

Abstract
Non-coding RNAs play important roles in regulation of gene expression. Specific recognition and inhibition of these biologically important RNAs that form complex double-helical structures will be highly useful for fundamental studies in biology and practical applications in medicine. This protocol describes a strategy developed in our laboratory for sequence-selective recognition of double-stranded RNA (dsRNA) using triple helix forming peptide nucleic acids (PNAs) that bind in the major grove of RNA helix. The strategy developed uses chemically modified nucleobases, such as 2-aminopyridine (M) that enables strong triple helical binding at physiologically relevant conditions, and 2-pyrimidinone (P) and 3-oxo-2,3-dihydropyridazine (E) that enable recognition of isolated pyrimidines in the purine rich strand of the RNA duplex. Detailed protocols for preparation of modified PNA monomers, solid-phase synthesis and HPLC purification of PNA oligomers, and measuring dsRNA binding affinity using isothermal titration calorimetry are included.
Keywords: double-stranded RNA, triple helix, peptide nucleic acids, PNA, isothermal titration calorimetry
Introduction
Since its discovery in 1991, PNA has been extensively studied and used for sequence-selective recognition of single-stranded DNA and RNA and double-stranded DNA (Corradini et al., 2011; Duca et al., 2008; Malnuit et al., 2011; Rozners, 2012; Wojciechowski and Hudson, 2007). Recognition of double-stranded RNA (dsRNA) structures has received relatively little attention. Herein we describe a new strategy of sequence-selective recognition of dsRNA using peptide nucleic acids (PNA) that form Hoogsteen triple helix (Figure 1) in the major groove of target RNA. Our studies (Li et al., 2010, Gupta et al., 2011, 2012; Muse et al., 2013; Zengeya et al., 2012) showed that PNAs carrying nucleobase modifications allow formation of stable and sequence-selective triple helices with dsRNA. Therapeutic potential of PNA has been proposed since early studies; however, in vivo applications of unmodified PNA are hindered by poor cell permeability and endosomal entrapment (Rozners, 2012). We demonstrated that nucleobase- and lysine-modified PNA was taken up efficiently by HEK293 cells (Muse et al., 2013).
Figure 1.

(A) Structure of nucleobase modified PNA. (B) The standard Hoogsteen C+*G-C and U*A-U triplets; (C) The nucleobase modifications designed to mimic protonated cytosine (M+*G-C) and to recognize isolated pyrimidine interruptions (P*C-G, E*U-A) using Hoogsteen hydrogen-bonding.
Additionally, nucleobase-modified PNA recognized biologically relevant dsRNA sequences with high affinity at physiologically relevant conditions (Zengeya et al., 2012). The protocols described herein allow sequence-selective recognition of purine rich strands of dsRNA, which are common motifs in biologically important non-coding RNAs. Here we summarize our knowledge about synthesis of monomers E, P and M (Figure 2), their incorporation into PNA, conjugation of PNA with lysine peptides, purification and quantification of a synthetic PNA, and isothermal titration calorimetry (ITC) experiments and data analysis for determining the affinity of PNA for dsRNA targets.
Figure 2.
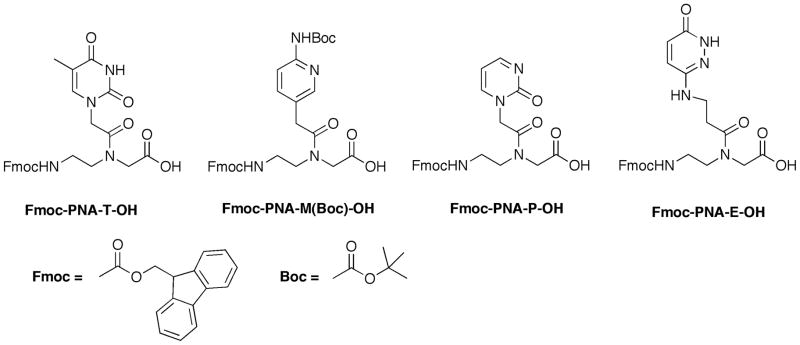
Structures of PNA monomers for solid-phase PNA synthesis: standard T (Link Technologies, UK) and modified E, P (Gupta et al., 2011, 2012) and M (Zengeya et al., 2012).
BASIC PROTOCOL 1: SYNTHESIS OF M, 2-AMINOPYRIDINE PNA MONOMER
This protocol provides a detailed description of chemical synthesis of 2-aminopyridine PNA monomer M starting from commercially available 2-(6-chloro-3-pyridinyl)acetonitrile in 5 steps (Figure 3). The strategy for preparation of M was based on N,N′-dicyclohexylcarbodiimide (DCC) mediated coupling of Fmoc-protected PNA backbone and carboxylic acid derived from 2-(6-Chloro-3-pyridinyl)acetonitrile followed by deprotection of allyl group as described below (Zengeya et al., 2012). Silica gel column chromatography is used to purify the intermediate products and the final monomer.
Figure 3.

Preparation of PNA monomer M. Abbreviations: EtOH, ethanol; DCC, N,N-dicyclohexycarbodiimide; HOBt, 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one; DMF, Dimethylformamide.
Note: The sets of steps labeled a–c within the protocol each generate an intermediate product needed in the synthesis of 2-aminopyridine PNA monomer M (steps d).
Materials
2-(6-Chloro-3-pyridinyl)acetic acid ethyl ester
Tert-butyl carbamate
9,9-Dimethyl-4,5-bis(diphenylphosphino)xanthene
Tris(dibenzylideneacetone)dipalladium (0)
Cesium carbonate
Ammonium acetate, 10% aqueous solution
Dry tetrahydrofuran
Ethyl acetate
Methanol
Dichlromethane (CH2Cl2)
Hexanes
Sodium hydroxide, 1M aqueous solution
3, 4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one, HOBt
N, N-dicyclohexycarbodiimide, DCC
Dry dimethylformamide
Potassium hydrogen sulfate (KHSO4), saturated aqueous solution
Sodium chloride (NaCl), saturated aqueous solution
Sodium hydrogen carbonate (NaHCO3), saturated aqueous solution
Protocol 1.a. Synthesis of ethyl 2-(6-((tert-butoxycarbonyl)amino)pyridin-3-yl)acetate (3)
2-(6-Chloro-3-pyridinyl) acetic acid ethyl ester was synthesized from commercially available 2-(6-chloro-3-pyridinyl) acetonitrile (Matrix Scientific, CAS 39891-09-3) using catalytic amount of concentrated sulfuric acid in ethanol as described by Burns et al. (Burns et al., 2010).
-
1a
Add 2-(6-chloro-3-pyridinyl)acetic acid ethyl ester (1.2 g, 6.1 mmols), tert-butyl carbamate (2.1 g, 18 mmols), 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene (0.73 g, 1.3 mmols), tris(dibenzylideneacetone)dipalladium (0.59 g, 0.67 mmols) and cesium carbonate (3.1 g, 9.2 mmols) to a 250 mL round bottom flask with a magnetic stirrer bar.
-
2a
Add anhydrous tetrahydrofuran (50 mL) and stir at room temperature under nitrogen gas until everything dissolves.
-
3a
Reflux the mixture (65 °C) under nitrogen for 20 hours.
-
4a
Cool the reaction to room temperature and quench with ammonium acetate solution (20 mL) and extract the organic product with ethyl acetate (3 × 30 mL).
-
5a
Combine the organic extracts and wash with water (30 mL), brine (30 mL) and dry over anhydrous sodium sulfate.
-
6a
Filter of sodium sulfate and remove the solvent under reduced pressure to obtain a greenish yellow product.
-
7a
Purify the product by silica gel column chromatography using a gradient of 0–3% methanol in CH2Cl2. Yellow product elutes at 2.5 -3 % of methanol. Yield: 1.24 g, 73%. Rf = 0.77 (methanol:CH2Cl2, 1:19, v/v).
-
8a
Characterize the compound by NMR. 1H NMR (600MHz, CDCl3): δ 8.20 (s, 1H), 7.96-7.94 (d, 1H), 7.62-7.60 (dd, 1H), 4.16-4.12 (q, 2H), 3.53 (s, 2H), 1.54 (s, 9H), 1.25-1.23 (t, 3H). 13C NMR (600 MHz, DMSO-d6): δ 171.0, 156.5, 151.6, 147.9, 139.1, 124.1, 112.2, 79.6, 61.0, 37.9, 28.2, 14.1.
Protocol 1.b. Synthesis of 2-(6-((tert-butoxycarbonyl)amino)pyridin-3-yl)acetic acid (4)
-
1b
Dissolve ethyl (6-tert-butoxycarbonylaminopyridin-3-yl)acetate (3) (1.63 g, 5.9 mmols) in methanol (6.0 mL)
-
2b
Prepare sodium hydroxide solution (0.5 g NaOH in 6.0 mL water) and add to the reaction mixture while stirring at room temperature.
-
3b
Reflux the solution (65 °C) for 2 hours.
-
4b
Cool the reaction mixture and remove methanol under reduced pressure.
-
5b
Precipitate the product by adding 20% citric acid to reach pH between 4 and 5. Caution: lower pH may cleave the Boc protecting group. Product precipitates as a pink yellow solid.
-
6b
Filter the product and wash with a mixture of CH2Cl2 and hexanes (v/v 1:1, 40 mL). Dry the product in vacuum. Yield: 1.15 g, 79%.
-
7b
Characterize the compound by NMR. 1H NMR (600MHz; DMSO-d6): δ 12.54 (s, 1H), 9.72 (s, 1H), 8.12 (s, 1H), 7.75-7.73 (d, 1H), 7.63-7.62 (d, 1H), 3.55 (s, 2H), 1.48 (s, 9H). 13C NMR (DMSO-d6, 90.5 MHz): δ 172.4, 152.7, 151.1, 148.1, 138.8, 125.1, 111.9, 79.5, 337.0, 28.1.
Protocol 1.c. Synthesis of allyl 2-(N-(2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)ethyl)-2-(6-((tert-butoxycarbonyl)amino) pyridin-3-yl)acetamido)acetate (5)
-
1c
Add 2-(6-((tert-butoxycarbonyl)amino)pyridin-3-yl)acetic acid (4) (200 mg, 0.79 mmols), PNA backbone (7) (Wojciechowski and Hudson, 2008) (275 mg, 0.72 mmols) and 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one (130 mg, 0.80 mmols) to a dry 25 mL round bottom flask.
-
2c
Add anhydrous dimethylformamide (5 mL) while stirring at room temperature to dissolve the solids.
-
3c
Cool the mixture in an ice bath and add N, N-dicyclohexycarbodiimide (180 mg, 0.88 mmols) and stir at 0 °C for 1hour. Allow the reaction to stir at room temperature overnight.
-
4c
Evaporate the solvent on rotary evaporator and re-dissolve the crude product in CH2Cl2 (20 mL) and wash with 5% NaHCO3 (2 × 20 mL).
-
5c
Dry the organic layer over anhydrous sodium sulfate. Filter of sodium sulfate and remove the solvent under reduced pressure. Re-dissolve the organic residue in acetonitrile (5 mL) and evaporate in vacuum.
-
6c
Purify the crude product by silica gel column chromatography using a gradient of 0–5% methanol in CH2Cl2. Yield: 270 mg, 55%. Rf = 0.60 (methanol:CH2Cl2, 1:19, v/v).
-
7c
Characterize the compound by NMR. 1H NMR (600 MHz, DMSO-d6): δ 8.02-8.00 (d, 1H), 7.83-7.80 (t, 1H), 7.69-7.67 (d, 2H), 7.56 (s, 1H), 7.51-7.50 (d, 2H), 7.48 (s, 1H), 7.33-7.31 (d, 2H), 7.24-7.23 (t, 2H), 5.87-5.80 (m, 1H), 5.65-6.34 (t, 1H), 5.29-5.26 (d, 1H), 5.23-5.21 (d, 1H), 5.20-5.18 (d, 1H), 4.59-58 (d, 1H), 4.56-4.55 (d, 1H), 4.36-4.35 (d, 1H), 4.29-4.28 (d, 2H), 4.14-4.12 (t, 1H), 4.04 (s, 1H), 3.96 (s, 2H), 3.55 (s, 2H), 3.51-3.49 (t, 2H), 3.45 (s, 2H), 3.31-3.30 (d, 2H), 3.28-3.28 (d, 2H), 1.43 (s, 9H). 13C NMR (90.5 MHz, DMSO-d6): δ 171.7, 156.6, 152.3, 151.0, 147.8, 143.8, 141.3, 139.0, 131.4, 131.1, 127.8, 127.1, 125.1, 125.0, 124.7, 120.0, 119.8, 119.1, 112.1, 81.0, 66.9, 66.2, 49.6, 39.5, 36.3, 28.3.
Protocol 1.d. Synthesis of 2-(N-(2-((((9H-fluoren-9-yl) methoxy) carbonyl) amino) ethyl)-2-(6-((tert-butoxycarbonyl) amino) pyridin-3-yl)acetamido)acetic acid (6)
-
1d
Dissolve compound 5 (677 mg, 1.1 mmols) in dry tertrahydrofuran (25 mL).
-
2d
Add N-ethylaniline (2.6 mL, 2.1 mmols), Pd(PPh3)4 (559 mg, 0.48 mmols) and stir the reaction mixture at room temperature for 1.5 hours.
-
3d
Evaporate the solvent and re-dissolve the residue in ethyl acetate (25 mL) using gentle warming.
-
4d
Wash the mixture with saturated aqueous KHSO4 (25 mL), water (25 mL) and brine (25 mL) dry over anhydrous Na2SO4
-
5d
Filter of sodium sulfate and remove the solvent under reduced pressure.
-
6d
Purify the product on a short (8 cm) silica gel column using a gradient of 0–5% methanol in CH2Cl2. Yield: 561 mg, 88%. Rf = 0.50 (methanol:CH2Cl2, 1:4, v/v)
-
7d
Characterize the compound by NMR. 1H NMR (360MHz, DMSO-d6): δ 9.95 (s, 1H), 8.02 (d, 1H), 7.72 (s, 1H), 7.65-7.63 (t, 2H), 7.52-7.50 (d, 2H), 7.47-7.45 (d, 1H), 7.23-7.19 (t, 2H), 5.60 (s, 1H), 4.45 (d, 2H), 4.37-4.36 (d, 1H), 4.29-4.27 (d, 1H), 4.12-4.10 (t, 2H), 3.94 (s, 2H), 3.59 (s, 2H), 3.56 (s, 1H), 3.48 (s, 1H) 3.39 (s, 1H), 3.32-3.31 (d, 2H), 3.24 (s, 1H), 1.40 (s, 9H). 13CNMR (90.5MHz, DMSO-d6): δ 171.0, 156.2, 152.7, 150.6, 148.2, 140.7, 139.5, 138.9, 128.9, 127.5, 127.3, 127.1, 126.5, 125.3, 125.1, 120.1, 111.8, 79.4, 65.6, 46.8, 38.3, 35.8, 28.2.
BASIC PROTOCOL 2: SYNTHESIS OF P, 2-PYRIMIDINONE PNA MONOMER
This protocol contains procedure for synthesis of 2-pyrimidinone (P) PNA monomer. Derivatives of pyrimidinone have been used as oligonucleotide modifications for recognition of C-G inversions that interrupt a polypurine sequence in DNA (Buchini and Leumann, 2004; Gildea and McLaughlin, 1989). Before our studies (Gupta et al., 2011, 2012), monomer P had not been used in PNA. P-modified PNAs allowed excellent sequence-selectivity for the target C-G base pair in short polypurine tract of dsRNA (Gupta et al., 2011). The synthetic pathway is shown in Figure 4. It starts with coupling of commercially available (2-oxo-1(2H)-pyrimidinyl)acetic acid 9 with PNA backbone 8 (Wojciechowski and Hudson, 2008) to give Fmoc-protected P PNA monomer 10. Coupling is followed by hydrogenation reaction to give PNA monomer P (11) that is used for solid-phase synthesis of PNA oligomers.
Figure 4.
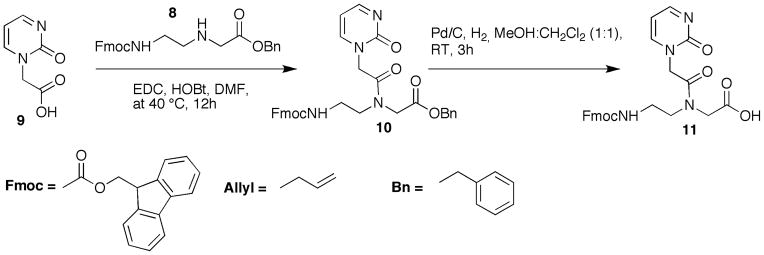
Preparation of PNA monomer P. Abbreviations: DMF, dimethylformamide; EDC, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; HOBt, 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one; MeOH, methanol; RT, room temperature, 25°C.
Note: The set of steps labeled “a” within the protocol generates an intermediate product needed in the synthesis of 2-pyrimidinone (P) PNA monomer (steps b).
Materials
(2-oxo-1(2H)-pyrimidinyl)acetic acid
3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one, HOBt
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide, EDC
Dry dimethylformamide
Methanol
Dichloromethane (CH2Cl2)
Ethyl acetate
Hydrochloric acid (HCl), 1M aqueous solution
Sodium bicarbonate (NaHCO3), saturated aqueous solution
Sodium chloride (NaCl), saturated aqueous solution (brine)
Sodium sulfate (Na2SO4), anhydrous
Deuterated dimethyl sulfoxide, DMSO-d6
Nitrogen gas, N2
Hydrogen gas, H2
Palladium on carbon, 10% Pd/C
Celite 545
Protocol 2.a. Synthesis of allyl 2-(N-(2-(((9H-fluoren-9-yl)methoxy)carbonylamino)ethyl)-2-(2-oxopyrimidin-1(2H)-yl)acetamido)acetate (10)
(2-oxo-1(2H)-pyrimidinyl)acetic acid (9) was purchased from ChemBridge. Backbone 8 was synthesized following the procedure by Wojciechowski and Hudson (Wojciechowski and Hudson, 2008).
-
1a
Dissolve backbone 8 (1.57 g, 3.64 mmol), (2-oxo-1(2H)-pyrimidinyl)acetic acid 9 (0.90 g, 5.83 mmol), and HOBt (1.19 g, 7.29 mmol) in DMF (100 mL) and warm to 40 °C. Add EDC (1.50 g, 7.84 mmol). Allow the reaction to stir at 40 °C overnight.
-
2a
Add water (150 mL).
-
3a
Extract the product with ethyl acetate (3 × 300 mL). Combine organic layers together.
-
4a
Wash the combined organic layers with 1 M aqueous HCl (400 mL), saturated aqueous NaHCO3 (400 mL), water (400 mL), brine (400 mL) and dry over Na2SO4.
-
5a
Filter off Na2SO4 and evaporate the solvent in vacuum using a rotary evaporator.
-
6a
Dry the residue in vacuum overnight.
-
7a
Purify the crude product by silica gel column chromatography using a gradient of 0–7% methanol in CH2Cl2 to afford 10 as a white solid material. Yield: 1.38 g, yield 67%. Rf = 0.36 (methanol:CH2Cl2, 7:93, v/v).
-
8a
Characterize the compound by NMR. Compound 10 exists in solution as a pair of slowly exchanging rotamers; the signals due to the major (ma.) and minor (mi.) rotamers are designated: 1H NMR (DMSO-d6, 360 MHz): δ 8.58 (t, 1H, J = 3.6 Hz), 8.01 and 7.96 (ddd, 1H, J = 3.6 Hz and 3.6 Hz), 7.88 (d, 2H, J = 7.6 Hz), 7.68 (m, 1H), 7.45-7.30 (m, 10H), 6.45 (t, 1H, J = 3.6 Hz), 5.22 (s, 0.5H, mi.), 5.13 (s, 1.5H, ma.), 4.92 (s, 1.5H, ma.), 4.74 (s, 0.5H, mi.), 4.45-4.23 (m, 3H), 4.16 (s, 1H), 3.51-3.31 (m, 5H). 13C NMR (DMSO-d6, 90.5 MHz): δ 169.3, 168.8, 167.2, 167.0, 166.6, 166.5, 156.3, 155.5, 150.7, 143.8, 140.7, 135.7, 128.4, 128.2, 128.1, 128.0, 127.9, 127.6, 127.0, 125.1, 120.1, 103.6, 66.6, 66.0, 65.5, 65.4, 50.6, 50.4, 49.2, 48.0, 47.1 46.9, 46.7, 40.2, 37.9.
Protocol 2.b. Synthesis of 2-(N-(2-(((9H-fluoren-9-yl)methoxy)carbonylamino)ethyl)-2-(2-oxopyrimidin-1(2H)-yl)acetamido)acetic acid (11)
-
1b
Dissolve compound 10 (1.20 g, 2.11 mmol) in a mixture of dry methanol and CH2Cl2 (1:1, 140 mL). Purge the mixture with N2.
-
2b
Add 10% Pd/C (420 mg).
-
3b
Purge the solution with H2 for 10 min and stir reaction mixture for 3 h under H2 atmosphere.
-
4b
Filter reaction mixture through a short pad of celite to remove Pd/C catalyst.
NOTE: Make sure that celite is pre-wet with methanol before pouring the reaction mixture.
-
5b
Wash the celite pad with methanol (50 mL).
-
6b
Evaporate the solvent in vacuum using a rotary evaporator.
-
7b
Dry the residue in vacuum overnight.
-
8b
Purify the crude product by silica gel column chromatography using a gradient of 0–15% methanol in CH2Cl2 to afford 11 as a white solid material. Yield: 910 mg, 91%. Rf = 0.43 (methanol:CH2Cl2, 4:6, v/v).
-
9b
Characterize the compound by NMR. 1H NMR (DMSO-d6, 360MHz): δ 7.87 (d, 2H, J = 7.2 Hz), 7.67 (d, 2H, J = 7.2 Hz), 7.43-7.31(m, 6H), 6.25 (d, 1H, J = 3.6 Hz), 4.32-4.10 (m, 4H), 3.95 (d, 2H, J = 10.8 Hz), 3.36-3.11(m, 6H), 1.76 (s, 1H). 13C NMR (DMSO-d6, 90.5 MHz): δ 171.1, 170.8, 169.5, 169.2, 156.3, 156.1, 155.6, 155.4, 148.6, 143.9, 140.7, 139.9, 127.6, 127.1, 125.1, 124.2, 120.1, 119.9, 65.5, 65.4, 54.9, 49.2, 48.2, 47.7, 47.4, 46.7, 46.0, 45.8, 41.8, 38.1, 21.9, 21.8.
BASIC PROTOCOL 3: SYNTHESIS OF E, 3-OXO-2,3-DIHYDROPYRIDAZINE PNA MONOMER
Dahl, Nielsen and coworkers have designed, synthesized and studied novel pyridazinone nucleobases, including the 3-oxo-2,3-dihydropyridazine E monomer for the potential recognition of T-A inversions in DNA (Eldrup et al., 1997; Olsen et al., 2004). Before our study (Gupta et al., 2011) E was not used for recognition of U-A inversions in polypurine tracts of dsRNA. In our hands, E showed excellent sequence selectivity for the target U-A base pair (Gupta et al., 2011). Synthesis of E monomer, shown in Figure 5, is achieved by N,N′-dicyclohexylcarbodiimide (DCC) and 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one (HOBt) mediated coupling of Fmoc-protected PNA backbone with the known 3-(6-(benzyloxy)pyridazin-3-ylamino)propanoic acid 12 (Eldrup et al., 1997) followed by hydrogenation.
Figure 5.
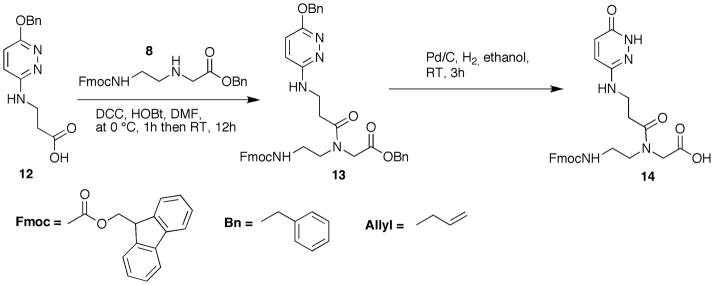
Preparation of PNA monomer E. Abbreviations: DCC, N,N-dicyclohexycarbodiimide; HOBt, 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazin-4(3H)-one; DMF, dimethylformamide; RT, room temperature, 25 °C.
Note: The set of steps labeled “a” within the protocol generates an intermediate product needed in the synthesis of PNA monomer E (steps b).
Materials (See Basic Protocols 1–2)
Protocol 3.a. Synthesis of allyl 2-(N-(2-(((9H-fluoren-9-yl)methoxy)carbonylamino)ethyl)-3-(6-(benzyloxy)pyridazin-3-ylamino)propanamido)acetate (13)
-
1a
Dissolve 8 (1.90 g, 4.41 mmol), 12 (1.53 g, 5.59 mmol) and HOBt (0.921 g, 5.66 mmol) in dry dimethylformamide (50 mL). Cool the reaction mixture on an ice-bath.
-
2a
Add dicyclohexylcarbodiimide (1.29 g, 6.22 mmol) and stir the reaction mixture on ice-bath for 1 h.
-
3a
Remove the ice-bath and stir the mixture overnight at room temperature.
-
4a
Evaporate the solvent in vacuum using a rotary evaporator.
-
5a
Dissolve the residue in CH2Cl2 (100 mL) and extract with 5% aqueous NaHCO3 (100 mL). Dry the organic layer over Na2SO4.
-
6a
Filter off Na2SO4 and evaporate the solvent in vacuum using a rotary evaporator.
-
7a
Dry the residue in vacuum overnight.
-
8a
Purify the crude product by silica gel column chromatography using a gradient of 0–3% methanol in CH2Cl2 to afford 13 as a white solid material. Yield: 2.7 g, 89%. Rf = 0.56 (methanol:CH2Cl2, 1:9, v/v).
-
9a
Characterize the compound by NMR. 1H NMR (DMSOd6, 360MHz): δ 8.59-7.30 (m, 15H), 6.44 (m, 1H), 5.13 (s, 2H), 4.92 (s, 1H), 4.74-4.16 (m, 5H), 3.51-3.13 (m, 6H). 13C NMR (90.5 MHz, CDCl3): δ 169.3, 168.8, 167.2, 167.0, 166.6, 166.5, 156.3, 155.5, 150.7, 143.8, 140.7, 135.7, 128.4, 128.2, 128.1, 128.0, 127.9, 127.6, 127.0, 125.1, 120.1, 103.6, 66.6, 66.0, 65.5, 65.4, 50.6, 50.4, 49.2, 48.0, 47.1 46.9, 46.7, 40.2, 37.9.
Protocol 3.b. Synthesis of 2-(N-(2-(((9H-fluoren-9-yl)methoxy)carbonylamino)ethyl)-3-(6-oxo-1,6-dihydropyridazin-3-ylamino)propanamido)acetic acid (14)
-
1b
Dissolve compound 13 (1.20 g, 2.11 mmol) in dry ethanol (100 mL) and purge with N2.
-
2b
Add 10% Pd/C (960 mg).
-
3b
Purge solution with H2 for 10 min and the stir reaction mixture for 3 h under H2 atmosphere.
-
4b
Filter the reaction mixture through a short pad of celite to remove Pd/C catalyst. NOTE: Make sure that celite is pre-wet with methanol before pouring the reaction mixture through.
-
5b
Wash the pad with methanol (50 mL).
-
6b
Evaporate the solvent in vacuum using a rotary evaporator.
-
7b
Dry the residue in vacuum overnight.
-
8b
Purify the crude product using a short silica gel column (3–4 cm) and CH2Cl2 first to wash out impurities and then using a gradient of 0–5% MeOH in CH2Cl2 to afford 14 as a white solid material. Yield: 650 mg, 73%. Rf = 0.15 (MeOH:CH2Cl2, 1:3, v/v).
-
9b
Characterize the compound by NMR. 1H NMR (DMSOd6, 600MHz): δ 9.94 (s br, 1H), 9.26 (s br, 1H), 8.08 (d, 2H, J = 9.0 Hz), 8.02 (d, 1H, J = 7.5), 7.75-7.69 (m, 3H), 7.58 (d, 2H, J = 7.5 Hz), 7.54 (d, 1H, J = 7.5), 7.38-7.31 (m, 1H), 7.25-7.22 (m, 2H), 4.43 (d, 2H, J = 6.8 Hz), 4.17 (t, 1H, J = 6.8 Hz), 4.13-4.10 (m, 2H), 4.02-4.00 (m, 2H), 3.61 (s, 1H), 3.55-3.46 (m, 2H), 3.40-3.32 (m, 2H), 1.27-1.24 (m, 1H). 13C NMR (90.5 MHz, CDCl3): δ 174.8, 171.7, 171.2, 171.0, 158.7, 156.1, 147.3, 143.9, 143.8, 140.7, 130.8, 130.7, 128.1, 127.6, 127.1, 125.2, 120.1, 65.4, 54.9, 47.5, 46.7, 46.6, 38.3, 37.3, 37.2, 31.5, 31.1.
BASIC PROTOCOL 4: SOLID-PHASE PNA SYNTHESIS
This protocol describes solid-phase synthesis of PNA oligomers on an Expedite 8909. Synthesis consists of four major steps: 1) resin loading, 2) deprotection, 3) coupling, and 4) cleavage. Sieber resin is placed in an empty synthesis column followed by deprotection step. Expedite 8909 synthesizer initiates activation and coupling of the first monomer to the deprotected resin support followed by capping of unreacted resin. Cycle of deprotection-activation-coupling-capping is repeated to couple all monomers of the PNA molecule. All steps described and chemicals used in this protocol were taken from Expedite 8900 PNA Chemistry User’s Guide.
Materials
Expedite 8909
Fmoc-PNA-T-OH (commercially available from Link Technologies)
Fmoc-PNA-M(Boc)-OH (Basic protocol 1)
Fmoc-PNA-E-OH (Basic protocol 2)
Fmoc-PNA-P-OH (Basic protocol 3)
Fmoc-Lys(Boc)-OH (commercially available from NovaBiochem)
NovaSyn TG Sieber resin (commercially available from NovaBiochem, functionalized at 0.2 mmol/g).
Synthesis columns for the Expedite 8909 (commercially available from Glen Research)
Anhydrous dimethylformamide
Piperidine (biotech grade, Aldrich)
2-(1H-7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU)
Diisopropylethylamine (DIPEA)
2,6-Lutidine
Acetic anhydride (reagent grade)
m-Cresol (reagent grade)
Trifluoroacetic acid (HPLC grade)
Anhydrous diethyl ether
Deblocking solution (Reagents and Solutions)
Activator solution (Reagents and Solutions)
Base solution (Reagents and Solutions)
Capping solution (Reagents and Solutions)
Cleavage Cocktail (Reagents and Solutions)
Solid-phase synthesis of PNA oligomers
Weigh 10 mg of Sieber resin (2 mmol/g) into an empty synthesis column.
-
Prepare 0.2 M solutions of PNA monomers in anhydrous N-methyl-2-pyrrolidone (NMP). Use Expedite 8909 monomer vials to weight solid monomers.
NOTE: Gentle warming may be required to dissolve the P and E monomers.
NOTE: To calculate amount of monomer needed for synthesis, use the following formula:For example, for a synthesis of PNA sequence with 5 M monomers that have the purity 95%, calculate m(mg) = 5 × 0.04 × 574.62/0.95 = 121 mg that must be dissolved in V(mL) = 5 × 0.2 = 1 mL to prepare 0.2 M solution of M monomer.
NOTE: Add additional three couplings to monomer calculation, which is necessary to allow for priming of reagents at the beginning of automated synthesis (see step 4 below).
CAUTION: The minimum volume that can be loaded onto Expedite 8909 is 0.5 mL.
if switching between standard nucleic acid reagents and PNA reagents follow proper flushing procedures in the Expedited PNA synthesis manual. Load the reagents and monomers onto Expedite 8909 synthesizer taking necessary precautions to minimize contact with atmosphere.
Prime the Expedite 8909 synthesizer with the reagents and monomers.
Program the sequences to be synthesized.
Install the synthesis columns loaded with Sieber resin (step 1 above). On Expedite 8909 two sequences can be synthesized simultaneous.
start automated synthesis.
Collect the Deblocking solution at each Fmoc deprotection. Dilute the collected solution to 10 mL with the Deblocking solution and measure the absorbance at 254 nm using a UV spectrophotometer. Compare the absorbance after each deprotection step to determine the efficiency of monomer coupling. In a typical synthesis the absorbace may gradually decrease from A = 1 to A = 0.4.
After the synthesis is complete perform the Final Deblocking on the synthesizer and then remove the synthesis column(s).
Manual coupling of D-Lysine
Lysine residues are attached to the carboxy end of PNA to increase the solubility and cellular uptake of PNA.
-
1
Weigh 10 mg of the Sieber resin (amino functionalized at 2 mmol/g) into an empty synthesis column.
-
1
Using two 1 mL syringes, one on each side of the column, swell the column in 1 mL of dimethylformamide for 5 min.
NOTE: This simple procedure uses one syringe to push the reagent into the column while the other syringe is used to pull reagents out at the other end. The procedure is described in detail for DNA deprotection in Expedite 8909 user’s manual.
-
2
Drain the solvent.
-
3
Deprotect Fmoc group using 1 mL of the Deblocking Solution. Agitate the reaction mixture for 10 min.
-
4
Drain the solvent.
-
5
Use another portion of fresh 1 mL of the Deblocking Solution and agitate the reaction mixture for an additional 10 min.
-
6
Drain the solvent. Collect and combine Fmoc-deblock from step 5 and 7. Measure Fmoc-deblock. (See step 8 in Solid-phase synthesis of PNA oligomers). Absorbance must be equal or above one.
-
7
Wash the column using 0.5 mL of DMF.
-
8
Drain the solvent.
-
9
Repeat steps 8 and 9 four more times.
-
10
In a small vial closed with a silicone rubber septa dissolve 6.8 mg of HATU in 50 μL of anhydrous dimethylformamide.
-
11
In a separate small vial closed with a silicone rubber septa dissolve 10.6 mg of Fmoc-Lys(Boc)-OH in 50 μL of anhydrous dimethylformamide.
-
12
Add 5 μL of diisopropylethylamine to the vial containing HATU. Solution must turn yellow.
-
13
Add lysine solution (step 11) to the vial containing HATU solution (step 10). Vortex the mixture for 2 min. Solution turns red.
-
14
Add the mixture obtained in step 13 to the synthesis column.
-
15
Vortex for 5 h.
-
16
Drain the solvent.
-
17
Wash the column as described in steps 7–9.
-
18
Add 1 mL of Capping Solution to the resin and agitate the reaction mixture for 5 min.
-
19
Drain the Capping Solution.
-
20
Repeat steps 18 and 19 once more.
-
21
Wash the column as described in steps 7–9.
-
22
Deprotect Fmoc group using 1 mL of the Deblocking Solution. Agitate the reaction mixture for 5 min.
-
23
Drain the solvent.
-
24
Use another portion of fresh 1 mL of the Deblocking Solution and agitate the reaction mixture for an additional 5 min.
-
25
Drain the solvent. Collect and combine Fmoc-deblock from step 23 and 24. Measure Fmoc-deblock. (See step 8 in Solid-phase synthesis of PNA oligomers). If absorbance is more than 0.75 do the next lysine coupling.
-
26
Wash the column as described in steps 7–9.
-
27
Repeat steps 10–26 two more times.
-
28
After attaching 3 lysine molecules proceed to step 5 in Solid-phase synthesis of PNA oligomers.
Cleavage from solid support and deprotection of PNA
-
Remove the synthesis column from synthesizer and attach two 1 mL syringes to each end of the column. Take 0.8 mL of the Cleavage Cocktail in one of the syringes and move it forth and back through the column several times. Leave the synthesis column with the Cleavage Cocktail solution for two hours occasionally moving the solution forth and back through the column.
NOTE: This simple procedure uses one syringe to push the reagent into the column while the other syringe is used to pull reagents out at the other end. The procedure is described in detail in Expedite 8909 user’s manual for DNA deprotection.
CAUTION: Trifluoroacetic acid is highly corrosive and toxic; m-Cresol has strong stench. Perform all operations in a fume hood and wear proper personal protection.
After two hours distribute the Cleavage Cocktail solution equally in four plastic 2 mL centrifuge vials.
Wash the synthesis column with fresh Cleavage Cocktail (0.2 mL). Collect the wash into a separate 2 ml centrifuge vial.
Add 1.8 mL of anhydrous diethyl ether to each of the five plastic centrifuge vials with cleavage cocktail solution and mix vigorously. The PNA should precipitate.
Centrifuge the mixtures for 15 minutes at 14500 rpm. Carefully, decant diethyl ether from the PNA precipitate. Add another 1 mL of diethyl ether to each vial, mix vigorously, centrifuge and decant the solvent.
Add HPLC grade water (0.3 mL) to each vial to dissolve PNA. Combine the solutions in one vial and evaporate using SpeedVac. The sample is ready for HPLC purification.
BASIC PROTOCOL 5: PURIFICATION AND QUANTIFICATION OF PNA AND RNA
Before ITC experiments PNA and target RNA must be purified using HPLC and quantified using UV spectrometry. PNA can be purified using a gradient of acetonitrile in water containing 1% trifluoroacetic acid on a reverse-phase C-18 column. RNA can be purified using a gradient of acetonitrile in triethylammonium acetate on a reverse-phase C-18 column. The products must be confirmed by ESI or MALDI TOF mass spectrometry. UV absorbance for PNA and RNA is recorded at 260 nm.
Equipment
HPLC system consisting of gradient-capable pumps, column oven and UV detector.
Waters XBridge Prep C-18 column (5 μm, 10 mm × 150 mm)
Freeze-Dry system with SpeedVac capability
Rotary Evaporator
UV spectrometer
Materials
Syringe filter, 13 mm with a 0.2 μm PTFE membrane.
Trifluoroacetic acid (HPLC grade)
Acetonitrile (HPLC grade)
Acetic acid (HPLC grade)
Triethylamine (HPLC grade)
Water (HPLC grade)
Mobile phase A (Reagents and Solutions)
Mobile phase B (Reagents and Solutions)
Mobile phase C (Reagents and Solutions)
Mobile phase D (Reagents and Solutions)
HPLC purification of synthetic PNA
Dissolve the PNA sample in 1 mL of HPLC water. Filter the sample through a 13 mm syringe filter.
Purify the PNA using a semipreparative reverse-phase HPLC using an XBridge Prep C-18 column at 65 °C, eluting with a linear gradient of 10 to 35% (v/v) of mobile phase B in mobile phase A over 45 min, flow rate 5 mL/min.
Collect the fractions corresponding to the major peak as detected by UV absorbance monitored at 254 and 280 nm.
Reduce the volume of the collected fractions by one half of the original volume using rotary evaporator. This is done to remove trifluoroacetic acid that is harmful to oil vacuum pump operating the freeze-dryer.
Freeze-dry the sample.
Confirm the correct sequence of PNA by ESI or MALDI TOF mass spectrometry.
Quantification of synthetic PNA
-
Dissolve the PNA sample in 1 mL of water (HPLC grade). Take 50 μL of the sample and dilute to 1 mL (dilution factor of 20).
NOTE: The dilution factor can be adjusted if the synthesis gives more or less of PNA than usual.
Measure the UV absorbance of PNA at 260 nm. If the absorbance is greater than one, dilute the sample further or repeat step 1 and increase the dilution factor. If the absorbance is below 0.2, repeat step 1 and decrease the dilution factor.
Calculate the molar extinction coefficient, ε(260) M−1cm−1 of PNA by summing up the individual extinction coefficients of all nucleobases: 8700 for T (Puglisi and Tinoco, 1989), 4776 for M, 5984 for E and 2150 for P (Gildea and McLaughlin, 1989).
Determine the molar concentration of PNA by dividing the UV absorbance of PNA at 260 nm with the extinction coefficient.
-
Multiply the molar concentration by the dilution factor (see above) to get the amount (in millimoles) of the PNA. Adjust the result to nanomoles by multiplying with 106.
NOTE: A successful 2 μmol scale synthesis may yield a couple of hundred nanomols of PNA after the HPLC purification.
Freeze-dry the sample.
Use HPLC grade water to prepare 0.24 mM stock solution of PNA. Store the stock solution frozen at −20 °C.
Preparation of the RNA target
-
If necessary, deprotect the crude RNA sample according to manufacturers’ recommendations.
NOTE: The exact method for RNA preparation may vary depending on the identity, length, commercial source and purity of the RNA sample. We describe our procedure for preparation of an RNA model target starting from crude synthetic RNA obtained from Thermo Fisher Scientific.
Dissolve the crude RNA sample in 400 μL of deprotection buffer (provided by the vendor). Make sure the RNA pellet is completely dissolved by vortexing or vigorous shaking. Incubate the solution at 60 °C for 30 minutes and evaporate the sample in a SpeedVac.
Dissolve the RNA sample in 1 mL of HPLC-grade water. Filter the sample through a 13 mm syringe filter.
Purify the RNA using a semipreparative reverse-phase HPLC using an XBridge Prep C-18 column at 60 °C, eluting with a linear gradient of 5 to 15% of mobile phase D in mobile phase C over 40 min with a flow rate 5 mL/min.
Collect the fractions corresponding to the major peak as detected by UV absorbance monitored at 254 and 280 nm.
Freeze-dry the collected fractions.
Dissolve the purified RNA in 2 mL of HPLC grade water and freeze-dry again.
Repeat step 7.
Quantify the RNA and prepare a 0.24 mM stock solution using the same procedure as described for PNA quantification above, except use the nearest-neighbor approximation (Puglisi and Tinoco, 1989) to obtain the molar extinction coefficient of RNA.
BASIC PROTOCOL 6: RNA BINDING AFFINITY BY ISOTHERMAL TITRATION CALORIMETRY
Isothermal Titration Calorimetry (ITC) is used to study binding affinity, binding mode and sequence selectivity of PNA carrying modified nucleobases to target dsRNA (Li et al., 2010, Gupta et al., 2011, Zengeya et al., 2012, Muse et al., 2013). For binding studies we use phosphate buffer that mimics physiological salt and pH at 37 °C. In our ITC experiments we use four equivalents of PNA to achieve 1:4 final ratio of RNA:PNA. In a typical experiment, the RNA sample is placed into the sample cell of ITC instrument and the PNA sample is loaded into the titration syringe. RNA solution is titrated by PNA solution and the heat of binding is recorded by the ITC instrument (Figure 6). TA Instruments software NanoAnalyze is used to analyze the titration data. Blank (linear) and Independent models are used to calculate affinity constant, enthalpy, and binding stoichiometry (Figure 7).
Figure 6.
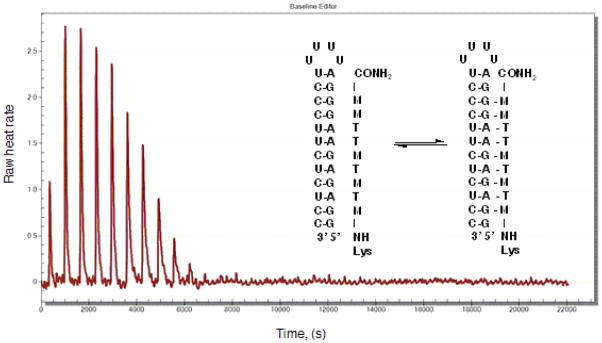
ITC titration trace of Lys-NH-MTMTMTTMM-CONH2 triple helical binding to an RNA hairpin at physiologically relevant conditions, pH = 7.4, 37 °C.
Figure 7.
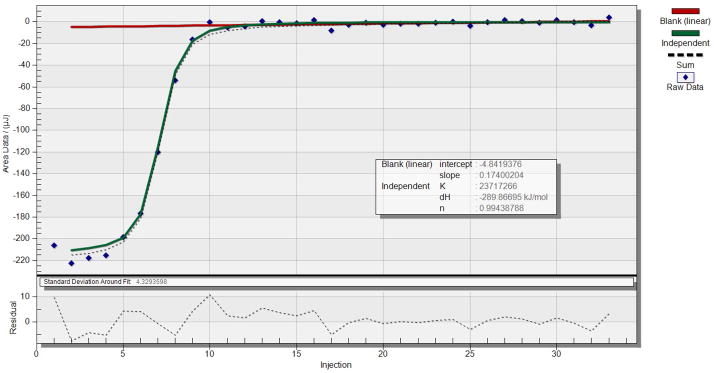
Analysis of ITC data for Lys-NH-MTMTMTTMM-CONH2 triple helical binding to an RNA hairpin. The red line shows the base line (dilution heat) fit using the Blank (linear) model. The green line fits the data using the Independent model. The results of this experiment were: association constant Ka = 2.4 × 107 M−1, binding enthalpy H = −289.9 kJ/mol, and binding stoichiometry n = 1.0.
Equipment
Isothermal titration microcalorimeter suitable for small biological samples (cell volume 1 mL or smaller and minimum detectable heat 0.1 μJ or smaller), for example, a TA instruments NanoITC 2G instrument.
Materials
Phosphate buffer, pH = 7.4 (Reagents and Solutions)
Isothermal Titration Calorimetry
Place 17.5 μL of RNA stock solution (0.24 mM) in a plastic 2 mL centrifuge vial and evaporate in a SpeedVac.
-
Dissolve the RNA sample in 1600 μL of degassed phosphate buffer.
NOTE: Add a small stir bar to the vial with ITC buffer and degas by stirring under moderate vacuum (~80 torr) for 10–15 min.
Place 70 μL of the PNA stock solution (0.24 mM) into a plastic 2 mL centrifuge vial and evaporate in SpeedVac.
Dissolve the PNA sample in 350 μL of degassed phosphate buffer.
Load the RNA sample (950 μL) into the sample cell of ITC instrument. Load HPLC-grade water into the reference cell of the ITC instrument.
Load the PNA sample (250 μL) into the titration syringe. Install the syringe into the ITC titration burette and install the burette onto the ITC instrument.
Program the experimental parameters in the TA Instruments software ITCRun: temperature (37 °C to mimic the physiological conditions), injection interval and number of injections. In a typical experiment we use 50 injections of 5 μL PNA and 600 second intervals between injections. The intervals can be either increased or decreased depending on the rate of the binding event. Start stirring (250 rpm) and set the equilibration time to one hour.
Start the ITC experiment and collect the titration data (Figure 6).
-
Analyze the data using TA Instruments software NanoAnalyze using the Blank (linear) and Independent models. An example of graphical analysis is shown in Figure 7.
NOTE: Blank (linear) model allows base line subtraction only if the last data points of the titration curve represent PNA dilution heats only, i.e., the receptor is completely saturated and there is no binding heat due to addition of more ligand. It is not uncommon that in cases of weak binding or complex equilibrium the receptor is not completely saturated at the end of titration curve. In such cases it is necessary to run a blank run, of the ligand (PNA) against the buffer solution. This is done in order to subtract out the heat of dilution from the actual binding heat. Perform the blank run as described above, except, omit the RNA receptor. In NanoAnalyze subtract the blank run from the actual binding experiment and analyze the data using the Independent model.
Reagents and Solutions
The following reagents should be prepared fresh before the synthesis.
Deblocking solution (20% (v/v) piperidine in dimethylformamide): mix 20 mL of piperidine (biotech grade, Aldrich) and 80 mL of anhydrous dimethylformamide.
Activator solution (0.19 M O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) in dimethylformamide): dissolve 1.089 g of HATU in 15 mL of anhydrous dimethylformamide.
Base solution (0.2 M diisopropylethylamine and 0.3 M 2,6-lutidine in dimethylformamide): dilute 1.92 mL of diisopropylethylamine (reagent grade distilled over CaH2) and 2.97 mL of lutidine (redistilled, Aldrich) to 100 mL using anhydrous dimethylformamide.
Capping solution (5% (v/v) acetic anhydride and 6% (v/v) 2,6-lutidine in dimethylformamide): mix (in the following order) 89 mL of anhydrous dimethylformamide, 6 mL of lutidine (redistilled, Aldrich), and 5 mL of acetic anhydride (reagent grade).
Cleavage Cocktail (20% (v/v) m-cresol in trifluoroacetic acid): mix 0.8 mL of trifluoroacetic acid (HPLC grade) and 0.2 mL of m-cresol (reagent grade).
The following buffers for HPLC can be stored at room temperature and used over a period of one week. Longer storage time is not recommended.
Mobile phase A (0.1% (v/v) trifluoroacetic acid in water): mix 1 mL of trifluoroacetic acid (HPLC grade) and 1 L of water (HPLC grade). Filter the solution through a 0.20 μm nylon membrane filter.
Mobile phase B (40% (v/v) acetonitrile and 0.1% (v/v) trifluoroacetic acid in water): mix 400 mL of acetonitrile (HPLC grade) and 600 mL of water (HPLC grade) and add 1 mL of trifluoroacetic acid (HPLC grade). Filter the solution through a 0.20 μm nylon membrane filter.
Mobile phase C (0.1 M triethylammonium acetate in water, pH 7.0): add 14 mL of triethylamine (HPLC grade) to 0.8 L of water (HPLC grade) followed by 5 mL of acetic acid (HPLC grade). In a separate vessel, mix 1 mL of acetic acid (HPLC grade) and 9 mL of water (HPLC grade); then use this solution to titrate mobile phase C until a pH 7.0 is obtained. Complete the preparation of mobile phase C by bringing the volume to 1 L by adding water (HPLC grade). Filter the solution through a 0.20 μm nylon membrane filter.
Mobile phase D (40% (v/v) acetonitrile and 0.1 M triethylammonium acetate in water, pH 7.0): mix 400 mL of acetonitrile (HPLC grade) and 600 mL of mobile phase C (see above). Filter the solution through a 0.20 μm nylon membrane filter.
Phosphate buffer (2 mM MgCl2, 90 mM KCl, 10 mM NaCl and 50 mM potassium phosphate, pH 7.4)
Prepare Solution A: dissolve 203 mg of MgCl2 (ACS grade), 3.355 g of KCl (ACS grade), 292 mg of NaCl (molecular biology grade) and 4.355 g of K2HPO4 (molecular biology grade) in 500 mL of water (HPLC grade). Prepare Solution B: dissolve 203 mg of MgCl2 (ACS grade), 3.355 g of KCl (ACS grade), 292 mg of NaCl (molecular biology grade) and 3.402 g of KH2PO4 (molecular biology grade) in 500 mL of water (HPLC grade). Adjust the pH of Solution A to 7.4 by slowly adding Solution B. The phosphate buffer can be stored in refrigerator (+4 °C) for up to a month.
COMMENTARY
Background Information
The central dogma of molecular biology (Crick, 1970) starts with transcription of double-stranded DNA (dsDNA) gene by RNA polymerase into single-stranded messenger RNA, which is then translated by the ribosome to produce a protein that corresponds to the given gene. For a long time RNA was believed to play a passive role in gene expression. The discovery of RNA interference (RNAi) by Andrew Fire, Craig Mello and colleagues (Fire et al., 1998) in 1998 has revealed a new pathway of gene regulation – sequence-specific gene knockdown by double-stranded RNA (dsRNA) molecules. RNAi plays an essential role in development, cell differentiation and proliferation, apoptosis, virus resistance and oncogenesis (Novina and Sharp, 2004).
Two important classes of small non-coding RNAs play a central role in RNAi – small interfering RNAs (siRNAs) and microRNAs (miRNAs). These two classes of silencing RNAs share similar central biogenesis and can perform interchangeable biochemical functions. Both siRNA and miRNA can direct the RNA-induced silencing complex (RISC) to bind target mRNA with full or partial sequence complementarity to promote mRNA cleavage or translational repression. The key difference between these two classes comes from their origin, evolutionary conservation and the types of genes that they downregulate (Bartel, 2004; Kusenda et al., 2006; Novina and Sharp, 2004). siRNAs are typically exogenous in mammalian cells, for example, introduced into the cell by an invading virus. They are processed from long RNA duplexes or extended hairpins and specify the silencing of the same locus from which they originate (Bartel, 2004, 2009). miRNAs are highly conserved endogenous RNA molecules that result from genomic loci distinct from other recognized genes and are believed to regulate expression of about 30% of protein-coding genes (Kusenda et al., 2006).
The important role that RNA plays in gene control makes it an attractive target for molecular recognition. Naturally occurring non-coding RNAs are either completely double-stranded or globular with double-stranded domains connected by short single-stranded regions. The most straightforward way of sequence-selective recognition of dsRNA would be a triple helix formation. The triple helix strategy has been extensively applied for the sequence-specific recognition of DNA double helix (Duca et al., 2008). This, so-called anti-gene approach uses triplex forming oligonucleotides (TFOs) that bind in the major groove of the DNA duplex through Hoogsteen or reversed Hoogsteen hydrogen bonds. Triplex formation has been shown to prevent protein binding to DNA, to alter gene expression during DNA transcription process, to inhibit DNA replication process, and to induce site-specific DNA damage (Duca et al., 2008; Malnuit et al., 2011). Peptide nucleic acid (PNA), first introduced by Nielsen and co-workers in 1991 (Nielsen et al., 1991), has the entire sugar-phosphate backbone replaced by a polyamide chain composed of N-2-aminoethylglycine repeating units. The PNA backbone is chemically stable, neutral and resistant to enzymatic degradation, which makes PNA an excellent oligonucleotide analogue for in vivo applications. PNAs have become important research tools for molecular recognition of dsDNA. However, compared to TFOs that normally bind the major groove of dsDNA and form triple helix, PNAs may bind dsDNA using different modes of recognition depending on the PNA and DNA sequence, concentration and reaction time. In many cases competition between duplex invasion and triplex formation is observed (Malnuit et al., 2011).
Compared to DNA, triple helical recognition of dsRNA has received relatively little attention. The RNA double helix has a shallow and wide minor groove and a narrow and deep major groove. The major groove of RNA is information rich as all the base pairs project their discriminatory edges into the major groove (Chow and Bogdan, 1997). However, the structural properties of the major groove make it, in general, inaccessible to many ligands. Practical application of triple helices are hindered by three problems: 1) slow binding kinetics and low stability of the triplex caused by electrostatic repulsion between the negatively charged helix and the incoming third strand of oligonucleotide; 2) the requirements for long homopurine tracts, as only standard Hoogsteen triplets (U*A-U and C+*G-C) can be used in recognition (Figure 1A); and 3) low stability of triple helices formed by unmodified oligonucleotides at physiological pH (Malnuit, 2011; Rozners, 2012).
Recently, we (Li et al., 2010) showed that PNAs as short as six nucleobases formed stable (Ka > 107) and sequence selective Hoogsteen triple helices with dsRNA at pH 5.5. We also found (Gupta et al., 2011, 2012) that single pyrimidine interruptions in a polypurine tract of dsRNA could be recognized using modified bases, 2-pyrimidinone P (Buchini and Leumann, 2004; Gildea and McLaughlin, 1989) and 3-oxo-2,3-dihydropyridazine E (Eldrup et al., 1997), that bind C-G and U-A inversions, respectively (Figure 1B).
Low stability of triple helices at physiological pH is caused by the need of cytosine protonation in order to form the standard Hoogsteen C+*G-C triplet. Having a pKa of 4.5, cytosine is hardly protonated under physiological conditions, which decreases stability of PNA-dsRNA complex. We found (Zengeya et al., 2012) that the more basic 2-aminopyridine (Cassidy et al., 1997; Hildbrand et al., 1997), monomer M (Figure 1B, pKa = 6.7) was an efficient analogue of protonated cytidine that enabled nanomolar binding at physiological conditions. Furthermore, conjugation of M-modified PNA with tetralysine peptide significantly improved RNA binding affinity without compromising sequence selectivity (Muse et al., 2013). PNA carrying M and lysine modifications showed good cellular uptake by HEK293 cells, whereas analogous PNA with no M-modifications (only lysines) showed little uptake (Muse et al., 2013). Cationic PNAs had at least two orders of magnitude higher affinity for dsRNA than for dsDNA, suggesting that the deep and narrow major groove of RNA was a better fit for PNA than the wider major groove of DNA (Muse et al., 2013; Zengeya et al., 2012). The results suggested that the triple-helix strategy might have the potential to recognize and interfere with the function of biologically important non-coding RNAs.
Critical Parameters and Troubleshooting
Monomer synthesis
All solvents should be freshly distilled over nitrogen atmosphere and appropriate drying agent. Alternatively, solvents may be dried using a Solvent Purification System, such as, MBraun MB-SPS. Commercial reagents are used as received without additional purification.
Care should be taken when using methanol:CH2Cl2 system for purification of PNA monomers by silica gel column chromatography. Using high concentrations of methanol (>10%) may dissolve some silica gel, causing accumulation of amorphous solid in collected fractions. Contamination of products with silica gel is difficult to detect by standard techniques (TLC and NMR) and may interfere with solid-phase PNA synthesis.
PNA synthesis
Lack of precipitate after addition of diethyl ether indicates that the synthesis was not successful. While there may be many reasons why a multistep procedure may fail, the most common problems we have encountered are blocked reagent delivery lines on the Expedite 8909, impure reagents and monomers, and moisture in reagent solutions. All solvents used for preparation of deblocking, activation, coupling and capping solutions must be dry. Solutions of PNA monomers should be freshly prepared just before solid-phase synthesis and used within 6 h.
ITC analysis
If poor signal to noise ratio is a problem, doubling the amount of PNA and RNA may be necessary to improve the quality of data.
Anticipated Results
Good to moderate yields (100–300 nmols) of Fmoc-M(Boc)-OH, Fmoc-P-OH and Fmoc-E-OH monomers are expected following the procedures in Basic Protocols 1–3.
A successful 2 μmol scale synthesis may yield a couple of hundred nanomols of PNA after the HPLC purification (Basic Protocols 4–5).
Time considerations
The synthesis of monomer M starting from 2-(6-Chloro-3-pyridinyl) acetonitrile can be accomplished in two weeks. The synthesis of monomers P and E can be accomplished in one week.
Automated synthesis of PNA usually requires three to four hours for preparation of a 9-mer. Manual synthesis of the same length PNA requires 12–14 hours.
Lyophilization of PNA molecules after HPLC purification or UV quantification is usually done overnight.
Acknowledgments
This work was funded by National Institutes of Health GM071461 grant.
Literature Cited
- Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchini S, Leumann CJ. Stable and selective recognition of three base pairs in the parallel triple-helical DNA binding motif. Angew Chem Int Ed. 2004;43:3925–3928. doi: 10.1002/anie.200460159. [DOI] [PubMed] [Google Scholar]
- Burns CJ, Goswami R, Jackson RW, Lessen T, Li W, Pevear D, Tirunahari PK, Xu H. Beta-lactamase inhibitors. W2010/130708 PCT/EP2010/056408: patent. 2010;2010:197. [Google Scholar]
- Cassidy SA, Slickers P, Trent JO, Capaldi DC, Roselt PD, Reese CB, Neidle S, Fox KR. Recognition of GC base pairs by triplex forming oligonucleotides containing nucleosides derived from 2-aminopyridine. Nucleic Acids Res. 1997;25:4891–4898. doi: 10.1093/nar/25.24.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CS, Bogdan FM. A Structural Basis for RNA-Ligand Interactions. Chem Rev. 1997;97:1489–1513. doi: 10.1021/cr960415w. [DOI] [PubMed] [Google Scholar]
- Corradini R, Sforza S, Tedeschi T, Totsingan F, Manicardi A, Marchelli R. peptide Nucleic Acids with a Structurally Biased Backbone. Updated review and Emerging Challenges. Curr Top Med Chem. 2011;11:1535–1554. doi: 10.2174/156802611795860979. [DOI] [PubMed] [Google Scholar]
- Crick F. Central Dogma of Molecular Biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. Survey and summary. The triple helix: 50 years later, the outcome. Necleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldrup AB, Dahl O, Nielsen PE. A novel peptide nucleic acid monomer for recognition of thymine in triple-helix structures. J Am Chem Soc. 1997;119:11116–11117. [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Gildea B, McLaughlin LW. The synthesis of 2-pyrimidinone nucleosides and their incorporation into oligodeoxynucleotides. Nucleic Acids Res. 1989;17:2261–2281. doi: 10.1093/nar/17.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Zengeya T, Rozners E. Triple helical recognition of pyrimidine inversions in polypurine tracts of RNA by nucleobase-modified PNA. Chem Commun. 2011;47:11125–11127. doi: 10.1039/c1cc14706d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Muse O, Rozners E. Recognition of Double-Stranded RNA by Guanidine-Modified Peptide Nucleic Acids. Biochemistry. 2012;51:63–73. doi: 10.1021/bi201570a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildbrand S, Blaser A, Parel SP, Leumann CJ. 5-Substituted 2-Aminopyridine C-Nucleosides as Protonated Cytidine Equivalents: Increasing Efficiency and Specificity in DNA Triple-Helix Formation. J Am Chem Soc. 1997;119:5499–5511. [Google Scholar]
- Li M, Zengeya T, Rozners E. Short Peptide Nucleic Acids Bind Strongly to Homopurine Tract of Double Helical RNA at pH 5.5. J Am Chem Soc. 2010;132:8676–8681. doi: 10.1021/ja101384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnuit V, Duca M, Benhida R. Targeting DNA base pair mismatch with artificial nucleobases. Advances and perspectives in triple helix strategy. Org Biomol Chem. 2011;9:326–336. doi: 10.1039/c0ob00418a. [DOI] [PubMed] [Google Scholar]
- Muse O, Zengeya T, Mwaura J, Hnedzko D, McGee DW, Grewer CT, Rozners E. Sequence Selective Recognition of Double-Stranded RNA at Physiologically Relevant Conditions Using PNA-Peptide Conjugates. ACS Chem Biol. 2013;8:1683–1686. doi: 10.1021/cb400144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- Novina CD, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- Olsen AG, Dahl O, Nielsen PE. Synthesis and evaluation of a conformationally constrained pyridazinone PNA-monomer for recognition of thymine in triple-helix structures. Bioorg Med Chem Lett. 2004;14:1551–1554. doi: 10.1016/j.bmcl.2003.12.093. [DOI] [PubMed] [Google Scholar]
- Puglisi JD, Tinoco I., Jr Absorbance melting curves of RNA. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- Rozners E. Recent Advances in Chemical Modification of Peptite Nucleic Acids. J Nucleic Acids. 2012;518162:8. doi: 10.1155/2012/518162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciechowski F, Hudson RHE. A Convenient Route to N-[2-(Fmoc)aminoethyl]glycine Esters and PNA Oligomerization Using a Bis-N-Boc Nucleobase Protecting Group Strategy. J Org Chem. 2008;73:3807–3816. doi: 10.1021/jo800195j. [DOI] [PubMed] [Google Scholar]
- Zengeya T, Gupta P, Rozners E. Triple-Helical Recognition of RNA Using 2-Aminopyridine-Modified PNA at Physiologically relevant Conditions. Angew Chem Int Ed. 2012;51:12593–12596. doi: 10.1002/anie.201207925. [DOI] [PMC free article] [PubMed] [Google Scholar]