

Abstract
Only a few percent of the 3 billion pairs of chemical letters in the human genome is responsible for protein-coding sequences. Recent advances in the field of epigenomics have helped us to understand how most of the remaining sequences are responsible for gene regulation at baseline and in disease conditions. Here we discuss recent advances in the area of epigenetics—specifically in cytosine modifications—and its application in the field of nephrology.
Keywords: chronic kidney disease, cytosine methylation, enhancers, epigenetics
INTRODUCTION
Epigenetics refers to heritable changes in gene expression patterns that are not caused by alterations in the nucleotide sequence itself. The epigenetic system therefore must be both heritable and reversible at the cellular level [1]. Cytosine modifications fulfill these criteria and are therefore considered the key epigenetic mechanism. Other factors including histone tail modifications, higher order chromatin organization and long and short non-coding RNA molecules are also often described as epigenetic mechanisms. The epigenetic information is stored in the form of chromatin, which is DNA wrapped around protein complexes and packaged into higher order structure [2]. Highly compacted chromatin is not readily accessible to the transcriptional machinery and represents mostly transcriptionally silent regions, called heterochromatin. Open chromatin regions, also called euchromatin, are transcriptionally competent regions. Nucleosomes are stretches of DNA (146 bp) wrapped around an octamer protein that consists of four dimers of histone proteins (H2A, H2B, H3, H4). Covalent modifications of histone tails constitute epigenetic marks as these modifications influence gene expression [3]. Upon transcription factor (TF) availability, the epigenome is the key determinant of transcriptional outcome, allowing certain genes to be expressed while others are not accessible to TFs. Understanding the epigenome is critical to comprehending cell-type-specific gene regulation at baseline and in disease conditions [3].
DETERMINING CELL-TYPE-SPECIFIC GENE REGULATORY REGIONS
Regulatory regions are characterized by TF binding. Unfortunately, it is often difficult to identify all TFs that are active in a given cell type. Recently, the DNase I hypersensitivity analysis followed by next-generation sequencing (DHS) has gained popularity to identify genome-wide regulatory regions [4]. The method takes advantage of the observation that TFs can only bind to DNA that is not wrapped around nucleosomes, so these regions are more sensitive to DNase I digestion. DNase I hypersensitivity therefore identifies regulatory regions, including promoter and enhancer regions in specific cell types with high precision.
Distinguishing the different types of regulatory elements imposes another challenge. Histone tail modifications are key regulators of gene expression. More than 100 different modifications have been described [5]. Methylation and acetylation of lysine residues on H3 histone (H3K4, H3K27, H3K9) are the most commonly studied modifications. In order to regulate gene expression, histones that are localized to nucleosomes right next to regulatory elements have specific tail alterations. These changes are so specific that histone tail modification patterns can be used to identify different cis-acting regulatory elements, including promoters, enhancers, silencers, insulators and locus control regions [6]. These cis-type gene regulatory regions are highly cell-type-specific and are very important for cell-type-specific transcription [7]. Enhancers are of critical importance for gene expression and for defining cell-type specificity, and accordingly, they are usually occupied by the cell-type-specific TFs. Enhancers are located within a few kilobases to hundreds of kilobases of the regulated gene, and they can be found on the opposite DNA strand, downstream or upstream of the regulated gene or commonly in intronic regions. Previously, characterization and localization of these important regulatory elements has proved to be difficult [8, 9]. Fortunately, it has been found that H4K3me1 and H3K27ac histone modifications can be used to define enhancers [10]. Using these distinct chromatin signatures, we can predict the location of the different regulatory elements with high sensitivity and specificity and construct cell-type-specific gene regulatory maps [11]. The most commonly used method for genome-wide characterization of chromatin regions is chromatin immunoprecipitation (ChIP) to pull down DNA with specific histone tail modifications, followed by next-generation sequencing.
CYTOSINE METHYLATION AND DEMETHYLATION; HOW DOES THIS WORK?
DNA methylation is a chemical mark that involves the addition of a methyl group to cytosines on the 5th carbon position (5mC) on CpG sites (-C-phosphate-G-) [12]. The majority of CpGs (70–80%) are methylated in the genome. CpG-rich regions, designated as CpG islands, are prevalent on gene promoter regions and are largely unmethylated (<10%). DNA methyltransferase 1 (DNMT1) maintains the methylation patterns of the adult tissues that were established during development. Since DNA polymerases cannot incorporate 5mC, DNMT1 co-localizes to the replication fork and is responsible for methylating hemimethylated cytosines (Figure 1). The DNMT1 knockout mice show developmental delay and embryonic lethality, indicating the importance of DNMT1 [13].
FIGURE 1:

Mechanisms of DNA methylation and active demethylation. (a) DNA cytosine methylation is established using DNMT3A and 3B. During DNA replication, cytosines on the nascent strand are not methylated. DNMT1 recognizes hemimethylated DNA, sits at the replication fork and methylates the cytosines according to the template methylation status. (b) Active demethylation. Global cytosine methylation levels have been shown to decrease with aging. The speed of losing methylation is higher than that of passive demethylation, and therefore, two major pathways have been discovered: one pathway is using the TET enzyme that hydroxylates methylated cytosine. The hydroxy-methyl cytosine is further recognized by DNA glycosylases and removed from the sugar-phosphate backbone. The second pathway involves proteins such as AID that induce a base pair mismatch. Different DNA repair mechanisms are involved after the base is removed.
DNA demethylation has several functions, including maintaining the fidelity of DNA methylation patterns and reactivating genes in somatic cells. DNA demethylation can be either an active or a passive process. Passive demethylation is mediated by reduced activity of methyltransferases during DNA replication and the subsequent loss of daughter strand methylation. Yet, the extent and dynamics of 5mC in the genome of mammals suggest that passive DNA demethylation alone cannot account for all changes. Two major processes have been described for active demethylation. The first process involves proteins such as thymine DNA glycosylase or activation-induced deaminase (AID) that induce a base pair mismatch [14], which is later recognized and excised by DNA glycosylase and different DNA repair mechanisms. The second process engages 5mC modification mainly by TET proteins [15], and is not directly associated with DNA repair (Figure 1).
TET proteins can oxidize 5 methyl cytosine (5mC) to 5 hydroxymethyl cytosine (5hmC), which then can be oxidized further to the 5 formyl-cytosine (5fC) and 5 carboxyl-cytosine (5caC) by TET [16]. The TET family has three members, TET1, TET2 and TET3, that are involved in many processes including gene regulation, development and cancer [17]. TET proteins take part both in passive and active demethylation. TET proteins likely have other proposed functions, including binding to CpG-rich promoters and transcription start sites, presumably to prevent DNMT binding and unwanted methylation and transcription repression through recruitment of Polycomb repressor complex 2 (protein complex associated with gene silencing through chromatin remodeling) [18, 19]. In summary, our understanding of cytosine methylation and demethylation is expanding; future work will be directed at determining how local and global cytosine methylation levels are maintained, keeping cell identity stable, yet still allowing environmental plasticity.
CYTOSINE METHYLATION AND DEVELOPMENT
The largest change in cytosine methylation levels is observed in the context of development. Most cytosines are methylated in adult tissues; however, after fertilization almost all methylation is removed and a new pattern is reestablished during implantation. There are few regions in the genome (imprinted loci) that keep their methylation pattern after fertilization, and these regions are likely responsible for the transgenerational epigenetic mechanism [20]. After implantation cytosine methylation is reestablished by de novo methylases, DNMT3a and DNMT3b [21]. What governs DNMT3 to methylate unmethylated cytosines during development, yet spare CpG islands, is still not clear. It is proposed that cell-type specific or developmental TFs might shield certain areas in the genome that will now be protected from cytosine methylation [21]. Global deletion of Dnmt3a/b in mice is associated with severe developmental defects indicating that establishing cytosine methylation is critical for development and differentiation [22, 23]. There are two demethylation waves in the embryo. The first wave occurs before implantation and consists of global demethylation, while the second one occurs later, as primordial germ cells (PGCs) develop and migrate to the genital ridge to later develop into gametes. The paternal pronucleus undergoes a relatively fast demethylation wave right after fertilization, sparing the maternal pronucleus, as well as imprinted regions in the paternal pronucleus. The maternal pronucleus demethylation occurs later in a slower kinetics. During embryonic development, TET3 is responsible for the active demethylation of the paternal genome, while the maternal genome loses its methylation through gradual passive demethylation. It has been suggested that TET1 and TET2 mediate PGCs passive demethylation.
The presumed goal of preimplantation demethylation is to reset the genome, which enables the expression of previously methylated pluripotency genes and genes responsible for extra-embryonic differentiation. In the post-implantation period, after de novo methylation is completed, there is targeted demethylation of tissue-specific genes that helps to maintain, and in some cases establish, the tissue-specific pattern [18].
Embryonic development and cell differentiation to specific cell lineages are regulated by epigenetic marks, including cytosine methylation and histone tail modifications. Xie et al. showed that genes that are active in the embryonic stem cell stage and genes that are active during lineage differentiation differ in promoter CpG content and in the epigenetic silencing patterns. Early developmental regulatory genes are mostly found in large genomic domains, and generally lack DNA methylation in most lineages. These domains were termed DNA methylation valleys by the Ren group [24].
Upon analyzing developmental mouse tissue samples, Hon et al. described that most differentially methylated regions (DMRs) occurred at distal cis-regulatory elements. Some tissue-specific DMRs marked enhancers that are dormant (hypomethylation and lack of active histone modifications) in adult tissues, but active (hypomethylation and active histone modifications) in embryonic development, and were termed ‘vestigial’ enhancers [25]. These results suggest that DNA methylation of tissue-specific enhancers may reflect the epigenetic memory of embryonic development in adult tissues. In summary, these studies indicate that epigenetic modifications are key regulators of embryonic development and maintain cell identity.
THE ROLE OF CYTOSINE METHYLATION: WHAT DO THEY DO?
In the human genome, over 70% of CpG sites are methylated at high levels (>85%). CpG islands (CGI), which are cytosine-rich regions that are prevalent on gene promoter regions, have low methylation levels (<10%) [26, 27]. Most mechanistic work describing the role of cytosine methylation comes from studies analyzing the role of cytosine methylation changes on gene promoter regions. Cytosine methylation of promoter regions is believed to be a general silencing mechanism, associated with reduced transcript levels. There are two basic models for promoter hypermethylation-induced transcriptional repression: DNA methylation can repress transcription directly by blocking transcriptional activators from binding to the DNA sequence, or indirectly by recruitment of co-repressors to methylated regions, and packaging of local chromatin to inaccessible inactive state.
DNA methylation is a key silencing mechanism in several physiological processes. These include X-chromosome inactivation (a process by which one of the two copies of the X chromosome present in female mammals is inactivated, as a dosage compensation), suppression of repetitive elements, and carcinogenesis (either by targeted de novo methylation, which represses tumor suppressor genes and inhibits differentiation and repair or apoptosis genes, or by general demethylation).
While promoter methylation has been studied most extensively, recent systematic analysis has finally provided us with a true genome-wide view of cytosine methylation patterns. After analyzing >40 different cell types, Ziller et al. [28] found that consistent with the previous observations, most cell types, except germ cells and preimplantation embryos, display relatively stable DNA methylation patterns. They found that 21.8% of CpGs show changes in their methylation state mostly during development. CpGs that show dynamic methylation changes, however, are mostly distal to transcription start sites. While they confirmed that promoters could change their methylation state, most DMRs are not promoters, but rather co-localized with gene regulatory elements, particularly enhancers and transcription-factor-binding sites. In addition, they showed that DMRs often contain single-nucleotide polymorphisms associated with cell-type-related diseases as determined by genome-wide association studies.
Cytosine methylation changes have also been evaluated in the context of cell differentiation, for example in the intestine and in the hematopoietic system. Using genome-wide bisulfite sequencing, Kaaij et al. found 50 DMRs in differentiating intestinal epithelial cells. These DMRs correlated with gene expression and represented enhancer-related chromatin marks. The binding of the transcription factor TCF4 close to DMRs correlated with hypomethylation, suggesting its contribution to the formation of DMR [29]. Hodges et al. described similar findings when hematopoietic stem cell differentiation was studied [30]. In summary, although in theory every CpG can change its methylation state, new genome-wide studies indicate the important role of cytosine methylation changes at enhancer regions.
Methylated cytosines can influence transcription by additional mechanisms as well. The methylated cytosine can influence the secondary structure of the DNA through enhanced binding of methyl-binding domain (MBD) proteins and repress binding of other DNA-binding proteins, which can modulate chromatin structure. Methylated cytosines can also affect the binding of histone deacetylases and polycomb proteins to regulate gene expression. The role of cytosine methylation of exonic regions is still debated. According to the classic model, methylation of cytosine in these regions would enhance gene expression as it could inhibit nascent RNA transcription and the unnecessary pausing of the RNA polymerase. Recent reports indicate that cytosine methylation of exonic regions is likely important for RNA alternative splicing, mediated by CTCF binding [31, 32]. In summary, the role of cytosine methylation changes is highly context dependent, and cytosine methylation of regulatory regions is associated with gene expression regulation (Figure 2).
FIGURE 2:
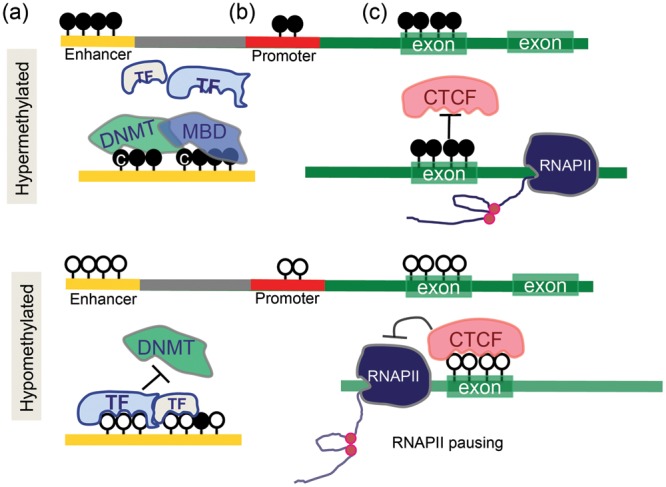
The role of cytosine methylation in gene regulation. (a) Cytosine methylation of enhancer regions. Enhancer regions contain TF binding sites. When these regions are unmethylated, TFs can bind and prevent DNMT from methylating the same region. When these regions are methylated by DNMTs, MBD can recognize the methylated sequences and further associate to other factors. (b) Promoter methylation. Mostly, inversely related to transcript levels. (c) Gene body methylation. Cytosine methylation of exonic regions can potentially increase the RNAPol II transcription efficiency by inhibiting CTCF binding. In contrast, when CTCF binds to unmethylated exonic regions, it hinders the RNAPol II transcription causing RNAPol II pausing.
EPIGENOME AS THE ENVIRONMENTAL FOOTPRINT
The epigenome is flexible and under constant environmental pressure, therefore theoretically it can be used to record long-term changes. It is clear that the magnitude of epigenetic ‘plasticity’ is much greater during development compared with what has been observed in different disease conditions. This observation raises the possibility that environmental factors have a much greater long-term effect on cell type and the epigenome profiles during development. The molecular mechanism of long-term environmental programming is not fully understood, yet it is known that cells adjust their metabolic state in response to nutrient availability. Since most chromatin-modifying enzymes require substrates and cofactors that are intermediates of cellular metabolism, fluctuations in metabolite levels can modulate enzymatic activity and affect chromatin dynamics. For example, acetyl groups generated following fatty acid metabolism are critical intermediates of histone acetylation. The universal methyl-donor SAM is the source for the methyl groups for cytosines and histones.
In this context, environmental factors-induced epigenetic alterations during development have been identified in several genes. The human glucocorticoid receptor has been heavily investigated. Its methylation status has been found to be influenced by maternal depression, abuse during pregnancy in humans and by maternal low-protein diet, uteroplacental insufficiency and maternal behavior in animal models [33]. Methylation of insulin growth factor 2 gene (IGF2) has been implicated in colon and breast cancer in offsprings after vitamin B or folic acid maternal diet both in humans and rodents [34]. In rats maternal choline intake has been related to increased histone methylation in the brain and liver [35].
The mechanistic link between maternal diet and epigenetic state is best demonstrated through the phenotypic effect of the methylation status of the agouti gene [36]. Since the epigenetic state of the murine Ay allele is highly variable and it exerts phenotypic effects (coat color), it is a convenient model to study the effect of methyl donor supplementation. Indeed, in mice methyl donors have been shown to change the methylation state of the Ay allele, resulting in characteristic coat color changes in offspring [37].
EPIGENETICS OF KIDNEY DISEASE
The field of epigenetics in kidney disease is still in its infancy [38], yet several lines of evidence are pointing to the fact that epigenetic differences might play a role in chronic kidney disease (CKD) development. First, hypertension and nephron endowment are significantly influenced by the in utero environment (programming). Animal model studies indicate that calorie, protein or oxygen restriction is associated with lower nephron number, hypertension and microalbuminuria [39]. In addition to developmental programming, transient metabolic changes appear to have long-term effect on kidney disease development as well. For example human clinical studies indicate that patients experiencing an episode of hyperglycemia still have increased incidence of diabetic kidney disease even after a quarter of a decade of normal metabolic control, a phenomenon called ‘metabolic memory’ [40].
Multiple studies have been published analyzing cytosine methylation changes of mixed peripheral blood samples obtained from control, diabetic or CKD patients. These studies indicated changes in cytosine methylation profiles of samples obtained from CKD cases. As the epigenome is cell-type-specific, the interpretation of results from mixed cell types that are not directly involved in disease pathology remains difficult [41].
The Zeisberg group conducted genome-wide methylation screening of fibroblasts taken from non-fibrotic and fibrotic kidney biopsies [42]. The analysis revealed 12 genes that were hypermethylated, of which RASAL1 was investigated further. RASAL1 hypermethylation and its resultant silencing lead to fibroblast activation through increased Ras-GTP activity. This hypermethylation is mediated by DNMT1, and indeed kidney fibrosis was ameliorated in heterozygous Dnmt1+/− mice.
Systematic analysis of the human methylome of the target tissue can provide invaluable insight into the mechanism of chronic human diseases, including CKD. To address this critical issue, our lab performed genome-wide cytosine methylation profiling of tubule epithelial cells obtained from CKD and control kidneys. The original dataset included 26 samples with diabetic and hypertensive CKD samples. The replication dataset used 86 samples with diabetic kidney disease cases. Using complex statistical analytical methods we identified 4751 DMRs when control and CKD kidney samples were compared [43]. We were able to confirm our initial results in the external replication dataset as well. We found that 70% of the DMRs showed lower methylation levels in CKD (Figure 3). Changes were observed only on a handful of promoter regions, and most DMRs were outside of the promoter regions. To understand the role of the observed cytosine methylation changes, we performed histone ChIP-seq analysis using a panel of histone tail-specific antibodies that recognize specific chromatin regions in the genome (promoters, enhancers and transcribed regions). Using the hidden Markov modeling based computational algorithm, we generated kidney-specific regulatory maps. Histone tail-based chromatin annotation maps indicated that CKD-associated DMRs localized to kidney-specific enhancer regions. These enhancer regions were also enriched for kidney-specific TF consensus sequences (including SIX2, PAX2, etc.) [44]. Functional annotation analysis indicated that most DMRs were in the proximity of developmental and profibrotic genes. DMRs also correlated with the expression and regulation of many profibrotic genes [45]. Epigenetic dysregulation therefore can be the basis for CKD progression, as cytosine methylation changes of core fibrosis genes are causally linked to transcript level and phenotype development (Figure 3). Enrichment of developmental genes and binding sites for developmental TFs also raise the possibility that the epigenome is the mechanistic connection between developmental programming and CKD development.
FIGURE 3:

Cytosine methylation changes in CKD. (a) DMRs between CKD and control human kidney tubules. In CKD, most DMRs are hypomethylated (∼70%). (b) Annotation of the DMRs using a chromatin annotation map, which was generated from histone tail modification ChIP-seq and the ChromHMM algorithm. Most of the DMRs are on enhancers. (c) A hypothetical model of our findings; enhancer DMRs might modify TF binding and thereby affect the expression of downstream transcript levels. The DMRs were located mainly at candidate enhancers. Several consensus TF-binding motifs were found in DMRs, including key renal TFs (HNF, TCFAP, SIX2). Cytosine methylation levels correlated with gene expression changes.
CONCLUSION
The technological progress in whole-genome sequencing and computational analysis is leading us into a new era in our understanding of epigenetic modifications, specifically cytosine methylation changes. Such analysis highlighted that CpGs that are located on cell-type-specific enhancers show variability in their methylation levels. The methylation of these cell-type-specific enhancers follows a very specific pattern during cell-type specification, development and differentiation. Methylation of enhancer regions also showed functionally significant differences when epithelial cells of control and CKD kidneys were analyzed. Future studies will be needed to determine the role of enhancer-methylation changes in control and disease conditions.
FUNDING
Work in the Susztaklab is supported by National Institute of Health- National Institute of Diabetes and Digestive and Kidney Diseases (R01DK08763).
CONFLICT OF INTEREST STATEMENT
The laboratory of Dr Susztak receives research support from Boehringer Ingelheim.
REFERENCES
- 1.Ko YA, Susztak K. Epigenomics: the science of no-longer-junk DNA. Why study it in chronic kidney disease? Semin Nephrol. 2013;33:354–362. doi: 10.1016/j.semnephrol.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fratkin E, Bercovici S, Stephan DA. The implications of ENCODE for diagnostics. Nat Biotechnol. 2012;30:1064–1065. doi: 10.1038/nbt.2418. [DOI] [PubMed] [Google Scholar]
- 3.Ernst J, Kellis M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res. 2013;23:1142–1154. doi: 10.1101/gr.144840.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neph S, Stergachis AB, Reynolds A, et al. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maston GA, Landt SG, Snyder M, et al. Characterization of enhancer function from genome-wide analyses. Annu Rev Genomics Hum Genet. 2012;13:29–57. doi: 10.1146/annurev-genom-090711-163723. [DOI] [PubMed] [Google Scholar]
- 9.Inoki K, Mori H, Wang J, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohtat D, Susztak K. Fine tuning gene expression: the epigenome. Semin Nephrol. 2010;30:468–476. doi: 10.1016/j.semnephrol.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee I, Bachman KE, Park BH, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 14.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cadet J, Wagner JR. TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat Res. 2014 doi: 10.1016/j.mrgentox.2013.09.001. in press. [DOI] [PubMed] [Google Scholar]
- 17.Vincent JJ, Huang Y, Chen PY, et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2012;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartolomei MS. Epigenetics: role of germ cell imprinting. Adv Exp Med Biol. 2003;518:239–245. doi: 10.1007/978-1-4419-9190-4_21. [DOI] [PubMed] [Google Scholar]
- 21.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 22.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodge JE, Okano M, Dick F, et al. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 24.Xie W, Schultz MD, Lister R, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hon GC, Rajagopal N, Shen Y, et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45:1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ziller MJ, Muller F, Liao J, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akopian V, Chan MM, Clement K, et al. Epigenomics and chromatin dynamics. Genome Biol. 2012;13:313. doi: 10.1186/gb-2012-13-2-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziller MJ, Gu H, Muller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaaij LT, van de Wetering M, Fang F, et al. DNA methylation dynamics during intestinal stem cell differentiation reveals enhancers driving gene expression in the villus. Genome Biol. 2013;14:R50. doi: 10.1186/gb-2013-14-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges E, Molaro A, Dos Santos CO, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maunakea AK, Chepelev I, Cui K, et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–1269. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Sullivan L, Little MH, Combes AN, et al. Epigenetics and developmental programming of adult onset diseases. Pediatr Nephrol. 2012;27:2175–2182. doi: 10.1007/s00467-012-2108-x. [DOI] [PubMed] [Google Scholar]
- 34.Bjornsson HT, Brown LJ, Fallin MD, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. 2007;99:1270–1273. doi: 10.1093/jnci/djm069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davison JM, Mellott TJ, Kovacheva VP, et al. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. 2009;284:1982–1989. doi: 10.1074/jbc.M807651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakyan VK, Preis J, Morgan HD, et al. The marks, mechanisms and memory of epigenetic states in mammals. Biochem J. 2001;356:1–10. doi: 10.1042/0264-6021:3560001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cropley JE, Suter CM, Beckman KB, et al. Germ-line epigenetic modification of the murine Avy allele by nutritional supplementation. Proc Natl Acad Sci USA. 2006;103:17308–17312. doi: 10.1073/pnas.0607090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Susztak K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J Am Soc Nephrol. 2014;25:10–17. doi: 10.1681/ASN.2013050461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoppe CC, Evans RG, Bertram JF, et al. Effects of dietary protein restriction on nephron number in the mouse. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1768–R1774. doi: 10.1152/ajpregu.00442.2006. [DOI] [PubMed] [Google Scholar]
- 40.De Boer IH, Rue TC, Cleary PA, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the diabetes control and complications trial/epidemiology of diabetes interventions and complications cohort. Arch Intern Med. 2011;171:412–420. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sapienza C, Lee J, Powell J, et al. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics. 2011;6:20–28. doi: 10.4161/epi.6.1.13362. [DOI] [PubMed] [Google Scholar]
- 42.Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ko YA MD, Suzuki M, Park1 ASD, et al. Cytosine methylation changes of enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013;14:R108. doi: 10.1186/gb-2013-14-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dressler GR, Deutsch U, Chowdhury K, et al. Pax2, a new murine paired-box-containing gene and its expression in the developing excretory system. Development. 1990;109:787–795. doi: 10.1242/dev.109.4.787. [DOI] [PubMed] [Google Scholar]
- 45.Woroniecka KI, Park AS, Mohtat D, et al. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]