

Abstract
ATR and ATM kinases are central to the checkpoint activation in response to DNA damage and replication stress. However, the nature of the signal, which initially activates these kinases in response to UV damage, is unclear. Here, we have shown that DDB2 and XPC, two early UV damage recognition factors, are required for the damage-specific ATR and ATM recruitment and phosphorylation. ATR and ATM physically interacted with XPC and promptly localized to the UV damage sites. ATR and ATM recruitment and their phosphorylation were negatively affected in cells defective in DDB2 or XPC functions. Consequently, the phosphorylation of ATR and ATM substrates, Chk1, Chk2, H2AX, and BRCA1 was significantly reduced or abrogated in mutant cells. Furthermore, UV exposure of cells defective in DDB2 or XPC resulted in a marked decrease in BRCA1 and Rad51 recruitment to the damage site. Conversely, ATR- and ATM-deficiency failed to affect the recruitment of DDB2 and XPC to the damage site, and therefore did not influence the NER efficiency. These findings demonstrate a novel function of DDB2 and XPC in maintaining a vital cross-talk with checkpoint proteins, and thereby coordinating subsequent repair and checkpoint activation.
Keywords: ATM, ATR, XPC, DDB2, UV irradiation, checkpoint activation, nucleotide excision repair
1. Introduction
The DNA damage response (DDR) pathway plays a critical role in maintaining genomic stability and preventing carcinogenesis [1]. DDR invoked by genotoxic stress results in cell cycle arrest, enhanced DNA repair, changes in transcription, and apoptosis. Activation of the checkpoint arrests the cell cycle to allow repair of the damaged DNA. If the damage is excessive and beyond repair, apoptosis is triggered. NER is a versatile DNA repair pathway that can remove a broad range of structurally unrelated lesions including UV-induced bulky DNA adducts cyclobutane pyrimidine dimers (CPD) and pyrimidine (6-4) pyrimidone photoproducts (6-4PP) [2]. One sub-pathway of NER, global genome NER (GG-NER), removes damage from the entire genome, whereas DNA damage in the transcribed strand of active genes is preferentially eliminated by transcription-coupled NER (TC-NER) [3]. In GG-NER, damage is recognized by the UV-DDB (DDB1 and DDB2) and XPC-RAD23B complexes [4, 5]. DDB1 participates in NER through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. The DDB1-CUL4-ROC1 complex ubiquitylates XPC, which may enhance DNA binding by XPC and promotes NER [6, 7]. The DDB complex initially recognizes the CPD lesions and recruits XPC [4, 5, 8, 9], whereas XPC can independently recognize 6-4PP lesions [5]. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates lesion recognition by XPC. In turn, XPC helps in recruiting XPA, XPG, and TFIIH components that enable opening of the DNA helix around the damage site to form a bubble [8]. XPA stabilizes the bubble and aids in positioning the XPF and XPG endonucleases for respective 5’ and 3’ incisions to excise out a 24-32 bp oligonucleotide containing damaged lesion. The resulting gap is filled by repair synthesis, and finally the nick is ligated to complete NER [2, 10]. Importantly, the defects in components of the NER pathway result in Xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy which are characterized by sensitivity to UV irradiation and predisposition to skin cancers [11, 12].
The phosphoinositide-3-kinase-like kinases family of protein kinases including ATR (Ataxia telangiectasia- and Rad3- related) and ATM (Ataxia telangiectasia mutated) are the principal checkpoint kinases activated by DNA damage [13, 14]. Seckel (ATR-defective) and AT (ATM-deficient) cells show impaired signaling due to the defects in checkpoint activation. Activation of ATR and ATM triggers a phosphorylation-mediated cascade of events that lead to cell cycle arrest and stimulation of DNA repair. ATR is the primary sensor of single-stranded (ssDNA) breaks (SSB) caused by UV damage and replication stress. It has been shown that DNA damage and replication intermediates increase the unwinding of DNA, leading to the accumulation of RPA-coated ss-DNA, which recruits ATR [15, 16]. ATR phosphorylates Chk1, which results in checkpoint activation during G1, S, and G2/M phases. Activated Chk1 phosphorylates Cdc25 phosphatases to inhibit their function, and the cells delay progression through the cell cycle [17]. Although DNA double strand breaks (DSB) primarily activates the ATM pathway, recent studies including ours have implicated a participatory role of ATM in the NER pathway [18, 19]. ATM phosphorylates the checkpoint kinase Chk2, which also triggers degradation of Cdc25A phosphatases to delay the cell cycle [20]. ATR and ATM phosphorylate histone H2AX, which spreads along the DNA up to 200-400 kb, and helps in the recruitment of proteins involved in DNA damage repair and checkpoint activation [21]. Moreover, ATR- and ATM-mediated phosphorylation of BRCA1 and H2AX [22, 23] is required for S and G2/M phase checkpoints and homologous recombination (HR)-mediated DNA repair during S and G2 phases. During DNA replication, other ssDNA gaps are generated by the stalling of replication forks at unrepaired damage sites. Repair of these gaps may involve post-replicative recombinational repair [24]. If not repaired, stalled fork gaps can evolve into DSB [24]. Besides BRCA1, BRCA2, and Rad51 are also required for HR-mediated DNA repair and replication fork maintenance [25, 26]. Both Chk1 and Chk2 regulate the functional associations between BRCA1, BRCA2, and Rad51 proteins in response to DNA damage, and thus promote HR-mediated repair of stalled replication forks [27, 28].
In response to DSB, the lesion recognition factor Mre11-Rad50-Nbs1 (MRN) complex helps the recruitment of ATM to the damage site and its activation by phosphorylation [29]. However, whether UV-damage recognition factors directly influence ATR and ATM recruitment and their phosphorylation is not clearly established. Jiang and Sancar showed direct binding of ATR to the damaged DNA without lesion processing, raising the possibility that ATR may activate the checkpoint signaling directly [30]. Furthermore, Vrouwe et al reported that UV-induced photolesions results in checkpoint activation in NER-dependent and -independent pathways [31]. Recently, Oh et. al. reported γH2AX foci formation after UV-irradiation in cells lacking NER [32]. In yeast, UV-induced DNA damage results in checkpoint activation independent of NER lesion processing [33, 34]. These results support that lesion processing is not essential for γH2AX formation and checkpoint activation. However, several studies reported that lesion processing by NER factors might be an essential step in γH2AX foci formation [35-39]. Even though these studies support that the checkpoint activation induced by UV irradiation requires a functional NER apparatus, these studies do not show how and when ATR and ATM are recruited to the damage site and result in phosphorylation of downstream substrates. It has been shown that in response to UV irradiation, RPA-coated single-stranded DNA (ssDNA) recruits ATR to the UV damage site [15, 16]. This supports the possibility of ATR and ATM recruitment after incision of the UV damage. However, in case of mismatch repair, ATR is recruited to the damage site by the lesion recognition factors and also by the RPA-coated ssDNA [40]. Additionally, in DSB repair pathway, the lesion recognition factor MRN complex influences ATM recruitment [41]. Furthermore, in response to cisplatin treatment, XPC physically interacts with ATM, and is involved in ATM activation [42]. Whether the NER proteins play any direct role in ATR and ATM recruitment, however, has not been shown. To further gain insight into the mechanism of ATR and ATM recruitment and activation, we examined the roles of DDB2 and XPC in the recruitment and activation of ATR and ATM. Here, we show that XPC physically interacts with ATR and ATM. Both DDB2 and XPC facilitate ATR and ATM recruitment to the damage site, and promote their phosphorylation. This eventually affects the recruitment and phosphorylation of their substrate proteins at the damage site. We propose that DDB2 and XPC help assemble the ATR and ATM complex at the UV damage site and facilitate their activation to provoke the downstream cascade constituting the DNA damage response pathway.
2. Materials and Methods
2.1 Cell lines and antibodies
XP-E (GM01389, DDB2-mutated), XP-C (GM02096, XPC-mutated), Seckel (GM18366, ATR-defective) and AT (M04405, ATM-deficient) cells were obtained from Coriell Institute for Medical Research, Camden, NJ. HeLa cells were from ATCC, Manassas, VA. HeLa-60 cells expressing FLAG-DDB2 and HA-XPC, and normal human fibroblasts (NHF, OSU-2) were generated in our laboratory [43]. The cells were cultured as described [44]. XPC, DDB2, CPD, antibodies were raised in our laboratory. Antibodies specific for phospho-ATR (Ser 428), phospho-ATM (Ser 1981), phospho-Chk2 (Thr68), phospho-Chk1 (Ser 296), phospho-BRCA1 (S1524), γ-H2AX (Ser139), Chk1 (2345) and Chk2 (2662) were from Cell Signaling Technology. H2AX (sc-54606), ATM (sc-23921), ATR (N-19) (sc-1887), BRCA1 (sc-642), p53 (FL-393), and p21 (sc-397) antibodies were from Santa Cruz Biotechnology. Anti-FLAG M2 (F3165) antibody is from Sigma Aldrich and 6-4PP antibody (64M-2) was obtained from Dr Toshio Mori, Nara Medical University, Nara, Japan. Goat anti-rabbit IgG IR Dye 800CW (926-32211) is from LI-COR biosciences.
2.2. UV irradiation, protein isolation, and Western blotting
These were performed as described [19]. Cells were washed with phosphate-buffered saline (PBS) and irradiated through a germicidal lamp (254 nM) at a dose rate of 1.0 J m2/s as measured with a Kettering model 65 radiometer (Cole-Palmer, Vernon Hills, IL, USA). Media was added to the cells, returned to the 37°C incubator to allow repair and harvested at the indicated post-UV irradiation times. Total protein was extracted from the cells using sodium dodecyl sulfate (SDS) lysis buffer (62 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol) with protease and phosphatase inhibitors followed by boiling for 8 min. Protein amount was estimated using Bio-Rad DCTM Protein assay kit, and the whole cell lysates were resolved by SDS–polyacrylamide gel electrophoresis (PAGE) using Novex Tris-Glycine gels (Invitrogen, Carlsbad, CA, USA) followed by Western blotting to detect specific proteins.
2.3. Isolation of chromatin-bound proteins
Fractionation of extracts, isolation of chromatin-bound proteins, and immunoprecipitation were performed essentially as described [19, 45, 46].
2.4. Gene silencing
ATR, DDB2, and XPC siRNAs were from Dharmacon, Chicago, IL. ATM shRNA was obtained from Sigma Aldrich. Transfections with various RNAs were conducted using Lipofectamine™ 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions.
2.5. Qualitative and quantitative detection of UV damage
Lesions of the genomic DNA in native cellular environment were induced by micro-pore local UV irradiation and their detection was performed by dual immunofluorescent staining by our established methods [47, 48]. Repair rates of damage were obtained from ISB quantitation of dimers in DNA isolated from cells at different post-irradiation times as described earlier [44, 45, 49].
3. Results
3.1 ATR and ATM localize to the UV damage site and interact with XPC
We have previously shown that in response to UV damage, ATR and ATM co-localize with XPC in normal human and cancer cells [19]. Here we have further confirmed the specific ATR and ATM localization to the UV damage sites via micropore immunofluorescence (Fig.1 A). Irradiation through the micropore filters generates sub-nuclear localized damaged spots as opposed to the global exposures which result in damage over the entire cellular genome [19]. These local damage sites would have both CPD, and 6-4PP and therefore could be marked using one of the lesion-specific antibodies. In this experiment, normal human fibroblast (NHF) cells were exposed to 100 J/m2 UV irradiation through micropore filters, and allowed for 1 h post-repair incubation prior to determining the co-localization of pATM, ATR, and γH2AX with CPD. The UV damaged foci exhibited the distinct phosphorylation of H2AX, a recognized molecular marker of damage response initiation [19, 32, 35-37]. ATR and ATM are principal kinases which phosphorylate H2AX upon DNA damage. The co-localization of γ-H2AX with CPD and 6-4PP has been used to demonstrate the participation of ATR to the UV damage site [35]. Therefore, our data revealed an obvious involvement of ATR and ATM kinases in response to UV damage. To examine if ATR and ATM signal transduction is also operating in response to 6-4PP, we determined the co-localization of pATM and γ-H2AX with 6-4PP at the UV damage sites. The 6-4PP also co-localized with pATM and γ-H2AX, demonstrating that the ATR/ATM signal transduction is also operating in response to 6-4PP, and not specific to CPD (Fig. 1B). More importantly, we showed that ATR and ATM localize to damage sites in G1 arrested cells (Fig. 1C). This data further supports the involvement of ATR and ATM kinases in response to UV damage, which is clearly independent of DNA replication. The co-localization of ATR and ATM with XPC at the UV damage site prompted us to examine if these factors also interact physically. We have earlier shown that XPC interacts with SNF5, and SNF5 in turn interacts with ATM and influences ATM recruitment at the UV damage site [19]. Thus, it is highly likely that XPC, SNF5, and ATM form a complex at the damage site. So, we determined the association of XPC with ATR and ATM by co-immunoprecipitation in the presence or absence of UV treatment. Chromatin fractions were used for immunoprecipitation with ATR or pATM antibodies, and XPC was detected by Western blotting. We observed that both ATR and ATM physically interacted with XPC only in response to UV damage (Fig. 1D). Even though we could pull down ATR in the absence of UV damage, no XPC was associated with it in the immunoprecipitated samples. We specifically used p-ATM antibody for immunoprecipitation since it is known that following irradiation chromatin-bound ATM exists in the phosphorylated state. As p-ATM is a low abundance protein, it generated a weaker signal than observed with ATR. Nevertheless, the combined results strongly indicated that XPC associates with ATR and ATM. In accord, XPC has been shown to associate with ATM after cisplatin treatment, where NER is also the predominant pathway of DNA repair [42]. Thus, XPC and ATR/ATM interaction appears to be a conserved response to the induction of a variety of bulky lesions in the genome.
Fig. 1. ATR and ATM localize to the UV damage site and interact with XPC.
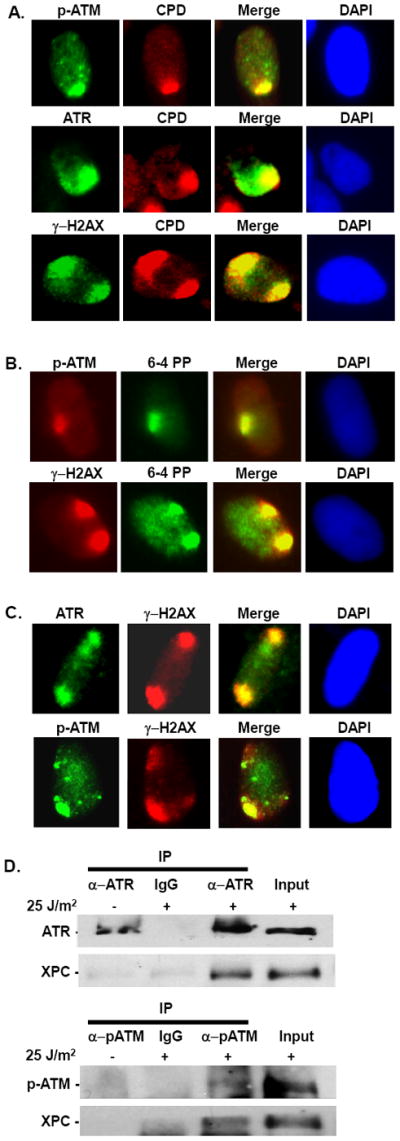
(A) ATR, ATM, and γH2AX co-localize with CPD at the UV damage site. NHF cells were arrested in G1 by serum starvation for 48 h. G1 cells were exposed to 100 J/m2 UV using micropore filter, and after 1 h post-treatment immunofluorescence was performed using ATR, pATM, γH2AX, and CPD antibodies. (B) ATM and γH2AX co-localize with 6-4PP at the UV damage site. Experiment was done as described in A, and immunofluorescence was performed using pATM, γH2AX, and 6-4PP antibodies. (C) Localization of ATR and ATM at the UV damage site in 48 h serum starved G1 arrested NHF cells. G1 cells were exposed to 100 J/m2 UV using micropore filter, and after 1 h post-treatment immunofluorescence was performed using ATR and pATM antibody. (D) Immunoprecipitation was performed using chromatin fractions from HeLa cells. Cells were treated with 25 J/m2 UV, or left untreated, and harvested 1 h post-treatment. Chromatin fractions were isolated as described in Methods, quantitated, and 1500 ug of chromatin fractions were used for immunoprecipitation with ATR or pATM (Ser 1981) antibodies. Immunoprecipitate was detected using XPC antibody. 10% of chromatin fractions were used as input.
3.2. DDB2 and XPC facilitate ATR and ATM recruitment and phosphorylation
While the lesion recognition NER factors as well as DDR kinases promptly congregate at the UV damage sites, it is unclear if the factors of two seemingly different pathways, co-recruited or cross-recruited to the damage site. Since XPC continuously scans and avidly binds to the UV damaged DNA, and more importantly, since XPC interacts with ATR and ATM, we speculated that XPC might influence ATR and ATM recruitment to the damage site. As DDB2 functions upstream of XPC in GG-NER pathway, we anticipated that DDB2 might also facilitate the recruitment of ATR and ATM to the UV damage site. To address this, we examined the ATR and ATM immunofluorescent localization to UV damage sites in NHF and patient-derived cells defective in DDB2 (XP-E) or XPC (XP-C) functions [50-52]. Foci formation via micropore UV-irradiation using ATR, pATM, and γH2AX antibodies was performed in asynchronous cells. The γH2AX foci were used as indicators and to score the sites of damage. About 100-200 cells were counted in each experiment to determine the percentage of cells containing the co-localized foci. Quantitative estimates of different foci formation revealed that ATR and ATM localization was dramatically affected in NER-defective XP-E and XP-C cells as compared to NHF cells (Fig. 2A). Moreover, even in the residual cells scored as positive for ATR, ATM, and γH2AX, the foci in fact exhibited a qualitatively diffused or dispersed signal as opposed to the well-defined foci of control NHF cells (Fig. 2B). Notably, we did not see a significant difference in the intensity using a high (100 J/m2) dose of radiation. The partial localization could be related to cells in different phases of the cell cycle. The decrease was coincident with the reduced H2AX phosphorylation seen in parallel in XP-E and XP-C cells. These data indicated that DDB2 and XPC recognize the damaged lesion and are also needed for the optimal level of recruitment of ATR and ATM to the damage site. To test whether DDB2 and XPC also regulate the activation of ATR and ATM by phosphorylation, we determined the phosphorylation levels of ATR and ATM in NHF, XP-E, and XP-C cells by Western blotting. Despite the essential role of ATR in the DDR pathway, the absence of suitable immunoanalytical tools has been an obstacle for its functional studies. Recently, Cell Signaling Technology has generated an antibody directed against phospho-ATR (Ser428). Unfortunately, this antibody also detects some non-specific signal in the absence of UV damage. In contrast, ATM phosphorylation at S1981 is strictly damage-dependent [53]. Using the available antibodies, we observed that the ATR phosphorylation at S428 and ATM phosphorylation at S1981 were markedly reduced or completely abrogated in XP-E and XP-C cells as compared to the vivid phosphorylation in NHF cells (Fig. 2C). In these experiments, the phosphorylated form of the protein was compared with the total cellular protein in each lane. These results were in agreement with the immunofluorescence data, showing that DDB2 and XPC facilitate ATR and ATM recruitment to the damage sites and affect their functional activation.
Fig. 2. DDB2 and XPC influence ATR and ATM recruitment and activation upon UV damage.

(A) ATR and ATM recruitment to the damage site is affected in XP-E and XP-C cells. Asynchronous NHF, XP-E, and XP-C cells were used for immunofluorescence using ATR, pATM, and γH2AX antibodies. In each experiment, at least 100-200 cells are counted to determine the % of cells containing foci. Error bars indicate the standard deviation (SD) from at least 3 independent experiments. (B) ATR and ATM phosphorylation is affected in XP-E and XP-C cells. NHF, XP-E, and XP-C cells were treated with 25 J/m2 UV, or left untreated, and harvested 1 h post-treatment. Total extracts were used to determine the phosphorylation of ATR (Ser 428) and ATM (Ser 1981) using phospho-specific antibodies. Total protein levels were determined using ATR and ATM specific antibodies. (C) Localization of ATR and ATM in XP-E and XP-C cells. XP-E or XP-C cells positive for ATR and ATM foci were selected from the above experiment to show the quality of the foci in these cells.
3.3. DDB2 and XPC recruitment to the UV damage site is unaffected in ATR- and ATM-compromised cells
To ascertain whether ATR and ATM serve an equivalent function and are required for DDB2 and XPC recruitment to the UV damage site, we knocked down ATR or ATM in HeLa cells using ATR siRNA or ATM shRNA, and determined the localization of DDB2 and XPC at the damage site. The extent of target knockdowns was established by Western blotting and immunofluorescence assays (Fig. 3A). Both ATR and ATM exhibited distinct co-localization with XPC in control siRNA/shRNA-treated cells, but ATR or ATM foci were significantly diminished in cells respectively treated with ATR siRNA or ATM shRNA, even though the XPC foci were highly prominent in these cells (Fig. 3B). Quantitative analysis showed that UV damage specific ATR foci were present in ~35% of control siRNA-treated cells, and only 5% of ATR siRNA-treated cells. Similarly, UV damage specific ATM foci were reduced from 35% in control shRNA-treated cells to only 2% in ATM shRNA-treated cells. We examined the localization of DDB2 and XPC to the UV damage site in ATR- and ATM-depleted cells through localized micropore UV irradiation assay. For this, we used HeLa cells stably expressing FLAG-DDB2 and HA-XPC. Following irradiation, DDB2 localization was detected using FLAG antibody, and XPC localization was detected using XPC antibody. The data showed that neither the DDB2 nor the XPC localization to the damage sites was impacted in ATR- or ATM-compromised cells (Fig. 3C). For instance, the estimation of damage co-localized foci indicated that about 30-35% cells showed DDB2 and XPC foci in control siRNA-, ATR siRNA-, or ATM shRNA-treated cells (Fig. 3D). Thus, DDB2 and XPC recruitment to the DNA damage sites was unaffected in the absence of ATR and ATM. This conclusion was further reaffirmed by the distinct and robust appearance of XPC at the DNA damage sites in ATR-defective Seckel and ATM-deficient AT cells (Fig. 3E).
Fig. 3. ATR and ATM do not influence DDB2 and XPC recruitment to the UV damage site.

(A) ATR and ATM knockdown in Hela-60 cells expressing FLAG-DDB2 and HA-XPC. ATR and ATM protein levels in Hela-60 cells upon siATR and shATM treatment. ATR and ATM were depleted using ATR siRNA or ATM shRNA for 48 h. Western blotting was done using ATR and ATM antibodies as described in Fig. 2C. (B) Defective localization of ATR and ATM at the UV damage site upon siATR and shATM treatment of Hela-60 cells. ATR and ATM were depleted as described in A. For ATM depletion, we used shRNA plasmid 39950 (#3 in Fig. 2C). Cells were UV exposed at 100 J/m2 using micropore filters, and after 1 h post-repair, immunofluorescence detection was performed using ATR, ATM (Ser1981), and XPC antibodies. (C) HeLa-60 cells expressing FLAG-DDB2 and HA-XPC were treated with Control siRNA or shRNA, ATR siRNA, or ATM shRNA for 48 h to knockdown ATR and ATM, respectively. The localization of DDB2 was determined by using FLAG antibody and XPC by using XPC antibody. A representative field of immunofluorescence was shown. Mock, ATR siRNA-, and ATM shRNA (vertical tracks). DDB2, XPC, Merge, and DAPI are presented in the horizontal tracks. (D) Foci formation was quantitated as described in Fig. 2A. (E) XPC localization is not affected in Seckel and AT cells. Immunofluorescence was performed as described in B.
3.4. DDB2 and XPC promote ATR and ATM substrate phosphorylation and influence checkpoint signaling in response to UV damage
To examine whether the reduced accumulation and activation of ATR and ATM in XP-E and XP-C cells affects phosphorylation of downstream substrate proteins, we examined the phosphorylation levels of ATR and ATM substrates in NHF, XP-E, and XP-C cells by Western blotting. Cells were exposed to 25 J/m2, harvested at 1 h post-treatment, and phosphorylation of ATR and ATM substrate proteins were determined using phospho-specific antibodies. As expected, the levels of phosphorylated forms of target proteins Chk1 (Ser 296), Chk2 (Thr68), BRCA1 (S1524), and γ-H2AX (Ser139) were either dramatically reduced or completely abrogated in the absence of functional DDB2 and XPC (Fig. 4A), indicating a defect in the ATR and ATM signaling pathways. Thus, defective DDB2 and XPC function caused an obvious impairment of checkpoint signal transduction cascade in response to UV damage. Interestingly, XP-E and XP-C cells did not exhibit a major difference in the attenuated levels of H2AX and phospho-Chk1, but the phospho-Chk2 levels were discernibly lower in XP-E as compared to XP-C cells. The reason for the difference in phospho-Chk2 levels between XP-E and XP-C cells is not fully clear, but it could be an effect of DDB2 on the ATM-Chk2 pathway, independent of its NER function. We also observed severely reduced levels of phospho-BRCA1 in both XP-E and XP-C cells. Interestingly, we found that the defect in the BRCA1 phosphorylation in XP-C cells was more prominent than in XP-E cells (Fig. 4A). Therefore, DDB2 and XPC might have distinct effects on phosphorylations of ATR-Chk1 and ATM-Chk2 signaling. Further experiments are needed to distinguish the basis of these subtleties. To confirm if the defects in ATR, ATM, and H2AX phosphorylation in XP-E and XP-C cells after UV irradiation were indeed caused by the innate defects of DDB2 and XPC function in these cells, we examined the upstream signaling pathway responses in NHF cells knocked down for DDB2 and XPC by target-specific siRNAs. Our data showed that NHF cells depleted of DDB2 and XPC proteins also had lower levels of ATR, ATM, and H2AX phosphorylation (Fig. 4B). Collectively, these results show that DDB2 and XPC regulate ATR-Chk1 and ATM-Chk2 checkpoint signaling pathways.
Fig. 4. DDB2 and XPC facilitate ATR and ATM, but not the p53-p21 pathway activation upon UV damage.

(A) ATR and ATM substrate phosphorylations are affected in XP-E and XP-C cells. NHF, XP-E, and XP-C cells were UV exposed at 25 J/m2, and after 1 h post-repair, total extracts were isolated, and the phosphorylation of BRCA1 (S1524), Chk1 (Ser296), Chk2 (Thr68), and H2AX (Ser139) were determined using phospho-specific antibodies. Total proteins were determined using BRCA1, Chk1, Chk2, and H2AX antibodies. (B) ATR, ATM, and H2AX phosphorylation in response to UV damage in siRNA-mediated DDB2-and XPC-depleted cells. DDB2 and XPC were depleted using their siRNAs for 48 h, UV exposed at 25 J/m2, and after 1 h post-repair total extracts were isolated, and the phosphorylation of ATR (Ser428), ATM (Ser1981), and H2AX (Ser139) were determined using phospho-specific antibodies. Total proteins were determined using ATR, ATM, and H2AX antibodies. (C) p53 and p21 regulation is not affected in XP-E and XP-C cells. Cells were irradiated at 25 J/m2 and total cell extracts, prepared at the indicated time points, were used to determine the p53 and p21 levels by Western blots. Band intensities of blots, determined by UNSCAN-IT software, are given as numbers relative to unirradiated control.
It has been shown that following damage induction p53 functions to arrest cells at either G1/S or G2/M boundary [54, 55]. In response to DNA damage, p53 is upregulated and activates expression of p21 [55]. In turn, p21 inhibits the activity of CDK complexes, resulting in cell cycle arrest [56]. To determine whether DDB2 and XPC also affect the p53-p21 pathway, we determined the levels of p53 and p21 in response to UV damage in cells defective in DDB2 or XPC function. It has been established that the induction patterns for p53 and p21 depend on cell lines, passage numbers, doses and post-repair times. As all our experiments were done at 25 J/m2, we performed a time-course experiment at this dose to determine the levels of p53 and p21 proteins in NHF, XP-E, and XP-C cells. As shown in Fig. 4C, p53 was promptly induced and continued to increase up to 8 hr post-irradiation in all three cell lines, indicating that p53-dependent checkpoint pathway is not influenced by the absence of DDB2 or XPC. In contrast, p21 levels decreased in NHF cells as well as XP-E and XP-C with a significant recovery by 8 hr post-irradiation in XP-C but not in NHF and XP-E cells. This is consistent with earlier studies showing that p21 degradation upon UV irradiation or low levels of p21 do not affect cell cycle checkpoint [57], and therefore we anticipate that checkpoint activation in XP-E or XP-C cells is intact.
3.5. DDB2 and XPC promote DNA repair through BRCA1- and Rad51- dependent HR pathway
It is well established that both ATR-Chk1 and ATM-Chk2 signaling help maintain DNA structural integrity during replication by resolving stalled forks through the HR-mediated repair pathway [27, 28], where both H2AX and BRCA1 phosphorylations have been known to play a facilitative role [22, 26]. Additionally, Rad51 foci form after stalled replication in S phase cells that have entered the HR pathway and contain functional recombination complexes [25, 26]. Since we observed a reduction in the phosphorylation levels of ATR-Chk1 and ATM-Chk2 in XP-E and XP-C cells, we speculated that DDB2 and XPC might also affect the S phase specific HR repair pathway. Our results showed that H2AX and BRCA1 phosphorylations were negatively affected in XP-E and XP-C cells (Fig. 5A). We further monitored the localization of BRCA1 and Rad51 to the UV damage sites using asynchronous NHF, XP-E, and XP-C cells. As expected, we noticed that phospho-BRCA1 and Rad51 exhibited lower intensities and diffused foci in XP-E and XP-C cells as compared to the pronounced foci of NHF cells. This indicated an obvious defect in their recruitment and/or phosphorylation in these cells (Fig. 5A). Quantitative analysis revealed a significant reduction in the localized foci of BRCA1 and Rad51 in both XP-E and XP-C cells as compared to NHF cells (Fig. 5B), indicating that DDB2 and XPC are required for optimal levels of recruitment of BRCA1 and Rad51. This demonstrated that DDB2 and XPC are involved in UV-induced damage signaling which leads to downstream BRCA1 and Rad51 phosphorylation.
Fig. 5. DDB2 and XPC facilitate BRCA1 and Rad51 recruitment to the UV damage site.
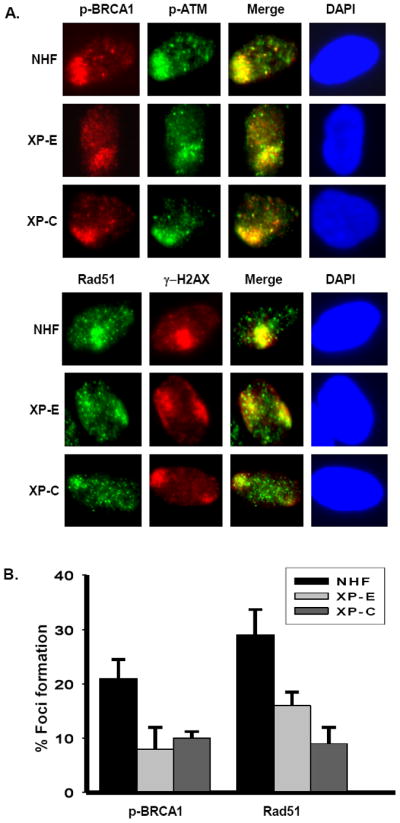
(A) Localization of BRCA1 and Rad51 to the UV damage site by immunofluorescence assay. Asynchronous NHF, XP-E, and XP-C cells were used to perform Immunofluorescence experiments. (B) Quantitation of BRCA1 and Rad51 foci formation in response to UV damage. Quantitation of foci formation was done as described in Fig. 2A.
3.6. ATR and ATM do not influence the NER efficiency
Based on the altered responses resulting from impaired transactions of NER and checkpoint components and the observed physical association of ATR and ATM with the pre-incision NER complex, it was tempting to speculate that these key transducer kinases might play a role in the execution of NER. To assess the possible influence on the NER of UV damage, we used the established immuno-slot blot assay [44, 49] to monitor the initial and repaired levels of CPD and 6-4PP lesions in the DNA of UV irradiated ATR- and ATM-depleted NHF cells. We used G1 arrested cells to determine the role of ATR and ATM in NER, and to avoid the interference of stalled replication forks. Upon ATR knockdown, the efficiency of NER did not change significantly as assessed by the extent of CPD and 6-4PP removal in normal and ATR-compromised cells (Fig. 6A). CPD remaining after 24 h in ATR-deficient cells was 39% compared to 37% in ATR-proficient cells (Fig. 6B). 6-4PP remaining after 8 h in ATR-deficient cells was 15% compared to 22% in ATR-proficient cells (Fig. 6B). Similarly, the rate of CPD and 6-4PP removal did not show a significant difference in ATM-deficient cells compared to ATM-proficient cells (Fig. 6A). The extent of CPD removal at 24 h was 19% in ATM-deficient cells as compared to 28% in ATM-proficient cells (Fig. 6C). The extent of 6-4PP removal at 8 h was 17% in ATM-deficient cells as compared to 29% in ATM-proficient cells. The results essentially support a model where ATR and ATM are exclusively involved in the checkpoint or DSB repair pathways through their influence on Chk1/Chk2 or BRCA1/Rad51 proteins, but do not play an accessory role in the NER pathway.
Fig. 6. ATR and ATM depletion do not affect the CPD removal efficiency upon UV irradiation.

(A) Control, siATR-, and shATM- treated OSU-2 cells were arrested in G1 by serum starvation, UV exposed (10 J/m2), and allowed to repair for indicated times. Identical amounts of isolated DNA were subjected to ISB analysis and the amount of CPD and 6-4PP were detected from binding of anti-CPD, anti-6-4PP, and goat anti-rabbit IgG IR Dye 800CW antibodies. (B) Quantitation of CPD and 6-4PP removal efficiency in siControl- and siATR-treated NHF samples. The percentage of CPD and 6-4PP remaining at different time points relative to the initial irradiated sample was calculated. Each data point represents an average of 3 independent experiments. Error bars indicate the standard deviation (±SD). (C) Quantitation of CPD and 6-4PP removal efficiency in shControl- and shATM-treated NHF samples. Quantitation was done as described in B.
4. Discussion
4.1. DDB2 and XPC are required for ATR and ATM recruitment to the UV damage site
Our study describes a novel upstream role of DDB2 and XPC in regulating ATR and ATM recruitment and activation following UV irradiation of mammalian cells. DDB2-defective GM01389 cells carry mutation in DDB2 [52], which affects complex formation with DDB1 [58], and consequently the formation of functional DDB-XPC complex. Similarly, XPC-defective cells are impaired in the functional DDB-XPC complex. Therefore, we predict that fully functional DDB1-DDB2-XPC complex formation at the damage site is required for optimal recruitment of ATR and ATM. Essentially, our work is built on the premise that DDB2/XPC complex represents the major sensor of UV-damage. Our results show that ATR and ATM associate with XPC in response to UV irradiation. Moreover, cells defective in XPC or DDB2 function exhibit a great reduction in the phosphorylation of ATR, ATM, and their substrate proteins (Fig. 2C, Fig. 4A, B), supporting a direct role of DDB2 and XPC in cell cycle checkpoint signaling. This is akin to the DSB repair pathway in which the damage recognition complex, Mre11-Rad50-Nbs1, enables checkpoint activation upstream of ATM recruitment to the damage site [41]. Similarly, in the mismatch repair pathway, ATR is recruited by the early damage recognition factor, MSH2, and the RPA-ATRIP complex. MSH2 interacts with ATR to form a signaling module and regulates the phosphorylation of Chk1 and SMC1 [40]. Apparently, DDB2/XPC act in DNA damage signaling through events similar to those provoked by the Mre11-Rad50-Nbs1 or MSH2 in activating ATR/ATM. In essence, some of the key protein factors of different DNA repair pathways physically associate with checkpoint sensors to coordinately execute DDR, and this seems to represent a conserved mechanism for activating signaling cascades in response to diverse DNA damage. As ATR is recruited by the RPA-ATRIP complex [15, 16] and influenced by DDB2 and XPC, it is possible that these NER factors also associate with the RPA-ATRIP complex, and thereby affect ATR and ATM recruitment. In such a situation, ATR and ATM might interact with both NER complex and RPA complex at the same time. Further dissection of the involvement of other proteins in ATR and ATM recruitment is necessary to distinguish between these possibilities.
4.2. DDB2 and XPC facilitate checkpoint activation through the Chk1-Chk2-Cdc25 pathway, but not the p53-p21 pathway
Our results showed that DDB2 and XPC affect both Chk1 and Chk2 phosphorylation in response to UV damage (Fig. 4A), which is required for cell cycle arrest by triggering Cdc25A degradation. On the other hand, we found that p53 upregulation is not affected in the cells defective in DDB2 and XPC function (Fig. 4C). As DNA damage triggers p53-dependent checkpoint arrest, we predict that p53-dependent cell cycle arrest is not affected in these cells. Interestingly, we observed the p21 level decreased significantly in NHF, XP-E and XP-C cells. Several studies have shown that p21 is up regulated in p53-mediated G1 arrest. Other studies have shown that p21 is degraded upon lower dose of UV irradiation even though this lower level does not affect the cell cycle checkpoint [57, 59]. Nonetheless, as the p53 level is up-regulated, we anticipate that the checkpoint is not affected in these cells. These observations suggest that DDB2 and XPC are required for efficient Chk1-Chk2-mediated checkpoint arrest, but not p53-mediated checkpoint arrest. Recently, Chung and Bunz have shown that Cdk2 is required for a p53-independent, but Chk1- and Chk2-dependent cell cycle arrest [60], raising the possibility that DDB2 and XPC might influence this axis of checkpoint signaling pathway. Future studies should help reveal if DDB2 and XPC might directly affect Cdk2-mediated cell cycle arrest.
4.3. DDB2 and XPC promote DNA repair through BRCA1-and Rad51-dependent pathway
It has been established that spontaneous HR is promoted by collapsed replication forks that are caused by endogenous DNA SSB [24, 28]. Unrepaired fork gaps can become frank DSB [24, 61, 62]. Furthermore, SSB can also form upon processing of UV-lesions [18, 19]. BRCA1, BRCA2, and Rad51 are known to participate in HR-mediated DNA repair and replication fork maintenance [25, 26]. Moreover, both the ATR-Chk1 and ATM-Chk2 pathways regulate HR-mediated repair of collapsed replication forks [27, 28]. Based on our results that DDB2 and XPC are required for the activation of both ATR-Chk1 and ATM-Chk2 pathways, we anticipate that the SSB and DSB will be repaired through ATR-Chk1- and ATM-Chk2 mediated HR pathway. Additionally, it is well established that ATR and ATM allow H2AX phosphorylation and spreading at the damage site, which changes the chromatin structure near the damage site and executes DNA repair through the HR pathway [21-23, 26, 63]. All these findings indicate that DDB2 and XPC might influence the HR pathway after introduction of UV damage. Indeed, we showed that DDB2 and XPC clearly play a role in the recruitment of BRCA1 and Rad51 proteins to the UV damage site (Fig. 5). Thus, our observations are intriguing because we clearly show that, besides their canonical function as the core repair factors of NER, DDB2 and XPC also play a definite role in regulating ATR-Chk1-BRCA1- and ATM-Chk2-BRCA1-dependent downstream signaling in the realm of UV damage response.
4.4. ATR and ATM do not influence recruitment of DDB2 and XPC to the damage site and do not affect NER efficiency
Our finding that ATR and ATM associate with XPC in response to UV damage is in agreement with others data showing ATR interacts with XPA upon irradiation [64], and phosphorylates XPA [65]. We also revealed that ATR and ATM do not facilitate recruitment of DDB2 and XPC to the UV damage site, and consequently fail to influence NER efficiency. It seems that ATR and ATM are mainly involved in establishing checkpoint arrest and DNA repair through the HR-mediated pathway in response to UV damage. Furthermore, it also confirms that DDB2 and XPC function upstream of ATR and ATM recruitment and are unique to ATR-Chk1-BRCA1 and ATM-Chk2-BRCA1 axis of checkpoint and repair.
Our cumulative results provide impetus for a clear cross-talk between the distinct factors of UV damage recognition and checkpoint response, which congregate in proximity of damage for invoking the critical signaling events. Based on different elements revealed by this work, we propose that DDB2 and XPC act as upstream damage sensors, and through their physical association with ATR and ATM play a role in their functional activation via the well-established phosphorylation of their target substrate proteins needed for the HR repair and checkpoint pathway (Fig. 7). Defects in these pathways are invoked as a key feature of several human cancers. Growing evidence suggests that ATR, ATM, Chk1, Chk2, and BRCA1 are multi-organ tumor suppressor genes found mutated in various cancers [66]. Interestingly, both DDB2 and XPC have also been identified as tumor suppressor genes. Patients deficient in XPA, XPB, XPC, XPD, XPF, XPG and DDB2 genes (XP) display over 2,000-fold increased incidence rates of skin cancer. Heterozygosity for XP is also a high risk factor for several cancers, including but not limited to lung, breast, prostate, squamous cell carcinoma, head and neck cancer, colorectal cancer, and leukemia [11, 12, 67-69]. The interactions described in this work herald a novel etiological link occurring through the dysregulated activation of two central kinases involved in tumorigenesis. Further understanding of the exact nature and the impact of DDB2- and XPC-mediated regulation of ATR-Chk1 and ATM-Chk2 pathways are expected to ultimately allow for tailoring personalized strategies for cancer therapy.
Fig. 7.

A mechanistic model depicting the factor association of DDB2 and XPC with ATR and ATM, influencing the DDR pathway in response to UV damage.
Highlights.
We assessed the upstream ATR foci formation in early NER factor-compromised cells.
We show that ATR/ATM activation is abrogated in these cells.
The pre-incision complex is necessary for the recruitment of ATR and ATM.
ATR and ATM recruitment is mediated by XPC and not the vice versa.
DDB2 and XPC influence HR mediated DNA repair pathway.
Acknowledgments
We would like to thank Dr. Qianzheng Zhu and Chesequa Blevins for careful reading of the manuscript. This work was supported by the National Institute of Health grant [ES2388, ES12991 & CA93413] to AAW, and Pelotonia postdoctoral fellowship to A.B.
Abbreviations
- 6-4PP
pyrimidine(6-4)pyrimidone photoproduct
- ATM
Ataxia telangiectasia mutated
- ATR
ATM and Rad3-related
- CPD
cyclobutane pyrimidine dimer
- DDB
damaged DNA binding protein
- NER
nucleotide excision repair
- UV
ultraviolet
- XPC
Xeroderma pigmentosum protein C
Footnotes
Conflict of interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 3.Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21:8949–8956. doi: 10.1038/sj.onc.1206096. [DOI] [PubMed] [Google Scholar]
- 4.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 5.Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003;278:46906–46910. doi: 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- 6.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, Wani AA. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 2006;66:8590–8597. doi: 10.1158/0008-5472.CAN-06-1115. [DOI] [PubMed] [Google Scholar]
- 8.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van DR. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci. 2007;120:2706–2716. doi: 10.1242/jcs.008367. [DOI] [PubMed] [Google Scholar]
- 10.Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 11.Reardon JT, Sancar A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 2003;17:2539–2551. doi: 10.1101/gad.1131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21:453–460. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- 13.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 14.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;119:71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 15.Cortez D. Unwind and slow down: checkpoint activation by helicase and polymerase uncoupling. Genes Dev. 2005;19:1007–1012. doi: 10.1101/gad.1316905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 17.Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- 18.Yajima H, Lee KJ, Zhang S, Kobayashi J, Chen BP. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J Mol Biol. 2009;385:800–810. doi: 10.1016/j.jmb.2008.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray A, Mir SN, Wani G, Zhao Q, Battu A, Zhu Q, Wang QE, Wani AA. Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol Cell Biol. 2009;29:6206–6219. doi: 10.1128/MCB.00503-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Savic V, B Yin, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP, Bassing CH. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell. 2009;34:298–310. doi: 10.1016/j.molcel.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie A, Odate S, Chandramouly G, Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9:3602–3610. doi: 10.4161/cc.9.17.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000;60:5037–5039. [PubMed] [Google Scholar]
- 24.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 25.Lundin C, Schultz N, Arnaudeau C, Mohindra A, Hansen LT, Helleday T. RAD51 is involved in repair of damage associated with DNA replication in mammalian cells. J Mol Biol. 2003;328:521–535. doi: 10.1016/s0022-2836(03)00313-9. [DOI] [PubMed] [Google Scholar]
- 26.Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999;59:1752s–1756s. [PubMed] [Google Scholar]
- 27.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 28.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 30.Jiang G, Sancar A. Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences. Mol Cell Biol. 2006;26:39–49. doi: 10.1128/MCB.26.1.39-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vrouwe MG, Pines A, Overmeer RM, Hanada K, Mullenders LH. UV-induced photolesions elicit ATR-kinase-dependent signaling in non-cycling cells through nucleotide excision repair-dependent and -independent pathways. J Cell Sci. 2011;124:435–446. doi: 10.1242/jcs.075325. [DOI] [PubMed] [Google Scholar]
- 32.Oh KS, Bustin M, Mazur SJ, Appella E, Kraemer KH. UV-induced histone H2AX phosphorylation and DNA damage related proteins accumulate and persist in nucleotide excision repair-deficient XP-B cells. DNA Repair (Amst) 2011;10:5–15. doi: 10.1016/j.dnarep.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J. 2004;23:429–438. doi: 10.1038/sj.emboj.7600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Taylor J, Siede W. Checkpoint arrest signaling in response to UV damage is independent of nucleotide excision repair in Saccharomyces cerevisiae. J Biol Chem. 2003;278:9382–9387. doi: 10.1074/jbc.M300061200. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto M, Yaginuma K, Igarashi A, Imura M, Hasegawa M, Iwabuchi K, Date T, Mori T, Ishizaki K, Yamashita K, Inobe M, Matsunaga T. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci. 2007;120:1104–1112. doi: 10.1242/jcs.03391. [DOI] [PubMed] [Google Scholar]
- 36.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28:2298–2304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 38.Marini F, Nardo T, Giannattasio M, Minuzzo M, Stefanini M, Plevani P, Muzi FM. DNA nucleotide excision repair-dependent signaling to checkpoint activation. Proc Natl Acad Sci U S A. 2000;103:17325–17330. doi: 10.1073/pnas.0605446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, Muzi-Falconi M. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc Natl Acad Sci U S A. 2011;108:13647–13652. doi: 10.1073/pnas.1108547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc Natl Acad Sci U S A. 2003;100:15387–15392. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grenon M, Gilbert C, Lowndes NF. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat Cell Biol. 2001;3:844–847. doi: 10.1038/ncb0901-844. [DOI] [PubMed] [Google Scholar]
- 42.Colton SL, Xu XS, Wang YA, Wang G. The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J Biol Chem. 2006;281:27117–27125. doi: 10.1074/jbc.M602826200. [DOI] [PubMed] [Google Scholar]
- 43.Venkatachalam S, Denissenko M, Wani AA. Modulation of (+/-)-anti-BPDE mediated p53 accumulation by inhibitors of protein kinase C and poly(ADP-ribose) polymerase. Oncogene. 1997;14:801–809. doi: 10.1038/sj.onc.1200890. [DOI] [PubMed] [Google Scholar]
- 44.Battu A, Ray A, Wani AA. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res. 2011;18:7931–7945. doi: 10.1093/nar/gkr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Q, Wang QE, Ray A, Wani G, Han C, Milum K, Wani AA. Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J Biol Chem. 2009;284:30424–30432. doi: 10.1074/jbc.M109.044982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28:386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 47.El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281:13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 48.Wang QE, Zhu Q, Wani MA, Wani G, Chen J, Wani AA. Tumor suppressor p53 dependent recruitment of nucleotide excision repair factors XPC and TFIIH to DNA damage. DNA Repair (Amst) 2003;2:483–499. doi: 10.1016/s1568-7864(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 49.Wani AA, D’Ambrosio SM, Alvi NK. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem Photobiol. 1987;46:477–482. doi: 10.1111/j.1751-1097.1987.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 50.Rapic-Otrin V, Navazza V, Nardo T, Botta E, McLenigan M, Bisi DC, Levine AS, Stefanini M. True XP group E patients have a defective UV-damaged DNA binding protein complex and mutations in DDB2 which reveal the functional domains of its p48 product. Hum Mol Genet. 2003;12:1507–1522. doi: 10.1093/hmg/ddg174. [DOI] [PubMed] [Google Scholar]
- 51.Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, Stefanini M. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res. 2000;60:1974–1982. [PubMed] [Google Scholar]
- 52.Nichols AF, Itoh T, Graham JA, Liu W, Yamaizumi M, Linn S. Human damage-specific DNA-binding protein p48 - Characterization of XPE mutations and regulation following UV irradiation. J Biol Chem. 2000;275:21422–21428. doi: 10.1074/jbc.M000960200. [DOI] [PubMed] [Google Scholar]
- 53.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 54.Bartek J, Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol. 2001;13:738–747. doi: 10.1016/s0955-0674(00)00280-5. [DOI] [PubMed] [Google Scholar]
- 55.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 56.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 57.Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Al-Khalaf HH, Hendrayani SF, Aboussekhra A. ATR controls the p21(WAF1/Cip1) protein up-regulation and apoptosis in response to low UV fluences. Mol Carcinog. 2012;51:930–938. doi: 10.1002/mc.20864. [DOI] [PubMed] [Google Scholar]
- 60.Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genet. 2010;6:e1000863. doi: 10.1371/journal.pgen.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proc Natl Acad Sci U S A. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 63.Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shell SM, Li Z, Shkriabai N, Kvaratskhelia M, Brosey C, Serrano MA, Chazin WJ, Musich PR, Zou Y. Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J Biol Chem. 2009;284:24213–24222. doi: 10.1074/jbc.M109.000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu X, Shell SM, Yang Z, Zou Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006;66:2997–3005. doi: 10.1158/0008-5472.CAN-05-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 67.Hernandez-Boluda JC, Pereira A, Cervantes F, Alvarez-Larran A, Collado M, Such E, Arilla MJ, Boque C, Xicoy B, Maffioli M, Bellosillo B, Marugan I, Amat P, Besses C, Guillem V. A polymorphism in the XPD gene predisposes to leukemic transformation and new nonmyeloid malignancies in essential thrombocythemia and polycythemia vera. Blood. 2012;119:5221–5228. doi: 10.1182/blood-2012-02-411215. [DOI] [PubMed] [Google Scholar]
- 68.Moraes MC, Neto JB, Menck CF. DNA repair mechanisms protect our genome from carcinogenesis. Front Biosci. 2012;17:1362–1388. doi: 10.2741/3992. [DOI] [PubMed] [Google Scholar]
- 69.Gil J, Ramsey D, Stembalska A, Karpinski P, Pesz KA, Laczmanska I, Leszczynski P, Grzebieniak Z, Sasiadek MM. The C/A polymorphism in intron 11 of the XPC gene plays a crucial role in the modulation of an individual’s susceptibility to sporadic colorectal cancer. Mol Biol Rep. 2012;39:527–534. doi: 10.1007/s11033-011-0767-5. [DOI] [PubMed] [Google Scholar]