

Abstract
Background
Tobacco cessation pharmacotherapies currently are limited to nicotine itself, the partial nicotine agonists varenicline and cytisine, and the antidepressant bupropion. Compared with agonists, nicotinic antagonists such as the noncompetitive, nonselective compound mecamylamine, and the competitive, α4β2-preferring antagonist dihydro-β-erythroidine (DHβE) may be a novel approach to the treatment of tobacco smoking as both are effective antagonists of nicotine’s central effects. Considering nicotinic acetylcholine receptors mediate critical peripheral effects of acetylcholine, such as cardiovascular effects, it is important to study how nicotinic antagonists would alter the cardiovascular system and the cardiovascular changes induced by nicotine.
Methods
The effects of several nicotinic agonists and antagonists on blood pressure and heart rate were measured in conscious, unrestrained rats following parenteral administration using a telemetry system.
Results
Nicotine and other nicotinic receptor agonists (epibatidine, varenicline, and cytisine) produced similar increases in blood pressure, whereas their effects on heart rate were biphasic. The cardiovascular changes were attenuated by the nonselective nicotine antagonist, mecamylamine, but the peripherally-restricted antagonist hexamethonium blocked only the agonist-induced changes in blood pressure. The α7-preferring antagonist, MLA, and the α4β2-preferring antagonist, DHβE, were much less effective in blocking the agonist-induced cardiovascular changes, indicating that nicotine’s cardiovascular effects, are due to activation at autonomic ganglia involving nicotinic receptor subtypes other than α4, α7, or β2.
Conclusions
The data indicate that the cardiovascular effects of nicotine and nicotine-like agents are mediated through receptor mechanisms that are distinct from those that mediate the central effects of nicotine.
Keywords: nicotine, nicotinic agonists, cardiovascular effects, rats
1. INTRODUCTION
Cigarette smoking in humans stimulates sympathetic activity, increasing heart rate and blood pressure (Tachmes et al., 1978; Spohr et al., 1979; Koch et al., 1980). Intravenous nicotine also increases blood pressure and heart rate in humans (Soria et al., 1996; Rose et al., 2000; Sofuoglu et al., 2008), but at least one report demonstrated that a large dose of intravenous nicotine produced a transient bradycardia in 50% of subjects followed by a subsequent tachycardia (Henningfield et al., 1985). In preclinical studies, the effects of nicotine on the cardiovascular system depended on the assay preparation and the route of nicotine administration. Much of the research in this area has been conducted in isolated heart or tissue preparations, in pithed animals, or in anesthetized animals, in which nicotine has typically been administered locally or by intra-arterial, intrathecal, or intracranial routes. Relatively little is known about the effects of parenterally administered nicotine or nicotinic ligands (in particular subcutaneous and intraperitoneal routes) on blood pressure and heart rate in intact, conscious rats. This makes it difficult to compare the cardiovascular effects of nicotinic receptor agonists with their behavioral and potential therapeutic effects, which are often evaluated in freely-moving animals. One aim of this study was to determine the effects of parenterally administered nicotine and nicotinic agonists (varenicline, cytisine, and epibatidine) in conscious, unrestrained rats. It is hypothesized that the presumed lower efficacy agonists and nicotinic receptor subtype-selective compounds produce minimal changes in hemodynamic responses as compared with nicotine.
Varenicline, arguably the most effective smoking cessation medication, is reported to act as a partial agonist at α4β2 receptors, which is its presumed mechanism of therapeutic action (Williams et al., 2011). Recently, varenicline has been reported to increase the risk of serious adverse cardiovascular effects compared with placebo in double-blind randomized controlled trials (Singh et al., 2011). It is not known whether these adverse effects are mediated through agonist actions at a nicotine receptor, and if so, whether they are due to actions at the α4β2 subtype of nicotine receptors or through other nicotine receptor subtypes. This issue, posed specifically for varenicline, is more general for the design of potential smoking-cessation medications. For example, what, if any, role do α4β2 receptors play in the cardiovascular effects of nicotinic ACh receptor ligands? The α4β2* nicotinic receptor subtype is thought to be critical to the central effects of nicotine, including the discriminative and reinforcing effects of nicotine (Corrigall et al., 1994; Stolerman et al., 1997; Watkins et al., 1999; Gommans et al., 2000; Mansbach et al., 2000; Shoaib et al., 2002; Zaniewska et al., 2006; Liu et al., 2007; Le Foll et al., 2009; for review see Smith and Stolerman, 2009; Jutkiewicz et al., 2011; but see Harvey et al., 1996; Harvey and Luetje, 1996; Chavez-Noriega et al., 1997); however, nicotinic acetylcholine receptors (nAChRs) are located in both the central and peripheral nervous systems (Gotti et al., 2006; Smith and Stolerman, 2009). Nicotine activates nAChRs in both the sympathetic and parasympathetic ganglia and thereby has the capacity to increase or decrease heart rate and blood pressure. Central nicotinic receptors in nuclei located primarily in the brainstem are also involved in cardiovascular control (Nadeau and James, 1967; Tseng et al., 1993; Haas and Kubler, 1996). Therefore, the cardiovascular effects of parenterally administered (e.g., smoked or injected) nicotine may be determined by multiple mechanisms.
The major nAChRs subtypes located in autonomic ganglia and brainstem nuclei contain α3 and β4 subunits, but other subunits, such as α4, β2, α5, and α7, are found in these areas and have been shown to contribute to cardiovascular regulation as well (Poth et al., 1997; Genzen et al., 2001; Perry et al., 2002; Del Signore et al., 2004; Mao et al., 2006). Another aim of this study was to determine, using receptor subtype-preferring nicotinic receptor antagonists, the receptor subtypes mediating the cardiovascular effects of parenterally-administered nicotine, epibatidine, varenicline, and cytisine in conscious, unrestrained rats. In addition, the cardiovascular effects of nicotinic antagonists alone were evaluated in order to determine the nAChR subtypes that may influence resting blood pressure and heart rate. It is hypothesized that the cardiovascular and the central effects, specifically the discriminative stimulus effects, of nicotine are mediated by different nicotinic receptor subtypes or populations of nicotinic receptors. Consistent with our recent drug discrimination study (Jutkiewicz et al., 2011), this report used the same drugs and routes of administration to determine the receptor subtypes through which the cardiovascular effects of nicotine and other nicotinic agonists are mediated, and whether these were distinct from those that mediate the discriminative stimulus effects of nicotine.
2. METHODS
2.1 Animals
Male Sprague-Dawley rats were obtained from Harlan, Inc. (Indianapolis, IN) and group-housed upon arrival. Food and water were freely available for all rats at all times. Housing and experimental rooms were maintained on a 12 h light/dark cycle with lights on at 7:00 AM with an average temperature of 21°C. Experiments described below occurred between the hours of 7:00AM and 3:00PM. The experimental protocols were approved by the University of Michigan University Committee on the Use and Care of Animals and conformed to the guidelines established by the NIH Guide for the Use of Laboratory Animals.
2.2 Surgical Procedures
To measure changes in heart rate and mean arterial pressure, rats were implanted with telemetric transmitters (TA11PA-C40 or TL11M2-C50-PXT, Data Sciences International, Transoma Medical Inc., St. Paul, MN, USA) under ketamine (90 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) anesthesia. An incision was made in the skin approximately 1 in to the left or right of the spine and over the left or right thigh to expose the femoral artery. The transmitter was placed into the incision on the dorsal side and into a subcutaneous pocket (made by separating the connective tissue between the skin and muscle) on the side of the abdomen. This positioning prevented the rats from manipulating the incision site and gaining access to the transmitter. The catheter extending from the base of the transmitter was threaded subcutaneously, placed 2-3 cm into the femoral artery, and secured with suture. Incisions were closed with 5.0 Ethilon nylon monofilament suture. Following surgery, rats were singly-housed and monitored for at least 7 days for signs of recovery (normal eating, drinking, and defecation patterns) prior to experimentation. All rats continued to have free access to food and water at all times.
2.3. Telemetry System
The system consisted of battery-operated transmitters implanted surgically (described above), Physiotel receivers, the DSI Data Exchange Matrix, and the Dataquest A.R.T. system, which collects and stores digital data from the receiver to a computer (Data Sciences International, Transoma Medical Inc., St. Paul, MN, USA). Blood pressure and heart rate data were compiled by the Dataquest A.R.T. Gold Analysis 3.01 software.
2.4. Experimental Design
At the start of an experiment, rats’ home cages were placed on top of the receivers, and baseline data were collected for at least 1 h to allow heart rate and blood pressure to return to resting levels. All rats were given a saline injection to assure they were habituated to the injection and handling procedure on the test day. After heart rate and blood pressure returned to resting levels, rats were injected with a treatment condition, consisting either of a single drug dose or a pretreatment drug administered 15 min prior to a test drug. Data were collected for at least 2 h following the last injection, with the exception of a few experiments in which data were collected for only 60-70 min following an injection of cytisine. All injections were administered subcutaneously (s.c.) in a volume of 1 ml/kg. Agonist dose ranges were selected from doses used in our behavioral studies for comparison purposes (Jutkiewicz et al., 2011) and below those that produced convulsions and/or lethality (unpublished observations).
Each rat was used to evaluate multiple experimental conditions, such that each rat received 4-6 different treatments, one per week with at least 6 d between drug exposures. Extensive preliminary experiments demonstrated that this procedure was sufficient to prevent the development of tolerance or sensitization to the effects of nicotine and to prevent changes in resting heart rate and blood pressure levels over the course of 6 weeks. Each experimental condition was evaluated in 6-7 different rats.
2.5. Data Analysis
As described earlier, data were collected throughout baseline, before, and after drug treatments. The analysis program calculated an average heart rate and mean arterial pressure (MAP) every 10 sec. These 10 sec epochs were then averaged over 1 min per rat, and data from at least 6 rats were averaged for each treatment group. Data points from mins 1, 2, 3, 5, 7, 10 and then every 5 min interval for 1 or 2 h post-injection are shown on the graphs. Individual subject data were calculated as percent change from resting mean arterial pressure (MAP) and heart rate (HR) and then averaged by treatment group. Data are also expressed as area under curve (AUC) over resting baseline (in Tables only). AUCs were calculated from time 0-60 or 0-90 min for MAP and from 0-30 and 60-90 min for HR, when the most robust drug effects were observed (Arslan et al., 1991; Houdi et al., 1995; Delaunois et al., 2009). AUCs were analyzed by one-way ANOVA with Tukey’s post hoc tests comparing saline control conditions to agonist alone or to antagonist + agonist tests in order to identify dose-dependent drug effects and to determine if antagonists statistically significantly attenuated the effects of nicotinic agonists. Data in Figure 2 (effects of antagonists alone) were analyzed by two-way ANOVA with Bonferroni’s post hoc tests and AUCs over resting baselines for antagonists alone are shown in Table 2. Values of F and pare reported in the text only if the effect was statistically significant (p<0.05).
Figure 2.
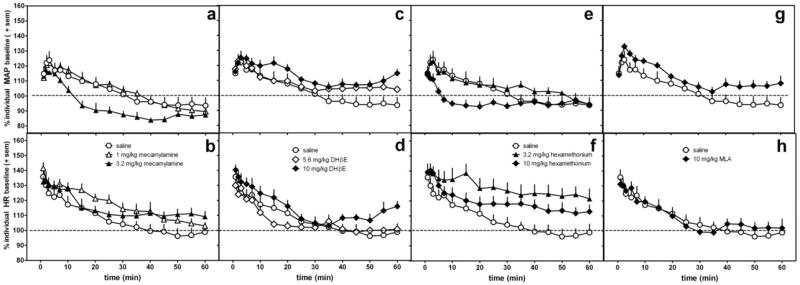
The effects of nicotinic antagonists alone on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats as compared with saline (same control data re-plotted in each graph). Doses of mecamylamine (a,b), DHbE (c,d), and hexamethonium (e,f) were evaluated at doses used in the antagonism experiments (figures 3-7) and at a dose ¼ to ½ log greater. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
Table 2.
Area under the curve (± SEM) over resting baseline for antagonists mecamylamine-, DHbE-, hexamethonium-, and MLA-induced changes in MAP (0-60 min) or HR (0-30 min)
| Treatment | MAP | HR
|
|
|---|---|---|---|
| 0-30 min | |||
|
| |||
| saline | 167.9 (259) | 429 (133) | |
|
| |||
| mecamylamine (mg/kg) | 1 | 222 (51) | 724 (73) |
| 3.2 | -467 (95)* | 551 (73) | |
|
| |||
| DHβE (mg/kg) | 5.6 | 474 (175) | 273 (72) |
| 10 | 762 (92) | 580 (97) | |
|
| |||
| hexamethonium (mg/kg) | 3.2 | 345 (113) | 953 (164) |
| 10 | -258 (164) | 662 (136) | |
|
| |||
| MLA (mg/kg) | 10 | 687 (111) | 410 (55) |
p<0.05 vs saline
2.6 Drugs
(-)-Nicotine hydrogen tartrate salt, (±)-epibatidine, mecamylamine, dihydro-beta-erythroidine (DHβE), hexamethonium, methyllycaconitine (MLA), and atropine were obtained from Sigma-Aldrich (St. Louis, MO). Cytisine was obtained from Xingcheng Chempharm Co., Ltd (Taizhou, Zhejiang, China) and then purified. Varenicline was obtained from the National Institutes of Drug Abuse Drug Supply Program. All drug doses were calculated based on the salt forms of the drugs and dissolved in saline. The doses of nicotine bitartrate salt used were 0.32, 1, and 1.78 mg/kg and are equivalent to 0.1, 0.325, and 0.58 mg/kg, respectively, of nicotine free base.
3. RESULTS
3.1. Agonist effects
The effects of saline or nicotine alone on mean arterial pressure (MAP) and heart rate (HR) are shown in Figure 1a and 1b, respectively, and Table 1. Data are presented as percent increase over resting baseline values, which ranged from 80-110 mmHg for MAP and 320-360 bpm for HR. A 2-h time course is shown in the figures, and longer duration effects, when they occurred and were measured, are described in the text. An injection of saline alone increased MAP and HR over resting baseline; resting baseline levels recovered within approximately 30-40 min. Nicotine produced a dose-dependent increase in MAP with the maximum effect occurring within 5 min following a subcutaneous injection (F(3,25)=23.5, p<0.0001) (Table 1, Fig 1a,b). The smallest nicotine dose (0.32 mg/kg) did not significantly increase MAP above saline control. The largest nicotine doses (1 and 1.78 mg/kg) significantly elevated MAP over saline (p<0.001 both doses) and above 0.32 mg/kg nicotine (p<0.01 and p<0.001 respectively). The largest nicotine dose tested increased MAP for more than 2 h.
Figure 1.
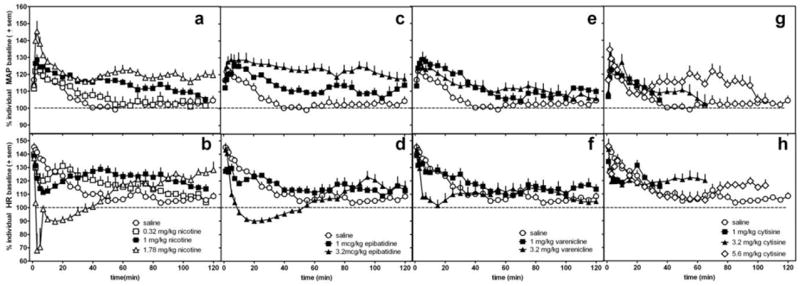
The effects of nicotine (a,b), epibatidine (c,d), varenicline (e,f), cytisine (g,h) or saline (same saline control data re-plotted in each graph) on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
Table 1.
Area under the curve (± SEM) over resting baseline for saline-, nicotine-, epibaitidine-, varenicline-, and cytisine-induced changes in MAP (0-90 min) or HR (2 periods)
| Treatment | MAP | HR
|
||
|---|---|---|---|---|
| 0-30 min | 60-90 min | |||
|
| ||||
| saline | 515 (117) | 775 (42) | 233 (76) | |
|
| ||||
| nicotine (mg/kg) | 0.32 | 695 (205) | 751 (135) | 446 (109) |
| 1 | 1474 (77)*** | 541 (97) | 712 (85)** | |
| 1.78 | 1896 (114)*** | -270 (46)*** | 551 (84) | |
|
| ||||
| epibatidine (mcg/kg) | 1 | 1307 (82)** | 545 (120) | 446 (96) |
| 3.2 | 2166 (245)*** | -21 (62)*** | 286 (81) | |
|
| ||||
| varenicline (mg/kg) | 1 | 1250 (171)** | 777 (91) | 339 (96) |
| 3.2 | 1271 (97)** | 274 (112)*** | 334 (51) | |
|
| ||||
| cytisine (mg/kg) | 1 | 544 (97) | 675 (41) | - |
| 3.2 | 818 (173) | 553 (53) | - | |
| 5.6 | 1427 (226)** | 679 (97) | 368 (75) | |
p<0.01 vs saline,
p<0.001 vs saline
single underline - p<0.01 vs smallest dose of specific drug tested
double underline - p<0.001 vs smallest dose of specific drug tested
All doses of nicotine produced a biphasic effect on HR; immediately after an injection, nicotine induced a decrease in HR (F(3,25)=33.5, p<0.0001) followed by an increase (F(3,20)=5.4, p=0.008) (Table 1, Fig 1a,b). The magnitude and duration of the initial bradycardia was dose-dependent. Acute administration of the largest nicotine dose produced a significant (p<0.001), but brief, suppression of HR that remained lower than resting HR levels for approximately 20-30 min. The time course, but not the magnitude, of subsequent increases in HR was dose-dependent. A dose of 1 mg/kg nicotine produced a significant (p<0.01, Table 1) increase in HR at 60-90 min after injection (Table 1, Fig 1b). The increase in HR produced by 1.78 mg/kg nicotine was delayed, tending to increase above saline levels at 60-90 min and remained elevated 120 min after administration (Fig 1b). Following approximately 150 min, the HR increase produced by 1.78 mg/kg nicotine returned to resting baseline values.
Similar to nicotine, epibatidine dose-dependently increased MAP (F(2,19)=28.8, p<0.0001) as compared with saline injection and produced a biphasic effect on HR (Table 1, Fig 1c and d). The largest dose (3.2 mcg/kg) of epibatidine induced significant increases in MAP (p<0.001) that remained elevated for more than 3 h. This dose of epibatidine also produced a significant (F(2,19) = 21.6; p<0.0001) decrease in HR followed by a small, but non-significant trend to increase in HR 90 min after injection.
Varenicline significantly increased MAP as compared with saline (F(2,19)= 11.8, p=0.0006) (Table 1, Fig 1e) for approximately 50-60 min post-injection. Varenicline did not significantly increase HR at any time point (Table 1, Fig 1f); however, the largest dose of varenicline tested decreased HR within 3 min of the injection (Table 1, F(2,19)= 12.6, p=0.0004) and the effect lasted for approximately 20-30 min (p<0.001).
Only the largest dose of cytisine, 5.6 mg/kg, significantly increased MAP (p<0.01, Table 1) with the peak effect occurring within the first 5 min post-injection (F(3,25)=7.2, p=0.002) (Fig 1g) and remaining elevated for approximately 80-90 min post-injection. Cytisine produced a non-significant trend to decrease HR as compared with saline (F(3,25)=2.4, p=0.09).
3.2. Antagonism of cardiovascular changes
The antagonists, given alone, differentially altered MAP and HR in the conscious, freely-moving rats (Fig 2, Table 2). As compared with saline, only the larger dose of mecamylamine (3.2 mg/kg) significantly decreased MAP (significant interaction for time course data: F(30,285)=2.3, p=0.0002,with p<0.01 at 15 and 20 min). Mecamylamine also altered HR (interaction for time course data: F(30,328)=1.7, p=0.02); however, this interaction was not significant at any specific dose or time point as determined by post-hoc analyses. At 10 mg/kg, hexamethonium significantly decreased MAP at 7 and 10 min post-injection (p<0.01 for both time points) as compared with saline control (Fig 2e, 2f) (interaction for time course data: F(30,255)=3.7, p<0.0001), and 3.2 mg/kg hexamethonium significantly increased HR at 15-60 min (significant interaction for time course data: F(30,255)=2.6, p<0.0001). DHβE and MLA did not alter significantly MAP or HR even at doses of 10 mg/kg (Fig 2c, 2d, 2g, 2h).
As expected, mecamylamine (0.32 and 1 mg/kg) antagonized the effects of nicotine (F(4,38)=22.5, p<0.0001), epibatidine (F(3,26)=28.7, p<0.0001), and varenicline (F(3,26)=20.7, p<0.0001), but did not significantly decrease the effects of cytisine on blood pressure (Fig 3; Table 3). The smallest mecamylamine dose (0.1 mg/kg) was ineffective at blocking nicotine-induced changes in MAP and, therefore, was not tested with the other nicotinic agonists. A dose of 0.32 mg/kg mecamylamine effectively attenuated the increase in MAP produced by nicotine (p<0.01) (Fig 3a) and varenicline (p<0.001) (Fig 3e), but this dose did not significantly alter the epibatidine (Fig 3c)- or cytisine-(Fig 3g) induced increases in MAP (also see table 3). However, a half-log larger dose of mecamylamine (1.0 mg/kg) not only attenuated increases in MAP produced by nicotine, epibatidine, and varenicline, but also had a tendency to decrease MAP below resting levels (only significant in varenicline-treated rats (p<0.05)). This exaggerated decrease in MAP was greater than any changes produced by 1.0 mg/kg mecamylamine alone (Fig. 2, Table 2).
Figure 3.
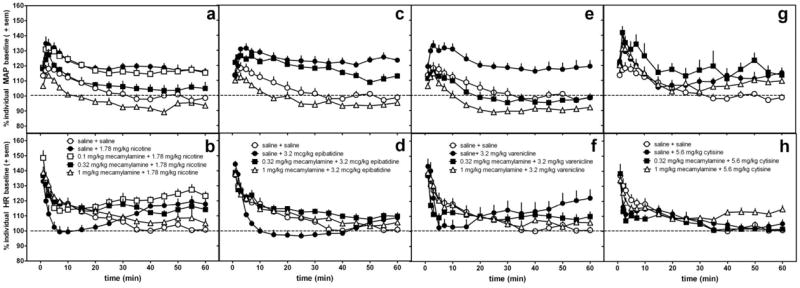
The effects of mecamylamine or saline pretreatment administered s.c. 15 min prior to 1.78 mg/kg nicotine (a,b), 3.2 mcg/kg epibatidine (c,d), 3.2 mg/kg varenicline (e,f), and 5.6 mg/kg cytisine (g,h) on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats as compared with saline control data (saline 15 min pretreatment to saline). The same saline control data are re-plotted in each graph. Data following the nicotinic agonist injection only are represented in the graph. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
Table 3.
Area under the curve (± SEM) over resting baseline for the effects of nicotinic antagonists on nicotine-, epibaitidine-, varenicline-, and cytisine-induced increases in MAP (0-90 min)
| nicotine | epibatidine | varenicline | cytisine | |
|---|---|---|---|---|
|
|
||||
| Antagonist | 1.78 mg/kg | 3.2 mcg/kg | 3.2 mg/kg | 5.6 mg/kg |
|
| ||||
| + saline | 1207 (130)*** | 1419 (127)*** | 1247 (183)*** | 933 (151)* |
|
| ||||
| + 0.1 mecamylamine | 1094 (150)*** | - | ||
| + 0.32 mecamylamine | 484 (141)## | 1058 (119)*** | 15 (111)### | 1912 (469)**# |
| + 1 mecamylamine | -238 (99)### | -146 (84)### | -376 (105)###* | 1246 (489) |
|
| ||||
| + 0.32 hexamethonium | 820 (55)* | - | - | - |
| + 3.2 hexamethonium | 741 (88)* | 960 (144)** | 135 (100)### | 579 (258) |
|
| ||||
| + 5.6 DHbE | 994 (103)** | 1026 (146)** | 883 (125)* | 1069 (113)** |
|
| ||||
| + 1 MLA | 1205 (213)*** | - | - | - |
| + 3.2 MLA | 1156 (99)*** | - | - | - |
| + 10 MLA | 1229 (107)*** | 970 (146)** | 1030 (163)* | 954 (217)* |
AUC for saline control = 243 (144)
p<0.05 vs saline control,
p<0.01 vs saline control,
p<0.001 vs saline control,
p<0.05 vs agonist alone,
p<0.01 vs agonist alone,
p<0.001 vs agonist alone single underline - p<0.01 vs other antagonist doses tested
Mecamylamine also significantly attenuated the changes in HR at 0-30 min produced by nicotine (F(4,37)=6.7, p=0.0004) and epibatidine (F(3,25)=7.9, p=0.0009) and produced a trend to attenuate varenicline-induced decreases in HR (F(3,24)=2.6, p=0.08)(Fig 3b,d,f,h, Table 4). Small doses of mecamylamine effectively blocked agonist-induced decreases in HR, but larger doses of mecamylamine were needed to attenuate increases in HR observed after 45 min (F(4,29)=11.6, p<0.0001, with 1 mg/kg mecamylamine being significantly different from saline pretreatment, p<0.01; data not shown).
Table 4.
Area under the curve (±SEM) over resting baseline for the effects of nicotinic antagonists on nicotine-, epibaitidine, varenicline-, and cytisine-induced decreases in HR (0-30 min)
| nicotine | epibatidine | varenicline | cytisine | |
|---|---|---|---|---|
|
|
||||
| Antagonist | 1.78 mg/kg | 3.2 mcg/kg | 3.2 mg/kg | 5.6 mg/kg |
|
| ||||
| + saline | 116 (74)** | 83 (61)** | 157 (98)* | 355 (88) |
|
| ||||
| + 0.1 mecamylamine | 514 (63)## | |||
| + 0.32 mecamylamine | 506 (24)## | 498 (32)## | 315 (90) | 259 (83) |
| + 1 mecamylamine | 474 (54)## | 435 (87)# | 409 (69) | 369 (22) |
|
| ||||
| + 0.32 hexamethonium | 292 (58) | - | - | - |
| + 3.2 hexamethonium | 332 (38) | -14 (75)*** | -1 (103)** | 70 (109)* |
|
| ||||
| + 5.6 DHbE | 351 (90) | 256 (60) | 329 (61) | 542 (46) |
|
| ||||
| + 1 MLA | 405 (121) | - | - | - |
| + 3.2 MLA | 198 (125) | - | - | - |
| + 10 MLA | 632 (100)## | 599 (162)# | 667 (161)# | 359 (132) |
AUC for saline control = 486 (84)
p<0.05 vs saline control,
p<0.01 vs saline control,
p<0.001 vs saline control
p<0.05 vs agonist alone,
p<0.01 vs agonist alone,
p<0.001 vs agonist alone
single underline - p<0.01 vs other antagonist doses tested
Hexamethonium (3.2 mg/kg) produced a trend to attenuate agonist-induced increases in MAP; however, post-hoc analyses revealed this effect was statistically significant only for varenicline (p<0.001) (Fig. 4, Table 3). One-way ANOVA results for each agonist in combination with saline pretreatment or antagonist pretreatment vs saline control reveal significant increases in MAP induced by agonists, but post-hoc analyses failed to identify significant attenuation by hexamethonium (except for varenicline): nicotine (F(3,29)=12.2, p<0.0001); epibatidine (F(2,20)=18.2, p<0.0001);varenicline (F(2,20)=15.3, p=0.0001); and cytisine (F(2,20)=3.7, p=0.04). In contrast to the effects of mecamylamine on MAP, hexamethonium failed to block changes in HR produced by nicotine, epibatidine, varenicline, or cytisine (Table 4) and in some instances, demonstrated a tendency to potentiate agonist-induced decreases in HR.
Figure 4.
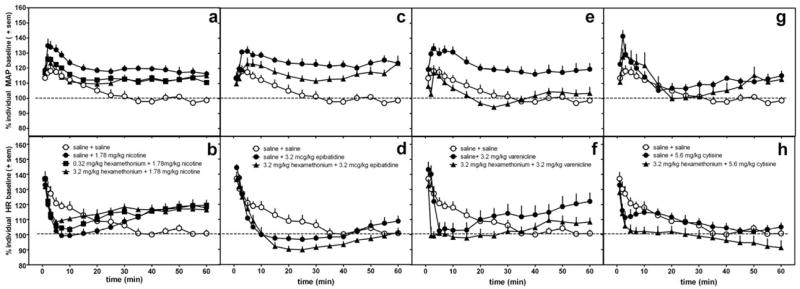
The effects of hexamethonium or saline pretreatment administered s.c. 15 min prior to 1.78 mg/kg nicotine (a,b), 3.2 mcg/kg epibatidine (c,d), 3.2 mg/kg varenicline (e,f), and 5.6 mg/kg cytisine (g,h) on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats. The same saline control data are re-plotted in each graph. Data following the nicotinic agonist injection only are represented in the graph. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
In contrast with mecamylamine or hexamethonium antagonism, DHβE did not significantly alter the changes in MAP produced by nicotine, epibatidine, varenicline, or cytisine (Fig 5, Table 3). The largest dose of DHβE had a tendency to lessen slightly the peak increase in MAP occurring 3-4 min after epibatidine administration without significantly altering overall magnitude of effect (AUC). There was a trend for DHβE to lessen the effects of nicotine, epibatidine, and varenicline on HR at 0-30 min after agonist administration; however, these effects were not statistically significant (Table 4). DHβE also did not alter later increases in HR observed with nicotine treatment.
Figure 5.
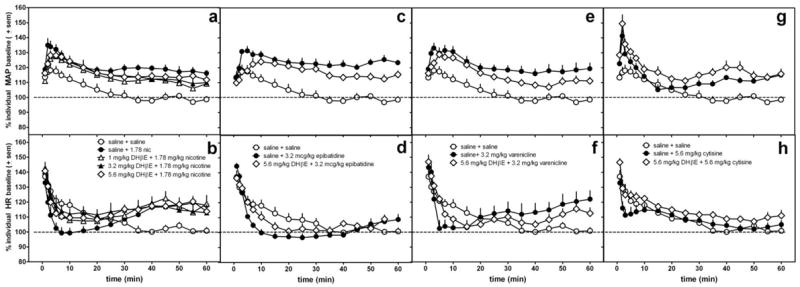
The effects of DHβE or saline pretreatment administered s.c. 15 min prior to 1.78 mg/kg nicotine (a,b), 3.2 mcg/kg epibatidine (c,d), 3.2 mg/kg varenicline (e,f), and 5.6 mg/kg cytisine (g,h) on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats. The same saline control data are re-plotted in each graph. Data following the nicotinic agonist injection only are represented in the graph. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
MLA failed to alter the effects of nicotine or cytisine on MAP (Fig 6a, Table 3). In epibatidine-and varenicline-treated rats, MLA had a tendency to reduce the initial increase in MAP; however, these effects were not statistically significant (Fig 6a, Table 3). For nicotine (p<0.01), epibatidine (p<0.05), varenicline (p<0.05), but not cytisine, MLA significantly attenuated the decrease in HR observed within the first 0-30 min after agonist administration (one-way ANOVA results for each agonist in combination with saline pretreatment or antagonist pretreatment compared with saline control: nicotine F(4,37)=4.6, p=0.005); epibatidine F(2,19)=6, p=0.01; varenicline F(2,18)=4.2, p=0.03) (Fig 6b, d, f, h; Table 4). However, it did not significantly alter the later increase in HR in nicotine-treated rats (Fig 6b). Similar to MLA, 0.1 mg/kg atropine pretreatment attenuated the initial nicotine-induced bradycardic effect, but did not alter the later positive chronotropic effect observed 60-120 min after injection (Fig 7d). Also, atropine failed to alter the nicotine-induced increase in MAP (Fig 7c). This dose of atropine produced few, if any, cardiovascular changes on its own (Fig 7a, b). At 0.1 mg/kg atropine, no changes in MAP were observed and small increases in HR occurred 20-50 min after injection (Fig 7a,b).
Figure 6.
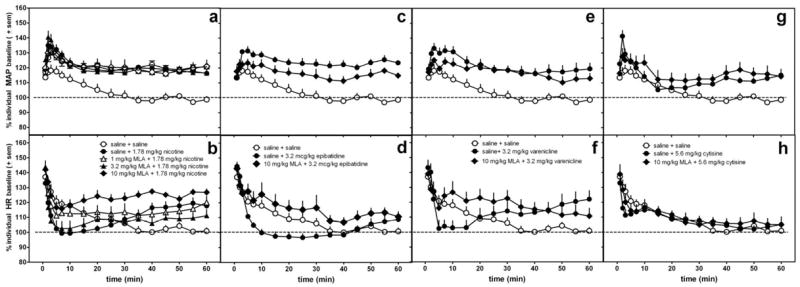
The effects of MLA or saline pretreatment administered s.c. 15 min prior to 1.78 mg/kg nicotine (a,b), 3.2 mcg/kg epibatidine (c,d), 3.2 mg/kg varenicline (e,f), and 5.6 mg/kg cytisine (g,h) on MAP (a,c,e,g) and heart rate (b,d,f,h) in conscious, freely-moving rats.The same saline control data are re-plotted in each graph. Data following the nicotinic agonist injection only are represented in the graph.Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
Figure 7.
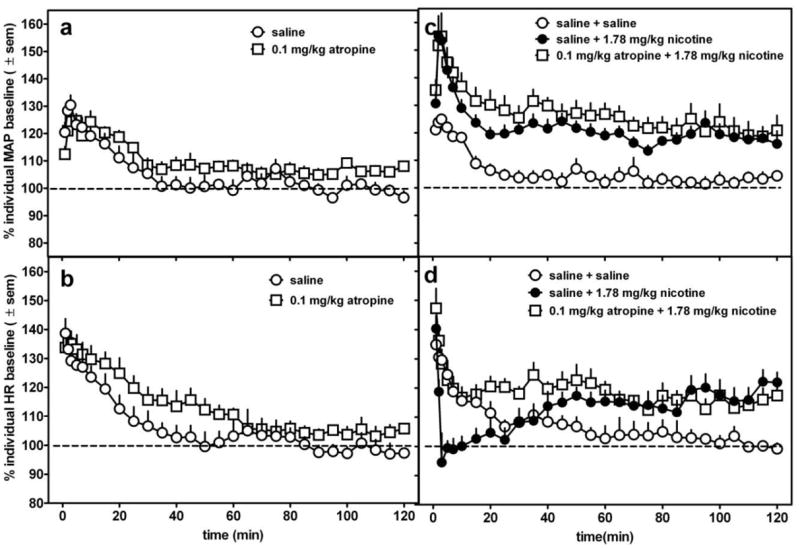
The effects of 0.1 mg/kg atropine (s.c.) alone (a,b) or as a 15 min pretreatment (s.c.) to 1.78 mg/kg nicotine (c,d) in conscious, freely-moving rats. In panels c and d, data following the nicotine injection only are represented in the graph. Data are presented as percent change over resting baseline for each subject and then averaged (± standard error of the mean (sem)) from 6-7 rats per treatment condition. The dotted line indicates the average resting MAP or heart rate (100%) for all rats.
4. DISCUSSION
In the present study, administration of nicotine or other nicotinic agonists in conscious, freely-moving rats produced changes in sympathetic and parasympathetic cardiovascular activity. Consistently across these agonists with varying receptor selectivities and efficacies (Buisson et al., 1996; Marks et al., 1996; Rollema et al., 2007; Jutkiewicz et al., 2011), nicotine, epibatidine, varenicline, and cytisine increased MAP and produced an immediate decrease in HR as compared with control. There was a delayed increase in HR, but this was inconsistent across agonists and only nicotine produced robust and prolonged delayed increases in HR. These data are consistent with a few other preclinical studies demonstrating that intravenous or subcutaneous administration of nicotine or cigarette smoke exposure in rodents increased MAP, but produced decreases or biphasic effects on HR (Henningfield et al., 1985; Houdi et al., 1995; Kaïdi et al., 2007; Donny et al., 2011). Microinjection of nicotine into the ventrolateral medulla also decreased HR (Aberger et al., 2001). However, not all preclinical studies report biphasic effects of nicotine on HR, and these differences may be due to different routes of administration, speed of sampling, and light restraining /tethering procedures (Khan et al., 1994; Marano et al., 1999).
Although these nicotinic agonists have been reported to differ in efficacy (Buisson et al., 1996; Eaton et al., 2003; Coe et al., 2005; Rollema et al., 2007; Smith et al., 2007; LeSage et al., 2009; Carroll et al., 2010), all agonists were capable of producing similar pressor responses with roughly similar increases in blood pressure (30-40% increase over resting baseline) at the doses evaluated in the present study. Epibatidine was the most potent agonist in producing hypertensive effects followed by nicotine, and there was little potency difference between varenicline and cytisine. This potency rank order is different from a previous report demonstrating that intrathecal cytisine was almost equipotent to nicotine (Khan et al., 1994), suggesting that following s.c. administration, cytisine is less likely to cross the blood brain barrier (Reavill et al., 1990). On the other hand, nicotine and epibatidine produced the most robust initial bradycardic effects at the doses studied. Unlike cytisine, the purported partial agonist varenicline also significantly decreased HR as compared with control. Overall, these data indicate that in conscious, unrestrained rats, s.c. administered nicotinic receptor agonists can produce both sympathetic (blood pressure and heart rate increases) and parasympathetic (heart rate decreases) responses and that parenterally-administered nicotine, epibatidine, and varenicline all engender similar cardiovascular responses, especially at large doses, irrespective of the purported receptor selectivity, potency, or efficacy.
The hypertensive effects of nicotinic agonists were attenuated by the general, non-competitive nicotine antagonist, mecamylamine, demonstrating that these effects were mediated through nicotinic receptors. However, mecamylamine was less effective at blocking the small cardiovascular changes produced by cytisine. Although previous findings have reported that the in vitro and in vivo effects of cytisine were partially or fully attenuated by mecamylamine (Stolerman et al., 1983; Hall et al., 1993; Chandler and Stolerman, 1997; Cunningham and McMahon et al., 2011; Cunningham et al., 2012), the current data suggest that the large doses of cytisine used in the present study have some non-nicotinic receptor-mediated effects.
The pressor effects of the nicotinic agonists were also fully or partially attenuated by nAChR blocker hexamethonium, which has little access to the central nervous system especially at the doses and with the route of administration used in the present study (Asghar and Roth 1971; Malin et al., 1997), indicating that peripheral nicotinic receptors mediate the effects of nicotinic agonists on blood pressure. The α7-preferring antagonist (but see, Baker et al., 2004; Salminen et al., 2005), MLA, was slightly effective in reducing varenicline- and epibatidine-induced elevations in MAP. This supports previous data indicating that varenicline has high efficacy agonist activity at α7 receptor subtypes (Mihalak et al., 2006), and that the α7 receptor may mediate nicotinic agonist-induced increases in sympathetic activity (Khan et al., 1994; Franceschini et al., 2000; Li et al., 2009, 2010; but see Deck et al., 2005). However, the hypertensive effects of the other nicotinic agonists were not greatly modified by MLA. In addition, the pressor effects of nicotinic agonists were not significantly reduced by administration of DHβE, demonstrating the blood pressure effects of most nicotinic agonists do not involve α7 or α4β2 receptor subtypes. This is in contrast to mediation of the discriminative stimulus effects of small doses of nicotine, where the α4β2 subtype is largely responsible for these effects (Stolerman et al., 1997; Gommans et al., 2000; Shoaib et al., 2002; Zaniewska et al., 2006; for review see Smith and Stolerman, 2009; Jutkiewicz et al., 2011).
The interaction of these agonists and antagonists on heart rate was somewhat different from their effects on blood pressure. The immediate parasympathetic activity (striking decreases in HR) observed with nicotinic receptor agonists was attenuated by mecamylamine and MLA, partially attenuated by DHβE, but not significantly altered by hexamethonium. Mecamylamine was the only antagonist that attenuated the biphasic actions of nicotine on HR. It therefore appears that centrally-acting nicotinic receptors, likely α7 and to a lesser extent α4β2 receptors subtypes and potentially other subtypes, contribute to agonist-induced bradycardia. These data are consistent with previous studies demonstrating that microinjection of nicotine into certain brain regions, specifically the rat nucleus tractus solitarius (NTS) or the caudal ventrolateral medulla (CVLM), decreased HR in anesthetized rats (Kubo and Misu, 1981; Robertson et al., 1988; Tseng et al., 1993; Ferreira et al., 2000; Aberger et al., 2001; Murota et al., 2008). DHβE also attenuated i.v. nicotine- and cytisine-induced bradycardia in anesthetized mice (Li et al., 2010). Additionally, atropine blocked nicotine- and nicotinic agonist-induced HR decrease (current data; Nadeau and James, 1967; Li et al., 2009, 2010) and vagotomy attenuated nicotine-induced bradycardia (Sloan et al., 1988), suggesting that muscarinic receptors located downstream of central nicotine receptor activation participate in producing the bradycardic effect.
Overall, these data demonstrate that parenteral administration of nicotine and other nicotinic agonists produce sympathetic and parasympathetic activity in conscious, unrestrained rats. Similar effects have been shown following acute nicotine ingestion by human smokers. The sympathetic activation produced by nicotinic agonists is likely due to nicotinic receptor activation at autonomic ganglia involving nicotinic receptor subtypes other than α4, α7, or β2. Parasympathetic activation and general nicotinic receptor regulation of HR is likely due to central nicotinic receptors involving multiple nicotinic receptor subtypes. The predominant receptors mediating the cardiovascular effects are most likely ganglionic α3β4 nicotinic receptor subtypes, as these are two of the most prominent subtypes in the ganglia (for review see Kirstein and Insel, 2004; Bibevski et al., 2000; Deck et al., 2005); however, there are few, if any, selective α3β4 ligands that are generally available to support or challenge this hypothesis.
We recently established the nicotine-like discriminative stimulus effects of several nicotinic agonists and partial agonists and evaluated the ability of several nicotinic antagonists to modify these stimulus effects (Jutkiewicz et al., 2011). One conclusion from that study was that the α4β2-preferring antagonist, dihydro-β-erythroidine (DHβE), produced a surmountable antagonism of the discriminative stimulus effects of nicotine (0.32 mg/kg), indicating that the discriminative stimulus effect of small nicotine doses was mediated by the α4β2 nicotinic receptor subtype, consistent with previous findings (Stolerman et al., 1997; Gommans et al., 2000; Shoaib et al., 2002; Zaniewska et al., 2006; for review see Smith and Stolerman, 2009, but see Harvey et al., 1996; Harvey and Luetje, 1996; Chavez-Noriega et al., 1997). The present data highlight the differential localization and receptor subtype regulation of cardiovascular responses produced by nicotine and nicotinic ligands and demonstrate that the nicotinic receptor subtypes mediating the discriminative stimulus effects of nicotine are different from subtypes mediating cardiovascular events. Therefore, drugs that target the α4β2 nicotine receptor selectively appear very promising for development for treatment of nicotine abuse because they block the discriminative stimulus functions of nicotine but have modest cardiovascular effects on their own and in combination with nicotine.
Acknowledgments
Role of Funding Source
This research and preparation of the manuscript was supported by grants from the University of Michigan Tobacco Research Network and National Institutes of Health National Institute of Drug Abuse [T32 DA007268]. NIDA had no further role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIDA. A portion of this work was also supported by the Intramural Research Programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism.
Footnotes
Dr. Jutkiewicz contributed to the design of the study, conducted experiments, performed data analysis, and wrote the manuscript. Dr. Woods contributed to the design of the study and to writing the manuscript. Drs. Carroll and Rice contributed compounds for the study. All authors contributed to and have approved the manuscript.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Emily M. Jutkiewicz, Department of Pharmacology, University of Michigan, 1150 W Medical Center Drive, Ann Arbor, MI 48109-5632
Kenner C. Rice, Chemical Biology Research Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, Room 4N03, MSC 9415, DHHS, Bethesda, MD, 20892
F. Ivy Carroll, Medicinal Chemistry, RTI International, Research Triangle Park, NC, 27709.
James H. Woods, Department of Pharmacology, University of Michigan, 1150 W Medical Center Drive, Ann Arbor, MI 48109-5632 Department of Psychology,University of Michigan.
References
- Aberger K, Chitravanshi VC, Sapru HN. Cardiovascular responses to microinjections of nicotine into the caudal ventrolateral medulla of the rat. Brain Res. 2001;892:138–146. doi: 10.1016/s0006-8993(00)03250-9. [DOI] [PubMed] [Google Scholar]
- Arslan BY, Ulus IH, Savci V, Kiran BK. Effects of intracerebroventricular injected choline on cardiovascular functions and sympathoadrenal activity. J Cardiovasc Pharmacol. 1991;17:814–821. doi: 10.1097/00005344-199105000-00018. [DOI] [PubMed] [Google Scholar]
- Asghar K, Roth LJ. Entry and distribution of hexamethonium in the central nervous system. Biochem Pharmacol. 1971;20:2787–2790. doi: 10.1016/0006-2952(71)90189-4. [DOI] [PubMed] [Google Scholar]
- Baker ER, Zwart R, Sher E, Millar NS. Pharmacological properties of alpha9alpha10 nicotinic acetylcholine receptor revealed by heterologous expression of subunit chimeras. Mol Pharmacol. 2004;65:453–460. doi: 10.1124/mol.65.2.453. [DOI] [PubMed] [Google Scholar]
- Bibevski S, Zhou Y, McIntosh JM, Zigmond RE, Dunlap ME. Functional nicotinic acetylcholine receptors that mediate ganglionic transmission in cardiac parasympathetic neurons. J Neurosci. 2000;20:5076–5082. doi: 10.1523/JNEUROSCI.20-13-05076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Ma W, Deng L, Navarro HA, Damaj MI, Martin BR. Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 3’-(substituted phenyl)epibatidine analogues. Nicotinic partial agonists. J Nat Prod. 2010;73:306–312. doi: 10.1021/np9006124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler CJ, Stolerman IP. Discriminative stimulus properties of the nicotinic agonist cytisine. Psychopharmacology. 1997;129:257–264. doi: 10.1007/s002130050188. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Manbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD, III, O’Neill BT. Varenicline: an a4b2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Cunningham CS, McMahon LR. The effects of nicotine, varenicline, and cytisine on schedule-controlled responding in mice: difference in a4b2 nicotinic receptor activation. Eur J Pharmacol. 2011;654:47–52. doi: 10.1016/j.ejphar.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CS, Javors MA, McMahon LR. Pharmacologic characterization of a nicotine-discriminative stimulus in rhesus monkeys. J Pharmacol Exp Ther. 2012;341:840–849. doi: 10.1124/jpet.112.193078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deck J, Bibevski S, Gnecchi-Ruscone T, Bellina V, Montano N, Dunlap ME. Alpha7-nicotinic acetylcholine receptor subunit is not required for parasympathetic control of the heart in the mouse. Physiol Genomics. 2005;22:86–92. doi: 10.1152/physiolgenomics.00085.2004. [DOI] [PubMed] [Google Scholar]
- Del Signore A, Gotti C, Rizzo A, Moretti M, Paggi P. Nicotinic acetylcholine receptor subtypes in the rat sympathetic ganglion: pharmacological characterization, subcellular distribution and effect of pre- and postganglionic nerve crush. J Neuropath Exp Neurology. 2004;63:138–150. doi: 10.1093/jnen/63.2.138. [DOI] [PubMed] [Google Scholar]
- Delaunois A, Dedoncker P, Hanon E, Guyaux M. Repeated assessment of cardiovascular and respiratory functions using combined telemetry and whole-body plethysmography in the rat. J Pharmacol Toxicol Methods. 2009;60:117–129. doi: 10.1016/j.vascn.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Sweitzer M, Chaudhri N, Gharib M, Sved AF. Self-administered and yoked nicotine produce robust increases in blood pressure and changes in heart rate with modest effects of behavioral contingency in rats. Pharmacol Biochem Behav. 2011;99:459–467. doi: 10.1016/j.pbb.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JB, Peng J-H, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo Y-P, Steinlein O, Lukas RJ. Characterization of human a4b2-nicotnic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- Ferreira M, Singh A, Dretchen KL, Kellar KJ, Gillis RA. Brainstem nicotinic receptor subtypes that influence intragastric and arterial blood pressures. J Pharmacol Exp Ther. 2000;294:230–238. [PubMed] [Google Scholar]
- Franceschini D, Orr-Urtreger A, Yu W, Mackey LY, Bond RA, Armstrong D, Patrick JW, Beaudet AL, De Biasi M. Altered baroreflex responses in alpha7 deficient mice. Behav Brain Res. 2000;113:3–10. doi: 10.1016/s0166-4328(00)00195-9. [DOI] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol. 2001;86:1773–1782. doi: 10.1152/jn.2001.86.4.1773. [DOI] [PubMed] [Google Scholar]
- Gommans J, Stolerman IP, Shoaib M. Antagonism of the discriminative and aversive stimulus properties of nicotine in C57BL/6J mice. Neuropharmacology. 2000;39:2840–2847. doi: 10.1016/s0028-3908(00)00130-1. [DOI] [PubMed] [Google Scholar]
- Gotti C, Riganti L, Vailati S, Clementi F. Brain neuronal nicotinic receptor as new targets for drug discovery. Curr Pharm Des. 2006;12:407–428. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- Haas M, Kubler W. Nicotine and sympathetic neurotransmission. Cardiovascular Drugs Ther. 1996;10:657–665. doi: 10.1007/BF00053022. [DOI] [PubMed] [Google Scholar]
- Hall M, Zerbe L, Leonard S, Freedman R. Characterization of [3H]cytisine binding to human brain membranes preparations. Brain Res. 1993;600:127–133. doi: 10.1016/0006-8993(93)90410-o. [DOI] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW. Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor beta subunits. J Neurosci. 1996;16:3798–3806. doi: 10.1523/JNEUROSCI.16-12-03798.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Maddoz FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem. 1996;67:1953–1959. doi: 10.1046/j.1471-4159.1996.67051953.x. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Miyasato K, Jasinski DR. Abuse liability and pharmacodynamic characteristics of intravenous and inhaled nicotine. J Pharmacol Exp Ther. 1985;234:1–12. [PubMed] [Google Scholar]
- Houdi AA, Dowell RT, Diana JN. Cardiovascular responses to cigarette smoke exposure in restrained conscious rats. J Pharmacol Exp Ther. 1995;275:646–653. [PubMed] [Google Scholar]
- Jutkiewicz EM, Brooks E, Kynaston A, Rice KC, Woods JH. Patterns of nicotinic receptor antagonism: nicotine discrimination studies. J Pharmacol Exp Ther. 2011;339:194–202. doi: 10.1124/jpet.111.182170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaïdi S, Brutel F, Van Deun F, Kramer K, Remie R, Dewe W, Remusat P, Delaunois A, Depelchin O. Comparison of two methods (left carotid artery and abdominal aorta) for surgical implantation of radiotelemetry devices in CD-1 mice. Lab Anim. 2007;41:388–402. doi: 10.1258/002367707781282839. [DOI] [PubMed] [Google Scholar]
- Khan IM, Printz MP, Yaksh TL, Taylor P. Augmented responses to intrathecal nicotinic agonists in spontaneous hypertension. Hypertension. 1994;24:611–619. doi: 10.1161/01.hyp.24.5.611. [DOI] [PubMed] [Google Scholar]
- Kirstein SL, Insel PA. Autonomic nervous system pharmacogenomics: a progress report. Pharmacol Rev. 2004;56:31–52. doi: 10.1124/pr.56.1.2. [DOI] [PubMed] [Google Scholar]
- Koch A, Hoffman K, Steck W, Horsch A, Hengen N, Mörl H, Harenberg J, Spohr U, Weber E. Acute cardiovascular reactions after cigarette smoking. Atherosclerosis. 1980;35:67–75. doi: 10.1016/0021-9150(80)90028-3. [DOI] [PubMed] [Google Scholar]
- Kubo T, Misu Y. Changes in arterial blood pressure after microinjection of nicotine into the dorsal area of the medulla oblongata of the rat. Neuropharmacology. 1981;20:521–524. doi: 10.1016/0028-3908(81)90188-x. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Chefer SI, Kimes AS, Shumway D, Stein EA, Mukhin AG, Goldberg SR. Baseline expression of alpha4beta2* nicotinic acetylcholine receptors predicts motivation to self-administer nicotine. Biol Psychiatry. 2009;65:714–716. doi: 10.1016/j.biopsych.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeSage MG, Shelley D, Ross JT, Carroll FI, Corrigall WA. Effects of the nicotinic receptor partial agonists varenicline and cytisine on the discriminative stimulus effects of nicotine in rats. Pharmacol Biochem Behav. 2009;91:461–467. doi: 10.1016/j.pbb.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, LaCroix C, Freeling J. Specific subtypes of nicotinic cholinergic receptors involved in sympathetic and parasympathetic cardiovascular responses. Neurosci Lett. 2009;462:20–23. doi: 10.1016/j.neulet.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, LaCroix C, Freeling J. Cytisine induced autonomic cardiovascular responses via activations of different nicotinic receptors. Autonomic Neurosci. 2010;154:14–19. doi: 10.1016/j.autneu.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Palmatier MI, Caggiula AR, Donny EC, Sved AF. Reinforcement enhancing effect of nicotine and its attenuation by nicotinic antagonists in rats. Psychopharmacology. 2007;194:463–473. doi: 10.1007/s00213-007-0863-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Schopen CK, Kirk JW, Sailer EE, Lawless BA, Upchurch TP, Shenoi M, Rajan N. Nicotine abstinence syndrome precipitated by central but not peripheral hexamethonium. Pharmacol Biochem Behav. 1997;58:695–699. doi: 10.1016/s0091-3057(97)90006-x. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Chambers LK, Rovetti CC. Effects of the competitive nicotinic antagonist erysodine on behavior occasioned or maintained by nicotine: comparison with mecamylamine. Psychopharmacology. 2000;148:234–242. doi: 10.1007/s002130050047. [DOI] [PubMed] [Google Scholar]
- Mao D, Yasuda RP, Fan H, Wolfe BB, Kellar KJ. Heterogeneity of nicotinic cholinergic receptors in rat superior cervical and nodose ganglia. Mol Pharmacol. 2006;70:1693–1699. doi: 10.1124/mol.106.027458. [DOI] [PubMed] [Google Scholar]
- Marano G, Ramirez A, Mori I, Ferrari AU. Sympathectomy inhibits the vasoactive effects of nicotine in conscious rats. Cardiovascular Res. 1999;42:201–205. doi: 10.1016/s0008-6363(98)00326-5. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Robinson SF, Collins AC. Nicotinic agonists differ in activation and desensitization of 86Rb+ efflux from mouse thalamic synaptosomes. J Pharmacol Exp Ther. 1996;277:1383–1396. [PubMed] [Google Scholar]
- Mihalak KB, Carroll FI, Leutje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- Murota Y, Fujii M, Sugiyama Y, Funabashi T, Takahashi T, Goshima Y. DOPA cyclohexyl ester, a DOPA antagonist, blocks the depressor responses elicited by microinjections of nicotine into the nucleus tractussolitarii of rats. Neurosci Lett. 2008;422:114–117. doi: 10.1016/j.neulet.2008.06.077. [DOI] [PubMed] [Google Scholar]
- Nadeau RA, James TN. Effects of nicotine on heart rate studied by direct perfusion of sinus node. Am J Physiol. 1967;212:911–916. doi: 10.1152/ajplegacy.1967.212.4.911. [DOI] [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Dávila-García MI, Kellar KJ. Measuring nicotinic receptors with characteristics of a4b2, a3b2 and a3b4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82:468–481. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- Poth K, Nutter TJ, Cuevas J, Parker MJ, Adams DJ, Luetje CW. Heterogeneity of nicotinic receptor class and subunit mRNA expression among individual parasympathetic neurons from rat intracardiac ganglia. J Neurosci. 1997;17:586–596. doi: 10.1523/JNEUROSCI.17-02-00586.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavill C, Walther B, Stolerman IP, Testa B. Behavioural and pharmacokinetic studies on nicotine, cytisine and lobeline. Neuropharmacology. 1990;29:619–624. doi: 10.1016/0028-3908(90)90022-j. [DOI] [PubMed] [Google Scholar]
- Robertson D, Tseng CJ, Appalsamy M. Smoking and mechanisms of cardiovascular control. Am Heart J. 1988;115:258–263. doi: 10.1016/0002-8703(88)90646-1. [DOI] [PubMed] [Google Scholar]
- Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, III, Williams KE. Pharmacological profile of the a4b2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Johnson M. Dissociating nicotine and nonnicotine components of cigarette smoking. Pharmacol Biochem Behav. 2000;67:71–81. doi: 10.1016/s0091-3057(00)00301-4. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Shoaib M, Gommans J, Morley A, Stolerman IP, Grailhe R, Changeux JP. The role of nicotinic receptor beta-2 subunits in nicotine discrimination and conditioned taste aversion. Neuropharmacology. 2002;42:530–539. doi: 10.1016/s0028-3908(01)00194-0. [DOI] [PubMed] [Google Scholar]
- Singh S, Loke YK, Spangler JG, Furberg CD. Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis. CMA J. 2011;183:1359–1366. doi: 10.1503/cmaj.110218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan JW, Martin WR, Bostwick M, Hook R, Wala E. The comparative binding characteristics of nicotinic ligands and their pharmacology. Pharmacol Biochem Behav. 1988;30:255–267. doi: 10.1016/0091-3057(88)90454-6. [DOI] [PubMed] [Google Scholar]
- Smith JW, Mogg A, Tafi E, Peacey E, Pullar IA, Szekeres P, Tricklebank M. Ligands selective for a4b2 but not a3b4 or a7 nicotinic receptors generalize to the nicotine discriminative stimulus in the rat. Psychopharmacology. 2007;190:157–170. doi: 10.1007/s00213-006-0596-8. [DOI] [PubMed] [Google Scholar]
- Smith JW, Stolerman IP. Recognising nicotine: the neurobiological basis of nicotine discrimination. Handb Exp Pharmacol. 2009;192:295–333. doi: 10.1007/978-3-540-69248-5_11. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Yoo S, Hill KP, Mooney M. Self-administration of intravenous nicotine in male and female cigarette smokers. Neuropsychopharmacology. 2008;33:715–720. doi: 10.1038/sj.npp.1301460. [DOI] [PubMed] [Google Scholar]
- Soria R, Stapleton JM, Gilson SF, Sampson-Cone A, Henningfield JE, London ED. Subjective and cardiovascular effects of intravenous nicotine in smokers and non-smokers. Psychopharmacol (Berl) 1996;128:221–226. doi: 10.1007/s002130050129. [DOI] [PubMed] [Google Scholar]
- Spohr U, Hofmann K, Steck W, Harenberg J, Walter E, Hengen N, Augustin J, Mörl H, Koch A, Horsch A, Weber E. Evaluation of smoking-induced effects on sympathetic, hemodynamic and metabolic variables with respect to plasma nicotine and COHb levels. Atherosclerosis. 1979;33:271–283. doi: 10.1016/0021-9150(79)90179-5. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Pratt JA, Garcha HS, Giardini V, Kumar R. Nicotine cue in rats analysed with drugs acting on cholinergic and 5-hydroxytryptamine mechanisms. Neuropharmacology. 1983;22:1029–1037. doi: 10.1016/0028-3908(83)90021-7. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Chandler CJ, Garcha HS, Newton JM. Selective antagonism of behavioural effects of nicotine by dihydro-b-erythroidine in rats. Psychopharmacol (Berl) 1997;129:390–397. doi: 10.1007/s002130050205. [DOI] [PubMed] [Google Scholar]
- Tachmes L, Fernandez RJ, Sackner MA. Hemodynamic effects of smoking cigarettes of high and low nicotine content. Chest. 1978;74:243–246. doi: 10.1378/chest.74.3.243. [DOI] [PubMed] [Google Scholar]
- Tseng CJ, Appalsamy M, Robertson D, Mosqueda-Garcia R. Effects of nicotine on brain stem mechanisms of cardiovascular control. J Pharmacol Exp Ther. 1993;265:1511–1518. [PubMed] [Google Scholar]
- Watkins SS, Epping-Jordan MP, Koob GF, Markou A. Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol Biochem Behav. 1999;62:743–751. doi: 10.1016/s0091-3057(98)00226-3. [DOI] [PubMed] [Google Scholar]
- Williams JM, Steinberg MB, Steinberg ML, Gandhi KK, Ulpe R, Foulds J. Varenicline for tobacco dependence: panacea or plight? Expert Opin Pharmacother. 2011;12:1799–1812. doi: 10.1517/14656566.2011.587121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaniewska M, McCreary AC, Przegaliński E, Filip M. Evaluation of the role of nicotinic acetylcholine receptor subtypes and cannabinoid system in the discriminative stimulus effects of nicotine in rats. Eur J Pharmacol. 2006;540:96–106. doi: 10.1016/j.ejphar.2006.04.034. [DOI] [PubMed] [Google Scholar]