

Abstract
BACKGROUND:
Although it is well established that African Americans (AA) experience greater social stressors than non-Hispanic Whites (NHW), the extent to which early life adversity and cumulative social stressors such as perceived discrimination, neighborhood violence, subjective social status, and socioeconomic status contribute to disparity in coronary heart disease (CHD) and stroke between AA and NHW are not well understood.
PURPOSE:
The purpose of this paper is to propose a conceptual model based upon McEwen’s Allostatic Load Model suggesting how the relationships among social context, early life adversity, psychological stress, inflammation, adaptation, and epigenetic signature may contribute to the development of CHD and ischemic stroke. We hypothesize that social context and prior life adversity are associated with genome-wide as well as gene-specific epigenetic modifications that confer a proinflammatory epigenetic signature that mediates an enhanced proinflammatory state. Exposure to early life adversity, coupled with an increased allostatic load places individuals at greater risk for inflammatory based diseases, such as CHD and ischemic stroke.
RESULTS:
Based on a review of the literature, we propose a novel model in which social context and psychological stress, particularly during early life, engenders a proinflammatory epigenetic signature, which drives a heightened inflammatory state that increases risk for CHD and stroke. In the proposed model, a proinflammatory epigenetic signature and adaptation serve as mediator variables.
CONCLUSIONS:
Understanding the extent to which epigenetic signature bridges the psycho-social environment with inflammation and risk for CHD may yield novel biomarkers that can be used to assess risk, development, and progression of CHD/stroke. Epigenetic biomarkers may be used to inform preventive and treatment strategies that can be targeted to those most vulnerable, or to those with early signs of CHD, such as endothelial dysfunction. Furthermore, epigenetic approaches, including lifestyle modification and stress reduction programs, such as mindfulness-based stress reduction, offer promise to reduce health inequity linked to social disadvantage, as emerging evidence demonstrates that adverse epigenetic marks can be reversed.
Keywords: Stroke, Cardiovascular Disease, Epigenetics, Epigenomics, Health Status Disparities, Cytokines
Coronary heart disease (CHD) and ischemic stroke account for more than one third of all U.S. deaths and are estimated to cost $444 billion per year [1]. Although death rates for CHD and ischemic stroke have decreased in recent years, disparities among African Americans (AA) and non-Hispanic Whites (NHW) are striking and remain a growing concern [2]. For example, the death rate for AA from CHD is 37% higher than for Whites and the risk of having a first-time stroke is almost two times greater for AA than for Whites [1,3,4]. Traditional risk factors related to CHD/stroke, such as obesity, hypertension, and diabetes do not fully explain the disparities found between AA and NHW individuals [5,6]. Evidence demonstrates that chronic stress contributes to inflammatory processes leading to CHD/stroke [7,8]. Identification of psychobiological determinants of disease acknowledges the individual as the center of a dynamic environment where multiple social, psychological, biological, and cultural factors mediate and moderate disease progression and outcomes over the life span [9]. Such a view is consistent with allostatic load, a concept derived from the term allostasis, meaning the maintenance of stability or “homeostasis.” [10]. Allostatic load refers to the fluctuations in health status based on response to stressors and can be thought of as the “wear and tear” on the body that results from acute and chronic stressors. McEwen [10] conceptualized a model of the relationship between perceived stress and physiologic responses and the resulting allostatic load on the individual. Allostatic load is considered to be cumulative exposure to stressors across the life span, including early life adversity (such as childhood maltreatment) and social stressors ranging from exposure to community violence, perceived discrimination and socioeconomic hardship. The three-hit theory (hit 1-genetic predisposition; hit 2-early-life environment; and hit 3-later-life environment) of vulnerability and resilience integrates the allostatic load model stating that “in a given context vulnerability is enhanced when failure to cope with adversity accumulates” [11 p.1859].
Studies demonstrate that social stressors contribute to health status through various biological pathways [12–16]. Furthermore, adverse early life experiences, may “prime” inflammatory pathways predisposing an individual to mount a greater proinflammatory response to future stressors in adulthood [17–19]. In addition, early life adversity, such as exposure to child abuse, emotional and physical neglect may epigenetically alter DNA methylation levels creating a proinflammatory epigenetic signature that influences acute stress reactivity and inflammatory cytokine production. This, in turn, predisposes such individuals to a heightened proinflammatory response to stress as an adult [20,21]. Exposure to early life adversity, coupled with an increased allostatic load (due to multiple social, psychological, biological, and cultural factors) places individuals at greater risk for inflammatory based diseases such as CHD.
Although it is well established that, as a group, AA experience greater social stressors than NHW [22], the extent to which early life adversity and cumulative social stressors such as perceived discrimination, neighborhood violence, subjective social status (SSS), and socioeconomic status (SES) contribute to disparity in CHD/stroke disease between AA and NHW are not well understood. Chronic stress doubles the risk of myocardial infarction and contributes to proinflammatory processes implicated in CHD and stroke [8]; and it is critical to clarify the underlying social-biological pathways. A conceptual model may assist in guiding future research explaining the mechanisms for how social stressors contribute to CHD disparities. Therefore, the purpose of this paper is to propose a conceptual model based upon McEwen’s Allostatic Load Model [10]. We propose social context and psychological stress, particularly during early life, engenders a proinflammatory epigenetic signature, which drives a heightened inflammatory state that increases risk for CHD and stroke.
Stress, Inflammation and CHD/Ischemic Stroke
Inflammation plays a key role in the development of CHD and ischemic stroke [23,24]. The most common mechanism for the development of CHD and ischemic stroke is atherogenesis, a complex process by which the artery becomes obstructed with plaque. The process appears to begin early in life with the deposit of small lipoprotein particles in the intima and progresses with leukocyte recruitment, formation of lipid-laden macrophages, and foam cells. Evidence continues to accumulate supporting a state of chronic, low-grade inflammation in the development of atherosclerosis [25]. Endothelial cells and leukocytes within atherosclerotic plaques produce a variety of inflammatory mediators, such as heat shock proteins (HSP) [26] and interleukin-6 (IL-6) [25]. Progression of an atherosclerotic lesion from an early fatty streak to an advanced fibroproliferative atherosclerotic lesion is believed to be related to infiltration of T cells and macrophages into the intimal wall. Activated T-cells present in atherosclerotic lesions can secret interferon (IFN)-γ which induces vascular cellular adhesion molecule (VCAM-1) [25] which is found to be associated with endothelial dysfunction and is a marker of early CHD [27].
Psychosocial stress triggers increases in several circulating proinflammatory cytokines [28,29]. Evidence demonstrates that elevations in circulating proinflammatory cytokines play a causal role in mediating stress-induced vascular inflammation and atherogenesis [30,31]. This may involve alterations in endothelial cell function, as proinflammatory cytokines induce endothelial cell dysfunction [31], likely by down regulating endothelial nitric oxide synthase (eNOS) and increasing vascular oxidative stress [30,32]. For example, treatment of porcine pulmonary artery endothelial cells with a combination of IFN-gamma, TNF-alpha, and IL-1 reduced eNOS mRNA and protein [33]. Also, TNF-alpha and IL-1 beta down-regulated eNOS mRNA expression in human coronary artery cells [34]. In contrast, the anti-inflammatory cytokine IL-10, decreased the effect of proinflammatory cytokine induced endothelial dysfunction [35]. Of note, glucocorticoids, may also contribute to stress-triggered endothelial dysfunction, as inhibition of adrenal glucocorticoid secretion was shown to prevent endothelial dysfunction in healthy volunteers exposed to stress [36].
Ample evidence demonstrates that both acute [37] and chronic psychological stress [38], as well as negative emotions, like anger [39], contribute to endothelial dysfunction. Acute psychosocial stress was shown to impair flow mediated endothelial dilation, an index of endothelial dysfunction [40,41]. Impaired endothelial flow mediated vasodilation occurs rapidly and is of long duration. For example, by 10 minutes following a 3-minute mental stress task, healthy subjects exhibited a 50% reduction in flow-mediated endothelium-dependent vasodilation, lasting for 45 minutes [37]. Also, in healthy subjects, a laboratory speech task (preparing and delivering a speech in front of an evaluative panel) produced a significant reduction in flow mediated vasodilation at 30 and 90 minutes post stress, with levels reduced to that comparable to diabetic subjects with chronic endothelial dysfunction [40]. Thus, these findings provide evidence that link acute stress exposure to both inflammation and endothelial dysfunction, an early indicator of CVD. Whether an increased allostatic load and a background of cumulative life stress exaggerate both the inflammatory and endothelial response to stress remains to be determined.
Social Context, Stress, and Inflammation
A pervasive feature common to those raised in an adverse environment with social disadvantage is exposure to a greater frequency and intensity of chronic stressors. These chronic stressors include economic hardship, discrimination, neighborhood violence and low perceived social status. It is well known that individuals exposed to chronic stress are vulnerable to anxiety and depression [42,43]. Epidemiological studies document that psychological stress may have a significant deleterious effect on the development and progression of atherosclerosis [44,45]. In fact, some contend that nearly 40% of patients with atherosclerosis have no other known risk factors, other than psychological stress [46]. Furthermore, an individual’s social context, especially during childhood and adolescence, is now recognized to be a powerful predictor of adult health [47] suggesting that the origins of health disparity are rooted in one’s early social environment and life experiences [48]. Provocative findings from the Adverse Childhood Experiences (ACE) study reveal that in a sample of 17,000 adults, those who were exposed to family violence, abuse, and neglect during childhood had a 1.5–2.0 fold greater rate of cardiovascular disease, autoimmune disorders, and premature death than those who did not experience such early life adversity [49]. Evidence derived from experimental animal models confirm these findings and suggest causal pathways of adverse early life experiences affecting adult health outcomes, especially CHD [50]. Such causal pathways in humans remain to be established.
Data substantiate an independent link between early adverse life events and elevated levels of proinflammatory cytokines [17,51–54]. Repeated exposure to psychological stress induces chronic inflammation that can facilitate atherosclerosis [28]. Moreover, chronic stress is associated with an increased risk of recurrent acute coronary events and mortality [55,56]. TNF-alpha and IL-6 are intimately involved in the pathogenesis of atherosclerosis and their association with chronic psychological stress is well-described [30]. Furthermore, evidence suggests that lower levels of subjective social status in AA are associated with higher levels of HSP [16]. As such, the association of psychological stress with low grade inflammation appears to be an important precursor to a variety of disease states and to reduce quality of life. However, the precise mechanism(s) by which stress stimulates low grade inflammation and precipitates or contributes to disease is unknown. Hence, future research is essential to clarify the role of interactions between psychological stress and genetic and epigenetic factors in the development and progression of cardiovascular disease [57,58].
Enduring and/or intense life stress, especially during early life, impacts lifelong emotionality and heightens stress reactivity, as evidenced by greater anxiety and cortisol response to future stressors in adulthood [59,60]. It is theorized that exposure to early life adverse events during critical windows of development, including childhood and adolescence, alters neuroendocrine-immune pathways to result in chronic low grade inflammation during adulthood that predisposes the individual to inflammatory disease [40]. Biological embedding posits that early life adversities and cumulative disadvantages “recalibrates” the physiological response to stress in a manner that results in a dysfunctional stress response pattern during adulthood that increases risk for disease [61,62]. Evidence demonstrates that adults exposed to early life adversities respond to acute stress with an exaggerated inflammatory response [17,63] and a reduced cortisol response [64], furthering their risk for developing inflammatory disease, such as CHD or stroke [17]. Although there is evidence demonstrating that individuals with CHD mount a greater response to an acute laboratory stress challenge than those without CHD [65], no studies were found that examined the influence of chronic and cumulative stress on the acute stress response in individuals at risk for CHD/stroke. A recent study demonstrated that adverse prior life events are associated with heightened inflammatory markers, such as IL-6, in AA but not for Whites [66]. It is well established that AAs experience significantly greater early adverse events compared to Whites [22]. Future research examining the link between adverse life events and inflammation in adulthood may shed light on health disparities found among AA.
Epigenetic Signature, Social Context, Stress, and Inflammation
Epigenetics signature refers to a variety of processes that affect gene expression independent of actual DNA sequence [59]. Epigenetic information provides instruction on how, where, and when genetic information will be used. Hence, the importance of epigenetic information is that it regulates gene expression. Epigenetics can refer to heritable effects on gene expression, or to the stable long-term alteration of the transcriptional potential of a cell, which may not necessarily be heritable. Most importantly, epigenetic information is susceptible to change, and represents an excellent target to understand how the environment may impact physiological function. The impact could be manifest as long as the environmental factor is present or could persist in its absence [20,67]. The effect could be transient (during the duration) or extended (subsequent) to the environmental impact and although not necessarily transmittable (mitotically and/or meiotically), could exert significant influence. While epigenetics refers to effects on single and/or sets of genes, epigenomics refers to global epigenetic modifications that encompass the entire genome. As such, genetic information provides the blueprint for the manufacture of the proteins necessary to cellular function; whereas, epigenetic information provides instruction for the use of that blueprint, permitting an ordered and regulated gene expression pattern.
Epigenetically regulated gene expression is a consequence of small covalent chemical modifications, which mark the genome and play a role in turning genes on or off [68]. DNA methylation is one such a mark. In this process, methyl groups attach to the backbone of the DNA molecule at cytosine rings found at CpG dinucleotides [69]. These methyl groups typically turn genes off by affecting the accessibility of DNA. Another type of mark, known as histone modifications, indirectly affects the accessibility of DNA. There are a variety of such chemical marks that modify the amino terminal tails of histones (e.g. acetylation, methylation, phosphorylation), changing how tightly or loosely DNA is packaged. If the wrapping is tight, a gene may be hidden from the cell’s transcription machinery, consequently less accessible and hence switched off. In contrast, if the wrapping is loosened, a gene that was formerly inaccessible can become accessible. For example, histone deacetylation results in transcriptional repression. Conversely, histone acetylation, which involves the covalent addition of acetyl groups to the lysine moieties in the amino terminal histone tails, results in an increase in gene expression. Other relevant epigenetic regulators of gene expression are non-coding RNAs (ncRNAs, i.e., transcripts that are not translated into protein), which can range in size from a few nucleotides to several kilobase. These ncRNAs can mediate both transcriptional and post-transcriptional gene silencing as well as activation [70].
One of the best characterized epigenetic processes is DNA methylation. Within recent years, the impact of epigenetic mechanisms in cardiovascular pathophysiology has become increasing recognized as a major factor in the interface between genotype and environment, which is largely responsible for phenotype variability. Compelling evidence demonstrates that the epigenome dynamically responds to changes in environment throughout an individual’s life, controlling normal development, homeostasis, aging, and mediating responses to environmental stimuli [71]. One’s epigenetic signature can be modulated by one’s environment over the lifespan, and thus the epigenome serves as an important bridge linking life experiences, phenotypic expression, and disease risk (Figure 1). Emerging evidence demonstrates that an individual’s behavior, stress response, disease susceptibility, and even longevity are influenced by their epigenome and their epigenetic signature. Importantly, both are potentially malleable and thus, potentially reversible, through lifestyle environmental factors, such as nutrition, exercise, behavior modification and stress reduction (e.g. mindfulness-based stress reduction [72–75]. Such behavioral modifications have the capacity to impact complex, multifactorial diseases such as those affecting the cardiovascular system.
Figure 1:
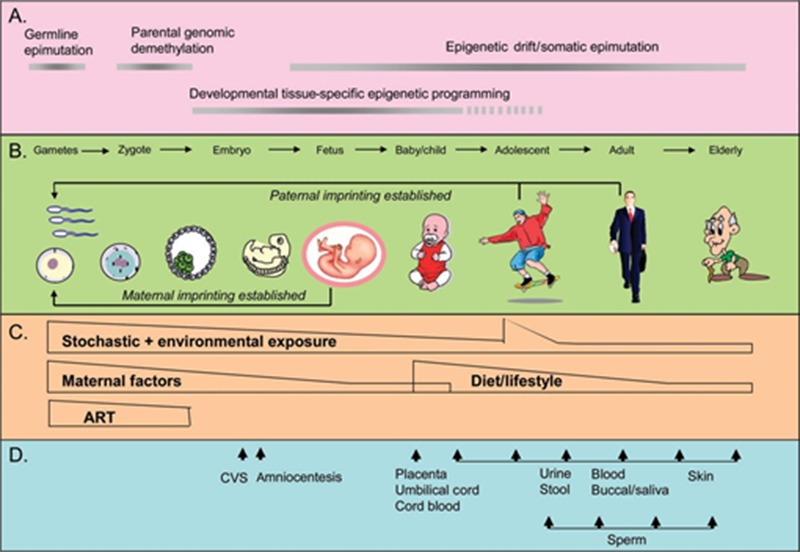
Epigenetics over the lifespan. Source: Figure is used with permission and is from “Prospects for Epigenetic Epidemiology” by D.L. Foley, J.M. Craig, R. Morely, C.A. Olsson, T. Dwyer, K. Smith et al. 2009, American Journal of Epidemiology, 169, pp. 389–400. Copyright 2009 by Oxford University Press [95].
Mathews and Janusek [60] demonstrated that epigenetic pattern and immune dysregulation are associated with psychosocial distress. Their work suggests that this may be mediated by exposure to cortisol [76]. The role of epigenetics in the development and progression of complex diseases, such as cancer [77], atherosclerosis [78], and stroke [71] continues to be elucidated through ongoing research. Animal studies provide causal evidence that stress modifies epigenetic signatures and plays a role in stress adaptation [79,80]. In primates, increased global DNA methylation was found to be associated with heightened behavioral stress reactivity following early life stress. Convincing evidence indicates that prior life adversity, such as low SES, contributes to DNA methylation levels [67,81,82] and may influence the regulation of the proinflammatory response to stress [79,80]. McGuinness et al. [67] demonstrated low SES to be associated with global DNA hypomethylation and IL-6 levels in a population-based study of men and women living in deprived areas of Glasgow. Results from animal studies, suggest that alterations in DNA methylation due to maternal maltreatment may exert lifelong and even trans-generational effects [20]. Our recent findings demonstrate both childhood emotional neglect and exposure to neighborhood violence in urban AA men (mean age 21 yrs) to result in a greater IL-6 response and a lower cortisol response to a laboratory paradigm of social evaluative stress. Moreover, the extent of DNA methylation of the IL-6 promoter in peripheral blood mononuclear cells moderated the association of childhood emotional neglect and exposure to violence. That is, AA men with lower DNA methylation and higher levels of emotional neglect or violence exposure produced the greatest amount of IL-6 in response to social evaluative stress [83]. Given that AA experience greater prior life adversity [22], alteration in the proinflammatory response may underlie CHD/stroke disparities among minorities and may relate to global DNA methylation levels. Understanding the linkages among social stressors, CVD risk, and epigenetic signature may yield novel biomarkers with clinical utility to assess risk, development, and progression of CHD/stroke disease [71].
As noted above, prior research demonstrates that early life abuse results in a life long burden of behavioral and pathophysiological problems [84,85] including an increased proinflammatory tone 20 years later [54]. Evidence in animal models suggests that such effects may emerge as a result of epigenetic modification. For example, findings derived using animal models demonstrate that early life stress or maltreatment result in DNA methylation modification of genes expressed in the brain, shaping HPA stress reactivity and behavioral stress responsiveness throughout life [86,87,88]. In humans, epigenetic modifications for the regulatory regions of NR3C1 (i.e., glucocorticoid receptor) have been observed in post-mortem brains of human suicide victims with a positive history of childhood abuse and likely relate to dysregulation of the HPA axis [89]. Moreover, such epigenetic modification are not limited to the brain in that prior life adversity was shown to produce epigenetic modifications in peripheral blood cells and those modifications related to increased inflammation [67,90,91]. Changes in global DNA methylation may be some of the earliest cellular events in disease onset [92] and such aberrant changes have been linked to a broad range of diseases, including CVD. Based on our review of the literature, we propose a conceptual model in which we hypothesize that social context and psychological stress engenders a proinflammatory epigenetic signature, which confers an enhanced proinflammatory status and increased CHD and ischemic stroke risk (Figure 2).
Figure 2.
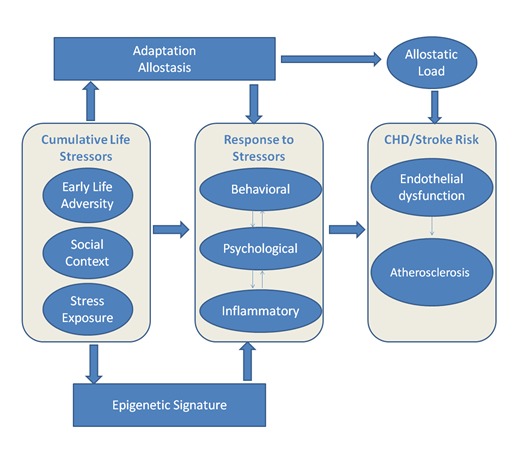
The Figure depicts the proposed conceptual model. The model is based on allostatic load theory, which posits that cumulative life stress results in an altered calibration of the physiological response to acute stress, which predisposes individuals to inflammatory diseases, such as CHD and ischemic stroke [61,96,97]. Behavioral (i.e. symptoms such as fatigue), psychological (i.e. depression and anxiety), and inflammatory (i.e. elevated proinflammatory cytokines) responses are driven by cumulative life stressors. We hypothesize that social context and prior life adversity are associated with genome-wide as well as gene-specific epigenetic modifications that confer a proinflammatory epigenetic signature that mediates an enhanced proinflammatory state. Furthermore, the degree of predisposition to disease subsequent to psychological stress and inflammatory response are modulated by an individual’s adaptive capacity [11].
IMPLICATIONS AND FUTURE RESEARCH
In the last decade, increasing knowledge in the field of epigenetics has dramatically changed our understanding of how one’s environment, including one’s social environment, affects gene regulation to influence one’s health over the lifespan and even the health of future generations. It is very likely that epigenetic processes are the missing link that explicates how one’s social context and early-life adverse experiences alter the stress-related inflammatory responses over the lifespan, which, in turn, contributes to social inequalities in risk for CHD/stroke. In the future, epigenetic biomarkers may be used to identify CHD/stroke risk, informing clinical decision making so that preventive and treatment strategies can be targeted to those most vulnerable. Furthermore, epigenetic approaches, including lifestyle modification offer promise to reduce health inequities linked to social disadvantage, as evidence demonstrates that adverse epigenetic marks attributed to early life adversity can be reversed [93]. Mind-body approaches, such as mindfulness based stress reduction, meditation, and yoga are emerging as highly promising interventions to reduce stress underlying pathophysiological pathways leading to CHD/stroke. Such innovative therapeutic approaches are only beginning to be investigated.
Advances in research that investigates linkages among social context, epigenetics, and inflammation in those at risk for CHD/stroke have potential to support health policies aimed at the social roots of disparity in CVD. For example, the recognition of aspects of the family and community that increase the risk of epigenetic modification may reveal aspects of the early environment that can be modified as part of community-level efforts to improve well being in those at risk. As well, the identification of protective factors (i.e., coping strategies, social support, resiliency) will yield information about malleable processes that can be targeted to maximize the efficacy of preventive and treatment strategies. This may include providing prenatal care and early childhood enrichment programs or interventions that may more effectively reduce risk for health disparity in CHD/stroke.
Research examining the interaction among social stressors, epigenetic mechanisms, and CHD/ischemic stroke is in its infancy. The continued development of new techniques to examine epigenetic mechanisms, including gene-epigene interactions, as well as epigenome-wide associations studies (EWAS) [94] will no doubt carry us into a new age of understanding how individuals adapt to their environments, ultimately leading to evidenced-based interventions that will eliminate health disparities in CHD/stroke.
Acknowledgments
The following grant support is acknowledged: NIH/NINR K01 training grant (K01 NR013478) and HSR&D NRI 12-413to KLS; NIH/RCA134736 to LWJ and HLM; and the Loyola University President’s Multidisciplinary grant to LWJ and HLM.
The views expressed do not necessarily reflect the position or policy of the U.S. Department of Veteran Affairs or the United States Government.
References
- [1].Centers for Disease Control and Prevention Heart disease and stroke prevention. 2011. [cited 2012 Feb 14] Available from www.cdc.gov/chronicdisease/resources/publications/AAG/dhdsp.htm.
- [2].Cruz-Flores S, Rabinstein A, Biller J, Elkind M, Griffith P, Gorelick P, et al. Racial-ethnic disparities in stroke care: The American experience. Stroke. 2011;42:2091–2116. doi: 10.1161/STR.0b013e3182213e24. [DOI] [PubMed] [Google Scholar]
- [3].National Heart Lung and Blood Institute . Incidence and Prevalence: 2006 Chart Book on Cardiovascular and Lung Diseases. Bethesda, MD: 2006. [Google Scholar]
- [4].Williams RA. Cardiovascular disease in African American women: a health care disparities issue. Journal of the National Medical Association. 2009;101(6):536–40. doi: 10.1016/s0027-9684(15)30938-x. [DOI] [PubMed] [Google Scholar]
- [5].Jolly S, Vittinghoff E, Chattopadhyay A, Bibbins-Domingo K. Higher cardiovascular disease prevalence and mortality among younger blacks compared to whites. Am.J.Med. 2010;123(9):811–8. doi: 10.1016/j.amjmed.2010.04.020. [DOI] [PubMed] [Google Scholar]
- [6].Kuzawa CW, Sweet E. Epigenetics and the embodiment of race: developmental origins of US racial disparities in cardiovascular health. American Journal of Human Biology. 2009;21(1):2–15. doi: 10.1002/ajhb.20822. [DOI] [PubMed] [Google Scholar]
- [7].Kornerup H, Osler M, Boysen G, Barefoot J, Schnohr P, Prescott E. Major life events increase the risk of stroke but not of myocardial infarction: results from the Copenhagen City Heart Study. European Journal of Cardiovascular Prevention & Rehabilitation 2010. 2010;17(1):113–8. doi: 10.1097/HJR.0b013e3283359c18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Park KE, Pepine CJ. Pathophysiologic mechanisms linking impaired cardiovascular health and neurologic dysfunction: the year in review. Cleveland Clinic Journal of Medicine. 2010;77(Suppl-5):S40–45. doi: 10.3949/ccjm.77.s3.07. [DOI] [PubMed] [Google Scholar]
- [9].DeVon HA, Saban KL. Psychosocial and Biological Stressors and the Pathogenesis of Cardiovascular Disease. In: Rice VH, editor. Handbook of Stress, Coping, and Health. 2nd ed. Thousand Oaks, CA: Sage; 2012. [Google Scholar]
- [10].McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Annals of the New York Academy of Sciences. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- [11].Daskalakis NP, Bagot RC, Parker KJ, Vinkers CH, de Kloet ER. The three-hit concept of vulnerability and resilience: toward understanding adaptation to early-life adversity outcome. Psychoneuroendocrinology. 2013;9:1858–73. doi: 10.1016/j.psyneuen.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thurston RC, Matthews KA. Racial and socioeconomic disparities in arterial stiffness and intima media thickness among adolescents. Social Science & Medicine. 2009;68(5):807–13. doi: 10.1016/j.socscimed.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ghaed SG, Gallo LC. Subjective social status, objective socioeconomic status, and cardiovascular risk in women. Health Psychol. 2007;26(6):668–74. doi: 10.1037/0278-6133.26.6.668. [DOI] [PubMed] [Google Scholar]
- [14].Friedman EM, Williams DR, Singer BH, Ryff CD. Chronic discrimination predicts higher circulating levels of E-selectin in a national sample: the MIDUS study. Brain, Behavior, & Immunity. 2009;23:684–92. doi: 10.1016/j.bbi.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stuller KA, Jarrett B, DeVries AC. Stress and social isolation increase vulnerability to stroke. Exper Neurol. 2012;233:33–9. doi: 10.1016/j.expneurol.2011.01.016. [DOI] [PubMed] [Google Scholar]
- [16].Saban KL, Hoppensteadt-Moorman D, Bryant FB, DeVon HA. Social predictors of inflammatory biomarkers among African American and Non-Hispanic White women with cardiovascular disease. Bio Res Nur. 2014;16:258–268. doi: 10.1177/1099800413491422. [DOI] [PubMed] [Google Scholar]
- [17].Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH. Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology. 2010;35:2617–23. doi: 10.1038/npp.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McEwen BS. Early life influences on life-long patterns of behavior and health. Mental Retardation & Developmental Disabilities Research Reviews. 2009;9:149–54. doi: 10.1002/mrdd.10074. [DOI] [PubMed] [Google Scholar]
- [19].Packard CJ, Bezlyak V, McLean JS, Batty GD, Ford I, Burns H, et al. Early life socioeconomic adversity is associated in adult life with chronic inflammation, carotid atherosclerosis, poorer lung function and decreased cognitive performance: a cross-sectional, population-based study. BMC Public Health. 2011;11:42. doi: 10.1186/1471-2458-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roth TL, Sweatt JD. Epigenetic marking of the BDNF gene by early-life adverse experiences. Hormones & Behavior. 2011;59:315–20. doi: 10.1016/j.yhbeh.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McEwen BS. Understanding the potency of stressful early life experiences on brain and body function. Metabolism: Clinical & Experimental. 2008;57(Suppl-5):S11–15. doi: 10.1016/j.metabol.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Roberts AL, Gilman SE, Breslau J, Breslau N, Koenen KC. Race/ethnic differences in exposure to traumatic events, development of post-traumatic stress disorder, and treatment-seeking for post-traumatic stress disorder in the United States. Psychol Med. 2011;41:71–83. doi: 10.1017/S0033291710000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pola R. Inflammatory markers for ischaemic stroke. Thrombosis & Haemostasis. 2009;101:800–1. [PubMed] [Google Scholar]
- [24].Guldiken B, Guldiken S, Turgut B, Turgut N, Demir M, Celik Y, Arikan E, Tugrul A. The roles of oxidized low-density lipoprotein and interleukin-6 levels in acute atherothrombotic and lacunar ischemic stroke. Angiology. 2008;59:224–9. doi: 10.1177/0003319707304134. [DOI] [PubMed] [Google Scholar]
- [25].Reiss AB, Glass AD. Atherosclerosis: immune and inflammatory aspects. Journal of Investigative Medicine. 2006;54(3):123–31. doi: 10.2310/6650.2006.05051. [DOI] [PubMed] [Google Scholar]
- [26].Xu Q. Role of heat shock proteins in atherosclerosis. Arteriosclerosis, Thrombosis & Vascular Biology. 2002;22:1547–59. doi: 10.1161/01.atv.0000029720.59649.50. [DOI] [PubMed] [Google Scholar]
- [27].Aragones G, Ferre R, Girona J, Plana N, Merino J, Heras M, Masana L, et al. Small artery dilation and endothelial markers in cardiovascular risk patients. European Journal of Clinical Investigation. 2012;42:34–41. doi: 10.1111/j.1365-2362.2011.02553.x. [DOI] [PubMed] [Google Scholar]
- [28].Hansel A, Hong S, Camara RJ, von KR. Inflammation as a psychophysiological biomarker in chronic psychosocial stress. Neuroscience & Biobehavioral Reviews. 2010;35(1):115–21. doi: 10.1016/j.neubiorev.2009.12.012. [DOI] [PubMed] [Google Scholar]
- [29].Yasui T, Maegawa M, Tomita J, Miyatani Y, Yamada M, Uemura H, Ueno S, et al. Association of serum cytokine concentrations with psychological symptoms in midlife women. Reprod Immunol. 2007;75:56–62. doi: 10.1016/j.jri.2007.02.004. [DOI] [PubMed] [Google Scholar]
- [30].Lu XT, Zhao YX, Zhang Y, Jiang F. Psychological stress, vascular inflammation, and atherogenesis: potential roles of circulating cytokines. Journal of Cardiovascular Pharmacology. 2013;62:6–12. doi: 10.1097/FJC.0b013e3182858fac. [DOI] [PubMed] [Google Scholar]
- [31].Toda N, Nakanishi-Toda M. How mental stress affects endothelial function. Pflugers Archiv - European Journal of Physiology. 2011;462:779–94. doi: 10.1007/s00424-011-1022-6. [DOI] [PubMed] [Google Scholar]
- [32].Bhagat K, Vallance P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation. 1997;96:3042–7. doi: 10.1161/01.cir.96.9.3042. [DOI] [PubMed] [Google Scholar]
- [33].Zhang J, Patel JM, Li YD, Block ER. Proinflammatory cytokines downregulate gene expression and activity of constitutive nitric oxide synthase in porcine pulmonary artery endothelial cells. Research Communications in Molecular Pathology & Pharmacology. 1997;96:71–87. [PubMed] [Google Scholar]
- [34].Seidel M, Billert H, Kurpisz M. Regulation of eNOS expression in HCAEC cell line treated with opioids and proinflammatory cytokines. Kardiolo Pols. 2006;64:153–8. [PubMed] [Google Scholar]
- [35].Zemse SM, Chiao CW, Hilgers RH, Webb RC. Interleukin-10 inhibits the in vivo and in vitro adverse effects of TNF-alpha on the endothelium of murine aorta. American Journal of Physiology - Heart & Circulatory Physiology. 2010;299(4):H1160–H1167. doi: 10.1152/ajpheart.00763.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Broadley AJ, Korszun A, Abdelaal E, Moskvina V, Jones CJ, Nash GB, Ray C, Deanfield J, Frenneaux MP. Inhibition of cortisol production with metyrapone prevents mental stress-induced endothelial dysfunction and baroreflex impairment. Journal of the American College of Cardiology. 2005;46(2):344–50. doi: 10.1016/j.jacc.2005.03.068. [DOI] [PubMed] [Google Scholar]
- [37].Spieker LE, Hurlimann D, Ruschitzka F, Corti R, Enseleit F, Shaw S, Hayoz D, Deanfield JE, Luscher TF, Noll G. Mental stress induces prolonged endothelial dysfunction via endothelin-A receptors. Circulation. 2002;105(24):2817–20. doi: 10.1161/01.cir.0000021598.15895.34. [DOI] [PubMed] [Google Scholar]
- [38].Mausbach BT, Roepke SK, Ziegler MG, Milic M, von KR, Dimsdale JE, Mills PJ, Patterson TL, Allison MA, Ancoli-Israel S, et al. Association between chronic caregiving stress and impaired endothelial function in the elderly. Journal of the American College of Cardiology. 2010;55(23):2599–606. doi: 10.1016/j.jacc.2009.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Burg MM, Soufer A, Lampert R, Collins D, Soufer R. Autonomic contribution to endothelin-1 increase during laboratory anger-recall stress in patients with coronary artery disease. Molecular Medicine. 2011;17(5–6):495–501. doi: 10.2119/molmed.2010.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O’Connor G, Betteridge J, Klein N, Steptoe A, et al. Mental stress induces transient endothelial dysfunction in humans. Circulation. 2000;102(20):2473–8. doi: 10.1161/01.cir.102.20.2473. [DOI] [PubMed] [Google Scholar]
- [41].Glasser SP, Selwyn AP, Ganz P. Atherosclerosis: risk factors and the vascular endothelium. Am.Heart J. 1996;131(2):379–84. doi: 10.1016/s0002-8703(96)90370-1. [DOI] [PubMed] [Google Scholar]
- [42].Kim P, Evans GW, Angstadt M, Ho SS, Sripada CS, Swain JE, Liberzon I, Phan KL. Effects of childhood poverty and chronic stress on emotion regulatory brain function in adulthood. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(46):18442–7. doi: 10.1073/pnas.1308240110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sweet E, Nandi A, Adam EK, McDade TW. The high price of debt: household financial debt and its impact on mental and physical health. Social Science & Medicine. 2013;91:94–100. doi: 10.1016/j.socscimed.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kamarck TW, Shiffman S, Sutton-Tyrrell K, Muldoon MF, Tepper P. Daily psychological demands are associated with 6-year progression of carotid artery atherosclerosis: the Pittsburgh Healthy Heart Project. Psychosom.Med. 2012;74(4):432–9. doi: 10.1097/PSY.0b013e3182572599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang HX, Leineweber C, Kirkeeide R, Svane B, Schenck-Gustafsson K, Theorell T, et al. Psychosocial stress and atherosclerosis: family and work stress accelerate progression of coronary disease in women. The Stockholm Female Coronary Angiography Study. Int Med. 2007;261(3):245–54. doi: 10.1111/j.1365-2796.2006.01759.x. [DOI] [PubMed] [Google Scholar]
- [46].Black PH, Garbutt LD. Stress, inflammation and cardiovascular disease. J.Psychosom.Res. 2002;52(1):1–23. doi: 10.1016/s0022-3999(01)00302-6. [DOI] [PubMed] [Google Scholar]
- [47].Cohen S, Janicki-Deverts D. Can we improve our physical health by altering our social networks? Perspectives on Psychological Science. 2009;4:375–8. doi: 10.1111/j.1745-6924.2009.01141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Steptoe A, Kivimaki M. Stress and cardiovascular disease. Nature Reviews Cardiology. 2012;9:360–70. doi: 10.1038/nrcardio.2012.45. [DOI] [PubMed] [Google Scholar]
- [49].Anda RF, Dong M, Brown DW, Felitti VJ, Giles WH, Perry GS, et al. The relationship of adverse childhood experiences to a history of premature death of family members. BMC Public Health. 2009;9:106. doi: 10.1186/1471-2458-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. JAMA. 2007;298:1685–7. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- [51].Danese A, Caspi A, Williams B, Ambler A, Sugden K, Mika J, Werts H, Freeman J, Pariante CM, Moffitt TE, et al. Biological embedding of stress through inflammation processes in childhood. Molecular Psychiatry. 2011;16:244–6. doi: 10.1038/mp.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Danese A, Moffitt TE, Harrington H, Milne BJ, Polanczyk G, Pariante CM, Poulton R, Caspi A. Adverse childhood experiences and adult risk factors for age-related disease: depression, inflammation, and clustering of metabolic risk markers. Archives of Pediatrics & Adolescent Medicine. 2009;163:1135–43. doi: 10.1001/archpediatrics.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Archives of General Psychiatry. 2008;65:409–15. doi: 10.1001/archpsyc.65.4.409. [Erratum appears in Arch Gen Psychiatry 2008 Jun;65(6):725] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(4):1319–24. doi: 10.1073/pnas.0610362104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Steptoe A, Kivimaki M. Stress and cardiovascular disease: an update on current knowledge. Annu Rev Public Health. 2013;34:337–54. doi: 10.1146/annurev-publhealth-031912-114452. [DOI] [PubMed] [Google Scholar]
- [56].Steptoe A, Kivimaki M. Stress and cardiovascular disease. Nature Reviews Cardiology. 2012;9:360–70. doi: 10.1038/nrcardio.2012.45. [DOI] [PubMed] [Google Scholar]
- [57].Williams RB. Psychosocial and biobehavioral factors and their interplay in coronary heart disease. Annual Review of Clinical Psychology. 2008;4:349–65. doi: 10.1146/annurev.clinpsy.4.022007.141237. [DOI] [PubMed] [Google Scholar]
- [58].Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychol.Bull. 2011;137:959–97. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mathews HL, Janusek LW. Epigenetics and psychoneuroimmunology: mechanisms and models. Brain, Behavior, & Immunity. 2011;25(1):25–39. doi: 10.1016/j.bbi.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mathews H, Konley T, Kosik K, Eddy J, Albuquerque K, Janusek LW. Epigenetic patterns associated with the immune dysregulation that accompanies psychosocial distress. Brain, Behavior, & Immunity. 2011;25(5):830–839. doi: 10.1016/j.bbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hertzman C. The biological embedding of early experience and its effects on health in adulthood. Annals of the New York Academy of Sciences. 1999;896:85–95. doi: 10.1111/j.1749-6632.1999.tb08107.x. [DOI] [PubMed] [Google Scholar]
- [62].Bock J, Rether K, Groger N, Xie L, Braun K. Perinatal programming of emotional brain circuits: an integrative view from systems to molecules. Frontiers in Neuroscience. 2014;8:11. doi: 10.3389/fnins.2014.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Witek JL, Tell D, Albuquerque K, Mathews HL. Childhood adversity increases vulnerability for behavioral symptoms and immune dysregulation in women with breast cancer. Brain, Behavior, & Immunity. 2013;30(Suppl-62) doi: 10.1016/j.bbi.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Carpenter LL, Tyrka AR, Ross NS, Khoury L, Anderson GM, Price LH. Effect of childhood emotional abuse and age on cortisol responsivity in adulthood. Biological Psychiatry. 2009;66:69–75. doi: 10.1016/j.biopsych.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kop WJ, Weissman NJ, Zhu J, Bonsall RW, Doyle M, Stretch MR, et al. Effects of acute mental stress and exercise on inflammatory markers in patients with coronary artery disease and healthy controls. American Journal of Cardiology. 2008;101:767–73. doi: 10.1016/j.amjcard.2007.11.006. [DOI] [PubMed] [Google Scholar]
- [66].Slopen N, Lewis TT, Gruenewald TL, Mujahid MS, Ryff CD, Albert MA, Williams DR. Early life adversity and inflammation in African Americans and whites in the midlife in the United States survey. PsychosomMed. 2010;72:694–701. doi: 10.1097/PSY.0b013e3181e9c16f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mcguinness D, McGlynn L, Johnson P, MacIntyre A, Batty D, Burns H, Cavanagh J, Deans K, Ford I, McConnachie A, et al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. International Journal of Epidemiology. 2012;1:1–10. doi: 10.1093/ije/dyr215. [DOI] [PubMed] [Google Scholar]
- [68].Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- [69].Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- [70].Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17. doi: 10.1016/j.gene.2007.12.008. [DOI] [PubMed] [Google Scholar]
- [71].Qureshi IA, Mehler MF. Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Archives of Neurology. 2010;67:1316–22. doi: 10.1001/archneurol.2010.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Niculescu MD. Nutritional epigenetics. Ilar Journal. 2012;53(3–4):270–8. doi: 10.1093/ilar.53.3-4.270. [DOI] [PubMed] [Google Scholar]
- [73].Abel JL, Rissman EF. Running-induced epigenetic and gene expression changes in the adolescent brain. International Journal of Developmental Neuroscience. 2013;3:382–90. doi: 10.1016/j.ijdevneu.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sanchis-Gomar F, Garcia-Gimenez JL, Perez-Quilis C, Gomez-Cabrera MC, Pallardo FV, Lippi G. Physical exercise as an epigenetic modulator: Eustress, the “positive stress” as an effector of gene expression. Journal of Strength& Conditioning Research. 2012;26:3469–72. doi: 10.1519/JSC.0b013e31825bb594. [DOI] [PubMed] [Google Scholar]
- [75].Burdge GC, Hoile SP, Lillycrop KA. Epigenetics: are there implications for personalised nutrition? Current Opinion in Clinical Nutrition & Metabolic Care. 2012;15:442–7. doi: 10.1097/MCO.0b013e3283567dd2. [DOI] [PubMed] [Google Scholar]
- [76].Krukowski K, Eddy J, Kosik KL, Konley T, Janusek LW, Mathews HL. Glucocorticoid dysregulation of natural killer cell function through epigenetic modification. Brain, Behavior, & Immunity. 2011;25:239–49. doi: 10.1016/j.bbi.2010.07.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Robertson KD. DNA methylation and human disease. Nature Reviews Genetics. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- [78].Gluckman PD, Hanson MA, Buklijas T, Low FM, Beedle AS. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nature Reviews Endocrinology. 2009;5:401–8. doi: 10.1038/nrendo.2009.102. [DOI] [PubMed] [Google Scholar]
- [79].Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;106:20912–7. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chandramohan Y, Droste SK, Arthur JS, Reul JM. The forced swimming-induced behavioural immobility response involves histone H3 phospho-acetylation and c-Fos induction in dentate gyrus granule neurons via activation of the N-methyl-D-aspartate/extracellular signal-regulated kinase/mitogen- and stress-activated kinase signalling pathway. European Journal of Neuroscience. 2008;27:2701–13. doi: 10.1111/j.1460-9568.2008.06230.x. [DOI] [PubMed] [Google Scholar]
- [81].Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. The Janus face of DNA methylation in aging. 2010;2:107–10. doi: 10.18632/aging.100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biological Psychiatry. 2009;65:760–9. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Janusek LW, Tell D, Fishe K, Gaylord-Harden N, Mathews HL. Influence of childhood adversity and neighborhood violence on inflammatory risk in young African American men. Brain, Behavior & Immunity. 2012;26:S33–S34. doi: 10.1016/j.bbi.2016.10.006. [DOI] [PubMed] [Google Scholar]
- [84].Fernandez-Botran R, Miller JJ, Burns VE, Newton TL. Correlations among inflammatory markers in plasma, saliva and oral mucosal transudate in post-menopausal women with past intimate partner violence. Brain, Behavior, & Immunity. 2011;25:314–21. doi: 10.1016/j.bbi.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biological Psychiatry. 2001;49:1023–39. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- [86].Koenen KC, Uddin M, Amstadter AB, Galea S. Incorporating the social environment in genotype environment interaction studies of mental disorders. International Journal of Clinical Practice. 2010;64:1489–92. doi: 10.1111/j.1742-1241.2010.02359.x. [DOI] [PubMed] [Google Scholar]
- [87].Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de los SR, Goldmann E, Galea S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2010;107:9470–5. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Uddin M, Koenen KC, Aiello AE, Wildman DE, de los SR, Galea S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychological Medicine. 2011;41:997–1007. doi: 10.1017/S0033291710001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12:342–8. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics: Official Journal of the DNA Methylation Society. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- [91].Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? Journal of Internal Medicine. 2007;261:488–99. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- [92].Chmielewski M, Lindholm B, Stenvinkel P, Ekstrom JT. The role of epigenetics in kidney diseases. Makedonska Akademija na Naukite i Umetnostite Oddelenie Za Bioloshki i Meditsinski Nauki Prilozi. 2011;32:45–54. [PubMed] [Google Scholar]
- [93].Szyf M. The early life environment and the epigenome. Biochimica et Biophysica Acta. 2009;1790:878–85. doi: 10.1016/j.bbagen.2009.01.009. [DOI] [PubMed] [Google Scholar]
- [94].Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nature Reviews Genetics. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, Saffery R. Prospects for epigenetic epidemiology. Am.J.Epidemiol. 2009;169:389–400. doi: 10.1093/aje/kwn380. [Erratum appears in Am J Epidemiol 2009 Jun 1;169(11):1409 Note: Olsson, Craig J [corrected to Olsson, Craig A]] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Juster RP, Bizik G, Picard M, Arsenault-Lapierre G, Sindi S, Trepanier L, et al. A transdisciplinary perspective of chronic stress in relation to psychopathology throughout life span development. Development & Psychopathology. 2011;23:725–76. doi: 10.1017/S0954579411000289. [DOI] [PubMed] [Google Scholar]
- [97].McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load Annals of the New York Academy of Sciences. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]