

Abstract
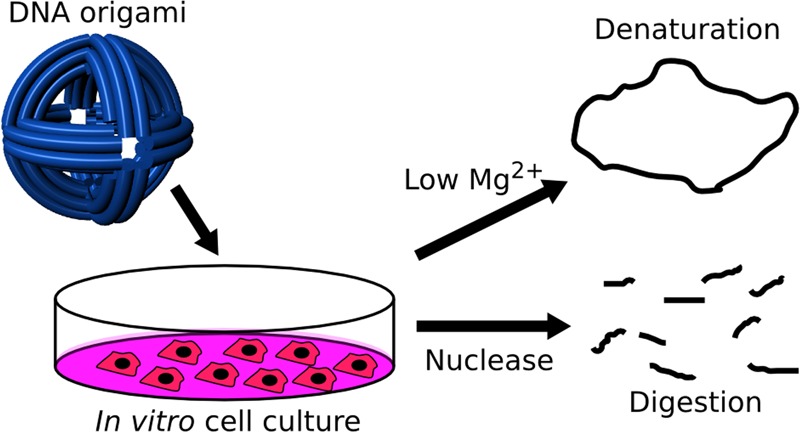
DNA nanotechnology is an advanced technique that could contribute diagnostic, therapeutic, and biomedical research devices to nanomedicine. Although such devices are often developed and demonstrated using in vitro tissue culture models, these conditions may not be compatible with DNA nanostructure integrity and function. The purpose of this study was to characterize the sensitivity of 3D DNA nanostructures produced via the origami method to the in vitro tissue culture environment and identify solutions to prevent loss of nanostructure integrity. We examined whether the physiological cation concentrations of cell culture medium and the nucleases present in fetal bovine serum (FBS) used as a medium supplement result in denaturation and digestion, respectively. DNA nanostructure denaturation due to cation depletion was design- and time-dependent, with one of four tested designs remaining intact after 24 h at 37 °C. Adjustment of medium by addition of MgSO4 prevented denaturation. Digestion of nanostructures by FBS nucleases in Mg2+-adjusted medium did not appear design-dependent and became significant within 24 h and when medium was supplemented with greater than 5% FBS. We estimated that medium supplemented with 10% FBS contains greater than 256 U/L equivalent of DNase I activity in digestion of DNA nanostructures. Heat inactivation at 75 °C and inclusion of actin protein in medium inactivated and inhibited nuclease activity, respectively. We examined the impact of medium adjustments on cell growth, viability, and phenotype. Adjustment of Mg2+ to 6 mM did not appear to have a detrimental impact on cells. Heat inactivation was found to be incompatible with in vitro tissue culture, whereas inclusion of actin had no observable effect on growth and viability. In two in vitro assays, immune cell activation and nanoparticle endocytosis, we show that using conditions compatible with cell phenotype and nanostructure integrity is critical for obtaining reliable experimental data. Our study thus describes considerations that are vital for researchers undertaking in vitro tissue culture studies with DNA nanostructures and some potential solutions for ensuring that nanostructure integrity and functions are maintained during experiments.
Keywords: DNA nanotechnology, DNA origami, cells, in vitro, tissue culture, nuclease, stability, structural integrity, cation, nanorobot
DNA nanotechnology1 is a rapidly progressing field owing to the ease of producing two- and three-dimensional nanostructures through methods such as DNA origami.2,3 DNA nanostructures can be designed with virtually any arbitrary geometry4,5 and to display sophisticated capabilities (e.g., mechanical,6 logic-gating7) unavailable to other nanoscale materials. These can act as platforms for precise spatial organization of functional molecular features such as fluorophores,8 biomolecules (e.g., aptamers,9 antibodies10), or synthetic nanomaterials11 (e.g., quantum dots) through coupling to an oligonucleotide that is integrated into a nanostructure’s design. These unique advantages have provided ample motivation for nanomedicine researchers to develop in vitro and in vivo biomedical applications for DNA nanotechnology. Recent studies have demonstrated a long-circulating virus-inspired DNA nanodevice,12 a nanocaliper for mediating cell ligand–receptor interactions,13 a computational molecular cascade for cell surface receptor characterization,14 a synthetic membrane ion pore,15 an immune-activating programmable adjuvant,16 and a logic-gated nanorobot for therapeutic delivery.17
In vitro tissue culture model systems are an obvious choice for prototyping new diagnostic and therapeutic devices, including those based on DNA nanotechnology. The conditions used for tissue culture are typically determined by what has been empirically established as appropriate for the cells of interest. For mammalian cells, this means incubation at 37 °C in a defined cell culture medium, such as RPMI or DMEM, containing amino acids, glucose, and vitamins, and at a physiological pH and concentration of ions. Maintaining cell growth and phenotype often requires such medium to be supplemented with 2–20% of mammalian serum (e.g., fetal bovine serum), which contributes essential but undefined factors. The compatibility of the in vitro tissue culture environment with DNA origami nanostructures has not been systematically tested but is of obvious importance for biomedical applications of these materials.
The synthesis or “folding” of DNA nanostructures involves thermal annealing in the presence of a minimum concentration of cations to overcome the negative charge-repulsion forces of the nucleic acid phosphodiester backbone. Compact and high-density 3D nanostructures produced via DNA origami are typically produced in a buffer containing the divalent cation Mg2+ at a concentration of ∼5–20 mM,3 ∼1 order of magnitude higher than its concentration in blood and tissue culture media (<1 mM). This discrepancy between the divalent cation concentration required by DNA nanostructures and that present in biological environments could cause nanostructure denaturation. Alternatively, synthesized nanostructures may have some capacity to sequester cations and may not therefore display sensitivity to dilute Mg2+ environments. As well, blood and tissue culture media contain high concentrations of the monovalent cation Na+ (∼140 mM) that would likely support nanostructure integrity. Nevertheless, the sensitivity of DNA nanostructures to cation depletion when transferred into tissue culture media could impact the performance of their engineered capabilities.
A second concern is in regards to the supplementation of tissue culture medium with fetal bovine serum, a blood product known to contain a variety of nucleases18−20 that have hindered translation of nucleic acid-based biotechnologies such as gene therapy.21,22 The digestion half-life of unprotected plasmid DNA has been measured at 20 min with freshly isolated plasma21 and 10 min after in vivo intravenous injection.22 Nuclease digestion of DNA nanostructures23,24 and the crossover motifs25 used in their architecture appear to be partially inhibited when compared to oligonucleotides or plasmid DNA. However, even partial digestion of DNA nanostructures could greatly affect their geometry and capabilities and could cause release of attached molecular features.
In this study, we present a systematic characterization of the sensitivity of various DNA nanostructures to typical mammalian tissue culture conditions. We examined the denaturation of nanostructures due to divalent cation depletion and their digestion by nucleases present in FBS. On the basis of our findings, we developed strategies to maintain DNA nanostructure stability by making adjustments to tissue culture medium. We then tested the impact of those adjustments on cell growth and viability. Finally, we carried out two typical tissue culture assays to measure the impact of nanostructure instability and medium adjustments on cell phenotype and experimental outcomes.
Results and Discussion
As test cases for this study, we synthesized three nanostructures that were designed using the DNA origami method (Figure 1a–c and Supporting Information Figures 1–3). These were produced by annealing three previously described nanostructure-specific oligonucleotide “staple” sets with single-stranded, modified26 M13 phage “scaffold” DNA of 7308 nucleotides. Owing to the sensitivity of mammalian cells to bacterial cell wall lipopolysaccharide or “endotoxin”, we developed a protocol for removal of this byproduct of M13 scaffold production with Triton X-114 (Supporting Information Figure 4).
Figure 1.

DNA nanostructure sensitivity to cation depletion in tissue culture medium. The three test nanostructures are (a) DNA nano-octahedron (DNO), (b) six-helix bundle nanotube (NT), and (c) 24-helix nanorod (NR). (d–f) Three nanostructures were incubated for 24 h at 37 °C in unmodified RPMI tissue culture medium containing 0.4 mM Mg2+ or adjusted to 0.7–10 mM Mg2+, and the products were analyzed by agarose gel electrophoresis (AGE). In comparison to the control lanes of stable sample, migration of the DNO and NR is retarded after low Mg2+ incubation, indicative of denaturation, whereas no obvious difference in migration is observed with the NT. (g–i) Transmission electron microscopy images of nanostructures incubated in unmodified medium, showing varying levels of denaturation. (j–l) With RPMI medium adjusted to 6 mM Mg2+, structural integrity is maintained in all three designs. M = molecular weight ladder, C = nanostructure in TE + 10 mM Mg2+, S = M13 scaffold. Scale bar = 100 nm.
Using a first set of 144 staple strands, we synthesized a ∼50 nm diameter wireframe DNA nano-octahedron (DNO).12 The DNO struts were each composed of a bundle of six DNA double helices27 connected by antiparallel crossover junctions.28,29 A ∼90° curvature was added to the struts via targeted insertions and deletions.4,12 The scaffold and staple strands were combined in Tris-EDTA (TE) buffer with 14 mM MgCl2, and the nanostructure was produced using a 15 h thermal annealing ramp. Using the same annealing ramp and two alternative sets of staple strands, we produced nanotubes26 (NT) composed of a single 400 nm long six-helix bundle (168 staple strands, 20 mM MgCl2) and a 16 × 89 nm nanorod30 (NR) composed of 24 interconnected helices (178 strands, 18 mM MgCl2). Correctly folded nanostructures were purified using a previously described gradient ultracentrifugation method.30 After purification of the DNO and NR, we annealed a fluorescent Cy5-labeled oligonucleotide to single-stranded DNA “handles” present on the nanostructure to represent molecular cargo typically added to nanostructures as functional features.
We then characterized the sensitivity of these DNA nanostructures to Mg2+ depletion in standard RPMI medium (132.9 mM Na+, 5.3 mM K+, 0.4 mM Mg2+, 0.4 mM Ca2+) without FBS supplementation. To further elucidate the dependence of nanostructure integrity on divalent cation concentration, we included a series of adjusted RPMI medium samples with Mg2+ concentrations of 0.7–10 mM and a control previously determined to maintain nanostructure integrity (TE buffer, 10 mM Mg2+). Following incubation at 37 °C for 24 h, the products were analyzed by agarose gel electrophoresis (AGE). In comparison to control samples, the DNO and NR displayed reduced electrophoretic migration after incubation in standard RPMI medium and adjusted medium with Mg2+ concentrations below ∼6 mM (Figure 1d,f). This suggested that some degree of denaturation had occurred, which would likely increase the dimensions of the nanostructures and slow their travel through the agarose matrix. Surprisingly, NT migration did not appear dependent on Mg2+ concentration over the tested range (Figure 1e), demonstrating design-dependent variability in the sensitivity of nanostructures to Mg2+ depletion. Because the DNO and NR were labeled with fluorescent Cy5 oligonucleotides, we were able to examine via AGE if low divalent cation conditions may cause a loss of molecular cargo attached to the nanostructures. We imaged the gels for Cy5 fluorescence and observed a partial Mg2+-concentration-dependent loss of Cy5 cargo from the DNO and NR, demonstrating that denaturation via divalent cation depletion can cause release of attached molecular cargo (Supporting Information Figure 5).
We further characterized the RPMI incubation products via negative-stain transmission electron microscopy (TEM) (Figure 1g–l). Imaging confirmed that the DNO and NR denature in RPMI, and that their structural integrity is maintained in medium adjusted to 6 mM Mg2+. Although we observed particles approximating the dimensions of the DNO and NR after incubation in standard RPMI (0.4 mM Mg2+), they were amorphous and lacked recognizable features. This suggests that the low divalent and high monovalent cation concentrations in the RPMI medium were unable to overcome charge repulsion of the DNA phosphodiester backbone, and that the Mg2+ ions associated with the nanostructures from the synthesis did not remain sequestered after transfer into RPMI. The impact of medium on NT integrity appeared less dramatic. Intact NTs were visible by TEM after incubation in 0.4 mM Mg2+ RPMI. Relative to the control sample, there were far fewer NTs present, but this may be a Mg2+-dependent artifact of sample preparation for TEM imaging.
Based on its stability in medium, the DNA NT may be an excellent choice for in vitro tissue culture studies. To characterize a nanostructure with relevance to biomedical applications, we also examined the logic-gated DNA nanorobot17 for sensitivity to divalent cation depletion using the same assay as described above. After incubation at 37 °C for 24 h, AGE analysis showed a difference in migration between controls and those incubated in standard RPMI medium and adjusted medium with Mg2+ concentrations below ∼2 mM (Supporting Information Figure 6a). This shift became apparent after 30 min of incubation (Supporting Information Figure 6b). Imaging of nanostructures incubated at 2 mM Mg2+ for 24 h revealed nanorobots that were intact but which appeared stressed, in comparison to the control 10 mM Mg2+ sample (Supporting Information Figure 6c,d). We were unable to find intact nanostructures after incubation in standard medium, but this might be a false negative, attributable to an artifact of TEM sample preparation. Relative to the NR and DNO, the nanorobot appears to be less sensitive to low Mg2+ concentrations. In light of the published functional data,17 it may be possible that electrostatic repulsion from divalent cation depletion causes some flexing of the nanorobot structure, without a loss of the logic-gating molecular features and open versus closed conformations. Further in-depth studies would be needed to fully elucidate these details.
The variability in nanostructure sensitivity to divalent cation depletion is intriguing. The designs tested here share a common scaffold strand and GC content. The design-specific oligonucleotide sets of the DNO, NT, and NR are comparable in mean (±standard deviation) length at 42.0 ± 2.5, 40.6 ± 4.6, and 41.9 ± 0.8 nucleotides, respectively, whereas the nanorobot set is shorter (33.3 ± 7.0). The secondary structure (six-helix bundle motif) is common between the DNO and NT, although curvature included in the DNO strut design might further stress the structure in a low-Mg2+ environment. The charge density is highest in the NR, in which 12 of the 24 double helices have three nearest-neighbors, compared to two for the double helices of the DNO, NT, and nanorobot. Based on a lack of any clear pattern, it is possible that nanostructure sensitivity to divalent cation depletion could be dependent on a variety of design parameters. Until this is better understood, the stability of nanostructures intended for use in tissue culture experiments should be tested in appropriate conditions.
Next, the various nanostructures were incubated in RPMI supplemented with 10% FBS to assess nuclease digestion kinetics. Note that the FBS used in these experiments had been heat-treated at 56 °C by the vendor for inactivation of immune factors, as is standard practice. For these experiments, the RPMI medium was adjusted to 6 mM Mg2+ using MgSO4 (a component of RPMI) to prevent nanostructure denaturation. After incubation of 5 nM DNO at 37 °C for 0.25–24 h, samples were analyzed by AGE and compared to a control incubated in RPMI + 6 mM Mg2+, but lacking FBS, for 24 h. The results show little change in migration of the DNO (Figure 2a), NT, and NR (Supporting Information Figure 7) over the first 8 h. However, there is an almost complete loss of the primary product band by 24 h. TEM imaging of the DNO control sample showed no apparent changes (Figure 2b), whereas nanostructures incubated in medium with serum for 2 h showed varying degrees of digestion (Figure 2c). Based on this, it appears that some amount of nuclease activity remains in the FBS typically used to supplement tissue culture medium. We also observed, over numerous repetitions of these experiments, that the level of nuclease activity in different FBS lots and frozen aliquots can be quite variable, with digestion progressing faster or much slower than that presented in Figure 2. As well, the nuclease activity was highest after initial thawing of an FBS stock and was lost over a time span of weeks when prepared medium was stored at 4 °C (not shown). The presence of nuclease in the medium could have a dramatic impact on the results of tissue culture studies that utilize DNA nanostructures, and care must be taken to ensure that it is absent during experiments.
Figure 2.

Kinetics of nanostructure digestion by nucleases present in serum and heat inactivation. (a) DNO nanostructure (5 nM) was incubated at 37 °C for 0.25–24 h in RPMI + 6 mM Mg2+ + 10% FBS. Analysis by AGE shows smearing and decreased intensity of the product band as digestion progresses. (b) TEM image of DNO control sample showing intact nanostructures. (c) Partially digested DNO sample after 2 h incubation in medium + 10% FBS. Top left: example of a partially digested DNO. (d) Aliquots of FBS were heat-treated for 0.25–10.0 min at 75 °C prior to medium preparation. DNO was incubated for 24 h at 37 °C and analyzed by AGE. Medium without nanostructures was analyzed on the right half of the gel, showing a change in appearance from 5 and 10 min heat treatment. (e) TEM of a control and (f) DNO incubated in RPMI medium prepared with FBS heat-inactivated for 5 min. M = molecular weight ladder, C = nanostructure in TE + 10 mM Mg2+, S = M13 scaffold. Scale bars = 100 nm.
To determine the relative nuclease activity present in tissue culture medium, we carried out a 12 h, 37 °C incubation of DNO in 6 mM Mg2+ RPMI medium supplemented with 1.25–20% FBS versus 1–4096 U/L of DNase I (Supporting Information Figure 8). Significant digestion was apparent at 10 and 20% FBS and at more than 256 U/L of DNase I, suggesting that typical tissue culture conditions may contain between 256 and 1024 U/L equivalent of DNase I activity. Note that digestion was not apparent by AGE for low concentrations of FBS (1.25–2.5%), suggesting one potential solution for circumventing medium nuclease activity when short incubation times and low serum concentrations are compatible with the assay and cells of interest.
Heat treatment of FBS is a potentially easy and scalable method for inactivation of nuclease activity. In order to determine the minimum heat treatment required, FBS was incubated at 75 °C for 0–10 min. This was carried out in 1 mL aliquots to allow for efficient heat transfer. We used this to prepare RPMI + 6 mM Mg2+ + 10% FBS, to which the DNO nanostructure was added at 5 nM and incubated at 37 °C for 24 h. Analysis of the incubation products by AGE showed that a minimum treatment of 5 min was required to prevent altered migration of the nanostructure (Figure 2d). TEM imaging confirmed that DNO (Figure 2e,f) incubated in FBS treated thusly remained intact, with no obvious difference in appearance compared to a control. Heat inactivation of FBS could therefore address the problem of nuclease activity in tissue culture experiments. However, the altered appearance of the 5 and 10 min treated FBS in our AGE analysis (Figure 2d) suggests that it has a more general effect on serum proteins, which could also impact cell growth and phenotype.
An alternative to heat inactivation of FBS is inclusion of a nuclease inhibitor, such as actin protein,31 in tissue culture medium. Actin inhibition occurs via competitive binding to nuclease and should therefore be concentration-dependent. To test this and determine an appropriate relative concentration of actin, we incubated 5 nM DNO in RPMI + 6 mM Mg2+ + 10% FBS and 0–2048 nM actin. After 24 h at 37 °C, we observed by AGE that a 10-fold or greater molar excess of actin appears to be sufficient to inhibit digestion of nanostructures (Supporting Information Figure 9). The corresponding TEM image of intact DNO is comparable to a control, and both the NT and NR (Supporting Information Figure 10) were similarly protected, providing a potential alternative to heat inactivation.
Next we aimed to determine the effect of our various medium adjustments on the growth of three different cell lines: mouse embryonic fibroblast cells (3T3), human embryonic cells (HEK-293), and human adenocarcinoma cells (H441). These were first established in standard medium + 10% FBS for a minimum of 32 population doublings. They were then seeded at low density in medium modified by (1) adjustment to 6 mM Mg2+, (2) use of 75 °C heat-inactivated FBS, (3) inclusion of 200 nM actin, or (4) a combination of Mg2+ adjustment and heat-inactivated FBS or (5) actin. In cases where the Mg2+ concentration of the medium was adjusted, osmolarity was maintained by an equivalent dilution (∼5%). Cell content was measured on days 0, 1, 3, 5, and 7 after seeding using the CyQuant cell proliferation assay, which measures fluorescence of a cell nucleus stain (Figure 3a–c), and the change over time was normalized to day 0. Heat treatment of FBS produced a consistent, detrimental impact on growth, with all three cell lines showing significantly reduced fluorescent signal on day 7 relative to standard medium (p < 0.05). Adjustment of medium to 6 mM Mg2+ had a cell-line-dependent impact on growth. The 3T3 cells produced a significantly lower signal by day 7 (p < 0.01), whereas HEK-293 and H441 fluorescence were similar to that of the control medium (p > 0.05). The combination of Mg2+ adjustment and FBS heat inactivation resulted in slower cell growth with only the 3T3 cells (p < 0.01). Inclusion of 200 nM actin with and without Mg2+ adjustment produced no significant differences in signal by day 7 for all three lines (p > 0.05). Taken together, our data suggest that in vitro mammalian cell growth may not be sensitive to adjustment of Mg2+ medium up to a concentration of at least 6 mM and to the presence of actin at 200 nM. This tolerance to a Mg2+ concentration higher than that found in blood (<1 mM) is surprising, but some established media such as EGM-2 used for human umbilical vein endothelial cell (HUVEC) tissue culture contain nonphysiological concentrations of Mg2+ (10 mM). Heat inactivation of FBS for nuclease removal does not appear to be compatible with normal cell growth. It likely causes denaturation of a substantial fraction of serum proteins, leading to a state of serum-starved quiescence or cell death. The impact of this would be greatest when cells are seeded at a low density, as they were in this experiment to avoid them reaching confluence before day 7. Primary cells such as 3T3 fibroblasts may also be more sensitive to a low nutrient environment than cancer cell lines (e.g., H441), which are generally more robust owing to autocrine signaling and previous selective pressure for nutrient-deprived survival.
Figure 3.

Measuring the impact of medium modifications on cell growth, viability and phenotype. (a) Mouse 3T3 fibroblasts, (b) human HEK-293 embryonic kidney, and (c) human H441 adenocarcinoma cell lines were seeded into standard medium (dark blue line), Mg2+-adjusted (red line), heat-inactivated FBS (green line), Mg2+ + heat-inactivated FBS (purple line), 200 nM actin (light blue line), or combination Mg2+ + 200 nM actin medium (orange line). Change in cell number was estimated by fluorescence of CyQuant stain over time and was normalized to signal on day 0. Cell viability and metabolism of (d) 3T3, (e) HEK-293, and (f) H441 cells were profiled after 24 h incubation with and without 5 nM DNO. (g) Primary mouse immune cells were isolated from spleens and incubated with 1 nM DNO, an equivalent mass of CpG phosphorothioate oligonucleotide adjuvant, or medium only, and the concentration of IL-6 cytokine released into the supernatant was determined by ELISA. (h) 3T3 fibroblast cells were incubated for 16 h with 1 nM fluorescently labeled DNO or transferrin, and endocytosis of the two agents was measured by flow cytometry after extensive washing to remove surface-bound particles. (d–h) Culture conditions are standard medium (blue), medium + 6 mM Mg2+ + heat-inactivated FBS (red), or medium + 6 mM Mg2+ + FBS + 200 nM actin (yellow). (a–h) Error bars represent standard error of the mean of n = 5 replicates.
To cross-validate our cell growth data, we used the CellTiter-Glo luminescence assay to quantitate ATP concentrations in medium following a 16 h incubation of the three cell lines (Figure 3d–f). The medium concentration of ATP and corresponding luminescent signal correlates linearly with the number of metabolically active, viable cells in culture. Measurements were taken after incubation in standard medium or in the two adjusted conditions that we have identified as supportive of nanostructure integrity: 6 mM Mg2+ + 10% heat-inactivated FBS or 6 mM Mg2+ + 10% FBS + 200 nM actin. For each condition, we included DNO-positive (5 nM) and -negative treatments to investigate whether the presence of DNA nanostructures may be toxic to these cell lines. Consistent with the cell growth CyQuant data, the 3T3 line displayed sensitivity to heat inactivation of FBS, with DNO-positive and -negative treatments reduced to ∼75% of the luminescent signal observed for control DNO-negative standard medium (p < 0.01). No difference was observed with the actin medium for the 3T3 cells. Neither the H441 nor HEK-293 cells showed reduced luminescence in the two adjusted medium conditions, suggesting that these combinations of medium and cells could be used for short assays without impacting cell metabolism and viability. As well, the presence of 5 nM DNO did not independently reduce luminescence in any of the medium conditions, suggesting that this combination of nanostructure and assay conditions may not be toxic to these specific cell lines.
We next aimed to measure the impact of medium adjustments on cell phenotype using two assays relevant to nanomedicine. In a first assay, we examined how medium adjustment and nanostructure integrity impact results of immune cell activation by DNA nanostructures (Figure 3g). Immune cells have previously been shown to elicit an inflammatory cytokine response to DNA origami nanostructures, including release of the cytokine interleukin-6 (IL-6).16 Primary immune cells isolated from mouse spleens were washed extensively to remove inherent tissue nucleases, and we verified that nuclease-free medium incubated with splenocytes for 24 h does not regain nuclease activity capable of nanostructure digestion (Supporting Information Figure 11). We then incubated splenocytes for 16 h with 1 nM DNO in standard medium or the two adjusted medium conditions that support nanostructure integrity and measured the concentration of IL-6 released into the tissue culture supernatant via an ELISA assay. As positive and negative controls, we measured response to a nuclease-resistant DNA adjuvant (CpG 1826) or no activating agent, respectively, in the same panel of medium conditions. These controls showed that the cells were phenotypically normal for IL-6 response in all three medium conditions. IL-6 was produced at high levels in response to the CpG adjuvant, and negative control cells were not activated in the presence of actin or heat-inactivated FBS. The level of IL-6 produced in response to DNO (37.7 ± 3.7 pg/mL) and CpG (743.9 ± 85.8) was significantly reduced in adjusted medium with heat-inactivated FBS, in comparison to standard medium (94.8 ± 6.5 pg/mL for DNO and 1013.4 ± 50.2 pg/mL for CpG, p < 0.01). No significant differences were observed for either agent in adjusted medium with actin (81.9 ± 5.4 pg/mL for DNO and 862.7 ± 49.5 pg/mL for CpG, p > 0.05), suggesting that this medium environment should produce reliable IL-6 response data. Yet having demonstrated that our DNO is not stable in standard medium, we might expect a meaningful difference in immune cell response when nanostructures are intact (i.e., in adjusted medium with actin) compared to standard medium where we expect nanostructure instability. Other work from our laboratory has found that the response of splenocytes to DNA nanostructures is dependent on the total mass of DNA present and not on their design or integrity (data not shown). Therefore, the results from this assay demonstrate that adjusted medium with actin may be a good choice for maintaining normal cell phenotype and nanostructure integrity. Perhaps more importantly, this assay shows that nanostructure integrity cannot be a priori assumed and indeed may lead to an incorrect interpretation of results.
In a second assay and final experiment, we determined how adjusted medium and nanostructure integrity can impact the results of particle endocytosis (Figure 3h). We incubated 3T3 fibroblast cells with 1 nM DNO fluorescently labeled by Cy5-coupled oligonucleotides. A 16 h incubation was carried out in standard medium or the two adjusted medium conditions that support nanostructure integrity, and uptake was measured via flow cytometry. Similar to the immune-response assay, we verified that medium enriched through incubation with 3T3 cells does not display nuclease activity capable of digesting the DNO (data not shown). As a positive control, we used fluorescently labeled transferrin. In comparison to standard culture medium (637.8 ± 10.4 afu), uptake of transferrin was similar in medium + 6 mM Mg2+ + actin (631.6 ± 6.8 afu, p > 0.05) but lower in medium + 6 mM Mg2+ + heat-inactivated FBS (520.2 ± 29.7 afu, p < 0.01). This was not surprising based on our earlier observation that 3T3 growth and metabolism are decreased in medium with heat-inactivated FBS. After the DNO was incubated, we observed that 3T3 fluorescence was lowest in medium + 6 mM Mg2+ + heat-inactivated FBS (446.6 ± 96.5 afu), but the difference relative to standard medium was not meaningful (704.8 ± 38.3 afu, p > 0.05). More remarkably, fluorescence of cells in medium + 6 mM Mg2+ + actin (1230.2 ± 73.8 afu) was 75% higher than in standard medium (p < 0.01). On the basis of our earlier analysis, we would expect the growth and metabolism of 3T3 cells to be similar in standard medium and adjusted medium with actin. This increase in uptake might therefore be due to a difference in DNO integrity between the two conditions and would suggest that uptake of intact nanostructures may be higher than Cy5-labeled debris that would result from denaturation and digestion of DNO in standard medium. Therefore, this endocytosis assay shows a drastic difference in experimental outcome when medium is supportive of normal cell phenotype and nanostructure integrity, unlike what was observed from immune activation.
DNA nanotechnology offers unique and exciting advantages that may contribute to important advances in nanomedicine. However, this in vitro study and our earlier in vivo(12) study have shown that DNA nanostructures produced via the origami method are susceptible to denaturation and digestion in biological environments. Previous studies have probed the stability of DNA nanostructures exposed to cell lysate32 and after injection into the lumen of Caenorhabditis elegans.33 DNA origami nanostructures exposed to cell lysate were found to remain largely intact, which is in contrast to our findings in cell culture medium. The intracellular compartment has high total Mg2+ content of 5–20 mM.34 Much of this is associated with proteins and so should be unavailable, but the cell lysate was generated using sodium dodecyl sulfate, which may have denatured proteins (including nucleases) and released ionic Mg2+ into solution. The nanostructures tested in C. elegans were smaller and so are not directly comparable to origami particles but may be less susceptible to Mg2+ depletion and denaturation. Significant nuclease activity was not observed in either case. This perhaps suggests that not all biomedical applications of DNA nanotechnology will face the same challenges to nanostructure integrity, and that systemic injection of origami-based particles into mammalian systems may be one of the most difficult.
Conclusions
Researchers designing nanodevices for biomedical applications should consider what degree of protection strategies, such as compartmentalization in a lipid membrane,12 is required to maintain integrity within the biological environment of interest. Even without protection strategies, in vitro tissue culture studies are highly useful for development and prototyping of DNA nanodevices and their functions, such as in the case of the nanorobot.17 On the basis of this rationale, we have identified and characterized the sources of DNA nanostructure instability within the tissue culture environment and demonstrated methods to overcome them. Our data show that DNA nanostructures can be sensitive to depletion of Mg2+ in tissue culture medium, causing denaturation in a design-dependent manner, and that they are digested by nucleases present in FBS used as a medium supplement.
Avoidance of FBS nuclease activity is perhaps the easier challenge to overcome. Our experience has been that the level of nuclease activity present in tissue culture FBS is variable and disappears over days to weeks when prepared medium is stored at 4 °C. High levels of nuclease activity may therefore only be a concern for a short period following preparation of medium from freshly thawed FBS. We found that medium supplemented with low volumes of FBS (1–2.5%) had little capacity to digest a 5 nM concentration of DNA nanostructure over 24 h, suggesting that assays requiring short incubations could make use of low serum concentrations to avoid significant nuclease activity. This may slow but not drastically alter cell metabolism and phenotype. Various serum-free media are also available from vendors for specific cell types. In any case where FBS nuclease activity is a concern, our data support the use of actin as an inhibitor. We did not observe any difference in cell growth, viability, or phenotype when actin was included in medium at 200 nM. In contrast, heat inactivation of nuclease activity using a 5 min, 75 °C treatment of FBS had a consistently detrimental impact on cells and on results of murine immune cell activation and 3T3 fibroblast endocytosis assays. Heat inactivation using these conditions does not therefore appear to be a viable option.
The denaturation of DNA origami nanostructures due to depletion of divalent cations is a major challenge to in vitro and in vivo applications. We observed that the sensitivity of nanostructures to cation depletion is design- and time-dependent. Of the four designs that we tested, only one (DNA nanotube) appeared to be stable in physiological cation concentrations. Adjustment of Mg2+ medium concentration to ∼1 order of magnitude higher than that of the physiological conditions was able to prevent denaturation of the DNO, NR, and DNA nanorobot. Surprisingly, we observed little impact of Mg2+ adjustment on the growth and viability of mouse 3T3 fibroblast cells, human H441 adenocarcinoma, and HEK-293 embryonic kidney cells. Similarly, the phenotype of primary mouse splenocyte immune cells and 3T3 fibroblast cells was not sensitive to higher Mg2+ concentrations during immune activation and uptake assays, respectively. This, and the fact that other examples of high Mg2+ standard medium are well-established (e.g., EGM-2 for HUVECs), suggests that many mammalian cell types may not be sensitive to higher than physiological extracellular Mg2+ concentrations. Addition of MgSO4 to tissue culture medium, and an equivalent dilution to prevent osmotic shock, thus appears to be a potentially viable and perhaps generalizable approach for maintaining DNA nanostructure integrity during in vitro experiments.
In any case of tissue culture medium modification, it is strongly recommended that extensive testing and appropriate controls with standard medium are used to ensure that the cell property being investigated is not sensitive to the adjustments. Similarly, the data presented in this study reinforce the need for more rigorous testing and verification of nanostructure integrity in applications involving biological environments.
The medium adjustments tested here will suffice for some in vitro prototyping studies and applications of DNA nanotechnology. However, ultimately, many DNA nanotechnology-based diagnostic and therapeutic devices will be developed toward in vivo use or in biological conditions (e.g., blood, urine) that may not be easily modified. Future studies could therefore aim to develop DNA nanostructure protection strategies that obviate the need to alter their environment.
Materials and Methods
Chemicals and Supplies
Endotoxin test cartridges (0.05–5.0 EU/mL) were purchased from Charles River. Accugene 10× TBE buffer, PCR tubes, and 96-well PCR plates (Axygen) were purchased from VWR. SYBR Safe stain and CyQuant direct cell proliferation assay were purchased from Life Technologies Corporation. Agarose was purchased from Lonza. Glycerol, Tris base, EDTA, Triton X-114, Tween20, magnesium chloride, magnesium sulfate, and sodium chloride were purchased from Sigma-Aldrich. Actin was purchased from Cytoskeleton Inc. RPMI, DMEM, PBS, FBS, and penicillin-streptomycin were purchased from Gibco. Carbon Formvar grids and uranyl formate were purchased from Electron Microscopy Sciences. Amicon Ultra filtration devices and Seton ultracentrifugation tubes were purchased from Fisher Scientific. DNA gel extraction spin column was purchased from Bio-Rad.
M13 Scaffold Prep, Endotoxin Removal, Nanostructure Synthesis, and Purification
The four sets of design-specific staple strands were purchased reverse-phase-purified from Bioneer or Life Technologies Corporation. The fluorescent Cy5-coupled oligonucleotide feature (5′-GTGAGTTGTGGTAGATAATTT-3′) was synthesized by IDT Technologies. The p7308 scaffold strand was produced from M13 phage replication in Escherichia coli, as described previously.26
The scaffold strand was endotoxin-purified using Triton X-114. In brief, surfactant was added to scaffold stock to a final concentration of 2% (v/v). This was incubated at 4 °C on an inversion mixer for 30 min to solubilize endotoxin. The solution was mixed at 37 °C for 5 min to cause phase separation and then centrifuged at 37 °C for 30 min at maximum speed. The top aqueous fraction was transferred to a new tube. This was repeated four times to reduce endotoxin in the scaffold stock solution to acceptable levels of less than 5 EU/mL and quantified using the Endosafe-PTS system and test cartridges (Charles River).
Nanostructure synthesis was carried out by mixing 50 nM M13 scaffold with a 5× excess of staples in TE + 14 mM (DNO), 20 mM (NT), 18 mM (NR), or 8 mM (nanorobot) MgCl2. The solutions were subjected to a thermal annealing ramp on a Tetrad 2 Peltier thermal cycler (Bio-Rad) according to the following schedule: 80 °C for 5 min, decrease to 65 °C at 5 min/°C, incubate at 65 °C for 20 min, and decrease to 25 °C at 20 min/°C. Solutions of folded DNA nanostructures were concentrated using a 30k MWCO Amicon Ultra centrifugal filter device (Millipore) and then purified by glycerol gradient ultracentrifugation.30 Following purification, the stock solution was diluted appropriately for TEM imaging to verify quality. The stock concentration was determined by UV absorbance at 260 nm on a Nanodrop spectrophotometer (Thermo Scientific) using disposable cuvettes (Sarstedt). Stock solutions were stored at 4 °C until use.
Agarose Gel Analysis
DNA nanostructures were analyzed by gel (1.5% agarose, 0.5× TBE 10 mM MgCl2, 1× SYBR Safe) electrophoresis with Thermo Scientific Owl B2 EasyCast Mini Gel System apparatus. The samples were loaded into the agarose gel and allowed to migrate for 4 h (running buffer: 0.5× TBE, 10 mM MgCl2; 4.3 V/cm). The gel was imaged with Typhoon FLA 9000 (GE Healthcare Life Sciences). To recover nanostructures, the bands were visualized with UV light and cut out from the gel. Extracted bands were crushed and placed into a DNA gel extraction spin column (Bio-Rad). Then, the nanostructure solution was recovered by centrifugation of the loaded column for 3 min at 4000g.
Negative-Stain Transmission Electron Microscopy
TEM imaging was carried out by dropping 3.5 μL of product onto a plasma-treated carbon Formvar grid (Electron Microscopy Sciences). This was incubated for 1 min. The solution was wicked away onto filter paper, and 3.5 μL of 2% uranyl formate (in H2O, w/v) was immediately added. This was incubated for 0.5 min and then wicked away by filter paper. Imaging was carried out on a JEOL 1400 transmission electron microscope.
Cation Depletion Assay
RPMI medium (Gibco) was modified to 0.7, 1.0, 2.0, 3.0, 4.0, 6.0, 8.0, and 10 mM Mg2+ by addition of MgSO4 from a 500 mM stock solution. Each nanostructure was adjusted to 150 ug/mL and then mixed with unmodified RPMI medium (0.4 mM Mg2+) or modified RPMI medium at a 1:10 ratio. The samples were incubated at 37 °C for 24 h on a Tetrad 2 Peltier thermal cycler (Bio-Rad) and analyzed using agarose gel electrophoresis (AGE) and TEM imaging.
Nuclease Digestion Kinetics Assay
RPMI medium (Gibco) was adjusted to 6 mM Mg2+ by addition of MgSO4 and supplemented with 10% freshly thawed FBS (Gibco; heat-inactivated at 56 °C by the vendor). Each nanostructure was adjusted to 150 μg/mL and mixed with modified RPMI at a 1:10 ratio. The samples were incubated at 37 °C for 0.25, 0.5, 1, 2, 4, 8, and 24 h on a Tetrad 2 Peltier thermal cycler (Bio-Rad) and crushed into ice to stop nuclease digestion. The nanostructures were immediately analyzed using AGE. Then, the bands were extracted to recover nanostructures, and the nanostructures were imaged under TEM.
Nuclease Digestion Quantitation Assay
RPMI medium (Gibco) was adjusted to 6 mM Mg2+ by addition of MgSO4 and supplemented with either 1.25–20% freshly thawed FBS (Gibco; heat-inactivated at 56 °C by the vendor) or 1–4096 U/L of DNase I (New England Biolabs). The DNO was adjusted to 150 μg/mL and mixed with each medium condition in a 1:10 ratio. These were incubated for 12 h at 37 °C, and the products were analyzed by AGE.
Serum Heat Inactvation Assay
Freshly thawed FBS (Gibco) was heat-treated at 75 °C for 0.25, 0.5. 1, 2, 5, and 10 min in 1 mL aliquots on an Eppendorf thermoshaker with rapid shaking. RPMI medium (Gibco) was adjusted to 6 mM Mg2+ by addition of MgSO4 and supplemented with 10% freshly thawed FBS or heat-treated FBS. Each nanostructure was adjusted to 150 μg/mL and mixed with modified RPMI at a 1:10 ratio. The samples were incubated at 37 °C for 24 h on a Tetrad 2 Peltier thermal cycler (Bio-Rad) and crushed into ice to stop nuclease digestion. The nanostructures were analyzed using AGE. Then, the bands were extracted to recover nanostructures, and the nanostructures were imaged under TEM.
Actin Nuclease Inhihbition Assay
RPMI medium (Gibco) was adjusted to 6 mM Mg2+ by addition of MgSO4 and supplemented with 10% freshly thawed FBS (Gibco; heat-inactivated at 56 °C by the vendor). A dilution series of actin protein was prepared and added to the medium. A 150 μg/mL solution of each nanostructure was prepared and mixed with modified RPMI + actin at a 1:10 ratio. The samples were incubated at 37 °C for 24 h on a Tetrad 2 Peltier thermal cycler (Bio-Rad) and crushed into ice to stop nuclease digestion. The nanostructures were analyzed using AGE. Then, the bands were extracted to recover nanostructures, and the nanostructures were imaged under TEM.
Cell Proliferation Assay
To prepare medium with 6 mM Mg2+, equi-osmolar MgSO4 solution was prepared and added to RPMI and DMEM media (149 and 175 mOsM, respectively). Heat-inactivated FBS was prepared by incubation at 75 °C for 5 min in 1 mL aliquots. 3T3 and HEK-293 cells were grown in DMEM medium, and H441 cells were grown in RPMI medium. Cells were grown to confluency in appropriate medium supplemented with 10% freshly thawed FBS (Gibco). 3T3 (2.5 × 103 cells/mL), HEK-293 (1 × 104 cells/mL), and H441 (1.5 × 104 cells/mL) cells were seeded by adding 200 μL of cell suspension into the wells (n = 5) of a 96-well plate for each time point (0, 1, 3, 5, or 7 days after seeding), in modified medium prepared as follows: medium + 10% FBS, medium + 10% FBS + 6 mM Mg2+, medium + 10% heat-inactivated FBS, medium + 10% heat-inactivated FBS + 6 mM Mg2+, medium + 10% FBS + 200 nM actin, and medium + 10% FBS + 200 nM actin + 6 mM Mg2+. At each time point, cell proliferation was measured by CyQuant Direct Cell proliferation assay (Life Technologies) following the vendor protocol exactly. Fluorescence was measured on a BioTek NEO HTS plate reader with excitation at 480 nm and emission at 530 nm.
Cell Viability Assay
The same methods from the cell proliferation assay were used to modify medium. Each cell line was seeded by adding 100 μL of 105 cells/mL cell suspension into wells (n = 5) of a 96-well plate for control or experimental groups in the following medium conditions: medium + 10% FBS, medium + 10% heat-inactivated FBS + 6 mM Mg2+, and medium + 10% FBS + 200 nM actin + 6 mM Mg2+. Five microliters of 100 ng/μL DNO was added to the experimental group after overnight incubation to allow cell attachment. Twenty-four hours following addition of the nanostructure, cell viability was measured by CellTiter-Glo Luminescent cell viability assay (Promega) following the vendor protocol exactly. Luminescence signal was measured on a BioTek NEO HTS plate reader.
Splenocyte Activation Assay
Standard and adjusted medium was prepared as described above. Spleens were obtained from female 8 week old C57Bl/6 mice (Charles River). Two spleens were processed for this experiment. They were transferred into 70 μm cell strainers (BD Falcon) and were dissociated using a sterile syringe plunger. The single cell suspension was washed with 25 mL of PBS (Gibco) into a Petri dish. The suspension was transferred to a 50 mL Falcon tube, centrifuged at 500g for 5 min, and the supernatant discarded. A total of three washes were used to remove nuclease activity carried over from the tissue. Two milliliters of ACK lysing buffer (Lonza) was added, and the cells were gently resuspended and incubated for 7 min. Following this, 20 mL of PBS and 20 mL of RPMI medium were added, and the suspension was centrifuged as above. Cell concentration was determined, and 1 × 106 live cells were transferred in 450 μL of the various media to 5× wells of a 48-well plate for each agent to be assayed (DNO, CpG adjuvant, nonactivated control). Fifty microliter of 10 nM DNO or 50 μg/mL CpG oligonucleotide 1826 (InvivoGen) was transferred into the wells (n = 5). These were incubated for 16 h in a CO2 incubator at 37 °C. Supernatants were removed and centrifuged for 10 min at 500g, transferred to new tubes, and assayed for IL-6 concentration immediately by ELISA (R&D Systems).
Endocytosis Assay
3T3 fibroblast cells were first grown in standard culture medium to establish normal growth. The cells were then trypsinized, collected, and washed three times via centrifugation at 500g for 5 min with PBS to remove any nuclease present in the culture medium. Then, 1 × 104 cells were transferred into the various medium conditions and seeded into wells of a 96-well plate. After 24 h to allow cell attachment, 1 nM of Cy5-labeled DNO or FITC-labeled transferrin (Life Technologies) was added. This was incubated for 16 h. Cells were then washed twice with ice-cold PBS and incubated with glycine-HCL (50 mM) in PBS (pH 2.8) for 10 min at room temperature to dissociate surface-bound particles. The cells were resuspended in standard PBS and analyzed by flow cytometry on a LSRFortessa (BD), measuring 10 000 events/sample (n = 5). Gating was performed on the cells by gating upon the forward scatter and side scatter plot. Identical gates were applied to all samples. After gating, a negative population was defined using the histogram obtained for medium-only negative control samples. Samples that showed a rightward peak shift were determined as positive, and median values of each sample population were reported.
Statistics
ANOVA with post-hoc Dunnet’s or Tukey’s tests were performed using an excel plug-in, inerSTAT-a v1.3 by Mario H. Vargas (Instituto Nacional de Enfermeda Respiratorias, Mexico).
Acknowledgments
J.H. holds a National Science Foundation Graduate Research Fellowship, S.D.P. holds a Canadian Institutes of Health Research Fellowship and a Wyss Institute Technology Development Fellowship, and this work was supported by an NIH grant to W.M.S. and by the Wyss Institute at Harvard. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. We thank the Wyss Institute for support of this project.
Supporting Information Available
Nanostructure design diagrams and staple sequences, as well as additional data on endotoxin removal from the M13 scaffold and nanostructure stability in various conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): SDP and WMS own a patent covering the protection of DNA nanostructures for use in biological environments.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Seeman N. C. Nanomaterials Based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA To Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Högberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H.; Douglas S. M.; Shih W. M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedl T.; Högberg B.; Tytell J.; Ingber D. E.; Shih W. M. Self-Assembly of Three-Dimensional Prestressed Tensegrity Structures from DNA. Nat. Nanotechnol. 2010, 5, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C.; Sun W.; Shen Z.; Seeman N. C. A Nanomechanical Device Based on the B-Z Transition of DNA. Nature 1999, 18, 2531–2537. [DOI] [PubMed] [Google Scholar]
- Seelig G.; Soloveichik D.; Zhang D. Y.; Winfree E. Enzyme-Free Nucleic Acid Logic Circuits. Science 2006, 314, 1585–1588. [DOI] [PubMed] [Google Scholar]
- Dutta P. K.; Varghese R.; Nangreave J.; Lin S.; Yan H.; Liu Y. DNA-Directed Artificial Light-Harvesting Antenna. J. Am. Chem. Soc. 2011, 133, 11985–11993. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Lin C.; Li H.; Yan H. Aptamer-Directed Self-Assembly of Protein Arrays on a DNA Nanostructure. Angew. Chem. 2005, 117, 4407–4412. [DOI] [PubMed] [Google Scholar]
- He Y.; Tian Y.; Ribbe A. E.; Mao C. Antibody Nanoarrays with a Pitch of 20 Nanometers. J. Am. Chem. Soc. 2006, 128, 12664–12665. [DOI] [PubMed] [Google Scholar]
- Schreiber R.; Do J.; Roller E.-M.; Zhang T.; Schüller V. J.; Nickels P. C.; Feldmann J.; Liedl T. Hierarchical Assembly of Metal Nanoparticles, Quantum Dots and Organic Dyes Using DNA Origami Scaffolds. Nat. Nanotechnol. 2013, 9, 74–78. [DOI] [PubMed] [Google Scholar]
- Perrault S. D.; Shih W. M. Virus-Inspired Membrane Encapsulation of DNA Nanostructures To Achieve In Vivo Stability. ACS Nano 2014, 8, 5132–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A.; Lundin V.; Petrova E.; Fördős F.; Benson E.; Al-Amin A.; Herland A.; Blokzijl A.; Högberg B.; Teixeira A. I. Spatial Control of Membrane Receptor Function Using Ligand Nanocalipers. Nat. Methods 2014, 11, 841–846. [DOI] [PubMed] [Google Scholar]
- Rudchenko M.; Taylor S.; Pallavi P.; Dechkovskaia A.; Khan S.; Butler V. P. Jr.; Rudchenko S.; Stojanovic M. N. Autonomous Molecular Cascades for Evaluation of Cell Surfaces. Nat. Nanotechnol. 2013, 8, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langecker M.; Arnaut V.; Martin T. G.; List J.; Renner S.; Mayer M.; Dietz H.; Simmel F. C. Synthetic Lipid Membrane Channels Formed by Designed DNA Nanostructures. Science 2012, 338, 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller V. J.; Heidegger S.; Sandholzer N.; Nickels P. C.; Suhartha N. A.; Endres S.; Bourquin C.; Liedl T. Cellular Immunostimulation by CpG-Sequence-Coated DNA Origami Structures. ACS Nano 2011, 5, 9696–9702. [DOI] [PubMed] [Google Scholar]
- Church G. M.; Douglas S. M.; Bachelet I. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 2012, 335, 831–834. [DOI] [PubMed] [Google Scholar]
- Miyauchi K.; Ogawa M.; Shibata T.; Matsuda K.; Mori T.; Ito K.; Minamiura N.; Yamamoto T. Development of a Radioimmunoassay for Human Deoxyribonuclease I. Clin. Chim. Acta 1986, 154, 115–123. [DOI] [PubMed] [Google Scholar]
- Koizumi T. Tissue Distribution of Deoxyribonuclease I (DNase I) Activity Level in Mice and Its Sexual Dimorphism. Exp. Anim. 1995, 44, 181–185. [DOI] [PubMed] [Google Scholar]
- Koizumi T. Deoxyribonuclease II (DNase II) Activity in Mouse Tissues and Body Fluids. Exp. Anim. 1995, 44, 169–171. [DOI] [PubMed] [Google Scholar]
- Houk B.; Hochhaus G.; Hughes J. Kinetic Modeling of Plasmid DNA Degradation in Rat Plasma. AAPS J. 1999, 1, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata K.; Takakura Y.; Hashida M. The Fate of Plasmid DNA after Intravenous Injection in Mice: Involvement of Scavenger Receptors in Its Hepatic Uptake. Pharm. Res. 1995, 12, 825–830. [DOI] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D.-N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8, 221–229. [DOI] [PubMed] [Google Scholar]
- Keum J.-W.; Bermudez H. Enhanced Resistance of DNA Nanostructures to Enzymatic Digestion. Chem. Commun. 2009, 7036–7038. [DOI] [PubMed] [Google Scholar]
- Lu M.; Guo Q.; Seeman N. C.; Kallenbach N. R. DNase I Cleavage of Branched DNA Molecules. J. Biol. Chem. 1989, 264, 20851. [PubMed] [Google Scholar]
- Douglas S. M.; Chou J. J.; Shih W. M. DNA-Nanotube-Induced Alignment of Membrane Proteins for NMR Structure Determination. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu F.; Liao S.; Kopatsch J.; Wang T.; Mao C.; Seeman N. C. Six-Helix Bundles Designed from DNA. Nano Lett. 2005, 5, 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu T. J.; Seeman N. C. DNA Double-Crossover Molecules. Biochemistry (Moscow) 1993, 32, 3211–3220. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang X.; Qi J.; Seeman N. C. Antiparallel DNA Double Crossover Molecules as Components for Nanoconstruction. J. Am. Chem. Soc. 1996, 118, 6131–6140. [Google Scholar]
- Lin C.; Perrault S. D.; Kwak M.; Graf F.; Shih W. M. Purification of DNA-Origami Nanostructures by Rate-Zonal Centrifugation. Nucleic Acids Res. 2013, 41, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarides E.; Lindberg U. Actin Is the Naturally Occurring Inhibitor of Deoxyribonuclease I. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 4742–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Q.; Wei X.; Su F.; Liu Y.; Youngbull C.; Johnson R.; Lindsay S.; Yan H.; Meldrum D. Stability of DNA Origami Nanoarrays in Cell Lysate. Nano Lett. 2011, 11, 1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana S.; Bhatia D.; Krishnan Y. A Method To Study In Vivo Stability of DNA Nanostructures. Methods 2013, 64, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnen-Dechent W.; Ketteler M. Magnesium Basics. Clin. Kidney J. 2012, 5, i3–i14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.