

The crystal structure of (Z)-N-(5-ethyl-2,3-dihydro-1,3,4-thiadiazol-2-ylidene)-4-methylbenzenesulfonamide contains an imine tautomer, rather than the previously reported amine tautomer. The tautomers can be distinguished using dispersion-corrected density functional theory calculations and by comparison of calculated and measured 13C solid-state NMR spectra.
Keywords: crystal structure, powder diffraction, NMR analysis, amine–imine tautomerism, dispersion-corrected DFT
Abstract
The crystal structure of the title compound, C11H13N3O2S2, has been determined previously on the basis of refinement against laboratory powder X-ray diffraction (PXRD) data, supported by comparison of measured and calculated 13C solid-state NMR spectra [Hangan et al. (2010 ▶). Acta Cryst. B66, 615–621]. The molecule is tautomeric, and was reported as an amine tautomer [systematic name: N-(5-ethyl-1,3,4-thiadiazol-2-yl)-p-toluenesulfonamide], rather than the correct imine tautomer. The protonation site on the molecule’s 1,3,4-thiadiazole ring is indicated by the intermolecular contacts in the crystal structure: N—H⋯O hydrogen bonds are established at the correct site, while the alternative protonation site does not establish any notable intermolecular interactions. The two tautomers provide essentially identical Rietveld fits to laboratory PXRD data, and therefore they cannot be directly distinguished in this way. However, the correct tautomer can be distinguished from the incorrect one by previously reported quantitative criteria based on the extent of structural distortion on optimization of the crystal structure using dispersion-corrected density functional theory (DFT-D) calculations. Calculation of the 13C SS-NMR spectrum based on the correct imine tautomer also provides considerably better agreement with the measured 13C SS-NMR spectrum.
Introduction
Determination of molecular crystal structures from powder X-ray diffraction (PXRD) data is now relatively common in the chemical literature (see, for example, Sanphui et al., 2014 ▶; Madusanka et al., 2014 ▶; Braun et al., 2013 ▶; Smart et al., 2013 ▶). On account of the compression of the diffraction data into one dimension in the powder pattern, it is frequently necessary, and in any case always of value, to supplement refinement against PXRD data with independent information that can establish the correctness of the structure or provide a more reliable indication of features that cannot be distinguished from the PXRD data alone. Energy minimization using quantum-chemical calculations provides one option, which can be especially useful for the determination of accurate positions for H atoms (Deringer et al., 2012 ▶). Energy minimization of a correct experimental crystal structure should lead to relatively smaller distortions compared to minimization of an incorrect structure, thereby providing possibilities for quantitative structure validation (Van de Streek & Neumann, 2010 ▶). Another possibility is ab initio calculation of solid-state nuclear magnetic resonance (SS-NMR) spectra for comparison to experimental spectra. Progress in this area is developing into the subfield of ‘NMR crystallography’ (Harris et al., 2009 ▶).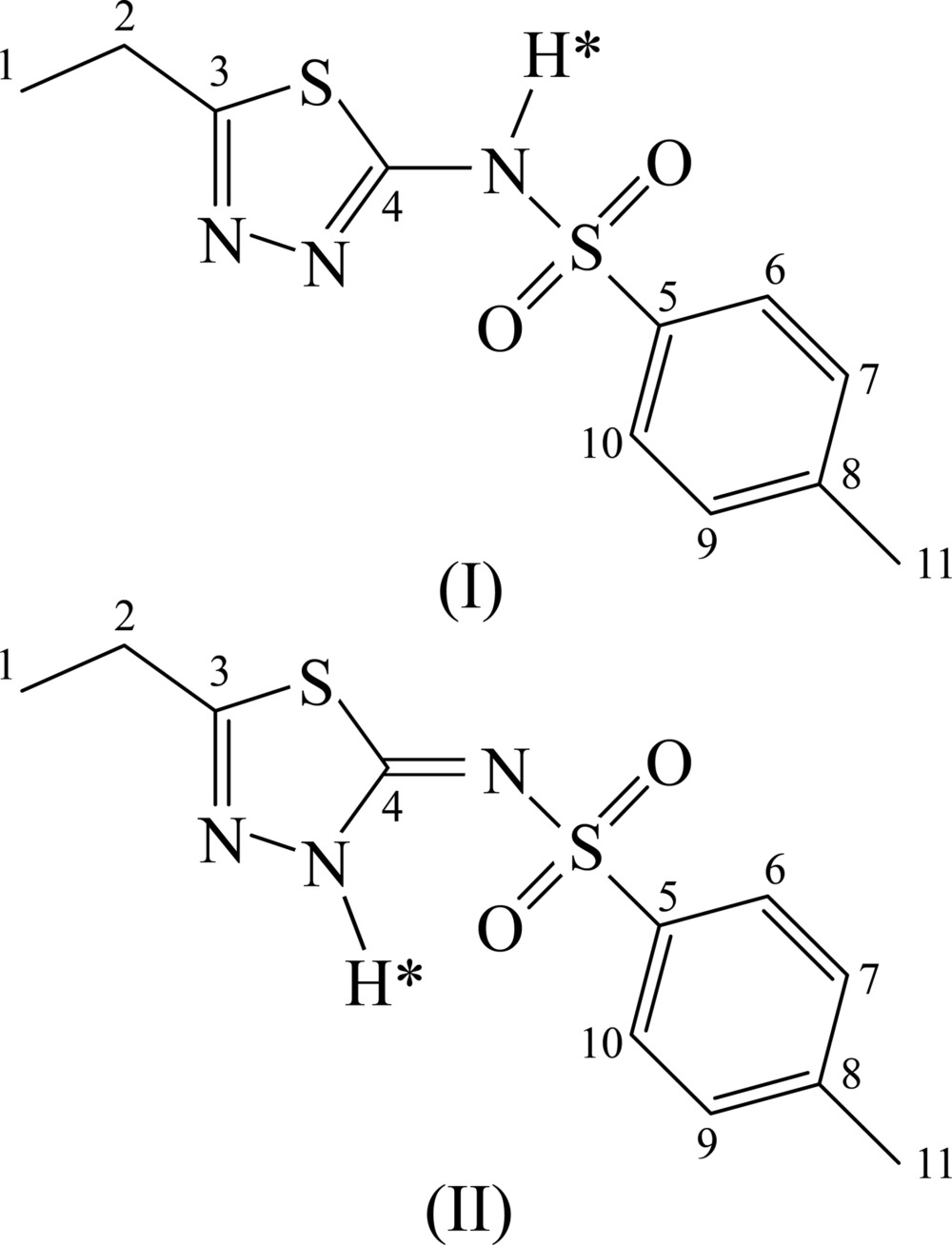
The crystal structure of the title compound has been reported by Hangan and co-workers [Hangan et al., 2010 ▶; Cambridge Structural Database (CSD; Allen, 2002 ▶) refcode UKIRAI] on the basis of refinement against laboratory PXRD data, supplemented by comparison of measured and calculated 13C SS-NMR spectra. The compound is tautomeric, with alternative H-atom positions on the N atom external to the 1,3,4-thiadiazole ring [referred to as the amine tautomer, (I)] or on one N atom of the 1,3,4-thiadiazole ring [referred to as the imine tautomer, (II)]. Hangan et al. reported the crystal structure and accompanying 13C SS-NMR calculations on the basis of the amine tautomer (I). Looking at the structure, there is an indication that this might not be correct: the postulated amine N—H group points directly towards a neighbouring methyl group (Fig. 1 ▶ a), while the N atom at the alternative protonation site on the 1,3,4-thiadiazole ring forms a contact of 2.78 Å to an O atom in a neighbouring molecule. Thus, it seems more likely that the imine tautomer (II) is present in the crystal structure, with the N—H group forming intermolecular N—H⋯O hydrogen bonds that generate ribbons along the a axis (Fig. 1 ▶ b).
Figure 1.

Intermolecular interactions for (a) amine tautomer (I), the N—H group points directly towards a neighbouring methyl group, and (b) imine tautomer (II), where intermolecular N—H⋯O hydrogen bonds generate ribbons along the a axis.
It seems unlikely that the H-atom positions could be determined explicitly by refinement against the laboratory PXRD data (vide infra), so it is interesting to examine the extent to which the tautomers can be distinguished by complementary methods, and especially the published 13C SS-NMR data. The discrepancy between observed and calculated 13C SS-NMR chemical shifts obtained by Hangan et al. for the amine tautomer (I) [mean absolute deviation (MAD) = 7.70 p.p.m. and root-mean-square deviation (RMSD) = 8.95 p.p.m.] is quite large compared to others in the literature (see, for example, Kuttatheyil et al., 2013 ▶; Dudenko et al., 2013 ▶; Filip et al., 2013 ▶) and the agreement could potentially be improved by considering the imine tautomer (II). In this paper, we reconsider the published experimental data (PXRD and 13C SS-NMR), together with some additional geometry optimizations and solid-state NMR calculations based on dispersion-corrected density functional theory (DFT-D) calculations. The aim is to establish the extent to which the tautomers might be distinguishable in the solid state.1 It is shown that the imine tautomer (II) is present in the crystal structure, and that tautomers (I) and (II) can be quantitatively distinguished by the results of DFT-D geometry optimizations and the published 13C SS-NMR spectra.
Experimental
Structure refinement
Crystal data, data collection and structure refinement details are summarized in Table 1 ▶. Structure refinements were carried out with TOPAS Academic (Coelho, 2012 ▶), using the PXRD data of Hangan et al. (2010 ▶). The published crystal structure was used as a starting point and H atoms were added in calculated positions. Two models were made, corresponding to tautomers (I) and (II), then both were subjected to preliminary DFT-D energy minimization with all atoms and unit-cell parameters free to vary within the constraints of the space group Pbca. This first step provides optimized models from which to extract restraints on the molecular geometry (Naelapää et al., 2012 ▶). The model structures for (I) and (II) were then subjected to Rietveld refinement against the published data, with restraints on the intramolecular bond distances and angles taken from the DFT-D calculations. The applied restraints are slightly different for each refinement, and the refined models therefore differ slightly where the bond lengths are influenced by the tautomeric form. Since the positions of the H atoms refined against the laboratory PXRD data are uncertain, a final CASTEP optimization (energy cut-off = 520 eV) was applied, with only the H atoms allowed to move. The final experimental structures for (I) and (II) have the unit cell and non-H-atom positions obtained from the Rietveld refinement, with the CASTEP-optimized positions for the H atoms.
Table 1. Experimental details.
The experimental data were taken from Hangan et al. (2010 ▶).
| (I) | (II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C11H13N3O2S2 | C11H13N3O2S2 |
| M r | 283.36 | 283.36 |
| Crystal system, space group | Orthorhombic, P b c a | Orthorhombic, P b c a |
| Temperature (K) | 298 | 298 |
| a, b, c (Å) | 8.53925 (14), 15.0207 (3), 21.3958 (3) | 8.53937 (13), 15.0206 (2), 21.3960 (3) |
| V (Å3) | 2744.33 (8) | 2744.39 (7) |
| Z | 8 | 8 |
| Radiation type | Cu Kα1, λ = 1.54056 Å | Cu Kα1, λ = 1.54056 Å |
| Specimen shape, size (mm) | Flat sheet, 25 × 1 | Flat sheet, 25 × 1 |
| Data collection | ||
| Diffractometer | Bruker D8 Advance diffractometer | Bruker D8 Advance diffractometer |
| Specimen mounting | Bruker sample cup | Bruker sample cup |
| Data collection mode | Reflection | Reflection |
| Scan method | Continuous | Continuous |
| 2θ values (°) | 2θmin = 3.54, 2θmax = 50.03, 2θstep = 0.005 | 2θmin = 3.54, 2θmax = 50.03, 2θstep = 0.005 |
| Refinement | ||
| R factors and goodness of fit | R p = 0.058, R wp = 0.081, R exp = 0.053, R(F) = 0.034, χ2 = 1.53 | R p = 0.058, R wp = 0.081, R exp = 0.053, R(F) = 0.033, χ2 = 1.53 |
| No. of data points | 9298 | 9298 |
| No. of parameters | 127 | 127 |
| No. of restraints | 88 | 88 |
| H-atom treatment | H-atom parameters not refined | H-atom parameters not refined |
DFT-D optimizations and calculation of 13C SS-NMR spectra
Geometry optimizations and solid-state NMR calculations were carried out using CASTEP (Academic Release 6.1; Clark et al., 2005 ▶), via the interface within Materials Studio (Accelrys, 2011 ▶). The Perdew, Burke and Ernzerhof (PBE) exchange-correlation functional (Perdew et al., 1996 ▶) was applied, with the Grimme-06 semi-empirical dispersion correction (Grimme, 2006 ▶). Integrals taken over the Brillouin zone were carried out on a Monkhorst–Pack grid (Monkhorst & Pack, 1976 ▶) with a maximum sample spacing of 0.05 Å−1. For both (I) and (II), the experimental structures were optimized and 13C SS-NMR spectra were calculated by following the flowchart shown in Fig. 2 ▶. In general, an optimization of the crystal structure is divided into three sequential steps with: (i) only H atoms allowed to move; (ii) all atoms allowed to move with unit-cell parameters fixed; (iii) all atoms allowed to move with unit-cell parameters free. The optimizations were carried out with an energy cut-off of 520 eV to permit comparison with the results of a published validation study (Van de Streek & Neumann, 2010 ▶). The optimized structure at 520 eV with the unit cell free was further optimized with an energy cut-off of 1200 eV and then used for 13C SS-NMR calculations at 1200 eV, with ultrasoft pseudopotentials generated on-the-fly (Yates et al., 2007 ▶). Optimization with the higher-quality basis set is more time-consuming, but provides a more accurate calculated 13C SS-NMR spectrum.
Figure 2.

Protocol for the structure optimizations and calculations of the 13C SS-NMR spectra.
In order to isolate the influence of the H-atom positions on the calculated 13C SS-NMR spectra, an ‘average structure’ was prepared from the experimental structures of (I) and (II), by averaging each corresponding unit-cell parameter and the atomic coordinates of the non-H atoms. The unit cell and non-H atoms of this model are not biased towards either tautomer. The H atoms were then placed so as to form either tautomer (I) or (II) and their positions were optimized with an energy cut-off of 1200 eV, with the unit cell and non-H-atom positions fixed. These two structures are referred to as ‘average (I)’ and ‘average (II)’, respectively. 13C solid-state NMR calculations were made for the two structures with an energy cut-off of 1200 eV.
Results and discussion
Structure refinement against PXRD data
Structure refinement against the PXRD data using either tautomer (I) or (II) produced essentially identical results (Table 1 ▶ and Fig. 3 ▶). The obtained figures-of-merit are moderately improved compared to the refinement of Hangan et al. (2010 ▶), principally due to improved anisotropic peak-shape modelling, and inclusion of a preferred-orientation correction [March, 1932 ▶; direction [100], refined parameter ca 1.08 for both (I) and (II)]. Nonetheless, the features in the difference curves reveal remaining problems with the peak shape, which possibly play some role in obscuring any differences that might have been evident between the two models. Using the present PXRD data, we conclude that it is not possible to distinguish directly the two tautomers.
Figure 3.

Rietveld plots for the refined experimental structures of (I) and (II). The PXRD data are taken from Hangan et al. (2010 ▶). Key: red crosses = measured data, blue line = calculated pattern and black line = difference curve.
Structure validation by DFT-D optimization
The results of DFT-D energy minimizations at 520 eV for the experimental structures of (I) and (II) are shown in Table 2 ▶. The input and optimized structures in CIF format are provided as Supporting information. On minimization, structure (I) undergoes significantly larger distortions compared to structure (II), both when the unit cell is fixed to the experimental one and when it is free to be optimized. The difference between (I) and (II) is visually evident in overlays of the experimental and optimized structures (Fig. 4 ▶). According to a previous validation study (Van de Streek & Neumann, 2010 ▶), an RMS Cartesian displacement for the non-H atoms greater than 0.30 Å when the unit-cell parameters are allowed to vary indicates either that the structure is incorrect, or that it undergoes some significant temperature-dependent variation. Values below 0.25 Å indicate that the structure is likely to be correct. These established geometrical criteria indicate that structure (II) is acceptable and identify structure (I) as suspicious (Table 2 ▶).
Table 2. Root-mean-square Cartesian displacements (Å) for the DFT-D optimizations of (I) and (II), compared to the experimental structures.
| Energy cut-off (eV) | Optimization protocol | All-atom RMS Cartesian displacement | Non-H-atom RMS Cartesian displacement | |
|---|---|---|---|---|
| (I) | 520 | Unit cell fixed | 0.4309 | 0.3487 |
| (I) | 520 | Unit cell free | 0.4356 | 0.3561 |
| (I) | 1200 | Unit cell free | 0.6972 | 0.5749 |
| (II) | 520 | Unit cell fixed | 0.1494 | 0.1283 |
| (II) | 520 | Unit cell free | 0.1569 | 0.1329 |
| (II) | 1200 | Unit cell free | 0.1626 | 0.1282 |
Figure 4.

Overlay of experimental structure (red) and optimized structure (blue) at 520 eV with the unit-cell dimensions free. The larger distortion for (I) can be seen clearly.
13C Solid-state NMR
The results of our 13C SS-NMR calculations are listed in Table 3 ▶. The resonance assignment is based on that of Hangan et al., which was made by comparison with the solution 13C spectrum. For the topologically-equivalent atom pairs C6/C10 and C7/C9, single resonances in the solution spectrum are split into two pairs in the solid state, and it is not possible to state a priori which resonance corresponds to which atom within each pair. In these cases, the assignment is made so as to provide the best fit with the experimental spectrum. For (I), and for the two average structures, the assignment corresponds to that of Hangan et al. For (II), the agreement with the experimental spectrum is improved by exchanging the assignment of C6 and C10 (see Table 3 ▶). Overall, the calculated chemical shifts of (II) give better agreement with the experimental chemical shifts than do the calculated shifts of (I). The RMSD value of 1.9 p.p.m. for (II) is identical to a mean value obtained for similar test calculations on 15 organic compounds (Salager et al., 2010 ▶), so it can be viewed as being compatible with established expectations. By contrast, the RMSD value of 4.0 p.p.m. for (I) is significantly larger than expectation. The largest single deviation for (II) is −3.4 p.p.m. for atom C11, compared to 10.7 p.p.m. for atom C3 in (I). The latter error is clearly substantial, and indicative of the incorrect tautomeric assignment. Thus, comparison of the published 13C SS-NMR data with our new calculations clearly distinguishes the two tautomers.
Table 3. Experimental and calculated 13C SS-NMR chemical shifts (p.p.m.).
Deviations compared to the experimental values are indicated in parentheses. All calculations are based on optimized structures at 1200 eV (as described in the text) and are carried out at 1200 eV.
| Experimentala | (I) | (II) | Average (I) | Average (II) | |||||
|---|---|---|---|---|---|---|---|---|---|
| C1 | 14.0 | 11.8 | (−2.2) | 11.8 | (−2.2) | 13.2 | (−0.8) | 10.9 | (−3.1) |
| C2 | 23.5 | 19.4 | (−4.1) | 20.9 | (−2.6) | 20.6 | (−2.9) | 22.1 | (−1.4) |
| C3 | 165.7 | 176.4 | (10.7) | 168.2 | (2.5) | 165.4 | (−0.3) | 161.4 | (−4.3) |
| C4 | 161.9 | 165.4 | (3.5) | 163.1 | (1.2) | 159.3 | (−2.6) | 162.6 | (0.7) |
| C5 | 138.9 | 138.9 | (0.0) | 140.3 | (1.4) | 140.4 | (1.5) | 140.6 | (1.7) |
| C6 | 127.6 | 125.5 | (−2.1) | 128.1 | (–0.2)b | 126.2 | (−1.4) | 128.4 | (0.8) |
| C7 | 130.5 | 128.7 | (−1.8) | 131.6 | (1.1) | 132.5 | (2.0) | 132.0 | (1.5) |
| C8 | 145.0 | 147.4 | (2.4) | 147.1 | (2.1) | 148.9 | (3.9) | 148.2 | (3.2) |
| C9 | 132.0 | 131.5 | (−0.5) | 133.3 | (1.3) | 136.6 | (4.6) | 135.0 | (3.0) |
| C10 | 128.3 | 125.9 | (−2.4) | 126.7 | (−0.9)b | 126.9 | (−1.4) | 128.5 | (0.2) |
| C11 | 21.3 | 17.8 | (−3.5) | 17.9 | (−3.4) | 18.9 | (−2.4) | 19.3 | (−2.0) |
| MAD | 3.0 | 1.7 | 2.2 | 2.0 | |||||
| RMSD | 4.0 | 1.9 | 2.5 | 2.3 | |||||
Notes: (a) experimental values and resonance assignments from Hangan et al. (2010 ▶); (b) the assignments of the topologically equivalent atoms C6 and C10 are exchanged: C6 is matched to the experimental value of C10, and vice versa (see main text).
For the models made by averaging the non-H-atom positions in the two experimental structures, the MAD and RMSD values for (I) and (II) are quite similar, and both have some atoms with relatively large deviations (> 4 p.p.m.) compared to the measured chemical shifts (Table 3 ▶). This demonstrates that the calculated 13C chemical shifts are very sensitive to the positions of all atoms, rather than just the H-atom positions in this tautomeric case. The averaged model provides a better fit compared to the fully optimized structure for the incorrect tautomer (I) but a worse fit for the correct tautomer (II). This is related to the ‘tautomer-dependent’ restraints that were applied in the Rietveld refinements, derived from preliminary DFT-D optimizations (see Experimental, §2.2). For (I), the preliminary DFT-D optimization was influenced by the incorrect choice of H atom positions, and therefore provided relatively inaccurate positions for the non-H atoms. These positions are carried over into the experimental structures through the applied restraints. For (II), the preliminary DFT-D optimization provided more accurate positions for the non-H atoms because of the correct choice for the H atoms. Averaging of the two experimental structures moves the relatively poor heavy-atom positions in the incorrect structure (I) towards the better heavy-atom positions in the correct structure (II), thereby giving progressively improved fits to the 13C SS-NMR data in the sequence (I) → average (I) → average (II) → (II).
Conclusions
The crystal structure of the title compound contains the imine tautomer (II) rather than the previously reported amine tautomer (I). The tautomers cannot be directly distinguished from the laboratory PXRD data (although the intermolecular contacts in the crystal structure are strongly indicative of the imine tautomer); so this is a case in which independent quantitative information becomes useful. The aim of this paper was to reconsider the published information, rather than to collect any new experimental data (e.g. 1H SS-NMR data), with the addition of some computational work. The incorrect tautomer is highlighted by DFT-D optimization, on the basis of criteria presented in an earlier validation study (Van de Streek & Neumann, 2010 ▶). This computational procedure is relatively accessible, and we suggest that it should always be used to support molecular crystal structures determined from PXRD data. Comparison of the calculated and published 13C solid-state NMR spectra also provides a clear quantitative distinction between the two tautomers.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2053229614015356/fa3347sup1.cif
Rietveld powder data: contains datablock(s) I. DOI: 10.1107/S2053229614015356/fa3347Isup2.rtv
Input and optimized structure for (I). DOI: 10.1107/S2053229614015356/fa3347Isup3.txt
Input and optimized structure for (II). DOI: 10.1107/S2053229614015356/fa3347Isup4.txt
CML file for (I). DOI: 10.1107/S2053229614015356/fa3347Isup5.cml
CCDC reference: 1011334
Acknowledgments
The Villum Foundation (Denmark) is gratefully acknowledged for financial support (project No. VKR023111). The Materials Studio software was provided by the Lundbeck Foundation (Denmark) (grant No. R49-A5604).
Footnotes
The paper of Hangan et al. does not contain a 1H SS-NMR spectrum. A 1H NMR spectrum in d 6-DMSO solution is mentioned, but caution should be applied when extrapolating solution data to the solid state, because protonation states and hydrogen bonding may be different (e.g. barbituric acid; Schmidt et al., 2011 ▶).
Supporting information for this paper is available from the IUCr electronic archives (Reference: FA3347).
References
- Accelrys (2011). Materials Studio Accelrys Inc., San Diego, California, USA.
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Braun, D. E., Bhardwaj, R. M., Florence, A. J., Tocher, D. A. & Price, S. L. (2013). Cryst. Growth Des. 13, 19–23. [DOI] [PMC free article] [PubMed]
- Clark, S. J., Segall, M. D., Pickard, C. J., Hasnip, P. J., Probert, M. J., Refson, K. & Payne, M. C. (2005). Z. Kristallogr. 220, 567–570.
- Coelho, A. A. (2012). TOPAS Academic Coelho Software, Brisbane, Australia.
- Deringer, V. L., Hoepfner, V. & Dronskowski, R. (2012). Cryst Growth Des. 12, 1014–1021.
- Dudenko, D. V., Williams, P. A., Hughes, C. E., Antzutkin, O. N., Velaga, S. P., Brown, S. P. & Harris, K. D. M. (2013). J. Phys. Chem. C, 117, 12258–12265. [DOI] [PMC free article] [PubMed]
- Filip, X., Grosu, I.-G., Miclăuş, M. & Filip, C. (2013). CrystEngComm, 15, 4131–4142.
- Grimme, S. (2006). J. Comput. Chem. 27, 1787–1799. [DOI] [PubMed]
- Hangan, A., Borodi, G., Filip, X., Tripon, C., Morari, C., Oprean, L. & Filip, C. (2010). Acta Cryst. B66, 615–621. [DOI] [PubMed]
- Harris, R. K., Wasylishen, R. E. & Duer, M. J. (2009). In NMR Crystallography Chichester: John Wiley & Sons.
- Kuttatheyil, A. V., Lässig, D., Lincke, J., Kobalz, M., Baias, M., König, K., Hofmann, J., Krautscheid, H., Pickard, C. J., Haase, J. & Bertmer, M. (2013). Inorg. Chem. 52, 4431–4442. [DOI] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Madusanka, N., Eddleston, M. D., Arhangelskis, M. & Jones, W. (2014). Acta Cryst. B70, 72–80. [DOI] [PubMed]
- March, A. (1932). Z. Kristallogr. 81, 285–297.
- Monkhorst, H. & Pack, J. (1976). Phys. Rev. B, 13, 5188–5192.
- Naelapää, K., Van de Streek, J., Rantanen, J. & Bond, A. D. (2012). J. Pharm. Sci. 101, 4214–4219. [DOI] [PubMed]
- Perdew, J. P., Burke, K. & Ernzerhof, M. (1996). Phys. Rev. Lett. 77, 3865–3868. [DOI] [PubMed]
- Salager, E., Day, G. M., Stein, R. S., Pickard, C. J., Elena, B. & Emsley, L. (2010). J. Am. Chem. Soc. 132, 2564–2566. [DOI] [PubMed]
- Sanphui, P., Bolla, G., Nangia, A. & Chernyshev, V. (2014). IUCrJ, 1, 136–150. [DOI] [PMC free article] [PubMed]
- Schmidt, M. U., Brüning, J., Glinnemann, J., Hützler, M. W., Mörschel, P., Ivashevskaya, S. N., Van de Streek, J., Braga, D., Maini, L., Chierotti, M. R. & Gobetto, R. (2011). Angew. Chem. Int. Ed. 50, 7924–7926. [DOI] [PubMed]
- Smart, P., Mason, C. A., Loader, J. R., Meijer, A. J. H. M., Florence, A. J., Shankland, K., Fletcher, A. J., Thompson, S. P., Brunelli, M., Hill, A. H. & Brammer, L. (2013). Chem. Eur. J. 19, 3552–3557. [DOI] [PubMed]
- Van de Streek, J. & Neumann, M. A. (2010). Acta Cryst. B66, 544–558. [DOI] [PMC free article] [PubMed]
- Yates, J., Pickard, C. & Mauri, F. (2007). Phys. Rev. B, 76, 024401.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2053229614015356/fa3347sup1.cif
Rietveld powder data: contains datablock(s) I. DOI: 10.1107/S2053229614015356/fa3347Isup2.rtv
Input and optimized structure for (I). DOI: 10.1107/S2053229614015356/fa3347Isup3.txt
Input and optimized structure for (II). DOI: 10.1107/S2053229614015356/fa3347Isup4.txt
CML file for (I). DOI: 10.1107/S2053229614015356/fa3347Isup5.cml
CCDC reference: 1011334