang W.; Chun B.-K.; Du J.; Nagarathnam D.; Rachakonda S.; Ross B. S.; Zhang H.-R.; Bansal S.; Espiritu C. L.; Keilman M.; Lam A. M.; Niu C.; Micholochick Steuer H.; Furman P. A.; Otto M. J.; Sofia M. J. Bioorg. Med. Chem. Lett. 2010, 20, 7376. [DOI] [PubMed] [Google Scholar]
- Reddy P. G.; Chun B.-K.; Zhang H.-R.; Rachakonda S.; Ross B. S.; Sofia M. J. J. Org. Chem. 2011, 76, 3782. [DOI] [PubMed] [Google Scholar]
- For a specific reviews about alkoxyalkyl prodrugs, see:; a Hostetler K. Y. Antiviral Res. 2009, 82, A84. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Painter G. R.; Hostetler K. Y. Trends Biotechnol. 2004, 22, 423. [DOI] [PubMed] [Google Scholar]
- Piantadosi C.; Marasco C. J. Jr.; Morris-Natschke S. L.; Meyer K. L.; Gumus F.; Surles J. R.; Ishaq K. S. J. Med. Chem. 1991, 34, 1408. [DOI] [PubMed] [Google Scholar]
- Mavromoustakos T.; Calogeropoulou T.; Koufaki M.; Kolocouris A.; Daliani I.; Demetzos C.; Meng Z.; Makriyannis A.; Balzarini J.; De Clercq E. J. Med. Chem. 2001, 44, 1702. [DOI] [PubMed] [Google Scholar]
- Hostetler K. Y.; Beadle J. R.; Kini G. D.; Gardner M. F.; Wright K. N.; Wu T.-H.; Korba B. A. Biochem. Pharmacol. 1997, 53, 1815. [DOI] [PubMed] [Google Scholar]
- a Beadle J. R.; Kini G. D.; Aldern K. A.; Gardner M. F.; Wright K. N.; Ryback R. J.; Kern E. R. Nucleosides Nucleotides 2000, 19, 471. [DOI] [PubMed] [Google Scholar]; b Hostetler K. Y.; Beadle J. R.; Hornbuckle W. E.; Bellezza C. A.; Tochkov I. A.; Cote P. J.; Gerin J. L.; Korba B. E.; Tennant B. C. Antimicrob. Agents Chemother. 2000, 44, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Narayanasamy J.; Schinazi R. F.; Chu C. K. Bioorg. Med. Chem. 2006, 14, 2178. [DOI] [PubMed] [Google Scholar]
- Ludwig P. S.; Schwendener R. A.; Schott H. Eur. J. Med. Chem. 2005, 40, 494. [DOI] [PubMed] [Google Scholar]
- Sigmund H.; Pfleiderer W. Helv. Chim. Acta 1996, 79, 426. [Google Scholar]
- Ciesla S. L.; Trahan J.; Winegarden K. L.; Aldern K. A.; Painter G. R.; Hostetler K. Y. Antiviral Res. 2003, 59, 163. [DOI] [PubMed] [Google Scholar]
- Kern E. R.; Hartline C.; Harden E.; Keith K.; Rodriguez N.; Beadle J. R.; Hostetler K. Y. Antimicrob. Agents Chemother. 2002, 46, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle J. R.; Rodriquez N.; Aldern K. A.; Hartline C.; Harden E.; Kern E. R.; Hostetler K. Y. Antimicrob. Agents Chemother. 2002, 46, 2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krecmerova M.; Holy A.; Piskala A.; Masojidkova M.; Balzarini J.; Andrei G.; Snoeck R.; Naesens L.; Neyts J.; De Clercq E. J. Med. Chem. 2007, 50, 1069. [DOI] [PubMed] [Google Scholar]
- Wan W. B.; Beadle J. R.; Hartline C. B.; Kern E. R.; Ciesla S. L.; Valiaeva N.; Hostetler K. Y. Antimicrob. Agents Chemother. 2005, 49, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler K. Y.; Beadle J. R.; Trahan J.; Aldern K. A.; Owens G.; Schriewer J.; Melman L.; Buller R. M. Antiviral Res. 2007, 73, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiaeva N.; Beadle J. R.; Aldern K. A.; Trahan J.; Hostetler K. Y. Antiviral Res. 2006, 72, 10. [DOI] [PubMed] [Google Scholar]
- Holy A.; Gunter J.; Dvorakova H.; Masojidkova M.; Andrei G.; Snoeck R.; Balzarini J.; De Clercq E. J. Med. Chem. 1999, 42, 2064. [DOI] [PubMed] [Google Scholar]
- Ruiz J. C.; Aldern K. A.; Beadle J. R.; Hartline C. B.; Kern E. R.; Hostetler K. Y. Antiviral Chem. Chemother. 2006, 17, 89. [DOI] [PubMed] [Google Scholar]
- Choo H.; Beadle J. R.; Chong Y.; Trahan J.; Hostetler K. Y. Bioorg. Med. Chem. 2007, 15, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadle J. R.; Wan W. B.; Ciesla S. L.; Keith K. A.; Hartline C. B.; Kern E. R.; Hostetler K. Y. J. Med. Chem. 2006, 49, 2010. [DOI] [PubMed] [Google Scholar]
- Vrbkova S.; Dracinsky M.; Holy A. Tetrahedron 2007, 63, 11391. [Google Scholar]
- Holy A. Synthesis 1998, 381. [Google Scholar]
- Valiaeva N.; wyles D. L.; Schooley R. T.; Hwu J. B.; Beadle J. R.; Prichard M. N.; Hostetler K. Y. Bioorg. Med. Chem. 2011, 19, 4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichy T.; Andrei G.; Snoeck R.; Balzarini J.; Dracinsky M.; Krecmerova M. Eur. J. Med. Chem. 2012, 55, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Humton R. N.; Jones A. S.; McGuigan C.; Walker R. T.; Balzarini J.; De Clercq E. J. Med. Chem. 1984, 27, 440. [DOI] [PubMed] [Google Scholar]; b Jones B. C. N. M.; McGuigan C.; Riley P. A. Nucleic Acids Res. 1989, 17, 7195. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McGuigan C.; Tollerfield S. M.; Riley P. A. Nucleic Acids Res. 1989, 17, 6065. [DOI] [PMC free article] [PubMed] [Google Scholar]; d McGuigan C.; Nicholls S. R.; O’Connor T. J.; Kinchington D. Antiviral Chem. Chemother. 1990, 1, 25. [Google Scholar]; e McGuigan C.; O’Connor T. J.; Nicholls S. R.; Nickson C.; Kinchington D. Antiviral Chem. Chemother. 1990, 1, 355. [Google Scholar]; f Devine K. G.; McGuigan C.; O’Connor T. J.; Nicholls S. R.; Kinchington D. AIDS 1990, 4, 371. [PubMed] [Google Scholar]; g McGuigan C.; Nickson C.; Petrik J.; Karpas A. FEBS Lett. 1992, 310, 171. [DOI] [PubMed] [Google Scholar]; h McGuigan C.; Kinchington D.; Wang M. F.; Nicholls S. R.; Nickson C.; Galpin S.; Jeffries D. J.; O’Connor T. J. FEBS Lett. 1993, 322, 249. [DOI] [PubMed] [Google Scholar]; i McGuigan C.; Kinchington D.; Nicholls S. R.; Nickson C.; O’Connor T. J. Bioorg. Med. Chem. Lett. 1993, 3, 1207. [Google Scholar]
- a McGuigan C.; Pathirana R. N.; Mahmood N.; Devine K. G.; Hay A. J. Antiviral Res. 1992, 17, 311. [DOI] [PubMed] [Google Scholar]; b McGuigan C.; Davies M.; Pathirana R.; Mahmood N.; Hay A. J. Antiviral Res. 1994, 24, 69. [DOI] [PubMed] [Google Scholar]; c McGuigan C.; Pathirana R. N.; Davies M. P. H.; Balzarini J.; De Clercq E. Bioorg. Med. Chem. Lett. 1994, 4, 427. [Google Scholar]
- a Jones A. S.; McGuigan C.; Walker R. T.; Balzarini J.; De Clercq E. J. Chem. Soc., Perkin Trans. 1 1984, 1471. [Google Scholar]; b Jones A. S.; McGuigan C.; Walker R. T. J. Chem. Soc., Perkin Trans. 1 1985, 199. [Google Scholar]
- a Curley D.; McGuigan C.; Devine K. G.; O’Connor T. J.; Jeffries D. J.; Kinchington D. Antiviral Res. 1990, 14, 345. [DOI] [PubMed] [Google Scholar]; b McGuigan C.; Devine K. G.; O’Connor T. J.; Galpin S. A.; Jeffries D. J.; Kinchington D. Antiviral Chem. Chemother. 1990, 1, 107. [DOI] [PubMed] [Google Scholar]; c McGuigan C.; Devine K. G.; O’Connor T. J.; Kinchington D. Antiviral Res. 1991, 15, 255. [DOI] [PubMed] [Google Scholar]; d McGuigan C.; Jones B. C. N. M.; Riley P. A. Bioorg. Med. Chem. Lett. 1991, 1, 607. [Google Scholar]; e McGuigan C.; Sheeka H. M.; Mahmood N.; Hay A. Bioorg. Med. Chem. Lett. 1993, 3, 1203. [Google Scholar]
- McGuigan C.; Jones B. C. N. M.; Devine K. G.; Nicholls S. R.; Kinchington D. Bioorg. Med. Chem. Lett. 1991, 1, 729. [Google Scholar]
- a Mehellou Y.; Balzarini J.; McGuigan C. ChemMedChem 2009, 4, 1779. [DOI] [PubMed] [Google Scholar]; b Cahard D.; McGuigan D.; Balzarini J. Mini-Rev. Med. Chem. 2004, 4, 371. [DOI] [PubMed] [Google Scholar]; c Zemlicka J. Biochim. Biophys. Acta 2002, 1587, 276. [DOI] [PubMed] [Google Scholar]
- Gilead, 2012. “Gilead Initiates Phase 2 Clinical Trial Evaluating GS-7340, A Low-Dose Novel Prodrug of Tenofovir for the Treatment of HIV”. Press release, retrieved from http://www.gilead.com/news/press-releases/2012/1/gilead-initiates-phase-2-clinical-trial-evaluating-gs7340-a-lowdose-novel-prodrug-of-tenofovir-for-the-treatment-of-hiv.
- Cihlar T. J. Acquired Immune Defic. Syndr. 2009, 51. [Google Scholar]
- Cahn P.; Rolon M. J.; Gun A. M.; Ferrari I.; Dibirdik I.; Qazi S.; D’Cruz O.; Sahin K.; Uckun F. J. AIDS Clin. Res. 2012, 3, 1. [Google Scholar]
- NewBiotics, 2002. “NewBiotics Presents Preliminary Data of Thymectacin Phase I Clinical Trial for Drug-Resistant Colon Cancer”. Press release, retrieved from http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=121432§ionID=&isEPVantage=no.
- Vernachio J. H.; Bleiman B.; Bryant K. D.; Chamberlain S.; Hunley D.; Hutchins J.; Ames B.; Gorovits E.; Ganguly B.; Hall A.; Kolykhalov A.; Liu Y.; Muhammad J.; Raja N.; Walters C. R.; Wang J.; Williams K.; Patti J. M.; Henson G.; Madela K.; Aljarah M.; Gilles A.; McGuigan C. Antimicrob. Agents Chemother. 2011, 55, 1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W.; Bao D.; Chun B. K.; Naduthambi D.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Zhang H. R.; Bansal S.; Espiritu C. L.; Keilman M.; Lam A. M.; Niu C.; Steuer H. M.; Furman P. A.; Otto M. J.; Sofia M. J. ACS Med. Chem. Lett. 2010, 2, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Balzarini J.; Karlsson A.; Aquaro S.; Perno C.-F.; Cahard D.; Naesens L.; De Clercq E.; McGuigan C. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 7295. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Balzarini J.; Egberink H.; Hartmann K.; Cahard D.; Vahlenkamp T.; Thormar H.; De Clercq E.; McGuigan C. Mol. Pharmacol. 1996, 50, 1207. [PubMed] [Google Scholar]; c McGuigan C.; Sutton P. W.; Cahard D.; Turner K.; O’Leary G.; Wang Y.; Gumbleton M.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 1998, 9, 473. [DOI] [PubMed] [Google Scholar]; d Egron D.; Lefebvre I.; Périaud C.; Beltran T.; Pompon A.; Gosselin G.; Aubertin A.-M.; Imbach J.-L. Bioorg. Med. Chem. Lett. 1998, 8, 1045. [DOI] [PubMed] [Google Scholar]; e Saboulard D.; Naesens L.; Cahard D.; Salgado A.; Pathirana R.; Valazquez S.; McGuigan C.; De Clercq E.; Balzarini J. Mol. Pharmacol. 1999, 56, 693. [PubMed] [Google Scholar]
- Birkus G.; Wang R.; Liu X.; Kutty N.; MacArthur H.; Cihlar T.; Gibbs C.; Swaminathan S.; Lee W.; McDermott M. Antimicrob. Agents Chemother. 2007, 51, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T. F.; Baraniak J.; Kaczmarek R.; Zhou X.; Cheng J.; Ghosh B.; Wagner C. R. Mol. Pharmacol. 2007, 4, 208. [DOI] [PubMed] [Google Scholar]
- a Abraham T. W.; Kalman T. I.; McIntee E. J.; Wagner C. R. J. Med. Chem. 1996, 39, 4569. [DOI] [PubMed] [Google Scholar]; b Kim J.; Chou T. F.; Griesgraber G. W.; Wagner C. R. Mol. Pharmacol. 2004, 1, 102–11. [DOI] [PubMed] [Google Scholar]; c Congiatu C.; Brancale A.; McGuigan C. Nucleosides Nucleotides 2007, 26, 1121. [DOI] [PubMed] [Google Scholar]; d Ora M.; Ojanperä J.; Lönnberg H. Chem.—Eur. J. 2007, 13, 8591. [DOI] [PubMed] [Google Scholar]
- Van Boom J. H.; Burgers P. M. J.; Crea R.; Luyten W. C. M. M.; Reese C. B. Tetrahedron 1975, 31, 2953. [Google Scholar]
- Uchiyama M.; Aso Y.; Noyori R.; Hayakawa Y. J. Org. Chem. 1993, 58, 373. [Google Scholar]
- We have to admit though that we found a paper describing the synthesis of several phosphoramidate prodrugs of ribavirin in high yield (69–78%) by reacting ribavirin and various chlorophosphoramidates in a mixture of THF/pyridine/TEA at 40–50 °C for 36 h. However, the unuasual high yields reported for the coupling reaction along with the lack of explanation why they did not apply one of the commonly used methods make these results questionable.Rao V. K.; Reddy S. S.; Babu K. R.; Kumar K. H.; Ghosh S. K.; Raju C. N. J. Korean Chem. Soc. 2011, 55, 952. [Google Scholar]
- Variation on the aryl group or on the nature of the amino acids simply implies their use in the general synthetic pathway instead of the phenol and the l-alanine. However, variations on the carboxyl ester terminus are chemically introduced from the free amino acid by chlorination (generally with SOCl2) and subsequent substitution by an alcohol or from a protected amino acid (generally Boc) by DCC-mediated coupling, or esterification with PTSA and subsequent deprotection. In a general manner, over the hundreds of combinations of aryloxyphosphoramidate, a large majority of these phosphorochloridate reagents can be prepared and used with the same protocol independently from their nature. Nevertheless, some exceptions can be found: (1) aspartic and glutamic acids bearing two carboxylic acid groups were used as their dimethyl ester, (2) cysteine thiol group has to be protected (S-methyl ether) to avoid spontaneous dimerization and synthetic problem during the coupling reaction to nucleoside, (3) an unexpected racemization of the amino acid chiral center (∼15%) was surprisingly noticed for the phenyl glycine, and (4) the coupling of dialkylglycine is really poor with standard method (∼2%). An alternative procedure based on a mesitylene nitrotriazolide (MSNT)-mediated coupling of d4T 5′-monophenyl phosphate and the dialkyl glycine had to be used.
- McGuigan C.; Salgado A.; Yarnold C.; Harries T. Y.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 1996, 7, 184. [Google Scholar]
- McGuigan C.; Bidois L.; Hiouni A.; Ballatore C.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 2000, 11, 111. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Tsang H.-W.; Cahard D.; Turner K.; Velazquez S.; Salgado A.; Bidois L.; Naesens L.; De Clercq E.; Balzarini J. Antiviral Res. 1997, 35, 195. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Cahard D.; Salgado A.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 1996, 7, 31. [Google Scholar]
- McGuigan C.; Tsang H.-W.; Sutton P.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 1998, 9, 109. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Pathirana R. N.; Mahmood N.; Hay A. J. Bioorg. Med. Chem. 1992, 2, 701. [Google Scholar]
- McGuigan C.; Pathirana R. N.; Balzarini J.; De Clercq E. J. Med. Chem. 1993, 36, 1048. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Cahard D.; Sheeka H. M.; De Clercq E.; Balzarini J. J. Med. Chem. 1996, 39, 1748. [DOI] [PubMed] [Google Scholar]
- a Siddiqui A. Q.; Ballatore C.; McGuigan C.; De Clercq E.; Balzarini J. J. Med. Chem. 1999, 42, 393. [DOI] [PubMed] [Google Scholar]; b Siddiqui A. Q.; McGuigan C.; Ballatore C.; Zuccotto F.; Gilbert I. H.; De Clercq E.; Balzarini J. J. Med. Chem. 1999, 42, 4122. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Salgado A.; Yarnold C.; Harries T. Y.; De Clercq E.; Balzarini J. Antiviral Chem. Chemother. 1996, 7, 184. [Google Scholar]
- McGuigan C.; Cahard D.; Sheeka H.-M.; De Clercq E.; Balzarini J. Bioorg. Med. Chem. Lett. 1996, 6, 1183. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Bellevergue P.; Sheekab H.; Mahmood N.; Hay A. J. FEBS Lett. 1994, 351, 11. [DOI] [PubMed] [Google Scholar]
- a Lehsten D. M.; Baehr D. N.; Lobl T. J.; Vaino A. R. Org. Process Res. Dev. 2002, 6, 819. [Google Scholar]; b Lackey D. B.; Groziak M. P.; Sergeeva M.; Beryt M.; Boyer C.; Stroud R. M.; Sayre P.; Park J. W.; Johnston P.; Slamon D.; Shepard H. M.; Pegram M. Biochem. Pharmacol. 2001, 61, 179. [DOI] [PubMed] [Google Scholar]; c Li Q.; Boyer C.; Lee J. Y.; Shepard H. M. Mol. Pharmacol. 2001, 59, 446. [DOI] [PubMed] [Google Scholar]; d McGuigan C.; Thiery J. C.; Daverio F.; Jiang W. G.; Davies G.; Mason M. Bioorg. Med. Chem. 2005, 13, 3219. [DOI] [PubMed] [Google Scholar]; e Congiatu C.; McGuigan C.; Jiang W. G.; Davies G.; Mason M. D. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 485. [DOI] [PubMed] [Google Scholar]; f Congiatu C.; Brancale A.; Mason M. D.; Jiang W. G.; McGuigan C. J. Med. Chem. 2006, 49, 452. [DOI] [PubMed] [Google Scholar]
- Wang P.; Rachakonda S.; Zennou V.; Keilman M.; Niu C.; Bao D.; Ross B. S.; Furman P. A.; Otto M. J.; Sofia M. J. Antiviral Chem. Chemother. 2012, 22, 217. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. Antiviral Chem. Chemother. 2010, 20, 153. [DOI] [PubMed] [Google Scholar]
- Guan H.-P.; Ksebati M. B.; Cheng Y.-C.; Drach J. C.; Kern E. R.; Zemlicka J. J. Org. Chem. 2000, 65, 1280. [DOI] [PubMed] [Google Scholar]
- Franchetti P.; Cappellacci L.; Grifantini M.; Messini L.; Sheikha G. A.; Loi A. G.; Tramontano E.; De Mantis A.; Spiga M. G.; La Colla P. J. Med. Chem. 1994, 37, 3534. [DOI] [PubMed] [Google Scholar]
- Liu M.-C.; Luo M.-Z.; Mozdziesz D. E.; Dutschman G. E.; Gullen E. A.; Cheng Y.-C.; Sartorelli A. C. Nucleosides Nucleotides 2005, 24, 135. [DOI] [PubMed] [Google Scholar]
- Shi J.; Zhou L.; Zhang H.; McBrayer T. R.; Detorio M. A.; Johns M.; Bassit L.; Powdrill M. H.; Whitaker T.; Coats S. J.; Götte M.; Schinazi R. F. Bioorg. Med. Chem. Lett. 2011, 21, 7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y.; Valente R.; Mottram H.; Walsby E.; Mills K. I.; Balzarini J.; McGuigan C. Bioorg. Med. Chem. 2010, 18, 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Du J.; Wang P.; Nagarathnam D.; Espiritu C. L.; Bao H.; Murakami E.; Furman P. A.; Sofia M. J. Nucleosides, Nucleotides Nucleic Acids 2012, 31, 277. [DOI] [PubMed] [Google Scholar]
- a Mehellou Y.; McGuigan C.; Brancale A.; Balzarini J. Bioorg. Med. Chem. Lett. 2007, 17, 3666. [DOI] [PubMed] [Google Scholar]; b Mehellou Y.; Balzarini J.; McGuigan C. Org. Biomol. Chem. 2009, 7, 2548. [DOI] [PubMed] [Google Scholar]
- a McGuigan C.; Wedgwood O. M.; De Clercq E.; Balzarini J. Bioorg. Med. Chem. Lett. 1996, 6, 2359. [DOI] [PubMed] [Google Scholar]; b Siddiqui A. Q.; McGuigan C.; Ballatore C.; Wedgwood O.; De Clercq E.; Balzarini J. Bioorg. Med. Chem. Lett. 1999, 9, 2555. [DOI] [PubMed] [Google Scholar]; c Balzarini Y.; Kruining J.; Wedgwood O.; Pannecouque C.; Aquaro S.; Perno C.-F.; Naesens L.; Witvrouw M.; Heijtink R.; De Clercq E.; McGuigan C. FEBS Lett. 1997, 410, 324. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Hassan-Abdallah A.; Srinivasan S.; Wang Y.; Siddiqui A.; Daluge S. M.; Gudmundsson K. S.; Zhou H.; McLean E. W.; Peckham J. P.; Burnette T. C.; Marr H.; Hazen R.; Condreay L. D.; Johnson L.; Balzarini J. J. Med. Chem. 2006, 49, 7215. [DOI] [PubMed] [Google Scholar]
- Toti K. S.; Derudas M.; McGuigan C.; Balzarini J.; Van Calenbergh S. Eur. J. Med. Chem. 2011, 46, 3704. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Wedgwood O.; Kruining J.; Pelemans H.; Heijtink R.; De Clercq E.; McGuigan C. Biochem. Biophys. Res. Commun. 1996, 225, 363. [DOI] [PubMed] [Google Scholar]
- Gudmundsson K. S.; Wang Z.; Daluge S. M.; Johnson L. C.; Hazen R.; Condreay L. D.; McGuigan C. Nucleosides Nucleotides 2004, 23, 1929. [DOI] [PubMed] [Google Scholar]
- Gavazza F.; Daverio F.; Chaytor A. T.; Griffith T. M.; McGuigan C. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 553. [DOI] [PubMed] [Google Scholar]
- Gudmundsson K. S.; Daluge S. M.; Johnson L. C.; Jansen R.; Hazen R.; Condreay L. D.; McGuigan C. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 1953. [DOI] [PubMed] [Google Scholar]
- Rawal R. K.; Singh U. S.; Chavre S.; Wang J.; Sugiyama M.; Hung W.; Govindarajan R.; Korba B.; Tanaka Y.; Chu C. K. Bioorg. Med. Chem. Lett. 2013, 23, 503. [DOI] [PubMed] [Google Scholar]
- Perrone P.; Luoni G.; Kelleher M. R.; Daverio F.; Angell A.; Mulready S.; Congiatu C.; Rajyaguru S.; Martin J.; Levêque V.; Le Pogam S.; Najera I.; Klumpp K.; Smith D. B.; McGuigan C. J. Med. Chem. 2007, 50, 1840. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Kelleher M. R.; Perrone P.; Mulready S.; Luoni G.; Daverio F.; Rajyaguru S.; Le Pogam S.; Najera I.; Martin J. A.; Klumpp K.; Smith D. B. Bioorg. Med. Chem. Lett. 2009, 19, 4250. [DOI] [PubMed] [Google Scholar]
- Perrone P.; Daverio F.; Valente R.; Rajyaguru S.; Martin J.; Levêque V.; Le Pogam S.; Najera I.; Klumpp K.; Smith D. B.; McGuigan C. J. Med. Chem. 2007, 50, 5463. [DOI] [PubMed] [Google Scholar]
- Rondla R.; Coats S. J.; McBrayer T.; Grier J.; Johns M.; Tharnish P. M.; Whitaker T.; Zhou L.; Schinazi R. Antiviral Chem. Chemother. 2009, 20, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Madela K.; Aljarah M.; Gilles A.; Battina S. K.; Ramamurty C. V. S.; Rao C. S.; Vernachio J.; Hutchins J.; Hall A.; Kolykhalov A.; Henson G.; Chamberlain S. Bioorg. Med. Chem. Lett. 2011, 21, 6007. [DOI] [PubMed] [Google Scholar]
- a Vande Voorde J.; Liekens S.; McGuigan C.; Murziani P. G. S.; Slusarczyk M.; Balzarini J. Biochem. Pharmacol. 2011, 82, 441. [DOI] [PubMed] [Google Scholar]; b McGuigan C.; Murziani P.; Slusarczyk M.; Gonczy B.; Vande Voorde J.; Liekens S.; Balzarini J. J. Med. Chem. 2011, 54, 7247. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Kanz B.; Mamiya B. M.; Kern J. T.; Kerwin S. M. Tetrahedron Lett. 2001, 42, 565. [Google Scholar]
- McGuigan C.; Harris S. A.; Daluge S. M.; Gudmundsson K. S.; McLean E. W.; Burnette T. C.; Marr H.; Hazen R.; Condreay L. D.; Johnson L.; De Clercq E.; Balzarini J. J. Med. Chem. 2005, 48, 3504. [DOI] [PubMed] [Google Scholar]
- Yoo C. B.; Valente R.; Congiatu C.; Gavazza F.; Angel A.; Siddiqui M. A.; Jones P. A.; McGuigan C.; Marquez V. E. J. Med. Chem. 2008, 51, 7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintiliani M.; Persoons L.; Solaroli N.; Karlsson A.; Andrei G.; Snoeck R.; Balzarini J.; McGuigan C. Bioorg. Med. Chem. 2011, 19, 4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Winter H.; Maeda Y.; Mitsuya H.; Zemlicka J. J. Med. Chem. 1996, 39, 3300. [DOI] [PubMed] [Google Scholar]; b Winter H.; Maeda Y.; Uchida H.; Mitsuya H.; Zemlicka J. J. Med. Chem. 1997, 40, 2191. [DOI] [PubMed] [Google Scholar]
- Qiu Y.-L.; Ptak R. G.; Breitenbach J. M.; Lin J.-S.; Cheng Y.-C.; Drach J. C.; Kern E. R.; Zemlicka J. Antiviral Res. 1999, 43, 37. [DOI] [PubMed] [Google Scholar]
- Manfredini S.; Giovanni P.; Durini E.; Vertuani S.; Balzarini J.; De Clercq E.; Karlson A.; Buzzoni V.; Thelander L. J. Med. Chem. 1999, 42, 3242. [DOI] [PubMed] [Google Scholar]
- For biological evaluations, see:; a Uchida H.; Kodama E. N.; Yoshimura K.; Maeda Y.; Kosalaraksa P.; Maroun V.; Qiu Y.-L.; Zemlicka J.; Mitsuya H. Antimicrob. Agents Chemother. 1999, 43, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoshimura K.; Feldman R.; Kodama E.; Kavlick M. F.; Qiu Y.-L.; Zemlicka J.; Mitsuya H. Antimicrob. Agents Chemother. 1999, 43, 2479. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rybak R. J.; Zemlicka J.; Qiu Y.-L.; Hartline C. B.; Kern E. R. Antiviral Res. 1999, 43, 175. [DOI] [PubMed] [Google Scholar]
- a Qiu Y.-L.; Ksebati M. B.; Ptak R. G.; Fan B. Y.; Breitenbach J. M.; Drach J. C.; Lin J.-S.; Cheng Y.-C.; Kern E. R.; Drach J. C.; Zemlicka J. J. Med. Chem. 1998, 41, 10. [DOI] [PubMed] [Google Scholar]; b Qiu Y.-L.; Zemlicka J. Synthesis 1998, 1447. [Google Scholar]
- Ambrose A.; Zemlicka J.; Kern E. R.; Drach J. C.; Gullen E.; Cheng Y.-C. Nucleosides Nucleotides 2005, 24, 1763. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Slater M. J.; Parry N. R.; Perry A.; Harris S. Bioorg. Med. Chem. Lett. 2000, 10, 645. [DOI] [PubMed] [Google Scholar]
- a McGuigan C.; Derudas M.; Bugert J. J.; Andrei G.; Snoeck R.; Balzarini J. Bioorg. Med. Chem. Lett. 2008, 18, 4364. [DOI] [PubMed] [Google Scholar]; b Derudas M.; Carta D.; Brancale A.; Vanpouille C.; Lisco A.; Margolis L.; Balzarini J.; McGuigan C. J. Med. Chem. 2009, 52, 5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M.; Kalayanov G.; Winqvist A.; Pinho P.; Sund C.; Zhou X.-X.; Wahling H.; Belfrage A.-K.; Pelcman M.; Agback T.; Benckestock K.; Wikstrom K.; Boothee M.; Lindqvist A.; Rydegard C.; Jonckers T. H. M.; Vandyck K.; Raboisson P.; Lin T.; Lachau-Durand S.; de Kock H.; Smith D. B.; Martin J. A.; Klumpp K.; Simmen K.; Vrang L.; Terelius Y.; Samuelsson B.; Rosenquist A.; Johansson N. G. Bioorg. Med. Chem. Lett. 2012, 22, 3265. [DOI] [PubMed] [Google Scholar]
- a McGuigan C.; Perrone P.; Madela K.; Neyts J. Bioorg. Med. Chem. Lett. 2009, 19, 4316. [DOI] [PubMed] [Google Scholar]; b McGuigan C.; Gilles A.; Madela K.; Aljarah M.; Holl S.; Jones S.; Vernachio J.; Hutchins J.; Ames B.; Bryant K. D.; Gorovits E.; Ganguly B.; Hunley D.; Hall A.; Kolykhalov A.; Liu Y.; Muhammad J.; Raja N.; Walters R.; Wang J.; Chamberlain S.; Henson G. J. Med. Chem. 2010, 53, 4949. [DOI] [PubMed] [Google Scholar]
- Meneghesso S.; Vanderlinden E.; Stevaert A.; McGuigan C.; Balzarini J.; Naesens L. Antiviral Res. 2012, 94, 35. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Daverio F.; Nájera I.; Martin J. A.; Klumpp K.; Smith D. B. Bioorg. Med. Chem. Lett. 2009, 19, 3122. [DOI] [PubMed] [Google Scholar]
- Gardelli C.; Attenni B.; Donghi M.; Meppen M.; Pacini B.; Harper S.; Di Marco A.; Fiore F.; Giuliano C.; Pucci V.; Laufer R.; Gennari N.; Marcucci I.; Leone J. F.; Olsen D. B.; MacCoss M.; Rowley M.; Narjes F. J. Med. Chem. 2009, 52, 5394. [DOI] [PubMed] [Google Scholar]
- Derudas M.; Brancale A.; Naesens L.; Neyts J.; Balzarini J.; McGuigan C. Bioorg. Med. Chem. 2010, 18, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J. H.; Amblard F.; Coats S. J.; Schinazi R. F. Tetrahedron 2011, 67, 5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J. H.; Coats S. J.; Schinazi R. F. Org. Lett. 2012, 14, 2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.; Kim J.-S.; Kish P. E.; Zhang J.; Mitchell S.; Gentry B. G.; Breitenbach J. M.; Drach J. C.; Hilfinger J. Bioorg. Med. Chem. Lett. 2009, 19, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.; Kim J.-S.; Hilfinger J. Synth. Commun. 2012, 358. [Google Scholar]
- Di Francesco M. E.; Avolio S.; Pompei M.; Pesci S.; Monteagudo E.; Pucci V.; Giuliano C.; Fiore F.; Rowley M.; Summa V. Bioorg. Med. Chem. 2012, 20, 4801. [DOI] [PubMed] [Google Scholar]
- Lehsten D. M.; Baehr D. N.; Lobl T. J.; Vaino A. R. Org. Process Res. Dev. 2002, 6, 819. [Google Scholar]
- Li X.; Fu H.; Jiang Y.; Zhao Y.; Liu J. Synlett 2004, 2600. [Google Scholar]
- a Walsh E. N. J. Am. Chem. Soc. 1959, 81, 3023. [Google Scholar]; b Stang S. L.; Meier R.; Rocaboy C.; Gladysz J. A. J. Fluorine Chem. 2003, 119, 141. [Google Scholar]
- The use of an excess of diaryl phosphite was reported to limit the formation of dinucleotides.
- Ora M.; Ojanpera J.; Lonnberg H. Chem.—Eur. J. 2007, 13, 8591. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Guo X.; Fu H.; Jiang Y.; Zhao Y. Synlett 2005, 2537. [Google Scholar]
- Verfuerth U.; Ugi I. Chem. Ber. 1991, 124, 1627. [Google Scholar]
- Leisvuori A.; Aiba Y.; Lonnberg T.; Poijarvi-Virta P.; Blatt L.; Beigelman L.; Lonnberg H. Org. Biomol. Chem. 2010, 8, 2131. [DOI] [PubMed] [Google Scholar]
- Donghi M.; Attenni B.; Gardelli C.; Di Marco A.; Fiore F.; Giuliano C.; Laufer R.; Leone J. F.; Pucci V.; Rowley M.; Narjes F. Bioorg. Med. Chem. Lett. 2009, 19, 1392. [DOI] [PubMed] [Google Scholar]
- Nillroth U.; Besidsky Y.; Classon B.; Chattopadhyaya J.; Ugi I.; Danielson U. H. Drug Des. Discovery 1995, 13, 43. [PubMed] [Google Scholar]
- Stavudine 5′-monophenyl phosphate was prepared by established methods:Jastorff B.; Hettler H. Tetrahedron Lett. 1969, 2543. [DOI] [PubMed] [Google Scholar]
- Román C. A.; Balzarini J.; Meier C. J. Med. Chem. 2010, 53, 7675. [DOI] [PubMed] [Google Scholar]
- Román C. A.; Wasserthal P.; Balzarini J.; Meier C. Eur. J. Org. Chem. 2011, 4899. [Google Scholar]
- Ross B. S.; Reddy P. G.; Zhang H.-R.; Rachakonda S.; Sofia M. J. J. Org. Chem. 2011, 76, 8311. [DOI] [PubMed] [Google Scholar]
- Velazquez S.; Tunon V.; Jimeno M. L.; Chamorro C.; De Clercq E.; Balzarini J.; Camarassa M. J. J. Med. Chem. 1999, 42, 5188. [DOI] [PubMed] [Google Scholar]
- Hatton W.; Hunault J.; Egorov M.; Len C.; Pipelier M.; Blot V.; Silvestre V.; Fargeas V.; Ané A.; McBrayer T.; Detorio M.; Cho J.-H.; Bourgougnon N.; Dubreuil D.; Schinazi R. F.; Lebreton J. Eur. J. Org. Chem. 2011, 7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlikova P.; Pohl R.; Votruba I.; Shih R.; Birkus G.; Cihlar T.; Hocek M. Bioorg. Med. Chem. 2011, 19, 229. [DOI] [PubMed] [Google Scholar]
- Ballatore C.; McGuigan C.; De Clercq E.; Balzarini J. Bioorg. Med. Chem. Lett. 2001, 11, 1053. [DOI] [PubMed] [Google Scholar]
- Chapman H.; Kernan M.; Prisbe E.; Rohloff J.; Sparacino M.; Terhorst T.; Yu R. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 621. [DOI] [PubMed] [Google Scholar]
- Meppen M.; Pacini B.; Bazzo R.; Koch U.; Leone J. F.; Koeplinger K. A.; Rowley M.; Altamura S.; Di Marco A.; Fiore F.; Giuliano C.; Gonzalez-Paz O.; Laufer R.; Pucci V.; Narjes F.; Gardelli C. Eur. J. Med. Chem. 2009, 44, 3765. [DOI] [PubMed] [Google Scholar]
- Jain H. V.; Kalman T. I. Bioorg. Med. Chem. Lett. 2012, 22, 4497. [DOI] [PubMed] [Google Scholar]
- For a review, see:Drontle D. P.; Wagner C. R. Mini-Rev. Med. Chem. 2004, 4, 409. [DOI] [PubMed] [Google Scholar]
- a Chang S. L.; Griesgraber G. W.; Southern P. J.; Wagner C. R. J. Med. Chem. 2001, 44, 223. [DOI] [PubMed] [Google Scholar]; b Kim J.; Drontle D. P.; Wagner C. R. Nucleosides, Nucleotides Nucleic Acids 2004, 23, 483. [DOI] [PubMed] [Google Scholar]; c Kim J.; Park S.; Tretyakova N. Y.; Wagner C. R. Mol. Pharmacol. 2005, 2, 233. [DOI] [PubMed] [Google Scholar]; d Chou T.-F.; Bieganowski P.; Shilinski K.; Cheng J.; Brenner C.; Wagner C. R. J. Biol. Chem. 2005, 280, 15356. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chou T.-F.; Baraniak J.; Kaczmarek R.; Zhou X.; Cheng J.; Ghosh B.; Wagner C. Mol. Pharmacol. 2007, 4, 208. [DOI] [PubMed] [Google Scholar]; f Cheng J.; Zhou X.; Chou T.-F.; Ghosh B.; Liu B.; Wagner C. W. Bioorg. Med. Chem. Lett. 2009, 19, 6379. [DOI] [PubMed] [Google Scholar]
- Michielssens S.; Maiti M.; Maiti M.; Dyubankova N.; Herdewijn P.; Ceulemans A. J. Phys. Chem. A 2012, 116, 644. [DOI] [PubMed] [Google Scholar]
- a Adelfinskaya O.; Herdewijn P. Angew. Chem., Int. Ed. 2007, 46, 4356. [DOI] [PubMed] [Google Scholar]; b Adelfinskaya O.; Terrazas M.; Froeyen M.; Marliere P.; Nauwelaerts K.; Herdewijn P. Nucleic Acids Res. 2007, 35, 5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrazas M.; Marliere P.; Herdewijn P. Chem. Biodiversity 2008, 5, 31. [DOI] [PubMed] [Google Scholar]
- a Giraut A.; Dyubankova N.; Song X. P.; Herdewijn P. ChemBioChem 2009, 10, 2246. [DOI] [PubMed] [Google Scholar]; b Zlatev I.; Giraut A.; Morvan F.; Herdewijn P.; Vasseur J.-J. Bioorg. Med. Chem. 2009, 17, 7008. [DOI] [PubMed] [Google Scholar]; c Giraut A.; Song X. P.; Froeyen M.; Marliere P.; Herdewijn P. Nucleic Acids Res. 2010, 38, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yang S.; Froeyen M.; Lescrinier E.; Marliere P.; Herdewijn P. Org. Biomol. Chem. 2011, 9, 111. [DOI] [PubMed] [Google Scholar]; e Yang S.; Herdewijn P. Nucleosides, Nucleotides Nucleic Acids 2011, 30, 597. [DOI] [PubMed] [Google Scholar]
- a Abraham T. W.; Wagner C. R. Nucleosides Nucleotides 1994, 13, 1891. [Google Scholar]; b Wagner C. R.; Chang S. L.; Griesgraber G. W.; Song H.; McIntee E. J.; Zimmerman C. L. Nucleosides Nucleotides 1999, 18, 913. [DOI] [PubMed] [Google Scholar]
- Abraham T. W.; McIntee E. J.; Vidhya V. I.; Schinazi R. F.; Wagner C. R. Nucleosides, Nucleotides Nucleic Acids 1997, 16, 2079. [Google Scholar]
- Abraham T. W.; Kalman T. I.; McIntee E. J.; Wagner C. R. J. Med. Chem. 1996, 39, 4569. [DOI] [PubMed] [Google Scholar]
- Maiti M.; Michielssens S.; Dyubankova N.; Maiti M.; Lescrinier E.; Ceulemans A.; Herdewijn P. Chem.—Eur. J. 2012, 18, 857. [DOI] [PubMed] [Google Scholar]
- Iyer V. I.; Griesgraber G. W.; McIntee E. J.; Wagner C. R. J. Med. Chem. 2000, 43, 2266. [DOI] [PubMed] [Google Scholar]
- Chang S. L.; Griesgraber G. W.; Wagner C. R. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1571. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Fu H.; Jiang Y.; Zhao Y. Synlett 2005, 1927. [Google Scholar]
- Gardelli C.; Attenni B.; Donghi M.; Meppen M.; Pacini B.; Harper S.; Di Marco A.; Fiore F.; Giuliano C.; Pucci V.; Laufer R.; Gennari N.; Marcucci I.; Leone J. F.; Olsen D. B.; MacCoss M.; Rowley M.; Narjes F. J. Med. Chem. 2009, 52, 5394. [DOI] [PubMed] [Google Scholar]
- Whalen L. J.; McEvoy K. A.; Halcomb R. L. Bioorg. Med. Chem. Lett. 2003, 13, 301. [DOI] [PubMed] [Google Scholar]
- Fu H.; Han B.; Zhao Y.-F Chem. Commun. 2003, 134. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Han B.; Fu H.; Jiang Y.; Zhao Y. J. Org. Chem. 2005, 70, 6676. [DOI] [PubMed] [Google Scholar]
- Nillroth U.; Vrang L.; Ahlsen G.; Besidsky Y.; Chattopadhyaya J.; Ugi I.; Danielson U. H. Antiviral Chem. Chemother. 1995, 6, 50. [Google Scholar]
- De Napoli L.; Di Fabio G.; D’Onofrio J.; Montesarchio D. Chem. Commun. 2005, 2586. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Kashemirov B. A.; Eriksson U.; Amidon G. L.; Kish P. E.; Mitchell S.; Kim J.-S.; Hilfinger J. M. J. Organomet. Chem. 2005, 690, 2673. [Google Scholar]
- a Fries K. M.; Joswig C.; Borch R. F. J. Med. Chem. 1995, 38, 2672. [DOI] [PubMed] [Google Scholar]; b Meyers C. L. F.; Hong L.; Joswig C.; Borch R. F. J. Med. Chem. 2000, 43, 4313. [DOI] [PubMed] [Google Scholar]
- Meyers C. L. F.; Borch R. F. J. Med. Chem. 2000, 43, 4319. [DOI] [PubMed] [Google Scholar]
- Tobias S. C.; Borch R. F. J. Med. Chem. 2001, 44, 4475. [DOI] [PubMed] [Google Scholar]
- Wu W.; Borch R. F. Mol. Pharmaceutics 2006, 3, 451. [DOI] [PubMed] [Google Scholar]
- Tobias S. C.; Borch R. F. Mol. Pharmaceutics 2004, 1, 112. [DOI] [PubMed] [Google Scholar]
- Wu W.; Sigmond J.; Peters G. J.; Borch R. F. J. Med. Chem. 2007, 50, 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Madela K.; Aljarah M.; Bourdin C.; Arrica M.; Barrett E.; Jones S.; Kolykhalov A.; Bleiman B.; Bryant K. D.; Ganguly B.; Gorovits E.; Henson G.; Hunley D.; Hutchins J.; Muhammad J.; Obikhod A.; Patti J.; Walters C. R.; Wang J.; Vernachio J.; Ramamurty C. V. S.; Battina S. K.; Chamberlain S. J. Med. Chem. 2011, 54, 8632. [DOI] [PubMed] [Google Scholar]
- Jones B. C. N. M.; McGuigan C.; O’Connor T. J.; Jeffries D. J.; Kinchington D. Antiviral Chem. Chemother. 1991, 2, 35. [Google Scholar]
- McGuigan C.; Jones B. C. N. M.; Devine K. G.; Nicholls S. R.; O’Connor T. J.; Kinchington D. Bioorg. Med. Chem. Lett. 1991, 1, 729. [Google Scholar]
- Korboukh I.; Hull-Ryde E. A.; Rittiner J. E.; Randhawa A. S.; Coleman J.; Fitzpatrick B. J.; Setola V.; Janzen W. P.; Frye S. V.; Zylka M. J.; Jin J. J. Med. Chem. 2012, 55, 6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafinowska H. T.; Ashton R. J.; Bailey S.; Harnden M. R.; Jackson S. M.; Sutton D. J. Med. Chem. 1995, 38, 1372. [DOI] [PubMed] [Google Scholar]
- Dang Q.; Kasibhatla S. R.; Jiang T.; Fan K.; Liu Y.; Taplin F.; Schulz W.; Cashion D. K.; Reddy K. R.; van Poelje P. D.; Fujitaki J. R.; Potter S. C.; Erion M. D. J. Med. Chem. 2008, 51, 4331. [DOI] [PubMed] [Google Scholar]
- Jansa P.; Baszczynski O.; Dracinsky M.; Votruba I.; Zidek Z.; Bahador G.; Stepan G.; Cihlar T.; Mackman R.; Holy A.; Janeba Z. Eur. J. Med. Chem. 2011, 46, 3748. [DOI] [PubMed] [Google Scholar]
- a Lavie A.; Schlichting I.; Vetter I. R.; Konrad M.; Reinstein J.; Goody R. S. Nat. Med. 1997, 3, 922. [DOI] [PubMed] [Google Scholar]; b Lavie A.; Vetter I. R.; Konrad M.; Goody R. S.; Reinstein J.; Schlichting I. Nat. Struct. Biol. 1997, 4, 601. [DOI] [PubMed] [Google Scholar]
- a Sommadossi J. P.; Carlisle R.; Zhou Z. Mol. Pharmacol. 1989, 36, 9. [PubMed] [Google Scholar]; b Harrington J. A.; Reardon J. E.; Spector T. Antimicrob. Agents Chemother. 1993, 37, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yan J. P.; Ilsley D. D.; Frohlick C.; Street R.; Hall E. T.; Kuchta R. D.; Melanco P. J. Biol. Chem. 1995, 270, 22836. [DOI] [PubMed] [Google Scholar]
- a van Wijk G. M. T.; Hostetler K. Y.; Schlame M.; van den Boch H. Biochim. Biophys. Acta 1991, 1086, 99. [DOI] [PubMed] [Google Scholar]; b van Wijk G. M. T; Hostetler K. Y.; van den Bosch H. Biochim. Biophys. Acta 1991, 1084, 307. [DOI] [PubMed] [Google Scholar]
- Paulus H.; Kennedy E. P. J. Biol. Chem. 1960, 235, 1303. [PubMed] [Google Scholar]
- Agranoff B. W.; Suomi W. D. Biochem. Prep. 1967, 10, 47. [Google Scholar]
- a Hostetler K. Y.; Stuhmiller L. M.; Lenting H. B. M.; van den Bosch H.; Richman D. D. J. Biol. Chem. 1990, 265, 6112. [PubMed] [Google Scholar]; b van Wijk G. M. T; Hostetler K. Y.; Suurmeijer C. N. S. P.; van den Bosch H. Biochim. Biophys. Acta 1992, 1165, 45. [DOI] [PubMed] [Google Scholar]
- a Hostetler K. Y.; Richman D. D.; Carson D. A.; Stuhmiller L. M.; van Wijk G. M. T; van den Bosch H. Antimicrob. Agents Chemother. 1992, 36, 2025. [DOI] [PMC free article] [PubMed] [Google Scholar]; b van Wijk G. M. T; Hostetler K. Y.; van den Bosch H. J. Lipid Res. 1992, 33, 1211. [PubMed] [Google Scholar]
- van Wijk G. M. T; Hostetler K. Y.; Kroneman E.; Richman D. D.; Sridhar C. N.; Kumar R.; van den Bosch H. Chem. Phys. Lipids 1994, 70, 213. [DOI] [PubMed] [Google Scholar]
- a Hostetler K. Y.; Parker S.; Sridhar C. N.; Martins M. J.; Li J.-L.; Stuhmiller L. M.; Van Wijk G. M. T.; Van Den Bosch H.; Gardner M. F.; Aldern K. A.; Richman D. D. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 11835. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shakiba S.; Freeman W. R.; Flores-Aguilar M.; Munguia D.; Tatebayashi M.; Besen G.; Amani R.; Wiley C. A.; Vuong C.; Aldern K. A.; Gardner M. F.; Hostetler K. Y. Antimicrob. Agents Chemother. 1995, 39, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu E. K.; Ross R. J.; Matsushita T.; MacCoss M.; Hong C. I.; West C. R. J. Med. Chem. 1982, 25, 1322. [DOI] [PubMed] [Google Scholar]
- a MacCoss M.; Ryu E. K.; Matsushita T. Biochem. Biophys. Res. Commun. 1978, 85, 714. [DOI] [PubMed] [Google Scholar]; b Turcotte J. G.; Srivastava S. P.; Meresak W. A.; Rizkalla B. A.; louzon F.; Wunz T. P. Biochim. Biophys. Acta 1980, 619, 604. [DOI] [PubMed] [Google Scholar]; c Turcotte J. G.; Srivastava S. P.; Steim J. M.; Calabresi P.; Tibbetts L. M.; Chu M. Y. Biochim. Biophys. Acta 1980, 619, 619. [DOI] [PubMed] [Google Scholar]; d Matsushita T.; Ryu E. K.; Hong C. I.; MacCoss M. Cancer Res. 1981, 41, 2707. [PubMed] [Google Scholar]
- a Piantadosi C.; Marasco C. J. Jr.; Morris-Natschke S. L.; Meyer K. L.; Gumus F.; Surles J. R.; Ishaq K. S. J. Med. Chem. 1991, 34, 1408. [DOI] [PubMed] [Google Scholar]; b Hong C. I.; Nechaev A.; Kirisits A. J.; Vig R.; West C. R.; Manouilov K. K.; Chu C. K. J. Med. Chem. 1996, 39, 1771. [DOI] [PubMed] [Google Scholar]
- a Hong C. I.; An S.-H.; Buchheit D. J.; Nechaev A.; Kirisits A. J.; West C. R.; Berdel W. E. J. Med. Chem. 1986, 29, 2038. [DOI] [PubMed] [Google Scholar]; b Hong C. I.; Kirisits A. J.; Nechaev A.; Buchheit D. J.; West C. R. J. Med. Chem. 1990, 33, 1380. [DOI] [PubMed] [Google Scholar]
- Bonnaffé D.; Dupraz D.; Ughetto-Monfrin J.; Namane A.; Henin Y.; Huynh-Dinh Y. J. Org. Chem. 1996, 61, 895. [Google Scholar]
- Bonnaffé D.; Dupraz B.; Ughetto-Monfrin J.; Namane A.; Huynh-Dinh T. Tetrahedron Lett. 1995, 36, 531. [Google Scholar]
- Kreimeyer A.; Ughetto-Monfrin J.; Namane A.; Huynh-Dinh T. Tetrahedron Lett. 1996, 37, 8739. [Google Scholar]
- Kreimeyer A.; Andre F.; Gouyette C.; Huynh-Dinh T. Angew. Chem., Int. Ed. 1998, 37, 2853. [DOI] [PubMed] [Google Scholar]
- Hong C. I.; Kirisits A. J.; Nechaev A.; Buchheit D. J.; West C. R. J. Med. Chem. 1985, 28, 171. [DOI] [PubMed] [Google Scholar]
- Brachwitz H.; Vollgraf C. Pharmacol. Ther. 1995, 66, 39. [DOI] [PubMed] [Google Scholar]
- a Brachwitz H.; Lachmann U.; Thomas Y.; Bergmann J.; Berdel W. E.; Langen P. Chem. Phys. Lipids 1996, 83, 77. [DOI] [PubMed] [Google Scholar]; b Brachwitz H.; Thomas Y.; Bergmann J.; Berdel W. E.; Langen P. Chem. Phys. Lipids 1997, 87, 31. [DOI] [PubMed] [Google Scholar]
- Brachwitz H.; Bergmann J.; Fichtner I.; Thomas Y.; Vollgraf C.; Langen P.; Berdel W. E. J. Lipid Res. 1998, 39, 162. [PubMed] [Google Scholar]
- Ruiz J. C.; Beadle J. R.; Aldern K. A.; Keith K. A.; Hartline C. B.; Kern E. R.; Hostetler K. Y. Antiviral Res. 2007, 75, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jessen H. J.; Schulz T.; Balzarini J.; Meier C. Angew. Chem., Int. Ed. 2008, 47, 8719. [DOI] [PubMed] [Google Scholar]; b Meier C.; Jessen H. J.; Balzarini J. Nucleic Acids Symp. Ser. 2008, 52, 83. [DOI] [PubMed] [Google Scholar]; c Schulz T.; Balzarini J.; Meier C. ChemMedChem 2014, 9, 762. [DOI] [PubMed] [Google Scholar]