

Abstract
Background
Perchlorate contamination has been detected in both ground water and drinking water. An attractive treatment option is the use of ion-exchange to remove and concentrate perchlorate in brine. Biological treatment can subsequently remove the perchlorate from the brine. When nitrate is present, it will also be concentrated in the brine and must also be removed by biological treatment. The primary objective was to obtain an in-depth characterization of the microbial populations of two salt-tolerant cultures each of which is capable of metabolizing perchlorate. The cultures were derived from a single ancestral culture and have been maintained in the laboratory for more than 10 years. One culture was fed perchlorate only, while the other was fed both perchlorate and nitrate.
Results
A metagenomic characterization was performed using Illumina DNA sequencing technology, and the 16S rDNA of several pure strains isolated from the mixed cultures were sequenced. In the absence of nitrate, members of the Rhodobacteraceae constituted the prevailing taxonomic group. Second in abundance were the Rhodocyclaceae. In the nitrate fed culture, the Rhodobacteraceae are essentially absent. They are replaced by a major expansion of the Rhodocyclaceae and the emergence of the Alteromonadaceae as a significant community member. Gene sequences exhibiting significant homology to known perchlorate and nitrate reduction enzymes were found in both cultures.
Conclusions
The structure of the two microbial ecosystems of interest has been established and some representative strains obtained in pure culture. The results illustrate that under favorable conditions a group of organisms can readily dominate an ecosystem and yet be effectively eliminated when their advantage is lost. Almost all known perchlorate-reducing organisms can also effectively reduce nitrate. This is certainly not the case for the Rhodobacteraceae that were found to dominate in the absence of nitrate, but effectively disappeared in its presence. This study is significant in that it reveals the existence of a novel group of organisms that play a role in the reduction of perchlorate under saline conditions. These Rhodobacteraceae especially, as well as other organisms present in these communities may be a promising source of unique salt-tolerant enzymes for perchlorate reduction.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0225-3) contains supplementary material, which is available to authorized users.
Keywords: Perchlorate contamination, Nitrate, Metagenomic sequencing, Community analysis, Salt-tolerance
Background
The perchlorate ion has been found to contaminate ground and surface water, as well as food, vitamins, and drinking water [1-5]. Perchlorate is known to inhibit the sodium-iodide symporter, which is responsible for the supply of iodine to the thyroid and to milk in the mammary glands [6].
The environmentally stable perchlorate ion can be removed from contaminated water using ion-exchange in combination with a microbial treatment processes. During bacterial respiration perchlorate acts as an electron acceptor and requires the addition of an electron donor [7-9]. Perchlorate is metabolized through a series of reductions to chlorite and then a dismutation to chloride and molecular oxygen [10]. In many cases, perchlorate is present in very low concentrations and must be treated to even lower levels, e.g., 6 ppb in California [11], and 2 ppb in Massachusetts [12]. This makes biological treatment very difficult as a stand-alone process since other electron acceptors such as nitrate and oxygen are often preferred.
Ion-exchange processes have been used to remove perchlorate to meet the required treatment goals, but the perchlorate and other co-contaminant anions such as nitrate are merely transferred to a concentrated brine stream, which in turn must be treated [13,14]. The combination of ion-exchange to remove and concentrate the perchlorate from the contaminated water source and biological treatment to remove the perchlorate from the brine is an attractive option for drinking water treatment. Since nitrate is often present with perchlorate and will also be concentrated in the ion-exchange brine, the biological system should also remove nitrate.
The cultures examined here were initially enriched from marine sediment in the year 2001. The culture designated as P30 has been maintained continuously in the laboratory in a defined 3% NaCl saline medium with perchlorate and acetate as the sole electron acceptor and donor respectively in a 1.5 L batch reactor [15]. In 2003, half of the P30 culture was transferred to a new reactor and fed with both nitrate and perchlorate as the electron acceptors and acetate as the sole electron donor. This culture was designated as NP30. The cultures have been used as the basis for many published studies over the last 10 years [13,15-23].
In order to better understand these cultures, an initial characterization of the microbial populations was obtained by cloning 16S rDNA genes and characterizing them by either DGGE or 16S rDNA sequencing [21]. In the work reported here, we further our understanding of the biota in these cultures by characterizing the total community DNA of both the P30 and NP30 cultures using Illumina DNA sequencing technology. This metagenome analysis is supplemented by sequencing the 16S rRNA genes of several strains that have been isolated from the cultures and successfully cultivated in the laboratory. The combination of these two approaches provides new insight into the major and minor components of the cultures and their contribution to perchlorate and nitrate removal from contaminated water.
Results
Pure culture isolation
Four pure strains were obtained using anaerobic incubations: NW (white colonies) and NY (yellow colonies), originated from NP30; PW (white colonies) and PY (yellow colonies), originated from P30. Another four pure strains were obtained from aerobically incubated plates and designated as NWO, NYO, PWO and PYO following the naming scheme for anaerobic isolates. When these new strains were transferred back into P medium, none of them could reduce perchlorate after 120 days’ incubation, although growth was observed. A single additional isolate that can reduce perchlorate was obtained from the NP30 culture and characterized in detail as reported elsewhere [24]. This strain was identified as Marinobacter sp. strain P4B1 a member of the Alteromonadaceae. 16S rDNA sequences were obtained for all the pure strains except the NW isolate, which produced heterogeneous sequencing patterns. Table 1 presents a summary of the results obtained by comparing the sequences obtained for the pure cultures with those from the NCBI nt database. All belong to the phylum Proteobacteria. Two of the NP30 strains were identified as members of the Rhodobacteraceae closely related to Stappia indica. The other was an Azoarcus like isolate close to the members of a recently discovered and vaguely defined genus Denitromonas (Rhodocyclaceae). Three of the five P30 isolates were identified as Hyphomonas jannaschiana. The remaining P30 isolate was identified as a Cellulomonas strain that belongs to the Cellulomonadaceae and is closely related to the C. hominis type strain.
Table 1.
Sequencing summary for 16S rDNA from pure cultures
| ID | Origin | Accession number | Length (bp) | Closest BLAST match in NCBI nt database*) | Identity (%) | Bit score | Taxonomic affiliation | |
|---|---|---|---|---|---|---|---|---|
| (excluding uncultured/environmental samples) | Family | Genus & Species | ||||||
| NWO | NP30 | KF135667 | 1340 | Stappia indica str. R32 (AB607882) | 99 | 2329 | Rhodobacteraceae | Stappia indica |
| NY | NP30 | KF135668 | 1379 | Denitromonas indolicum str. MPKc (AY972852), Denitromonas aromaticus (AB049763) | 95 | 2183 | Rhodocyclaceae | Denitromonas sp.*) |
| NYO | NP30 | KF135669 | 1343 | Stappia indica str. R32 (AB607882) | 99 | 2334 | Rhodobacteraceae | Stappia indica |
| PW | P30 | KF135670 | 1307 | Hyphomonas jannaschiana W6-15 (DQ659446) | 99 | 2331 | Hyphomonadaceae | Hyphomonas jannaschiana |
| PWO | P30 | KF135671 | 1297 | Hyphomonas jannaschiana W6-15 (DQ659446) | 99 | 2313 | Hyphomonadaceae | Hyphomonas jannaschiana |
| PY | P30 | KF135672 | 1373 | Cellulomonas sp. SJH-002 (KC335136) | 99 | 2435 | Cellulomonadaceae | Cellulomonas sp. |
| PYO | P30 | KF135673 | 1297 | Hyphomonas jannaschiana W6-15 (DQ659446) | 99 | 2313 | Hyphomonadaceae | Hyphomonas jannaschiana |
| P4B1 | NP30 | JN861074 | 1502 | Marinobacter sp. SCSWE03 (FJ461458) | 99 | 2513 | Alteromonadaceae | Marinobacter sp. |
*)“Denitromonas” is not a validly published genus; the closest properly defined genus is Azoarcus.
Comparative metagenomic analysis
Illumina sequencing of the two cultures yielded approximately 7.5 million reads for P30 and 6.3 million for NP30. Table 2 summarizes the assembly of the metagenomic contigs. The data analysis strategy yielded converging estimates for the taxonomic composition of the P30 and NP30 microbial communities at the family level and above (Additional files 1, 2 and 3). As shown in Figure 1, both mixed cultures were found to be dominated by Alpha, Beta, and Gamma Proteobacteria, while the proportion of other phyla is substantially lower. As shown in Figure 2, Alphaproteobacteria from the family Rhodobacteraceae constitute a prevailing taxonomic group in the P30 culture (Figure 2, Additional file 4). The second most abundant fraction is formed by Betaproteobacteria belonging to Rhodocyclaceae, which is followed by Gammaproteobacteria from families Chromatiaceae, Ectothiorhodospiraceae and Pseudomonadaceae. In the NP30 culture, Figure 2, the Rhodobacteraceae have effectively completely disappeared and are now replaced by an expanding representation from the Rhodocyclaceae and the emergence of the Alteromonadaceae at a much lower but increasingly significant fraction of the community. Other taxonomic groups found in the NP30 community include Gammaproteobacteria from the families Pseudomonadaceae, Ectothiorhodospiraceae and Chromatiaceae and Betaproteobacteria from families Burkholderiaceae and Comamonadaceae.
Table 2.
Summary of metagenomic contig assembly
| P30 culture | NP30 culture | |
|---|---|---|
| Number of 36-mer reads collected | 7,551,046 | 6,307,535 |
| Number of assembled contigs | 42,437 | 27,725 |
| Number of nucleotides in contigs > 100 nt | 9,850,121 | 5,030,930 |
| Longest sequence, nt | 3,421 | 5,099 |
| Mean, nt | 232 | 181 |
| N50, nt | 258 | 183 |
| Shortest sequence, nt | 100 | 100 |
Figure 1.
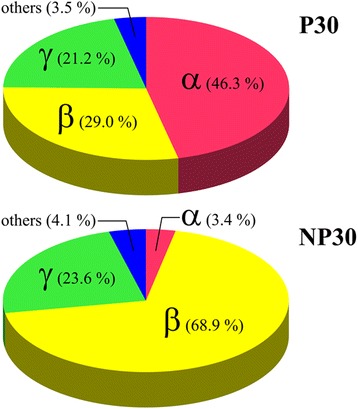
The relative abundance of members of the Alpha, Beta, and Gamma Proteobacter are shown in a pie chart with approximate percentages indicated. The dramatic decrease of the Alphaproteobacter in the NP30 culture and accompanying increase in the Betaproteobacter is clearly illustrated.
Figure 2.
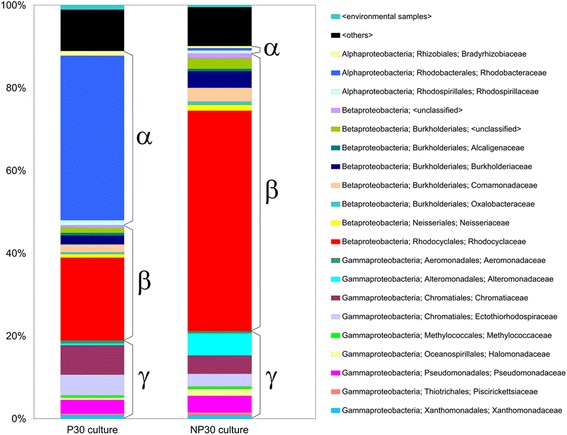
Relative abundance of major bacterial families in P30 and NP30 communities. Relative abundance was estimated using the ContigEval analysis pipeline. Height of the colored boxes represents a number of identities in all contigs assigned to a given taxon related to a number of identities in all contigs assigned to all taxa. The boxes are clustered by class. Names associated with each color are provided that list Class, Order, and Family. For each culture, only the most abundant families contributing to the 90% of cumulative percentage are shown. An alternative representation in which they are ordered by taxon abundance averaged for P30 and NP30 cultures is provided as Additional file 4.
Estimates of the P30 culture composition at the genus level vary depending on the employed data processing procedure. When the contigs were evaluated against NCBI nt nucleotide database (MEGAN, ContigEval), the highest scoring taxon was the genus Ruegeria (Rhodobacteraceae). However, when contig-derived protein sequences were searched against protein databases, the major portion of the hits was assigned to other members of the same family, either Roseovarius or Roseobacter. This discrepancy may reflect an influence of non-coding genomic sequences or codon usage pattern on the taxonomic assignment of the contigs. Other highly represented genera included Rhodobacter, Dinoroseobacter and Phaeobacter (Rhodobacteraceae), Azoarcus and Thauera (Rhodocyclaceae), Burkholderia (Burkholderiaceae), Pseudomonas (Pseudomonadaceae), Allochromatium (Chromatiaceae) and Thioalkalivibrio (Ectothiorhodospiraceae). While the abundance rank of the mentioned taxons depends on the contig binning method used, they can often be found among the top genera. The composition of the NP30 culture exhibits less ambiguity. The most abundant genera are Azoarcus and Thauera. They are followed by Marinobacter, Pseudomonas and, in some reconstructions, the Rhodocyclaceae genera Azospira and Dechloromonas. The Alphaproteobacteria genera that dominate the P30 culture are remarkably absent in the NP30 culture.
In addition to binning the Metavelvet-assembled contigs by similarity search in global sequence databases, taxonomic composition of the perchlorate-degrading cultures was assessed by cataloging 16S rRNA gene sequences extracted from Metavelvet and Oases assemblies of the metagenomic reads (Additional file 5, Additional file 6). The P30 culture featured 16S rDNA signatures related to the Alphaproteobacteria genus Roseovarius (Rhodobacteraceae), the Betaproteobacteria genera Azoarcus, Azospira, Thauera and Rhodocyclus (Rhodocyclaceae), the Gammaproteobacteria genus Marinobacter (Alteromonadaceae), and several Gammaproteobacteria genera of unclear phylogenetic relatedness (Sedimenticola, Thiobios and Thiodictyon). In the NP30 culture, 16S rDNA sequences belonging to the Rhodobacteraceae specifically and the Alphaproteobacteria in general were not observed. The Betaproteobacteria family Rhodocyclaceae was represented by all the above-mentioned genera with the addition of the genus Azonexus while the list of Gammaproteobacteria was expanded by addition of the genus Halomonas. The NP30 culture also exhibited signatures of Bacteroidetes and Spirochaetes, which were not seen in the P30 culture.
Searches of the metagenomic contigs for genes involved in perchlorate degradation revealed the presence of identical perchlorate reductase gene clusters, pcrABCD, and chlorite dismutase genes, cld, in both P30 and NP30 cultures. The recovered perchlorate reductase sequence showed highest similarity to an enzyme from Dechloromonas aromatica RCB, which is a member of the Rhodocyclaceae family. There is 64% sequence identity between the molybdopterin-bearing catalytic subunits (pcrA), 67% identity between the iron-sulfur subunits (pcrB), and 47% identity between the cytochrome-type subunits (pcrC). The closest homolog for the chlorite dismutase found in these cultures is the cld gene found in the Betaproteobacterium Ideonella dechloratans (47% identity). In view of the negative effect of nitrate addition on the abundance of members of the Rhodobacteraceae in the NP30 culture, the metagenomic contigs were searched for genes encoding nitrate-metabolyzing enzymes (Table 3, Additional file 7). It was found that only a few of the functionally annotated P30 and NP30 contigs with highest similarity to nitrate reductase genes are taxonomically affiliated with Alphaproteobacteria taxa including the family Rhodobacteraceae. On the other hand, the contigs similar to respiratory and dissimilatory nitrate reductase genes from Beta- and Gammaproteobacterial sources were observed in substantially larger numbers in both P30 and NP30 communities. This observation points to a relatively low nitrate-metabolizing potential of the Alphaproteobacteria fraction of the mixed cultures compared with that of the other taxonomic groups.
Table 3.
Taxonomic distribution of metagenomic contigs exhibiting highest similarity to nitrate reductase genes
| Annotation software | MG-RAST | MEGAN | ContigEval | |||
|---|---|---|---|---|---|---|
| Similarity search tool | Protein BLAT | BLASTX | BLASTN | |||
| Functional annotation source | GenBank | SEED | GenBank | |||
| P30 | NP30 | P30 | NP30 | P30 | NP30 | |
| Alphaproteobacteria | 5 | - | 4 | 1 | - | 1 |
| Betaproteobacteria | 15 | 16 | 12 | 7 | 28 | 18 |
| Gammaproteobacteria | 12 | 5 | 28 | 12 | 9 | 7 |
| Bacilli | 1 | - | - | - | - | - |
| Negativicutes | 1 | - | - | - | - | - |
| Undefined | - | - | 2 | 1 | 1 | - |
| Total number of contigs | 34 | 21 | 46 | 21 | 38 | 26 |
Discussion
The stability of microbial populations and function of cultures used for industrial wastewater treatment is an important consideration. It is therefore important to understand what the key organisms are in the P30 and NP30 cultures and ultimately how these populations are effected by changes in the feed brine. The ability to characterize and subsequently monitor culture composition is important for early recognition and treatment of undesirable changes. In this regard, the metagenomic characterization described here is much more informative than earlier methods such as DGGE [21]. Moreover, in light of ongoing advances in high throughput sequencing such as multiplexing and the use of paired end reads, the metagenomics approach is competitive in time and cost potentially allowing routine comprehensive monitoring of the communities at regular intervals in the future.
The communities examined here differ from those used in most other studies of perchlorate and nitrate reducing cultures due to their salt tolerance and longevity. They thus represent unique ecosystems. Comparing the results obtained in this study with the earlier characterization obtained with DGGE [21], it is noticed that the diversity of the cultures has likely decreased with time. Some of the prevailing genera of Gammaproteobacteria, such as Dechloromarinus in P30 and Acholeplasma-like species of the phylum Firmicutes in both P30 and NP30 observed in 2004 [21] have effectively disappeared in the present study. This change was probably due to the high salt concentrations (6% NaCl) occasionally fed to the two cultures, since similar changes were observed when the cultures were fed with ion-exchange brines [21,22]. The groups of Rhodocyclaceae and Rhodobacteraceae in P30 had been dominating since 2007 [21] and continue to dominate in this study (sampled in 2010). Members of the Gammaproteobateria (Marinobacter and Halomonas) observed in the NP30 culture in 2007 [21] are still observed in this study. However, their abundance decreased with members of the Betaproteobacteria (Azoarcus/Denitromonas) making up the difference. This may be because of the increased perchlorate concentrations in the NP30 feed that began in 2008. Consistent with this interpretation is the observation that the genus Denitromonas emerged to be an abundant member of the NP30 community when the community was fed with only perchlorate for one month [21]. The very defined media fed to the cultures has likely helped to maintain the long-term stability of the cultures, although, the change of the relative ratio of major electron acceptors could greatly change the community. Nevertheless, on the whole, given that the the P30 and NP30 communities are almost exclusively comprised of alpha, beta, and gamma proteobacteria they are far less diverse than what is encountered in natural environments such as soils.
Obtaining key organisms in pure culture will facilitate our understanding of the changes in biochemistry required to effectively utilize perchlorate in the presence or absence of nitrate in saline environments. In addition, such pure cultures could be used in practice to augment the mixed culture should the perchlorate-reducing bacteria be lost or diminished. The metagenomics approach has clarified the likely properties and known relatives of the key organisms in each culture. Thus, to the extent individual organisms can be isolated from the P30 and NP30 cultures, it is now possible to understand their significance in the context of the larger community.
Further effort is needed to obtain a complete set of representative cultures. Previously, a species of Marinobacter, a member of the Alteromonadaceae, was isolated from the NP30 culture and found to utilize perchlorate in the presence of nitrate in saline environments [24]. The metagenomic results confirm that this organism is likely a significant part of the NP30 community. In the present study, several additional isolates have been obtained from both cultures, but none degraded perchlorate.
An Azoarcus like Betaproteobacterium was isolated from the NP30 culture. It belongs to the Rhodocyclaceae that dominate the NP30 culture and are highly represented in the P30 culture. On the other hand, Hyphomonas sp. was not found in significant numbers in either culture in the metagenomic analysis. However, its distinct cellular morphology allowed it to be observed microscopically as a minor component of both cultures. The other two organisms obtained in pure culture, Stappia sp. and Cellulomonas sp., do not appear to be present in large amounts in either culture and may have been enriched from small numbers on the solid medium due to their ability to grow in low nutrient conditions. Thus, the isolation of the pure cultures was seemingly influenced by an ability of individual strains to proliferate under selected cultivation conditions in the absence of other members of the microbial communities rather than by their abundance in the mixed cultures.
The Marinobacter strain is the only isolate obtained to date that is capable of degrading perchlorate and nitrate simultaneously, although it prefers perchlorate as the electron acceptor [24]. In other studies, a Marinobacter species was found as the dominant organism in hydrogenotrophic membrane biofilm reactors (MBfRs) treating ion-exchange brines containing perchlorate [25,26]. Van Ginkel et al. [26] examined the microbial community in four H2-based MBfRs treating ion-exchange brines. Four different inocula including NP30 were used in that study. Except the freshwater inoculum, the other three salt-tolerant inocula in the reactors resulted in similar consortia, with members of the Gammaroteobacteria representing 95% and Alphaproteobacteria representing 0-4% of the total 16S rRNA gene clones. Among them, Marinobacter constituted 38-81% of the total clones. Even in the freshwater inoculated MBfR, Marinobacter represented 34% of the total clones. Since the nitrate fed to NP30 was more concentrated than perchlorate, Marinobacter would have a competitive advantage over organisms that could only degrade perchlorate or preferred nitrate to perchlorate. Its significant elevation in numbers in the NP30 cultures relative to the P30 cultures supports this argument. Less clear is why the Marinobacter are not present in large numbers in the P30 culture. Although Marinobacter can utilize perchlorate under saline conditions the simplest explanation is that the yet to be cultured Rhodobacteraceae species are simply better adapted to the saline environment. Previous studies [24] showed that Marinobacter sp. P4B1 grew much better at low salt (1.8%) than in the 3% concentration used here.
The genera Azoarcus and Thauera were consistently found in the metagenome studies as representatives of the Rhodocyclaceae that are major components of both cultures. These organisms are phylogenetically similar to each other and both are close relatives of the widely-known perchlorate-reducers of the Betaproteobacteria genera Dechloromonas and Azospira [27]. Members of Azoarcus and Thauera are known as denitrifiers capable of halobenzoate degradation [28]. In another study, Azoarcus was one of the major organisms in a toluene-degrading compost biofilter [29]. In a previous study of the NP30 microbial community by fluorescent in situ hybridization (FISH) [22], Azoarcus was proposed to be a major perchlorate reducer in NP30. However, the pure Azoarcus strain NY isolated from the NP30 culture in this study was not able to reduce perchlorate after over ~120 days’ incubation. Since nitrate was not available in the P30 mixed culture, the Azoarcus strain that was isolated need not be a denitrifier. Two ecologically different groups of Azoarcus exist: 1) soil-borne strains which can degrade aromatic hydrocarbons under denitrifying conditions, and 2) “plant-associated” strains which can fix nitrogen and require microaerobic conditions for growth on N2 [30]. If strain NY is truly representative of the community, then the members of the Azoarcus in P30 and NP30 may be the plant-associated type that are growing under microaerophilic conditions produced during perchlorate degradation.
Matches to Roseovarius, Rhodobacter, or Ruegeria were frequently found in the metagenome studies. Likewise, Rhodobacter sequences were consistently observed in DGGE analysis of the P30 culture as well [21]. Unfortunately, no members of the Rhodobacteraceae that dominate the P30 cultures have yet been obtained in pure culture. In the course of earlier FISH studies, Roseobacter accounted for only 0-5% of the total organisms in the NP30 culture [22]. This is consistent with the much reduced representation of the Rhodobacteraceae seen in the metagenome analysis of the NP30 culture described here.
The genus Stappia is also in the Rhodobacteraceae. Two Stappia strains NOW and NYO were isolated in the pure culture studies, but they were recovered unexpectedly from the NP30 culture where Rhodobacteraceae are present in small numbers. The genus was created from reclassification of marine Agrobacterium [31] and is closely related to Ruegeria. They usually form star-shaped-aggregates [32], which were observed in the isolated Stappia strain NW. The type species of Ruegeria, Ruegeria atlantica, could reduce nitrate to nitrite and to N2 gas [32], while some species of Ruegeria could only reduce nitrate to nitrite but not to N2 gas [31]. Neither of the pure strains, NWO and NYO, could reduce perchlorate.
Previous attempts to use real time PCR and other biochemical techniques from the literature to quantify and or isolate and purify enzymes associated with perchlorate and/or nitrate reduction were unsuccessful. The metagenome data allowed a preliminary search for such genes. Many, e.g. the perchlorate reductase genes belonging to the pcrABCD cluster and the chorite dismutase genes, were in fact found in both the P30 and NP30 cultures. However, the matches to known sequences were moderate to low, which likely explains earlier difficulty in amplifying them. This difference may be due to the saline environment.
Conclusions
The major components of the P30 and NP30 microbial communities have been established and several representative strains obtained in pure culture. Members of the Rhodobacteraceae dominate the P30 culture, but their numbers dramatically decrease in the NP-30 culture that is fed both nitrate and perchlorate. The Rhodobacteraceae are largely replaced in the NP30 culture with members of the Rhodocyclaceae and to a lesser extent the Altermonadaceae. Consistent with their common ancestry, the rest of the community remains similar in the two cultures. With the community structure established at the Family level, it will now be possible to attempt to culture additional key community members by utilizing published recipes and growth conditions of known members of the same families.
Although almost all known perchlorate-reducing organisms can effectively reduce nitrate, the Rhodobacteraceae that dominate the P30 culture apparently can’t and thus may represent a novel group of perchlorate reducers. These Rhodobacteraceae, as well as other organisms in these cultures, may be a promising source of unique salt-tolerant enzymes for perchlorate reduction.
Methods
Maintenance of P30 and NP30 cultures
The cultures were continuously maintained in the laboratory by spiking the electron acceptor and donor once a day along with weekly medium replacements using SBR procedures as described previously [22]. This provided a concentration in the culture of 100 mg/L sodium perchlorate (P30) or 100 mg/L perchlorate and 500 mg/L sodium nitrate (NP30). Beginning in 2008, the perchlorate fed to NP30 was increased to 500 mg/L. The cultures typically removed nitrate within 3–4 hours and perchlorate within 8 hours. The biomass concentration varied due to use as inoculum for experiments, but was typically 2000 mg/L as VSS.
Pure culture isolation
The selective agar medium for isolation of the pure cultures contained 0.62 g/L NaClO4, 30 g/L NaCl, and 12.5 g/L agar. The basal perchlorate (P) medium for cultivation contained 0.62 g/L NaClO4, 1.24 g/L CH3COONa · 3H2O, 0.25 g/L yeast extract, and 0.25 g/L peptone, 11 g/L MgCl2 · 6H2O, 1.4 g/L CaCl2 · 2H2O, 0.72 g/L KCl, 30 g/L NaCl, 0.3 mL/L resazurin (0.1%), 200 mg/L NaHCO3 (*), 5.0 mL/L of 67 mM Na2S · 9H2O (*), 1.0 mL/L of 50 g/L KH2PO4, and 1.0 mL/L of mineral solution. The asterisk (*) indicates items added to the media after boiling and cooling to room temperature. The mineral solution contained: 10 g (NH4)6Mo7O24 · 4H2O, 0.05 g ZnCl2, 0.3 g H3BO3, 1.5 g FeCl2 · 4H2O, 10 g CoCl2 · 6H2O, 0.03 g MnCl2 · 6H2O, and 0.03 g NiCl2 · 6H2O in 1 L deionized water.
The basal nitrate (N) medium was identical to the P medium except that 0.62 g/L NaClO4 was replaced by 0.68 g/L NaNO3. All media were sterilized by autoclaving at 121°C for 30 min. Nitrate and perchlorate were analyzed with an ion chromatograph (ICS-3000, Dionex, Sunnyvale, CA) equipped with AS16 and AS20 columns using dual system chromatography. The detection limits for NO3− and ClO4− of this method were 10 μg/L and 1 μg/L, respectively.
Serial dilutions of the mixed cultures NP30 and P30 were streaked on selective agar plates and incubated in an anaerobic glove box maintained under oxygen-free atmosphere (80% N2, 10% CO2 and 10% H2). When single colonies appeared, they were picked, streaked on fresh plates and incubated again either in the anaerobic glove box or in the air. After 5 days’ incubation, colonies were picked off the plates and transferred into Hungate tubes containing 10 mL P or N medium. In total, eight isolates were obtained. The growth of these isolates in the presence of perchlorate, nitrate and oxygen was examined using the measurement of optical density at 520 nm (OD520).
DNA extraction, preparation and sequencing
DNA isolation
The Promega Wizard® Genomic DNA Purification kit (Promega Corp. Madison, WI) was used to extract the DNA from the cultures from 1 mL culture as per the directions with the kit. The DNA was stored at 4°C prior to use.
-
(2)
16S rDNA Sequencing
Fragments of the 16S rRNA gene were amplified by PCR from cellular DNA obtained from each pure culture using 16S rDNA-specific primers 27 F, 5’-AGAGTTTGATCMTGGCTCAG [33], and U1510R, 5’-GGTTACCTTGTTACGACTT [34], purchased from Eurofins MWG (Huntsville, AL). The reaction was performed with 20 units/ml Taq DNA polymerase in 1 × ThermoPol buffer (New England Biolabs, Ipswich, MA) supplemented with 0.2 mM of each dNTP and 5 μM of forward and reverse primers. The PCR conditions were as follows: initial denaturation at 94°C for 5 min, 32 cycles consisting of denaturation at 94°C for 0.5 min, annealing at 52°C for 0.5 min, and extension at 72°C for 1 min, and final elongation at 72°C for 10 min. PCR products were column-purified [35] and sequenced bi-directionally by the dye-terminator method with the same primers used for amplification. Sequencing was performed by SeqWright, Inc. (Houston, TX).
-
(3)
Metagenome Sequencing
DNA samples for shotgun sequencing were prepared according to the protocols provided by Illumina Inc. (San Diego, CA). In brief, purified metagenomic DNA was nebulized to produce fragments of less than 800 bp. The resulting oligonucleotide mixtures were end-repaired, A-tailed and converted to dsDNA libraries tagged with Illumina adapters for single end sequencing. The libraries were amplified, gel purified, and finally dissolved in water. Quantification of the libraries was done using PicoGreen fluorimetric assay [36]. Sequencing was performed on Illumina Genome Analyzer IIx System according to the manufacturer’s specifications.
Data analysis
16S rRNA
The closest relatives of the partial 16S rRNA gene sequences were identified in the NCBI nt database (ftp://ftp.ncbi.nih.gov/blast/db) using BLAST 2.2.27 + [37]. Taxonomic affiliations were derived from the best hits excluding matches to uncultured or unidentified organisms.
-
(2)
Metagenome Analysis
The overall strategy of metagenomic datasets analysis is presented on Figure 3. The quality of the collected sequencing reads was assessed with the fastQC 0.10.1 tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The reads were processed with the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit) to remove terminal low-quality nucleotides, and then assembled using Metavelvet 1.2.02 which is an extension of the Velvet assembler designed to do de novo metagenome assembly from short sequence reads [38]. It is bundled with Velvet 1.2.08 [39]. The k-mer length and the minimum contig size were set to 19 and 100, respectively. The assembled contigs were submitted to the MG-RAST 3.3 server [40] for taxonomic assignment and functional annotation. Alternatively, the contigs were evaluated batchwise using BLASTN searches against the Silva rRNA database (version 111) [41] and/or the NCBI nt and 16SMicrobial databases (ftp://ftp.ncbi.nih.gov/blast/db). BLASTX searches against the NCBI nr database (ftp://ftp.ncbi.nih.gov/blast/db) were also conducted. Primary sorting of BLAST search results was performed using BlastParser 1.2 (http://geneproject.altervista.org). Contigs that matched database entries with E-value < 10−10 (NCBI nt and nr databases) or < 10−5 (NCBI 16SMicrobial and Silva rRNA databases) were selected for further analysis. Taxonomic classification of BLASTX hits was performed with MEGAN 4.70.4 [42] using the lowest common ancestor (LCA) algorithm. The same approach was used to evaluate BLASTN matches with the Silva rRNA and NCBI 16SMicrobial databases. Annotation of BLASTN hits in NCBI nt database was performed using proprietary Perl scripts (ContigEval pipeline) for sorting the BLAST output, retrieval of taxonomic information from NCBI nodes.dmp and names.dmp files (ftp://ftp.ncbi.nih.gov/pub/taxonomy), and identification of the hit genes on subject nucleotide sequences by parsing NCBI gene2accession and gene_info files (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA). Contigs mapped to sequences of particular interest were further extended using targeted assembly software, Mapsembler 1.3.16 [43]. In addition, some contig lengthening was achieved by running a transcriptome assembler, Oases 0.2.08 [44] in metagenomic context with k-mers ranging from 19 to 31 and coverage cut-off set to 3. Oases was used only in the reconstruction of rRNA and perchlorate reductase gene sequences. Due to the significant probability of chimeric assembly, the Oasis-produced contigs were extensively filtered using the Uchime tool from the Usearch 6.0 software package [45] and ChimeraSlayer release 20110519 [46].
-
(3)
GenBank Accession Numbers
Figure 3.
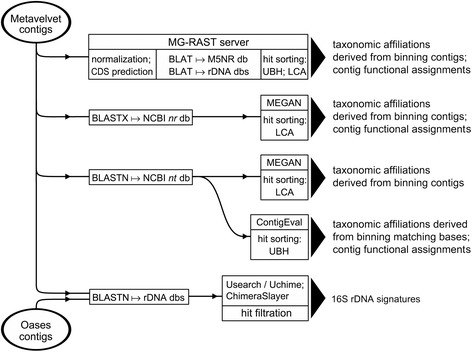
Flowchart of metagenome data analysis. The assembled contigs were evaluated against global sequence databases (NCBI nt and nr, MG-RAST M5NR) and rDNA databases (Silva, Greengenes, RDP, NCBI 16SMicrobial). Taxonomic binning was performed using either Unique Best Hit (UBH) or Lowest Common Ancestor (LCA) algorithm.
Partial 16S rRNA gene sequences of isolated strains NWO, NY, NYO, PW, PWO, PY and PYO were deposited in GenBank under accession numbers KF135667-KF135673, respectively. Nucleotide sequences encompassing pcrABCD gene clusters recovered from NP30 and P30 mixed cultures are assigned accession numbers KF135674 and KF135675, respectively. Chlorite dismutase (cld) gene sequences from NP30 and P30 mixed cultures are placed under accession numbers KF135676 and KF135677,
Acknowledgements
The P30 and NP30 cultures are maintained in the Biological Solutions Laboratory at the University of British Columbia by Dr. Y Xiao through funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant Program. This work was supported in part by a subcontract to GEF from the NASA Center for Ribosome Evolution and Adaptation at the Georgia Institute of Technology (NNA09DA78A). QT was supported in part by a NASA Earth and Space Science Fellowship (10-Planet10R-0025). We also wish to acknowledge partial support for RCW from the Welch Foundation under grant E-1264.
Additional files
Taxonomic composition of P30 and NP30 microbial communities estimated using MG-RAST server.
Taxonomic composition of P30 and NP30 microbial communities estimated using MEGAN program.
Taxonomic composition of P30 and NP30 microbial communities estimated using ContigEval pipeline.
Relative abundance of major bacterial families in P30 and NP30 communities shown ordered by taxon abundance averaged for P30 and NP30 cultures.
Identification and taxonomic assignment of 16S rRNA gene sequences in P30 and NP30 metagenomic contigs.
16S rDNA signature sequences identified in P30 and NP30 metagenomic contigs.
Identification and taxonomic assignment of nitrate reductase gene sequences in P30 and NP30 metagenomic contigs.
Footnotes
Victor G Stepanov and Yeyuan Xiao contributed equally to this manuscript.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VS conducted 16S rRNA sequence studies, analyzed metagenome data, and prepared tables and figures for the manuscript. YX maintained the P30 and NP30 cultures, isolated pure cultures, and assisted in manuscript development. QT constructed contigs to obtain initial population structure. MR processed raw sequencing data to obtain high quality filtered reads. YF and GF established procedures for data analysis . GF, RW, and DR conceived of the study, and participated in its design and coordination. VS, YX, GF, and DR drafted the initial manuscript and all authors read, offered revisions as appropriate, and approved the final manuscript.
Contributor Information
Victor G Stepanov, Email: vstepanov@uh.edu.
Yeyuan Xiao, Email: rogeryyxiao@gmail.com.
Quyen Tran, Email: rdirective@gmail.com.
Mark Rojas, Email: mmrojas@utmb.edu.
Richard C Willson, Email: rcwwww@gmail.com.
Yuriy Fofanov, Email: yufofano@utmb.edu.
George E Fox, Email: fox@uh.edu.
Deborah J Roberts, Email: Deborah.roberts@ubc.ca.
References
- 1.Kirk AB, Smith EE, Tian K, Anderson TA, Dasgupta PK. Perchlorate in milk. Environ Sci Technol. 2003;37:4979–4981. doi: 10.1021/es034735q. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez CA, Crump KS, Krieger RI, Khandaker NR, Gibbs JP. Perchlorate and nitrate in leafy vegetables of North America. Environ Sci Technol. 2005;39:9391–9397. doi: 10.1021/es050804k. [DOI] [PubMed] [Google Scholar]
- 3.Dasgupta PK, Dyke JV, Kirk AB, Jackson WA. Perchlorate in the United States. Analysis of relative source contributions to the food chain. Environ Sci Technol. 2006;40:6608–6614. doi: 10.1021/es061321z. [DOI] [PubMed] [Google Scholar]
- 4.Rajagopalan S, Anderson TA, Fahlquist L, Rainwater KA, Ridley M, Jackson WA. Widespread presence of naturally occurring perchlorate in high plains of Texas and New Mexico. Environ Sci Technol. 2006;40:3156–3162. doi: 10.1021/es052155i. [DOI] [PubMed] [Google Scholar]
- 5.Snyder SA, Pleus RC, Vanderford BJ, Holady JC. Perchlorate and chlorate in dietary supplements and flavor enhancing ingredients. Anal Chim Acta. 2006;567:26–32. doi: 10.1016/j.aca.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 6.Clark JJJ. Toxicology of Perchlorate. In: Urbansky ET, editor. Perchlorate in the Environment. New York, USA: Kluwer Academic/Plenum Publishers; 2000. [Google Scholar]
- 7.Kim K, Logan BE. Microbial reduction of perchlorate in pure and mixed culture packed-bed bioreactors. Water Res. 2001;35:3071–3076. doi: 10.1016/S0043-1354(01)00014-8. [DOI] [PubMed] [Google Scholar]
- 8.Nerenberg R, Rittmann BE. Perchlorate as a secondary substrate in a denitrifying, hollow-fiber membrane biofilm reactor. Water Sci Technol. 2002;2:259–265. [Google Scholar]
- 9.Shrout JD, Parkin GF. Influence of electron donor, oxygen, and redox potential on becterial perchlorate degradation. Water Res. 2006;40:1191–1199. doi: 10.1016/j.watres.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Logan BE. Measurement of chlorite dismutase activities in perchlorate respiring bacteria. J Microbiol Methods. 2003;54:239–247. doi: 10.1016/S0167-7012(03)00058-7. [DOI] [PubMed] [Google Scholar]
- 11.CDPH . History of Perchlorate in California Drinking Water. CA, USA: The California Department of Public Health; 2007. [Google Scholar]
- 12.MassDEP . Final Changes to the Massachusetts Contingency Plan – 310 CMR 40.0000. USA: Massachusetts Department of Environmental Protection; 2013. [Google Scholar]
- 13.Aldridge L, Gillogly T, Lehman G, Clifford DA, Roberts DJ, Lin X. Treatability of Perchlorate in Groundwater by ion-Exchange Technology, Phase II Pilot Plant Study. Denver, CO, USA: AWWA Research Foundation; 2004. [Google Scholar]
- 14.Tripp AR, Clifford DA, Roberts DJ, Cang Y, Aldridge L, Gillogly T, Boulos L. Treatment of Perchlorate in Groundwater by Ion-Exchange Technology. Denver, CO, USA: AWWA Research Foundation; 2003. [Google Scholar]
- 15.Cang Y, Roberts DJ, Clifford DA. Development of cultures capable of reducing perchlorate and nitrate in high salt solutions. Water Res. 2004;38:3322–3330. doi: 10.1016/j.watres.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 16.Hiremath T, Roberts DJ, Lin X, Clifford DA, Gillogly T, Lehman G. Biological treatment of perchlorate in spent ISEP ion-exchange brine. Environ Eng Sci. 2006;23:1009–1016. doi: 10.1089/ees.2006.23.1009. [DOI] [Google Scholar]
- 17.Lin X, Roberts DJ, Hiremath T, Clifford DA, Gillogly T, Lehman G: Divalent cation addition (Ca2+ or Mg2+) stabilizes biological treatment of perchlorate and nitrate in ion-exchange spent brine.Environ Eng Sci 2007, 24:725–735.
- 18.Lehman SG, Badruzzaman M, Adham S, Roberts DJ, Clifford DA. Perchlorate and nitrate treatment by ion exchange integrated with biological brine treatment. Water Res. 2008;42:969–976. doi: 10.1016/j.watres.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Patel A, Zuo G, Lehman SG, Badruzzaman M, Clifford DA, Roberts DJ. Fluidized bed reactor for the biological treatment of ion-exchange brine containing perchlorate and nitrate. Water Res. 2008;42:4291–4298. doi: 10.1016/j.watres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Van Ginkel SW, Ahn CH, Badruzzaman M, Roberts DJ, Lehman SG, Adham SS, Rittmann BE. Kinetics of nitrate and perchlorate reduction in ion-exchange brine using the membrane biofilm reactor (MBfR) Water Res. 2008;42:4197–4205. doi: 10.1016/j.watres.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Zuo G, Roberts DJ, Lehman SG, Jackson GW, Fox GE, Willson RC. Molecular assessment of salt-tolerant, perchlorate- and nitrate-reducing microbial cultures. Water Sci Technol. 2009;60:1745–1756. doi: 10.2166/wst.2009.635. [DOI] [PubMed] [Google Scholar]
- 22.Xiao Y, Roberts DJ, Zuo G, Badruzzaman M, Lehman GS. Characterization of microbial populations in pilot-scale fluidized-bed reactors treating perchlorate- and nitrate-laden brine. Water Res. 2010;44:4029–4036. doi: 10.1016/j.watres.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Y, Basu A, Kashyap V, Roberts DJ. Experimental and numerical analysis of biological regeneration of perchlorate laden Ion-exchange resins in batch reactors. Environ Eng Sci. 2010;27:75–84. doi: 10.1089/ees.2009.0207. [DOI] [Google Scholar]
- 24.Xiao Y, Roberts DJ. Kinetics analysis of a salt-tolerant perchlorate-reducing bacterium: effects of sodium, magnesium, and nitrate. Environ Sci Technol. 2013;47:8666–8673. doi: 10.1021/es304501n. [DOI] [PubMed] [Google Scholar]
- 25.Ahn C, Oh H, Ki D, Van Ginkel S, Rittmann B, Park J. Bacterial biofilm-community selection during autohydrogenotrophic reduction of nitrate and perchlorate in ion-exchange brine. Appl Microbiol Biotechnol. 2009;81:1169–1177. doi: 10.1007/s00253-008-1797-3. [DOI] [PubMed] [Google Scholar]
- 26.Van Ginkel SW, Lamendella R, Kovacik WP, Jr, Santo Domingo JW, Rittmann BE. Microbial community structure during nitrate and perchlorate reduction in ion-exchange brine using the hydrogen-based membrane biofilm reactor (MBfR) Bioresour Technol. 2010;101:3747–3750. doi: 10.1016/j.biortech.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 27.Achenbach LA, Michaelidou U, Bruce RA, Fryman J, Coates JD. Dechloromonas agitata gen. nov., sp. nov. and Dechlorosoma suillum gen. nov., sp. nov., two novel environmentally dominant (per)chlorate-reducing bacteria and their phylogenetic position. Int J Syst Evol Microbiol. 2001;51:527–533. doi: 10.1099/00207713-51-2-527. [DOI] [PubMed] [Google Scholar]
- 28.Song B, Palleroni N, Kerkhof L, Haggblom M. Characterization of halobenzoate-degrading, denitrifying Azoarcus and Thauera isolates and description of Thauera chlorobenzoica sp. nov. Int J Syst Evol Microbiol. 2001;51:589–602. doi: 10.1099/00207713-51-2-589. [DOI] [PubMed] [Google Scholar]
- 29.Juteau P, Larocque R, Rho D, LeDuy A. Analysis of the relative abundance of different types of bacteria capable of toluene degradation in a compost biofilter. Appl Microbiol Biotechnol. 1999;52:863–868. doi: 10.1007/s002530051604. [DOI] [PubMed] [Google Scholar]
- 30.Reinhold-Hurek B, Hurek T. The Genera Azoarcus, Azovibrio, Azospira and Azonexus. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K, Stackebrandt E, editors. The Prokaryotes. New York, USA: Springer; 2006. pp. 873–891. [Google Scholar]
- 31.Uchino Y, Hirata A, Yokota A, Sugiyama J. Reclassification of marine Agrobacterium species: Proposals of Stappia stellulata gen. nov., comb. nov., Stappia aggregata sp. nov., nom. rev., Ruegeria atlantica gen. nov., comb. nov., Ruegeria gelatinovora comb. nov., Ruegeria algicola comb. nov., and Ahrensia kieliense gen. nov., sp. nov., nom. rev. J Gen Appl Microbiol. 1998;44:201–210. doi: 10.2323/jgam.44.201. [DOI] [PubMed] [Google Scholar]
- 32.Rüger H, Höfle MG. Marine Star-Shaped-Aggregate-Forming Bacteria:Agrobacterium atlanticumsp. nov.; Agrobacterium meteorisp. nov.; Agrobacterium ferrugineum sp. nov., nom. rev.; Agrobacterium gelatinovorum sp. nov., nom. rev.; and Agrobacterium stellulatum sp. nov., nom. rev. Int J Syst Bacteriol. 1992;42:133–143. doi: 10.1099/00207713-42-1-133. [DOI] [PubMed] [Google Scholar]
- 33.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008;74:2461–2470. doi: 10.1128/AEM.02272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. J Microbiol Methods. 2003;55:541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Borodina TA, Lehrach H, Soldatov AV. DNA purification on homemade silica spin-columns. Anal Biochem. 2003;321:135–137. doi: 10.1016/S0003-2697(03)00403-2. [DOI] [PubMed] [Google Scholar]
- 36.The Lab Rat: Protocol for DNA quantitation using PicoGreen. [http://www.thelabrat.com/protocols/DNA-RNAQuantitation7.shtml]
- 37.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Namiki T, Hachiya T, Tanaka H, Sakakibara Y. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012;40:e155–e155. doi: 10.1093/nar/gks678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer F, Paarmann D, D’Souza M, Olson R, Glass E, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards R. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2011;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huson DH, Mitra S, Ruscheweyh H, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterlongo P, Chikhi R. Mapsembler, targeted and micro assembly of large NGS datasets on a desktop computer. BMC Bioinformatics. 2012;13:48. doi: 10.1186/1471-2105-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;2012(28):1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 46.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methe B, DeSantis TZ, The Human Microbiome Consortium. Petrosino JF, Knight R, Birren BW. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]