

Abstract
The genome of the rice blast fungus Magnaporthe oryzae codes for two proteins with N-terminal dioxygenase (DOX) and C-terminal cytochrome P450 (CYP) domains, respectively. One of them, MGG_13239, was confirmed as 7,8-linoleate diol synthase by prokaryotic expression. The other recombinant protein (MGG_10859) possessed prominent 10R-DOX and epoxy alcohol synthase (EAS) activities. This enzyme, 10R-DOX-EAS, transformed 18:2n-6 sequentially to 10(R)-hydroperoxy-8(E),12(Z)-octadecadienoic acid (10R-HPODE) and to 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid as the end product. Oxygenation at C-10 occurred by retention of the pro-R hydrogen of C-8 of 18:2n-6, suggesting antarafacial hydrogen abstraction and oxygenation. Experiments with 18O2 and 16O2 gas confirmed that the epoxy alcohol was formed from 10R-HPODE, likely by heterolytic cleavage of the dioxygen bond with formation of P450 compound I, and subsequent intramolecular epoxidation of the 12(Z) double bond. Site-directed mutagenesis demonstrated that the cysteinyl heme ligand of the P450 domain was required for the EAS activity. Replacement of Asn965 with Val in the conserved AsnGlnXaaGln sequence revealed that Asn965 supported formation of the epoxy alcohol. 10R-DOX-EAS is the first member of a novel subfamily of DOX-CYP fusion proteins of devastating plant pathogens.
Keywords: enzymology/enzyme mechanisms, fatty acid/oxidation, mass spectrometry, oxidized lipids, protein kinases/protein kinase A, heme peroxidase, linoleate diol synthase, mutagenesis/site-specific, oxylipin/biosynthesis
Oxylipins designate oxygenated metabolites of unsaturated fatty acids, and they are mainly formed by eukaryotic organisms (1, 2). Arachidonic acid is oxygenated in human cells by cyclooxygenases and cytochrome P450 (CYP) to prostaglandins, thromboxanes, prostacyclin, and related compounds and by lipoxygenases to hydroperoxides, leukotrienes, and epoxy alcohols (3, 4). These eicosanoids exert a plethora of both physiological and pathophysiological functions (e.g., in control of reproduction, cancer development, skin water permeability, fever, and inflammation). Homologous enzymes are found in plants and fungi with functions in reproduction, development, and infection, and they usually use linoleic and linolenic acids as substrates (5–7).
Plant lipoxygenases and allene oxide synthases (AOSs) catalyze the first steps during biosynthesis of jasmonic acid, which is a potent regulator of growth and defense with structural similarities to prostaglandins (7, 8). Pathogens can induce expression of lipoxygenases and fatty acid α-dioxygenase (DOX) in plants with formation of jasmonic acid and other oxylipins, which act as defense molecules. Oxylipins are also formed by fungi, which are human and plant pathogens. These fungi express lipoxygenases and DOX-CYP fusion proteins (5). The DOX domains are homologs to animal heme peroxidases, which include cyclooxygenases and α-DOX, and their CYP domains take advantage of the peroxide shunt pathway in analogy with thromboxane and prostacyclin synthases (9).
The fungal gene family of DOX-CYP fusion proteins contains five subfamilies, and an overview of their oxygenation products is shown in Fig. 1A. DOX-CYP fusion enzymes are self-sufficient, and the two domains are usually functionally linked. The first two described subfamilies were 5,8- and 7,8-linoleate diol synthase (LDS) of the genera Aspergillus and Gaeumannomyces. LDSs oxidize linoleic acid sequentially to 8(R)-hydroperoxy-9(Z),12(Z)-octadecadienoic acid (8R-HPODE) and to 5,8- or 7,8-diols, by their DOX and CYP domains, respectively (10–13). A third subfamily comprises 10R-DOX of aspergilli, and these proteins lack the functional heme thiolate ligand in the CYP domain (14, 15). Recent additions are the 9R-DOX-AOS subfamily of certain aspergilli and the 9S-DOX-AOS subfamily of the genera Fusarium and Colletotrichum (16, 17).
Fig. 1.

Overview of oxylipin biosynthesis by the genera Aspergillus, Gaeumannomyces, Magnaporthe, Colletotrichum, and Fusarium and phylogenetic tree of DOX-CYP subfamilies. A: Linoleic acid is oxidized by 5,8-LDS, 10R-DOX, and 9R-DOX-AOS of A. terreus and other aspergilli (top). Oxylipins formed from 18:2n-6 by 7,8-LDS of Gaeumannomyces and Magnaporthe (e.g., G. graminis, M. oryzae) and by 9S-DOX-AOS of F. oxysporum and C. graminicola (bottom). B: Phylogenetic tree of MGG_10859 (M. oryzae) and three homologous sequences (EJT82559, EFQ36272, and FOXB_03425, respectively) with >60% amino acid identity covering >90% of the sequence. The next sequences are from top to bottom: 10R-DOX (ACL14177, ATEG_04755, and AAT36614), 7,8-LDS (MGG_13239, Q9UUS2), 5,8-LDS (ACO55067, ATEG_03992), 9S-DOX-AOS (EFQ27323, FOXB_01332), and 9R-(DOX)-AOS [ATEG_02036, EHA25900 (Hoffman and Oliw, unpublished observation)]; the parentheses in 9R-(DOX)-AOS emphasize that recombinant ATEG_02036, in contrast to recombinant EHA25900, lacks the 9R-DOX activities.
Genome sequences of pathogenic fungi are being published at an increasing rate (18, 19). The catalytic activities of emerging DOX-CYP fusion proteins have not yet been fully determined. Putative new DOX-CYP subfamilies can be described by amino acid identity and by the linked and specific dual catalytic activities. In contrast, mammalian P450 families and subfamilies are defined by >40% and 55% amino acid identity, respectively, and not by their catalytic activities due to the functional redundancy within subfamilies. DOX-CYP fusion enzymes of different subfamilies can generally be aligned with 35% to 50% sequence identity, whereas enzymes of subfamilies with identical catalytic activities usually align with 60% or higher sequence identity. Based on the latter, a tentative new DOX-CYP subfamily could be identified in several genera of the top 10 plant pathogens (e.g., Magnaporthe, Colletotrichum, and Fusarium) (Fig. 1B) (20).
Wheat, rice, maize, and banana are staple crops, and rice feeds half of the world’s population. Fungi constitute a constant threat to this food supply. Magnaporthe oryza3 causes rice blast disease, which annually reduces the rice harvest by 10% to 30% (21). Colletotrichum graminicola infects maize at annual costs in Northern America on the order of 1 billion US$. Both fungi share a common infection process. Their conidia develop into an injection apparatus, the appressorium, required for blast cuticle penetration (21, 22). M. oryzae is the prototype organism for studying this process. Its appressorium accumulates lipid bodies, trehalose, and glycerol from the conidium, and the latter generates the turgor pressure for blast penetration. The catalytic subunit of cAMP-dependent protein kinase A (PKA), mitogen-activated protein kinase PMK-1 (PMK1), and triacylglycerol lipases take part in mobilization of fatty acids of lipid bodies in support of the infection process (23–27).
The genome of M. oryzae is sequenced (28). It is estimated to contain about 11,000 genes, and 2 of them code for DOX-CYP fusion proteins. One of them, MGG_13239,4 was previously identified as 7,8-LDS by gene deletion (29). The catalytic function of the second gene, MGG_10859,5 is unknown, but MGG_10859 is upregulated during appressorium formation and downregulated by gene deletion of PMK16 (30). Circumstantial evidence suggests that MGG_10859 may also be regulated by PKA. A mutant strain of M. oryzae (DA-99) with constitutive PKA activity (Δmac1 sum1-99) oxidized linoleic acid to a new profile of metabolites, likely due to expression of linoleate 10R-DOX with MGG_10859 as a possible but unproven candidate gene (29). The PMK1-controlled expression of MGG_10859 and the distribution of MGG_10859 homologs in devastating plant pathogens [Fig. 1B; Gaeumannomyces graminis (Take-all of wheat), C. graminicola (Anthracnose of maize), and Fusarium oxysporum (Panama disease of banana)] indicate a pathophysiological function, which is noteworthy.
The first objective of the present report was to express the two putative DOX-CYP fusion proteins of M. oryzae, MGG_10859 and MGG_13239, because the former could be the first member of a new subfamily (Fig. 1B). The second objective was to reexamine the products formed by the mutant strain DA-99 with constitutive PKA activity (Δmac1 sum1-99) of M. oryzae with regard to the catalytic activities of MGG_10859 and MGG_13239. Finally, we also expressed the MGG_10859 homolog of F. oxysporum, FOXB_03425,7 to determine whether MGG_10859 and this protein possess the same enzyme activities.
MATERIALS AND METHODS
Materials
Fatty acids were dissolved in ethanol and stored in stock solutions (50–100 mM) at −20°C. The 18:2n-6 (99%), 18:3n-6 (99%), and 18:3n-3 (99%) were from VWR. The 18:1n-9, 20:2n-6, 20:3n-3 (98%–99%), and [13C18]18:2n-6 (98%) were from Larodan (Malmö, Sweden). The [8R-2H]18:2n-6 was prepared as described (13, 31). M. oryzae strain DA-99 (Δmac1 sum1-99) (25) was provided by Dr. A. Sesma, Universidad Politécnica de Madrid, Spain. Phusion DNA polymerase and chemically competent Escherichia coli (NEB5α) were from New England BioLabs. Restriction enzymes were from New England BioLabs and Fermentas. Champion pET Directional TOPO Kit was from Invitrogen. Gel extraction kit and Pfu DNA polymerase were from Fermentas. RNaseA, ampicillin, and methylene blue were from Sigma. Sequencing was performed at Uppsala Genome Center (Biomedical Center, Uppsala University). The open reading frames of MGG_13239 (3,527 bp), MGG_10859 (3,462 bp), and FOXB_03425 (3,306 bp) were ordered in pUC57 vectors from GenScript (Piscatawy, NJ). PCR primers were ordered from TIB Molbiol (Berlin, Germany). The (+)- and (−)-vernolic acids were from Lipidox, and photooxidation of vernolic acids was performed with methylene blue (32). 18O2 gas (97%) was obtained from Isotec (Sigma-Aldrich). SepPak columns (silicic acid or C18) were from Waters.
Expression of recombinant proteins
The open reading frames of MGG_13239, MGG_10859, and FOXB_03425 in pUC57 vectors were transferred to pET101D-TOPO vectors by PCR technology according to Invitrogen’s instructions (all primers are listed in supplementary Table I). Competent E. coli (BL21) cells were transformed with the expression constructs by heat-shocking. Cells were grown until A600 of 0.6–0.8 in 2xYT medium prior to addition of 0.1 mM isopropyl β-d-1-thiogalactopyranoside to induce protein expression. After 5 h under moderate shaking (∼100 rpm) at room temperature, the cells were harvested by centrifugation (13,000 rpm, 4°C, 25 min) and sonicated (Bioruptor Next Gen, 10 × 30 s, 4°C). Cell debris was removed by centrifugation, and the supernatants were used immediately or frozen at −80°C until needed. Each protein was expressed in at least three independent expression experiments.
Site-directed mutagenesis of recombinant proteins
Site-directed mutagenesis was performed according to the Quick Change protocol (Stratagene) with 10 ng of the pUC57 constructs as templates and Pfu DNA polymerase (16 cycles). PCR products were incubated with DpnI (37°C, 2 h) to digest maternal DNA. Gel electrophoresis confirmed amplification of one distinct PCR product, which was then used for transformation of E. coli (NEB5α) cells by heat-shocking. All mutations were confirmed by sequencing before subcloning to pET101D-TOPO vectors described previously. All primers are listed in supplementary Table II.
Enzyme assays
Recombinant proteins were incubated with 100 µM 18:2n-6 or other fatty acids for 30 min on ice. The reaction (0.3–0.5 ml) was terminated with methanol (2–4 vol), and proteins were removed by centrifugation. The metabolites were extracted on octadecyl silica (SepPak/C18), evaporated to dryness, and diluted in ethanol (40–60 µl), and 7–10 µl was subject to LC/MS2 analysis.
Nitrogen powder of ground fungus was homogenized (glass-Teflon, 10 passes; 4°C) in 10 vol (w/v) of 0.1 mM KHPO4 buffer [(pH 7.3)/2 mM EDTA/0.04% Tween-20], centrifuged at 13,000g (10 min, 4°C), and immediately used for enzyme assay. An aliquot (0.5–1 ml) of the supernatant was incubated with 100 µM of fatty acids for 30–40 min on ice. The products were extracted with ethyl acetate or on a cartridge with octadecyl silica (SepPak/C18). Triphenylphosphine or NaBH4 was used to reduce hydroperoxides to alcohols.
Photooxidation of vernolic acids
Ten milligrams of 12S(13R)-epoxy-9(Z)-octadecenoic acid [(+)-vernolic acid] was photooxidized in 4 ml methanol under oxygen gas at room temperature with 0.01 molar equivalent methylene blue for 15 h (32). The reaction was followed by TLC until almost all substrate was oxidized. The reaction was evaporated to 1 ml and cooled on ice, and hydroperoxides were reduced to alcohols by addition of NaBH4. Nine milliliters of 0.2 M sodium acetate buffer (pH 4.0) was added, and the material was extracted on a cartridge of octadecyl silica (SepPak/C18). The products were then separated on a silicic acid cartridge (SepPak). Unreacted (+)-vernolic acid was eluted with 50 ml diethyl ether-hexane-acetic acid, 5:95:0.1, and the epoxy alcohols were eluted with diethyl ether-hexane-acetic acid, 50:50:0.1. The fractions with 12S(13R)-epoxy-10(S/R)-hydroxy-8(E)-octadecenoic acids were combined, and the syn and anti stereoisomers were separated by normal phase (NP)-HPLC (250 × 4.6 mm; Nucleosil 50-5) with hexane-isopropyl alcohol-acetic acid, 97:3:0.1, as eluent.
The 12R(13S)-epoxy-9(Z)-octadecenoic acid [(−)-vernolic acid] was oxidized by the same procedure, and the syn and anti isomers were separated in the same way (NP-HPLC).
Growth of M. oryzae (Guy11) and strain DA-99
M. oryzae Guy11 was grown in liquid culture for 5–16 days in complete medium at 22°C with a cycle of 16 h of fluorescent light (True-light) followed by 8 h of darkness (29). The genetically modified strain DA-99 of M. oryzae [Δmac1 sum1-99 (25)] was grown in the same way. Mycelia (0.5–20 g wet weight) were ground to a powder in liquid nitrogen and stored at −80°C. Aliquots were thawed and homogenized, and the supernatants were assayed for enzyme activity with 18:2n-6 as substrate as described previously. Nitrogen powders of both strains were assayed in parallel at different time points of growth. Each experiment was performed at least in duplicate. Conidia were harvested from mycelia, which had grown for 8–12 days on complete medium agar plates with a glass spreader and by rinsing with distilled water.
LC/MS analysis
Reversed phase (RP)-HPLC with MS2 analysis was performed with a Surveyor MS pump (ThermoFisher) and an octadecyl silica column (5 µm; 2.0 × 150 mm; Phenomenex), which was eluted at 0.3 ml/min with methanol-water-acetic acid, 800:200:0.05, or 750:250:0.05. The effluent was subject to ESI in a linear ion trap mass spectrometer (LTQ, ThermoFisher). The heated transfer capillary was set at 315°C, the ion isolation width at 1.5 amu, the collision energy at 35 (arbitrary scale), and the tube lens varied between 90 and 120 V. Prostaglandin F1α was infused for tuning. Samples were injected manually (Rheodyne 7510) or by an autosampler (Surveyor Autosampler Plus, ThermoFisher).
NP-HPLC with MS2 analysis was performed with a silicic acid column (5 µm; Kromasil 100SI, 250 × 2 mm, Dalco Chromtech, or 250 × 4.6 mm; Nucleosil 50-5) using 3% isopropyl alcohol in hexane for separation of hydroxy fatty acids and epoxy alcohols (0.3–0.5 ml/min; Constrametric 3200 pump, LDC/MiltonRoy). The effluent was combined with isopropyl alcohol-water (3:2; 0.2–0.3 ml/min) from a second pump (Surveyor MS pump). The combined effluents were introduced by ESI into the ion trap mass spectrometer (LTQ, ThermoFisher). Steric analysis of 8-hydroperoxyoctadecamonoenoic acid (HPOME) and 10-hydroperoxyoctadecadienoic acid (HPODE) was performed by chiral phase (CP)-HPLC/MS2 analysis on Reprosil Chiral NR as described (33).
Hydrogenation was performed with Pd/C in methanol and a small stream of hydrogen for 90 s. The catalyst was removed by filtration through Na2SO4. Chlorohydrin adducts of epoxides were prepared by reaction with 40 μl of methoxamine HCl in pyridine (20 mg/ml; 60°C, 1 h) (34). Water was added, and the products were purified on a C18 cartridge (SepPack/C18) column before analysis by HPLC/MS2.
Bioinformatics
The ClustalW algorithm was used for sequence alignments (Lasergene, DNASTAR Inc.), and the MEGA6 software for construction of phylogenetic trees with bootstrap tests of the resulting nodes (35). The distance between the branches is indicative of the expected number of substitutions per amino acid position.
RESULTS
Expression and oxidation of 18:2n-6 and 18:3n-3 by MGG_10859 (10R-DOX-EAS)
Recombinant MGG_10859 was obtained by expression in E. coli BL21 cells. This protein readily oxidized 18:2n-6 and 18:3n-3 as shown in Fig. 2. The18:2n-6 was oxidized to an epoxy alcohol as main metabolite and to smaller amounts of 10(R)-hydroperoxy-8(E),12(Z)-octadecadienoic acid (10R-HPODE), 8-HPODE, and their corresponding alcohols (Fig. 2A). The 18:3n-3 was oxygenated in an analogous way as 18:2n-6 with oxidation at C-8, C-10, and at C-10 with formation of an epoxy alcohol (Fig. 2B).
Fig. 2.

LC/MS analysis of oxygenation products formed from 18:2n-6 and 18:3n-3 by recombinant MGG_10859 (10R-DOX-EAS) and the mechanism of biosynthesis of hydroperoxides and epoxy alcohols. A: Products formed from 18:2n-6. Peak I contains 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid (see text). TIC, total ion current. B: Oxygenation products from 18:3n-3. Peak II contains 12(13)-epoxy-10(R)-hydroxy-8(E),15(Z)-octadecadienoic acid (see text). C: CP-HPLC of 10-HPODE. The R enantiomer of 10-HPODE is formed with high stereoselectivity. D: Molecular anions of the epoxy alcohol formed from 18:2n-6 under an atmosphere of both 16O2 and 18O2 were noted at m/z 311 {[10,12(13)-16O2]labeled epoxy alcohol anion} and 315 {[10,12(13)-18O2]labeled epoxy alcohol anion (A−)}. E: MS2 spectrum (m/z 295–296 → full scan) of HODEs, which were obtained after incubation of recombinant 10R-DOX-EAS with [8R-2H]18:2n-6 (64% 2H) and reduction to alcohols. The 8- and 10-HODE are formed with retention of the deuterium label as indicated by the values m/z 158 [−OOC-(CH2)6-C2HO] and 184 [−OOC-(CH2)6-CH = CH-C2HO], respectively.
Steric analysis showed that 10R-HPODE was formed with high stereoselectivity (Fig. 2C). This metabolite was apparently transformed to an epoxy alcohol, which was identified as 12S(13R)-epoxy-10R-hydroxy-8(E)-octadecenoic acid as described subsequently. The epoxy alcohol was formed from 10R-HPODE by an intramolecular rearrangement mechanism as judged by experiments performed under a mixture of 16O2 and 18O2 gas. The anions of 12(13)-epoxy-10-hydroxy-8(E)-octadecenoic acid, which were obtained under this condition, had m/z values of either 311 or 315, but not 313 (Fig. 2D). This demonstrates that the oxygen atom in the epoxy group of one molecule must have evolved from the hydroperoxy group of the same molecule. MGG_10859 will therefore be referred to as 10R-DOX-EAS.
MGG_10859 oxidized [8R-2H]18:2n-6 at C-10 and at C-8, and this occurred with retention of the pro-R deuterium label as shown in Fig. 2E. The retention of the deuterium label suggested that the hydroperoxides were formed by antarafacial hydrogen abstraction and oxygen insertion in analogy with hydroperoxides formed by 7,8-LDS of G. graminis and by 10R-DOX and 5,8-LDS of Aspergillus fumigatus (11, 13).
LC/MS analysis of the epoxy alcohols
The MS2 spectra of the epoxy alcohols formed from 18:2n-6 and 18:3n-3 are shown in Fig. 3. Both spectra contain unexpected rearrangement ions.
Fig. 3.

MS2 spectra of the epoxy alcohols formed from 18:2n-6 and 18:3n-3 by recombinant 10R-DOX-EAS, analysis of chlorohydrin derivatives, and separation of isotopes and stereoisomers. A: MS2 spectrum of the epoxy alcohol [12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid] formed from 18:2n-6. B, C: LC/MS analysis (m/z 347–349 full scan) of chlorohydrin derivatives, which were obtained by treatment of products formed from 18:2n-6 by recombinant MGG_10589 with methoxime HCl. The two main peaks, I and II, in B yielded the same MS2 and MS3 spectra. The molecular anions at m/z 347 and 349 with incorporation of one chlorine atom (35Cl or 37Cl). D: Products formed during oxidation of 12R(13S)-epoxy-(9Z)-octadecenoic acid by MGG_10859 and separation of the syn and anti isomers of 12R(13S)-epoxy-10-hydroxy-8(E)-octadecenoic acid by NP-HPLC (peaks marked I and II). The partial chromatogram of the inset was obtained after addition of the epoxy alcohol formed from 18:2n-6 by MGG_10859 to the products formed from 12R(13S)-epoxy-(9Z)-octadecenoic acid, illustrating that biological product formed from 18:n-6 coelutes with the first eluting stereoisomer in peak I [syn; 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid; see text for details]. E: MS2 spectrum of 12(13)-epoxy-10(R)-hydroxy-8(E),15(Z)-octadecenoic acid formed from 18:3n-3 by 10R-DOX-EAS.
12(13)-Epoxy-10-hydroxy-8(E)-octadecenoic acid.
LC/MS analysis revealed a complex fragmentation of the epoxy alcohol formed from 10R-HPODE. The MS2 spectrum (m/z 311 → full scan) is shown in Fig. 3A. The spectrum shows two signals, m/z 183 [−OOC-(CH2)6-CH = CH-CHO] and m/z 155 [183–28; −OOC-(CH2)6-CH = CH2], which are formed by α-cleavage at both sides of C-10. Signals were also noted at m/z 211 (A−-100), 193, and 181, likely due to rearrangements. This spectrum suggested that the epoxide group could possibly be positioned at C-12 and C-13, but not at C-8 and C-9 (cf. Ref. 36).
We next examined the fragmentation of the epoxy alcohol with regard to the signals at m/z 211, 193, and 181 by recording the MS2 spectra of three derivatives: i) the hydrogenated epoxy alcohol, ii) the uniformly 13C-labeled epoxy alcohol, and iii) and the epoxy alcohol formed under 18O2. These mass spectra are shown in the supplementary Figs. I–III.
The MS2 spectrum of the hydrogenated epoxy alcohol showed the rearrangement ions at m/z 183 and 211 (supplementary Fig. I). The MS2 spectrum of the uniformly labeled epoxy alcohol showed that the rearrangement ions at m/z 211 and 181 contained 12 and 11 carbon atoms, respectively, as they were increased to m/z 223 and 192, respectively (supplementary Fig. II). The MS2 spectrum of the epoxy alcohol with 18O-labeling of the hydroxy and the epoxide groups suggested that the ion at m/z 211 retained both oxygen atoms, as this ion shifted to m/z 215 (supplementary Fig. III). These results suggest that the signal at m/z 211 (A−-(18+82)) could be due to loss of water and C6H10 from the carboxylate anion (A−). This mechanism was also supported by analysis of the corresponding rearrangement ions in the MS2 spectra of the epoxy alcohol formed from 18:3n-3, 20:2n-6, and 20:3n-3 described subsequently. The precise fragmentation mechanism remains to be determined.
The complex spectra of the epoxy alcohol discussed previously did not clearly show the presence of an epoxide. We therefore prepared chlorohydrin adducts, and they were partly separated by RP-HPLC (Fig. 3B). The resulting molecular anions confirmed opening of an epoxide with incorporation of 35Cl and 37Cl and formation of a hydroxyl group (Fig. 3C). The MS2 spectrum of the chlorohydrin derivatives showed an intense signal at m/z 311 [A−-(36 or 38), loss of HCl], and the MS3 spectrum (m/z 347–349 → 311 → full scan) yielded the same mass spectrum as recorded for the original epoxy alcohol (Fig. 3A). We finally confirmed the structure of the epoxy alcohol by comparison with chemical standards.
Steric analysis of 12(13)-epoxy-10-hydroxy-8(E)-octadecenoic acid.
The epoxy alcohol formed from 10R-HPODE was identified as the syn stereoisomer by its retention time on NP-HPLC in comparison with synthetic and biological standards. Photooxidation of (+)- and (−)-vernolic acids, 12S(13R)-epoxy- and 12R(13S)-epoxy-9(Z)-octadecenoic acids, respectively, yielded two pairs of syn and anti stereoisomers with the same MS2 spectrum as discussed previously (Fig. 3A). The syn and anti stereoisomers were separated by NP-HPLC, but the two syn and the two anti stereoisomers were mirror images, and they were not separated by NP-HPLC.
Two stereoisomers of the epoxy alcohol were also obtained by oxidation of (−)-vernolic acid [12R(13S)-epoxy-9(Z)-octadecenoic acid] with 10R-DOX-EAS (after reduction of the hydroperoxides to alcohols) at C-10 in a 1:10 ratio (Fig. 3D). Products were also formed by oxidation at C-8 [12R(13S)-epoxy-8-hydroxy-9(Z)-octadecenoic acids; data not shown]. Based on the stereoselective oxidation at C-10, we identified the major metabolite, which eluted with the longest retention time, as the anti stereoisomer [2R(13S)-epoxy-10(R)-hydroxy-9(Z)-octadecenoic acid]. The epoxy alcohol formed from 18:2n-6 eluted as a single peak, which coeluted with the first eluting stereoisomer of these two biological standards on NP-HPLC (insert in Fig. 3D). It was thus identified as the syn stereoisomer, 12S(13R)-epoxy-10(R)-hydroxy-9(Z)-octadecenoic acid.
12(13)-Epoxy-10-hydroxy-8(E),15(Z)-octadecadienoic acid.
The LC/MS spectrum of the epoxy alcohol formed from 18:3n-3 is shown in Fig. 3E. The presence of an alcohol at C-10 was evident from the strong fragments at m/z 183 and 155 formed by α-cleavage at both sides of C-10. The rearrangement ion at m/z 209 (A−-100) was apparently formed by the same mechanism as the m/z 211 ion discussed previously. Importantly, this spectrum shows that the rearrangement cannot be formed by cleavage at C-11, which would yield a loss of 98 (OCH-CH2-CH = CH-CH2-CH3) and not a loss of 100.
Oxidation of 18:1n-9, 18:3n-6, 20:2n-6, and 20:3n-3 by MGG_10859 (10R-DOX-EAS)
18:1n-9.
NP-HPLC/MS2 analysis showed that oleic acid was oxidized by the 10R-DOX activity to 8-H(P)OME and 10-H(P)OME in a ratio of >5:1. CP-HPLC analysis showed that 8-HPOME consisted of the 8R stereoisomer to >95%.
18:3n-6.
This fatty acid was a poor substrate, and it was not oxidized to specific products.
20:2n-6.
The 20:2n-6 was oxidized at C-10 and C-12 as judged from NP-HPLC/MS analysis of hydroperoxides before and after reduction to alcohols. Both hydroperoxides were also converted to epoxy alcohols. The MS2 spectrum of one epoxy alcohol was consistent with 10(11)-epoxy-12-hydroxy-14(Z)-eicosenoic acid (data not shown), possibly formed by homolytic cleavage of the 10-hydroperoxy metabolite. The MS2 spectrum of the other epoxy alcohol, 14(15)-epoxy-12-hydroxy-10(E)-eicosenoic acid, is shown in supplementary Fig. IVA, and it allows a comparison with the spectrum in Fig. 3A. This illustrates that many fragments differed by 28, which supports the rearrangement mechanism discussed previously with loss of C6H10 and water from the carboxylate anion.
20:3n-3.
The 20:3n-3 was oxidized in analogy with 20:2n-6 at C-10 and C-12. The hydroperoxide at C-10 was transformed to significant amounts of 10(11)-epoxy-12-hydroxy-14(Z),17(Z)-eicosadienoic acid and the hydroperoxide at C-12 to 14(15)-epoxy-12-hydroxy-14(Z),17(Z)-eicosadienoic acid. The MS2 spectrum of the latter is shown in supplementary Fig. IVB for comparison with Fig. 3B.
Conversion of other hydroperoxides by the EAS activity
The capability to convert hydroperoxides other than 10R-HPODE was evaluated by incubations with 10S-, 9R-, 9S-, 13R-, and 13S-HPODE, respectively. Only 13R- and 13S-HPODE were converted to epoxy alcohols in significant amounts. Both formed the threo stereoisomer of 12(13)-epoxy-11-hydroxy-9(Z)-octadecenoic acids as the main metabolite (erythro/threo, 1:4) as judged by NP-HPLC analysis (supplementary Fig. V). Because the hydroperoxides were mostly converted to the threo isomers, it is possible that this occurred enzymatically.
Crucial amino acid residues of the EAS domain of MGG_10859
Catalytically important amino acid residues of the EAS domains are likely conserved residues of the I-helices and the heme thiolate pocket, as outlined by partial alignment of the CYP domains of 10R-DOX-EAS with three putative DOX-EAS and two 7,8-LDS sequences (Fig. 4A).
Fig. 4.

Comparison of partial sequences of four members of the 10R-DOX-EAS subfamily with 7,8-LDS of M. oryzae and G. graminis and effects of site-directed mutagenesis of Cys and Asn residues. A: The alignment covers the conserved AsnGlnXaaGln sequences, which are believed to be important for substrate positioning, the GluXaaXaaArg (“EXXR”) sequences, functioning as a salt bridge, and the motif with the heme thiolate ligand. The latter two sequences are typical of P450s. Asterisks indicate the three amino acid residues of MGG_10589, which were subject to site-directed mutagenesis. The sequence identity within the putative 10R-DOX-EAS subfamily is 60% or higher. B: RP-HPLC analysis of products formed from 18:2n-6 by 10R-DOX-EAS (MGG_10859). C: RP-HPLC/MS analysis of products formed from 18:2n-6 by 10R-DOX-EAS•C1086S. D: RP-HPLC/MS analysis of products formed from 18:2n-6 by 10R-DOX-EAS•N965V. Peak I designates 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid, which was identified by MS2 analysis in all three chromatograms.
The Cys1086Ser replacement of MGG_10859 fully supported the oxidation of 18:2n-6 by the 10R-DOX activities, but only traces of the epoxy alcohol were noted in comparison with native 10R-DOX-EAS (Fig. 4B, C). These small amounts were presumably formed by the heme group of the 10R-DOX domain in analogy with 10R-DOX of A. fumigatus and Aspergillus nidulans, which lack the heme thiolate ligand (14, 15).
We next examined two amide residues in the conserved Asn965GlnXaaGln motif of 10R-DOX-EAS, which is present in three homologs of 10R-DOX-EAS and in two 7,8-LDS sequences (Fig. 4A). The Gln968Leu replacement of 10R-DOX-EAS did not appear to alter the biosynthesis of the epoxy alcohol from 18:2n-6, but it was strongly influenced by the Asn965Val replacement (Fig. 4D). It is difficult to exactly quantify this reduction in formation of the epoxy alcohol, but the relative amount of epoxy alcohol to 10-HPODE was altered from 92% in the wild-type enzyme to 29% in the Asn965Val mutant. Asn965 thus supports the EAS activity, but it is not absolutely required. This is also the case with the catalytically important Asn residues of CYP74 and 9R-(DOX)-AOS of A. terreus (17, 37); the parentheses indicate that the latter appears to lack 9R-DOX activity.
Reanalysis of oxylipins formed by M. oryzae strain DA-99 with constitutive PKA activities
The oxidation of 18:2n-6 by nitrogen powder of mycelia of DA-99 (Δmac1 sum1-99) yielded a different profile of metabolites than the native strain (Guy11) as reported previously (29). DA-99 consistently formed 10R-HPODE and 8R-HPODE as main metabolites along with variable but relatively small amounts of 7,8-dihydroxy-9(Z),12(Z)-octadecadienoic acid (DiHODE) and 5,8-DiHODE (Fig. 5A), whereas M. oryzae (Guy11) transformed 18:2n-6 to 7,8-DiHODE and 8-HPODE as the main products (insert in Fig. 5A). We decided to reexamine these findings in the light of the properties of recombinant 10R-DOX-EAS and 7,8-LDS (MGG_13239).
Fig. 5.

LC/MS analysis of the oxidation of 18:2n-6 by M. oryzae DA-99, Guy11, and recombinant 7,8-LDS (MGG_13239). A: RP-HPLC analysis of products formed from nitrogen powder of DA-99. The inset shows separation by RP-HPLC of metabolites formed by nitrogen powder of M. oryzae Guy11. B: NP-HPLC analysis of metabolites, which were formed by recombinant 7,8-LDS (MGG_13239). The chromatograms show the TIC. Products were identified by their MS2 spectra as indicated.
A time curve covering a growth period of DA-99 for up to 16 days showed a variable formation of 5,8- and 7,8-DiHODE and a prominent biosynthesis of 10R-HPODE and 8R-HPODE (cf. Ref. 29). The P450 activity of 7,8-LDS thus appeared to be reduced in comparison with M. oryzae Guy11. 12(13)-Epoxy-10-hydroxy-8(E)-octadecenoic acid could be detected at some time points by its characteristic MS2 spectrum (Fig. 3A). The metabolite eluted on the right shoulder of 5,8-DiHODE (supplementary Fig. VI). NP-HPLC analysis of additional experiments showed that the epoxide was a racemic mixture of syn and anti stereoisomers. This showed that 10R-DOX-EAS did not form these epoxy alcohols.
Recombinant expression of 7,8-LDS of M. oryzae
We examined recombinant 7,8-LDS for biosynthesis of major and minor metabolites, which might contribute to the oxylipin profile of DA-99. The recombinant protein transformed 18:2n-6 sequentially to 8-HPODE and 7,8-DiHODE as major products. This enzyme was therefore confirmed to possess 7,8-LDS activities, as previously indicated by gene deletion (29). NP-HPLC/MS2 analysis also revealed significant biosynthesis of several minor products as illustrated in Fig. 5B. Trace amounts of 10-H(P)ODE were also formed, but 5,8-DiHODE could not be detected.
The protein sequence of 7,8-LDS contains several predicted phosphorylation sites with Ser and Thr residues (38). Single residues of five of these sites were altered by replacements with Asp or Glu residues (Ser350Asp in the 8R-DOX domain, and Ser753Glu, Ser975Asp, Thr998Glu, and Thr1098Asp in the CYP domain), but this did not seem to affect the enzymatic oxidation of 18:2n-6 (data not shown).
Site-directed mutagenesis studies revealed that Asn946, which is present in the AsnGlnXaaGln motif mentioned previously (Fig. 4A), facilitates the conversion of 8-HPODE to 7,8-DiHODE. MGG_13239•Asn946Val only formed small amounts of the 7,8-diol (supplementary Fig. VII) compared with the native enzyme (Fig. 5B).
Recombinant expression of FOXB_03425 of F. oxysporum
Recombinant FOXB_03425 converted 18:2n-6 to 10-HPODE, 8-HPODE, and an epoxy alcohol, as shown in Fig. 6A. LC/MS analysis of the epoxy alcohol yielded the same MS2 spectrum as 12(13)-epoxy-10-hydroxy-8(E)-octadecenoic acid (Fig. 3A). This epoxy alcohol was also formed from 10R-HPODE, but not from 10S-HPODE (Fig. 6B). The relative amounts of HPODE and the epoxy alcohol varied. CP-HPLC/MS2 analysis confirmed that 18:2n-6 was oxidized mainly to the R stereoisomer of 10-HPODE (data not shown). We conclude that FOXB_03425 is a 10R-DOX-EAS fusion enzyme. It aligns with 64% amino acid identity to MGG_10859.
Fig. 6.
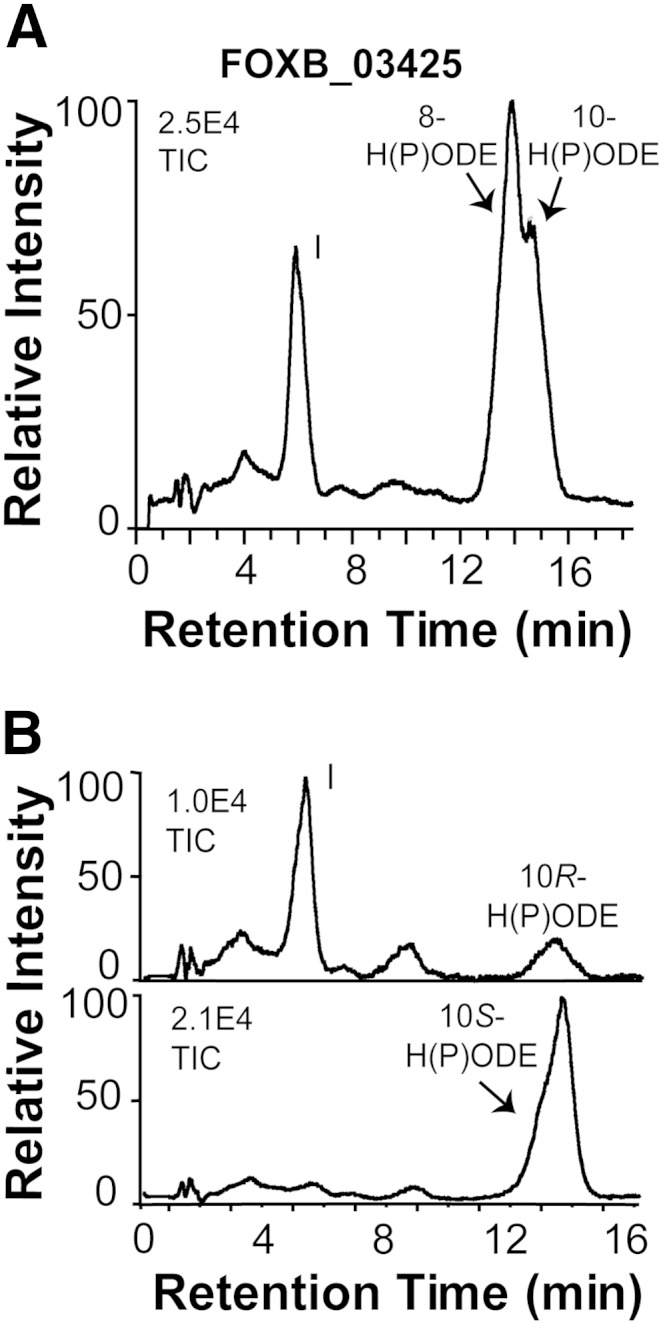
RP-HPLC/MS2 analysis of oxygenation products of 18:2n-6 formed by recombinant FOXB_03425 and transformation of 10-HPODE by this enzyme. A: RP-HPLC/MS2 analysis revealed biosynthesis of 8- and 10-H(P)ODE, and 12(13)-epoxy-10-hydroxy-(8E)-octadecenoic acid (peak I) from 18:2n-6. B: The top chromatogram shows transformation of 10R-HPODE by FOXB_03425 to 12(13)-epoxy-10(R)-hydroxy-(8E)-octadecenoic acid (peak I), and the bottom chromatogram shows that 10S-HPODE was a poor substrate.
DISCUSSION
The discovery of the first fungal EAS with its unique self-sufficient reaction mechanism is our main finding. The dual enzyme activities are combined in a DOX-CYP fusion protein, MGG_10859, of the rice blast fungus M. oryzae. The enzyme is designated 10R-DOX-EAS because it sequentially converts linoleic and α-linolenic acids to 10R-hydroperoxides and to 12S(13R)-epoxy-10R-hydroxy metabolites as end products by intramolecular oxygen transfer (Fig. 7).
Fig. 7.

Enzymatic conversion of 18:2n-6 by recombinant 10R-DOX-EAS. Abstraction of the pro-S hydrogen of C-8 is followed by antarafacial oxygen insertion mainly at C-10 and to some extent at C-8, yielding 10R- and 8R-HPODE, respectively. 10R-HPODE is further converted by the EAS activity to 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid by heterolytic scission of the O-O bond with formation of P450 compound I in a process supported by Asn965 (cf. Fig. 4).
Epoxy alcohols can be formed from fatty acid hydroperoxides by two main mechanisms: homolytic or heterolytic cleavage of the hydroperoxide oxygen-oxygen bond. Hematin, hemoproteins, and many enzymes (e.g., lipoxygenases and P450) catalyze homolytic cleavage with formation of epoxy alcohols (36, 39, 40). 10R-DOX-EAS likely forms epoxy alcohols by heterolytic cleavage of the O-O bond of 10R-HPODE with formation of a hydroxyl anion and P450 compound I for subsequent epoxidation (Fig. 7). To the best of our knowledge, 10R-DOX-EAS is the first described EAS with an intramolecular and position-specific oxygenation mechanism associated with heterolytic scission of the dioxygen bond. Heterolytic scission of dioxygen bonds are also catalyzed by heme peroxidases and presumably also by the secreted EAS of Magnaporthe salvinii (41, 42). Intramolecular oxygen transfer has also been described by arachidonate 12-Lipoxygenase linked to diol synthase activities in the red alga Gracilariopsis lemaneiformis and by 15-Lipoxygenase linked to epoxygenase activities of the fungus Saprolegnia parasitica (1, 43).
10R-DOX-EAS contains the conserved Asn965GlnXaaGln motif, presumably located in the I-helix of the EAS domain (Fig. 4A). The Asn residue of this sequence supports catalysis of both 7,8-LDS of G. graminis and M. oryzae (cf. Fig. 6 and supplementary Fig. VII) (9). In analogy, 10R-DOX-EAS•N965V restrained the conversion of 10R-HPODE to 12S(13R)-epoxy-10(R)-hydroxy-8(E)-octadecenoic acid (Fig. 3). It is therefore possible that Asn965 is involved in positioning of 10R-HPODE for heterolytic scission of the oxygen-oxygen bond in analogy with the homologous Asn residues of 7,8-LDS. The position of the heme iron in the active sites appears to differ in relation to the hydroperoxides at C-8 and C-10. 7,8-LDS catalyzes hydroxylation at C-7 of 8R-HPODE, whereas 10R-DOX-EAS catalyzes epoxidation of the 12(Z) double bond of 10R-HPODE. Future work will reveal whether 8R- and 10R-HPODE bind the active sites with their conserved residues in opposite directions.
10R-DOX-EAS of M. oryzae is the first member of a distinct subfamily of DOX-CYP fusion proteins (Fig. 1B). The amino acid identity of this enzyme with 10R-DOX of A. fumigatus and A. nidulans with nonfunctional CYP domains is ∼47% over the entire sequence and only slightly higher (∼51%) over the 10R-DOX domains (∼680 N-terminal amino acids), whereas these two aspergilli enzymes can be aligned with 78% amino acid identity (cf. Fig. 1B). The n-6 double bond is essential for oxygenation at C-10, as linoleate 10R-DOX-EAS transforms 18:1n-9 to 8-HPOME as main metabolite in analogy with 10R-DOX of aspergilli (14, 15). Alignments of 10R-DOX and 10R-DOX-EAS sequences clearly indicate two distinct subfamilies, and 10R-DOX does not appear to be an inactive form of 10R-DOX-EAS (Fig. 1B and supplementary Fig. VIII). The fact that two 10R-HPODE-producing subfamilies have evolved in parallel strengthens previous reports that 10R-HPODE is of biological importance (44).
A second 10R-DOX-EAS (FOXB_034257) was identified in F. oxysporum by recombinant expression. Sequence homologs are present in at least two other devastating fungal pathogens (Fig. 1B), and it seems likely that the latter also possess 10R-DOX-EAS activities. Virtually identical are the sequences of the homologs around the proximal and distal heme ligands of the10R-DOX domains (not shown) and critical regions (I-helices) of the EAS domains (Fig. 4A). Two sequences with five conserved amino acids are found in P450s with relatively few exceptions, namely Glu and Arg residues in the salt bridge (“EXXR”) and Cys and two Gly residues in the heme thiolate pocket (“GXXXCXG”) (19). The other amino acids in these two sequences are characteristic of fungal P450 families (19).
The PMK1 and cAMP-activated PKA constitute two important pathways for the infectious process of M. oryzae (25–27, 45). The biological function of 10R-DOX-EAS is unknown, but constitutive PKA expression alters the oxylipin profile of M. oryzae, and PMK1 affects expression of MGG_10859 mRNA. The latter is upregulated during the early and late phases of appressoria formation and downregulated by gene deletion of PMK16 (30). It raises the question of whether 10R-DOX-EAS also could be regulated by cAMP-activated PKA.
The strain DA-99 of M. oryzae with the suppressor mutant Δmac1 sum1-99 (23) has constitutive PKA activities. Nitrogen powder of this fungus formed 8R-HPODE and 10R-HPODE as main metabolites, but without significant further transformation by EAS and only with modest biosynthesis of 7,8-DiHODE in comparison with M. oryzae Guy11 (Fig. 5). There are no other known gene candidates for these 8R and 10R activities than 10R-DOX-EAS and 7,8-LDS of M. oryzae. The altered oxylipin profile is therefore likely related to these enzymes, either by direct effects of PKA on these proteins or by indirect effects on their gene expression.
Phosphorylation, in particular by PKA, has previously been found to downregulate the catalytic activity as a functional switch of several isoforms of the CYP2 family (46). 7,8-LDS and 10R-DOX-EAS contain several phosphorylation sites. We hypothesized that phosphorylation of 7,8-LDS might alter the catalysis. However, replacement of Ser and Thr with Asp or Glu residues in five predicted phosphorylation sites did not change the oxylipin profile (cf. Ref 47). PKA influences gene expression by phosphorylation of transcription factors and by alternative splicing of pre-mRNA (48). A. terreus forms a splice variant of 5,8-LDS with retention of the last intron, leading to a premature stop codon with complete loss of the P450 catalysis (17). Alternative splicing of 7,8-LDS and 10R-DOX-EAS in this way could lead to accumulation of 8R- and 10R-HPODE, and this hypothesis merits further investigation.
CONCLUSION
We have discovered and characterized catalytic properties of the first 10R-DOX-EAS. This fusion enzyme catalyzes “oxygen transfer” epoxidation and belongs to a novel DOX-CYP subfamily with homologs in several of the top 10 fungal plant pathogens (20). Previous work suggests that the underlying gene, MGG_10859, could be regulated by the gene PMK1, which is a key regulator of the infectious process (30). Based on these results, the biological function of 10R-DOX-EAS and its enzymatic products can now be assessed.
Supplementary Material
Acknowledgments
The authors thank Dr. Mats Hamberg, Karolinska Institutet, Sweden, for generous advice and for the gift of vernolic acids and deuterated 18:2n-6. M. oryzae strains Guy11 and DA-99 were kindly provided by Dr. Ane Sesma, Universidad Politécnica de Madrid, Spain.
Footnotes
Abbreviations:
- AOS
- allene oxide synthase
- CP
- chiral phase
- CYP
- cytochrome P450
- DiHODE
- dihydroxy-9(Z),12(Z)-octadecadienoic acid
- DOX
- dioxygenase
- EAS
- epoxy alcohol synthase
- HPODE
- hydroperoxyoctadecadienoic acid
- 8R-HPODE
- 8(R)-hydroperoxy-9(Z),12(Z)-octadecadienoic acid
- 10R-HPODE
- 10(R)-hydroperoxy-8(E),12(Z)-octadecadienoic acid
- HPOME
- hydroperoxyoctadecamonoenoic acid
- LDS
- linoleate diol synthase
- NP
- normal phase
- PKA
- protein kinase A
- PMK1
- mitogen-activated protein kinase PMK-1
- RP
- reversed phase
This work was supported by Vetenskapsrådet, the Knut and Alice Wallenberg Foundation/Grant KAW 2004.0123, and Uppsala University. Conflict-of-interest disclosure: The authors have no conflicts of interest to report.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables and eight figures.
M. oryzae and M. grisea are morphologically indistinguishable, and the former designates the rice-infecting species (49).
7,8-LDS of M. oryzae at gene locus MGG_13239 has the GenBank accession number EHA52010; open reading frame XM_003711769.
10R-DOX-EAS of M. oryzae at gene locus MGG_10859 has several GenBank accession numbers (e.g., EHA53428 and ELQ59178); open reading frame XM_003713187.
Database (http://cogeme.ex.ac.uk/supersage) provides expression levels of MGG_10859 mRNA, which increased 7- to 10-fold (and decreased 7-fold by deletion of PMK1) during early and late phase of appressoria formation. MGG_13239 mRNA was strongly reduced at all of these time points.
REFERENCES
- 1.Gerwick W. H., Moghaddam M., Hamberg M. 1991. Oxylipin metabolism in the red alga Gracilariopsis lemaneiformis: mechanism of formation of vicinal dihydroxy fatty acids. Arch. Biochem. Biophys. 290: 436–444. [DOI] [PubMed] [Google Scholar]
- 2.Andreou A., Brodhun F., Feussner I. 2009. Biosynthesis of oxylipins in non-mammals. Prog. Lipid Res. 48: 148–170. [DOI] [PubMed] [Google Scholar]
- 3.Smith W. L., Urade Y., Jakobsson P. J. 2011. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111: 5821–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haeggström J. Z., Funk C. D. 2011. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 111: 5866–5898. [DOI] [PubMed] [Google Scholar]
- 5.Brodhun F., Feussner I. 2011. Oxylipins in fungi. FEBS J. 278: 1047–1063. [DOI] [PubMed] [Google Scholar]
- 6.Andreou A., Feussner I. 2009. Lipoxygenases - structure and reaction mechanism. Phytochemistry. 70: 1504–1510. [DOI] [PubMed] [Google Scholar]
- 7.Hamberg M., Gardner H. W. 1992. Oxylipin pathway to jasmonates: biochemistry and biological significance. Biochim. Biophys. Acta. 1165: 1–18. [DOI] [PubMed] [Google Scholar]
- 8.Wasternack C., Kombrink E. 2010. Jasmonates: structural requirements for lipid-derived signals active in plant stress responses and development. ACS Chem. Biol. 5: 63–77. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann I., Oliw E. H. 2013. 7,8- and 5,8-Linoleate diol synthases support the heterolytic scission of oxygen-oxygen bonds by different amide residues. Arch. Biochem. Biophys. 539: 87–91. [DOI] [PubMed] [Google Scholar]
- 10.Champe S. P., el-Zayat A. A. 1989. Isolation of a sexual sporulation hormone from Aspergillus nidulans. J. Bacteriol. 171: 3982–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brodowsky I. D., Hamberg M., Oliw E. H. 1992. A linoleic acid (8R)-dioxygenase and hydroperoxide isomerase of the fungus Gaeumannomyces graminis. Biosynthesis of (8R)-hydroxylinoleic acid and (7S,8S)-dihydroxylinoleic acid from (8R)-hydroperoxylinoleic acid. J. Biol. Chem. 267: 14738–14745. [PubMed] [Google Scholar]
- 12.Su C., Sahlin M., Oliw E. H. 1998. A protein radical and ferryl intermediates are generated by linoleate diol synthase, a ferric hemeprotein with dioxygenase and hydroperoxide isomerase activities. J. Biol. Chem. 273: 20744–20751. [DOI] [PubMed] [Google Scholar]
- 13.Garscha U., Jernerén F., Chung D., Keller N. P., Hamberg M., Oliw E. H. 2007. Identification of dioxygenases required for Aspergillus development. Studies of products, stereochemistry, and the reaction mechanism. J. Biol. Chem. 282: 34707–34718. [DOI] [PubMed] [Google Scholar]
- 14.Garscha U., Oliw E. H. 2009. Leucine/valine residues direct oxygenation of linoleic acid by (10R)- and (8R)-dioxygenases: expression and site-directed mutagenesis of (10R)-dioxygenase with epoxyalcohol synthase activity. J. Biol. Chem. 284: 13755–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brodhun F., Schneider S., Göbel C., Hornung E., Feussner I. 2010. PpoC from Aspergillus nidulans is a fusion protein with one active heme. Biochem. J. 425: 553–565. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmann I., Oliw E. H. 2013. Discovery of a linoleate 9S-dioxygenase and an allene oxide synthase in a fusion protein of Fusarium oxysporum. J. Lipid Res. 54: 3471–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann I., Jerneren F., Oliw E. H. 2013. Expression of fusion proteins of Aspergillus terreus reveals a novel allene oxide synthase. J. Biol. Chem. 288: 11459–11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson R. A., Talbot N. J. 2009. Fungal physiology - a future perspective. Microbiology. 155: 3810–3815. [DOI] [PubMed] [Google Scholar]
- 19.Syed K., Mashele S. S. 2014. Comparative analysis of P450 signature motifs EXXR and CXG in the large and diverse kingdom of fungi: identification of evolutionarily conserved amino acid patterns characteristic of P450 family. PLoS ONE. 9: e95616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean R., Van Kan J. A., Pretorius Z. A., Hammond-Kosack K. E., Di Pietro A., Spanu P. D., Rudd J. J., Dickman M., Kahmann R., Ellis J., et al. 2012. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13: 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson R. A., Talbot N. J. 2009. Under pressure: investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol. 7: 185–195. [DOI] [PubMed] [Google Scholar]
- 22.Bechinger C., Giebel K. F., Schnell M., Leiderer P., Deising H. B., Bastmeyer M. 1999. Optical measurements of invasive forces exerted by appressoria of a plant pathogenic fungus. Science. 285: 1896–1899. [DOI] [PubMed] [Google Scholar]
- 23.Thines E., Weber R. W., Talbot N. J. 2000. MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell. 12: 1703–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z. Y., Soanes D. M., Kershaw M. J., Talbot N. J. 2007. Functional analysis of lipid metabolism in Magnaporthe grisea reveals a requirement for peroxisomal fatty acid beta-oxidation during appressorium-mediated plant infection. Mol. Plant Microbe Interact. 20: 475–491. [DOI] [PubMed] [Google Scholar]
- 25.Adachi K., Hamer J. E. 1998. Divergent cAMP signaling pathways regulate growth and pathogenesis in the rice blast fungus Magnaporthe grisea. Plant Cell. 10: 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell T. K., Dean R. A. 1995. The cAMP-dependent protein kinase catalytic subunit is required for appressorium formation and pathogenesis by the rice blast pathogen Magnaporthe grisea. Plant Cell. 7: 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J. R., Hamer J. E. 1996. MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes Dev. 10: 2696–2706. [DOI] [PubMed] [Google Scholar]
- 28.Dean R. A., Talbot N. J., Ebbole D. J., Farman M. L., Mitchell T. K., Orbach M. J., Thon M., Kulkarni R., Xu J. R., Pan H., et al. 2005. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 434: 980–986. [DOI] [PubMed] [Google Scholar]
- 29.Jernerén F., Sesma A., Franceschetti M., Hamberg M., Oliw E. H. 2010. Gene deletion of 7,8-linoleate diol synthase of the rice blast fungus: studies on pathogenicity, stereochemistry, and oxygenation mechanisms. J. Biol. Chem. 285: 5308–5316. [Erratum. 2010. J. Biol. Chem. 285: 20422.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soanes D. M., Chakrabarti A., Paszkiewicz K. H., Dawe A. L., Talbot N. J. 2012. Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 8: e1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamberg M., Zhang L. Y., Brodowsky I. D., Oliw E. H. 1994. Sequential oxygenation of linoleic acid in the fungus Gaeumannomyces graminis: stereochemistry of dioxygenase and hydroperoxide isomerase reactions. Arch. Biochem. Biophys. 309: 77–80. [DOI] [PubMed] [Google Scholar]
- 32.Chacón J. N., Gaggini P., Sinclair R. S., Smith F. J. 2000. Photo- and thermal-oxidation studies on methyl and phenyl linoleate: anti-oxidant behaviour and rates of reaction. Chem. Phys. Lipids. 107: 107–120. [DOI] [PubMed] [Google Scholar]
- 33.Garscha U., Nilsson T., Oliw E. H. 2008. Enantiomeric separation and analysis of unsaturated hydroperoxy fatty acids by chiral column chromatography-mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 872: 90–98. [DOI] [PubMed] [Google Scholar]
- 34.Oliw E. H. 1985. Analysis of epoxyeicosatrienoic acids by gas chromatography–mass spectrometry using chlorohydrin adducts. J. Chromatogr. B Biomed. Sci. Appl. 339: 175–181. [Google Scholar]
- 35.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliw E. H., Garscha U., Nilsson T., Cristea M. 2006. Payne rearrangement during analysis of epoxyalcohols of linoleic and alpha-linolenic acids by normal phase liquid chromatography with tandem mass spectrometry. Anal. Biochem. 354: 111–126. [DOI] [PubMed] [Google Scholar]
- 37.Lee D. S., Nioche P., Hamberg M., Raman C. S. 2008. Structural insights into the evolutionary paths of oxylipin biosynthetic enzymes. Nature. 455: 363–368. [DOI] [PubMed] [Google Scholar]
- 38.Blom N., Gammeltoft S., Brunak S. 1999. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294: 1351–1362. [DOI] [PubMed] [Google Scholar]
- 39.Brash A. R., Yu Z., Boeglin W. E., Schneider C. 2007. The hepoxilin connection in the epidermis. FEBS J. 274: 3494–3502. [DOI] [PubMed] [Google Scholar]
- 40.Chang M. S., Boeglin W. E., Guengerich F. P., Brash A. R. 1996. Cytochrome P450-dependent transformations of 15R- and 15S-hydroperoxyeicosatetraenoic acids: stereoselective formation of epoxy alcohol products. Biochemistry. 35: 464–471. [DOI] [PubMed] [Google Scholar]
- 41.Poulos T. L. 2010. Thirty years of heme peroxidase structural biology. Arch. Biochem. Biophys. 500: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wennman A., Oliw E. H. 2013. Secretion of two novel enzymes, manganese 9S-lipoxygenase and epoxy alcohol synthase, by the rice pathogen Magnaporthe salvinii. J. Lipid Res. 54: 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamberg M. 1996. Stereochemical aspects of fatty acid oxidation: hydroperoxide isomerases. Acta Chem. Scand. 50: 219–224. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez J. F., Somoza A. D., Keller N. P., Wang C. C. 2012. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat. Prod. Rep. 29: 351–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu J-R., Urban M., Sweigard J. A., Hamer J. E. 1997. The CPKA gene of Magnaporthe grisea is essential for appressorial penetration. Mol. Plant-Microbe Interact. 10: 187–194. [Google Scholar]
- 46.Oesch-Bartlomowicz B., Oesch F. 2003. Cytochrome-P450 phosphorylation as a functional switch. Arch. Biochem. Biophys. 409: 228–234. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert N. C., Rui Z., Neau D. B., Waight M. T., Bartlett S. G., Boeglin W. E., Brash A. R., Newcomer M. E. 2012. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 26: 3222–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kvissel A. K., Persson H. T., Aksaas A. K., Akusjärvi G., Skålhegg B. S., 2014. Regulation of adenovirus alternative RNA splicing by PKA, DNA-PK, PP2A, and SR proteins. iConcept Press, Hong Kong. [Google Scholar]
- 49.Couch B. C., Kohn L. M. 2002. A multilocus gene genealogy concordant with host preference indicates segregation of a new species, Magnaporthe oryzae, from M. grisea. Mycologia. 94: 683–693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.