

Abstract
Background
The Department of Health and Human Services recently called for public comment on human subjects research protections.
Objective
(1) To assess variability in reviews across Institutional Review Boards (IRBs) for a multisite minimal risk trial of financial incentives for evidence-based hypertension care. (2) To quantify the impact of review determinations on site participation, budget, and timeline.
Design
A natural experiment occurring from multiple IRBs reviewing the same protocol for a multicenter trial (May 2005–October 2007).
Participants
25 Veterans Affairs Medical Centers (VAMCs).
Measurements
Number of submissions, time to approval, and costs were evaluated. We compared patient complexity, academic affiliation, size, and location (urban/rural) between participating and non-participating VAMCs.
Results
Of 25 eligible VAMCs, 6 did not meet requirements for IRB review, and 2 declined participation. Of 17 applications, 14 were approved. This process required 115 submissions, lasted 27 months, and cost close to $170,000 in staff salaries. One IRB’s concern about incentivizing a particular medication recommended by national guidelines prompted a change in our design to broaden our inclusion criteria beyond uncomplicated hypertension. The change required amending the protocol at 14 sites to preserve internal validity. The IRBs that approved the protocol classified it as “minimal risk”. The 12 sites that ultimately participated in the trial were more likely to be urban, academically affiliated, and care for more complex patients, limiting the external validity of the trial’s findings.
Limitations
Because data came from a single multisite trial in the VA, which uses a two-stage review process, generalizability is limited.
Conclusions
Complying with IRB requirements for this minimal risk study required substantial resources and threatened the study’s internal and external validity. The current review of regulatory requirements may address some of the problems.
Primary Funding Sources
Veterans Affairs Health Services Research & Development and National Institutes of Health, National Heart, Lung, and Blood Institute
INTRODUCTION
A number of authors have documented variability in the process of obtaining human subjects approval from local Institutional Review Boards (IRBs) for multisite studies. This variability includes standards of review (1–4), consent documents and requirements (1–7), and the time from submission to approval (2, 3, 8). For example, one observational health services research protocol at 43 sites noted that the time from submission to approval ranged from 52 to 798 days (3). In a well-known case, a quality improvement study led by Pronovost, et al. (9), illustrated how regulations meant to protect human subjects were interpreted by the Office of Human Research Protections in a way that appeared contrary to their intent (10). A recent systematic review of evidence from 52 studies concluded that some decisions made by IRBs are not consistent with federal policy (11).
The gold standard for generalizable research is a multisite randomized controlled trial. However, such trials are relatively rare in health services research, and IRBs may lack experience in reviewing them. In this article, we focus upon the variability in review determinations across IRBs for a multisite trial that sought to improve the delivery of evidence-based hypertension care, and we seek to quantify the impact on the type of site ultimately participating, budget, timeline, and project staff. To our knowledge, this is the first study to evaluate the IRB approval process for this type of research and to highlight the impact on both the internal and external validity of the study’s findings. Our intent is that our findings will help to inform current efforts to solicit public comment regarding the need to revise the Common Rule (12).
METHODS
This study is a natural experiment occurring from multiple IRBs reviewing the same protocol for a multicenter trial. We reviewed records detailing the IRB approval process from May 2005 through mid-October 2007.
Description of the Trial
The trial was designed to test whether explicit financial incentives (also termed pay for performance) (13) improved hypertension guideline adherence (14). The study methods are described elsewhere (15). Briefly, 12 Department of Veterans Affairs (VA) medical centers were randomized to one of four study arms, differentiated by the type of incentive rewarded: (1) physician-level incentives; (2) health care provider group-level incentives; (3) physician- and group-level incentives; and (4) no incentives (control). Participants in all four arms received audit and feedback on their performance. Primary care physicians who worked at least 0.6 full-time equivalents (approximately three days per week related to clinical activities) or had a panel size of at least 500 patients were eligible to participate. At the six study sites randomized to the group-level incentive, the physicians invited up to 15 non-physician colleagues, including other clinicians (e.g., nurses and pharmacists) and administrative support staff (e.g., clerks), to participate.
Procedures for Obtaining IRB Approval
Multisite research studies conducted within the VA system are required to designate a local principal investigator (PI) and then to obtain approval from both the local IRB and the local VA Research and Development (R&D) committee. In this trial, the process of identifying a local PI at each site included contacting local leadership to identify potential site investigators, obtaining site PI assent to participate, educating the site PI about the project and his/her roles, and ensuring the site PI had an academic appointment and current research trainings. After identifying a site PI, research staff from the coordinating center in Houston prepared the IRB and R&D applications for each site. A certified IRB professional was hired eight months after beginning the submission process to help submit the regulatory paperwork; the need for such was not initially anticipated.
Site Selection
Leaders of five VA Networks (consisting of 45 facilities) agreed to participate (Figure 1). The study power calculations required at least five physicians per site. We pursued the 25 facilities with at least eight full-time physicians to account for potential attrition and intended to randomly select 12 facilities prior to study arm randomization in order to achieve a representative sample. Of the 25 facilities with at least eight full-time physicians, three did not have an on-site or affiliated IRB. At two of the 22 facilities with IRBs, the local hospital directors declined to participate. At another facility, we were unable to recruit a site PI. We prepared IRB and R&D applications for the 19 remaining facilities. However, at two of these, the site PI was unable to complete the academic credentialing or research certification process. Applications therefore were submitted to and reviewed at only 17 of the 25 eligible facilities.
Figure 1.
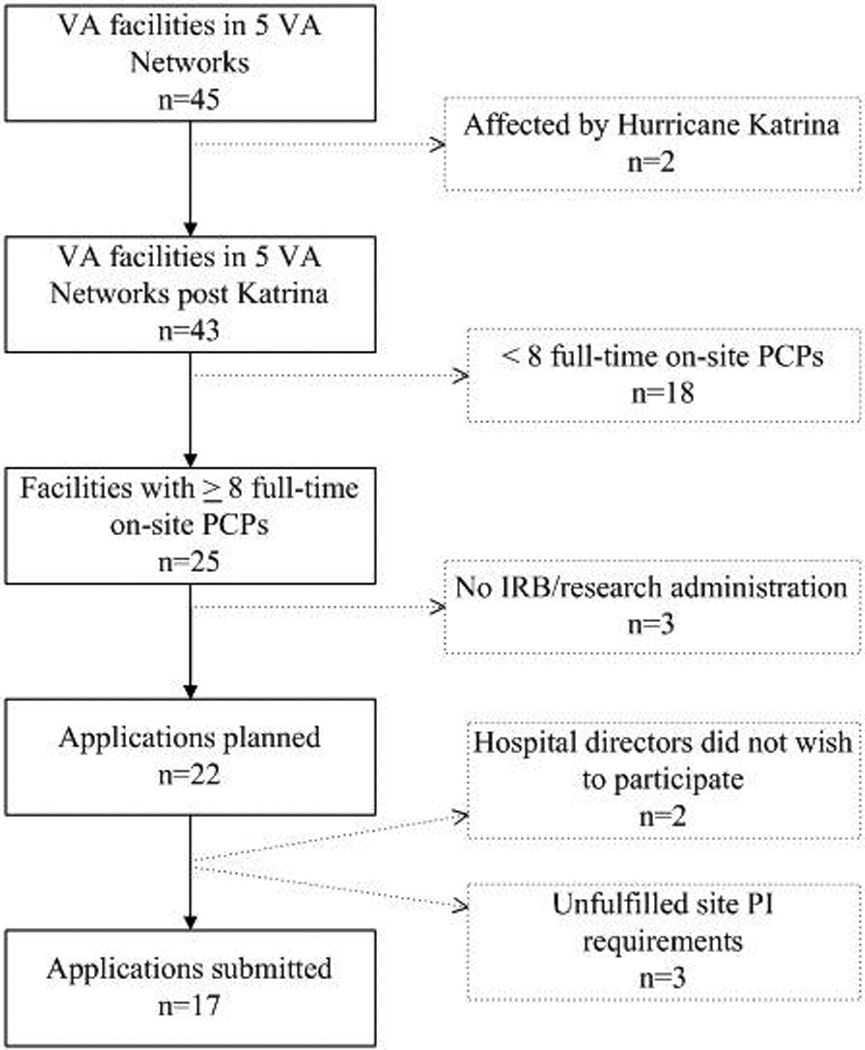
Site Identification to Submission of Applications
VA = Veterans Affairs; PCPs = primary care physicians; IRB = Institutional Review Board; PI = principal investigator
Record Review and Measures
At least two authors independently reviewed the regulatory submission materials and correspondences at each site to gather data on submission requirements, board structure, study review category, and submission and approval dates. When their observations disagreed, they consulted with a third author.
Time from initial submission to approval was calculated as the number of calendar days from the date the application was submitted to a site’s PI until the date of the initial approval letter from that site. One site’s IRB had a concern that could only be addressed by changing the study methods, necessitating a modification of the protocol at all sites to preserve the internal validity of the study (specifics of this change are described below). Because this delayed the commencement of the project, we also calculated the time from the submission of this modification request to its approval. For the 13 sites that approved the study before the modification was submitted, we evaluated the relationship between the date when the application was submitted and the number of days to initial approval. Site IDs reflect the order in which applications were submitted (applications submitted to Site 1 first and to Site 17 last).
We enumerated the submissions the team made to each site’s regulatory boards from the submission date of the initial application until the date of the initial approval letter, and from the submission date of the protocol modification until the date of its approval letter. We considered a submission to be any of the following: initial application; protocol modification; renewal; a response to an IRB or R&D committee decision requiring application modifications; and a response to any IRB/R&D request that involved a substantial amount of team effort.
We estimated the amount of staff time involved in the IRB approval process. In addition to the certified IRB professional, we employed a team of three other project coordinators (Master degree level) and two other research assistants (Bachelor degree level) who spent a portion of their overall work effort on IRB and R&D related tasks. To calculate the cost of these human resources, we multiplied staff time by staff salary, including benefits.
We compared characteristics between participating and non-participating facilities using Mann-Whitney tests. Using methods published elsewhere (16), we summarized the complexity of the patients cared for at each study site (where a higher complexity index corresponds to more a complex patient); number of resident slots per 10,000 patients to assess each facility’s academic mission; number of hospital operating beds to quantify facility size; and number of hospital beds in the community to distinguish between urban and rural areas. Analyses of facility characteristics were performed using Stata, version 11.2 (StataCorp LP, College Station, TX).
Role of the Funding Source
VA Health Services Research & Development and the National Institutes of Health, National Heart, Lung, and Blood Institute, provided funding for this study. The study sponsors played no role in the design, conduct, and analysis of the parent trial or this record review, nor did they have any role in the preparation, review, or approval of the manuscript.
RESULTS
The original premise of the study was to determine whether financial incentives to physicians could improve the translation of the findings from the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) into outpatient practice. Specifically, the study planned to incentivize the ALLHAT findings about the effectiveness of thiazide diuretics in lowering blood pressure in most hypertensive patients and the recommendations that blood pressure readings <140/90 mmHg or <130/80 mmHg in diabetics be considered controlled. In the summer of 2006, one IRB stated that their Office of General Counsel was concerned that the study provided incentives to employees “for their increased utilization of a particular drug”. Following several unsuccessful attempts to resolve the issue with the site directly, we consulted with the VA legal counsel. In November 2006, we modified the study premise from “translation of ALLHAT findings into practice” to “provision of care according to guidelines in the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7)”, rewarding physicians not just for prescription of thiazides in uncomplicated patients, but for prescription of any JNC 7 guideline-recommended antihypertensive medications. In order for the performance measures to remain consistent across sites, thereby preserving the internal validity of the study, we submitted this modification as a protocol amendment to all 13 sites that had approved the study, and we revised and resubmitted the initial application to one site where the study was pending a review decision. We chose to make the changes as we were concerned that these issues might arise at other sites after the intervention began, such as during a protocol renewal, when changing the methods would have threatened the entire project.
Of the 17 IRBs where we submitted the application, 15 required full board review, and 2 granted expedited review. IRB and R&D committee reactions to the study varied markedly by site. Some sites appreciated the novelty and timeliness of the proposal. The IRB at Site 8 noted, “It is well known that significant (and expensive) research such as the ALLHAT trial often fails to translate into changes in provider behavior. Financial incentives are a proposed mechanism for facilitating this translation, and it is important to evaluate them prior to wide-spread adoption.” The Site 4 R&D committee noted that they found this to be “an interesting and exciting project”. The intervention’s novelty caused concern at some sites, however. The Site 9 IRB questioned, “Is this legal for a research study? If legal, it seems to lead to unethical behavior similar to paying finder fees.” The IRB at Site 2 said, “Offering money to people to do what is expected of them is not ethical.” The study team responded to these concerns by noting that several public and private health plans already are implementing “pay-for-performance” models, yet there are few empirical studies of their effectiveness (13). Sites 2 and 9 ultimately approved the proposal.
At sites 11 and 13, the IRB granted approval via expedited review. At Site 13, the R&D committee then tabled the protocol, stating, “Address why patients are not being told (through written consent) that their physicians were being paid to follow a specific protocol for their care.” The study team explained that the physician was being incentivized for providing high quality care in accordance with national guidelines, and that, because each assessment of the physician’s care delivery was based on a random subset of his/her hypertensive patients, it was not feasible to obtain patient consent beforehand. The IRBs at several other sites expressed similar concerns about patient awareness of the study, so the team agreed to place in the clinic area at these sites a flyer notifying patients that their physician may be participating in this study. The Site 3 IRB insisted that patients be informed individually if their physician was participating, despite our concerns about breaching physician confidentiality and introducing bias through patient activation (where a patient questioning his/her physician about treatment affects the care provided). This IRB ultimately disapproved the study, stating, “The potential risk to hypertensive patients is too great to justify their involvement.” The Site 13 R&D committee also ultimately disapproved the study; ironically, despite an expedited IRB approval at this site, the application had to be formally closed. Despite the variability in initial reactions to the study protocol, amongst the 15 sites at which the IRB approved the study, 14 categorized it as minimal risk; the remaining site’s IRB did not determine its risk category.
Fourteen (82%) of the 17 submitted applications received the IRB and R&D approvals required for implementation. Additional submissions were required at all 17 sites, for a total of 80 submissions before the study application was either approved at or, if not approved, withdrawn from all 17 sites (median number of submissions per site, 4; mean, 5; range, 2 to 10). Among the 14 sites where the application received full approval, 35 additional submissions were required to approve the protocol modification, resulting in a total of 115 submissions before the study could be implemented (median number of submission per site, 6; mean, 7; range, 4 to 14). Among the 14 sites that received full approval, the number of days required for initial approval, plus the number of days required for approval of the protocol modification, ranged from 57 to 400 days per site (median, 168; mean, 181). There were no significant differences between VA- and university-affiliated IRBs in the average number of submissions per site, the average time from submission to approval, the percent of sites receiving IRB approvals, the percent of sites receiving R&D committee approvals, or the percent of sites where the protocol received expedited review (Table 1). Most IRBs required paper submissions; all 3 IRBs with electronic submissions were university-affiliated.
Table 1.
Submission Process and Results by IRB Structure
| IRB Structure | |||
|---|---|---|---|
| VA | University Affiliated |
P Value* | |
| Sites where applications submitted | |||
| Total, n | 8 | 9 | |
| Received expedited IRB review, n (%) | 0 | 2 (22) | .471 |
| Approved by IRB, n (%) | 8 (100) | 7 (78) | .471 |
| Approved by IRB and R&D, n (%) | 8 (100) | 6 (67) | .206 |
| Sites approved by IRB and R&D | |||
| Total, n | 8 | 6 | |
| Number of submissions required †, mean (SD) | 7 (3) | 6 (1) | .421 |
| Number of days to approval ‡, mean (SD) | 179 (117) | 184 (75) | .919 |
VA = Veterans Affairs; IRB = Institutional Review Board; R&D = VA Research & Development committee; SD = standard deviation
From two-tailed Fisher’s exact test for binomial data; from two-tailed Student’s t-test for continuous data
Number of submissions required for the initial approval of the application plus the number required for the approval of the protocol modification
Number of days from the initial submission to the approval of the application at each site plus the number from the submission of the protocol modification to its approval
The number of days from initial submission to approval by date of initial submission is displayed in Figure 2 for the 13 sites that approved the study before the modification was submitted. In July 2005 we submitted the first application, to Site 1, which approved the study in 36 days. We submitted the application to the final site, Site 17, in August 2006; it was approved in 76 days. The shortest time to approval, 36 days, occurred at Sites 1 and 7. Sites 9 and 10, the ninth and tenth applications submitted, took the longest to approve the study, at 198 and 230 days, respectively, suggesting that the protracted approval process was not due to a “learning curve” on the part of the study team. The total time spent in the IRB approval process before the study could be implemented, from the initial submission to the first site to the approval of the protocol modification at the final site, was 827 days, or over 27 months. This is 21 months longer than we had proposed and 23 months longer than that for which we had received a budget (Figure 3). Staff spent an estimated 6,729 hours working on IRB and R&D related tasks, costing approximately $168,229 in salaries. This estimate does not include the salary for the PI or site PIs.
Figure 2.
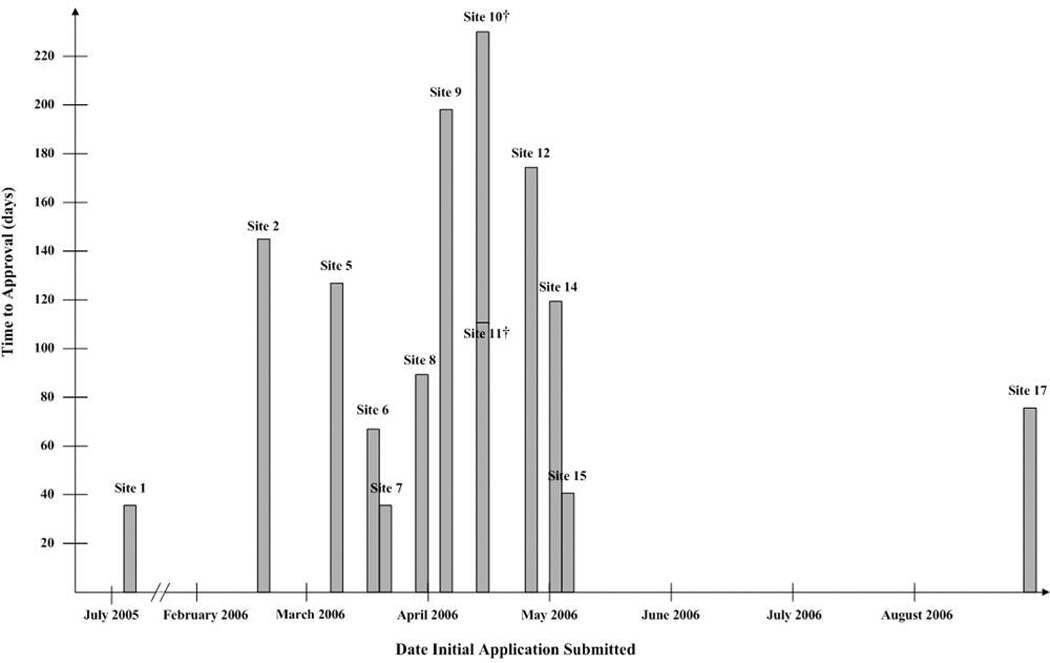
Number of Days from Initial Submission to Approval by Date of Initial Submission*
* For the 13 sites that approved the study before the modification was submitted. Sites 3, 4, and 13 did not approve the study, and the modification was incorporated into the application at Site 16 before their IRB had made an initial review decision.
† Applications were submitted to Sites 10 and 11 on the same date, April 14, 2006. Site 10 approved the application in 230 days, and Site 11 approved it in 111 days.
Figure 3.
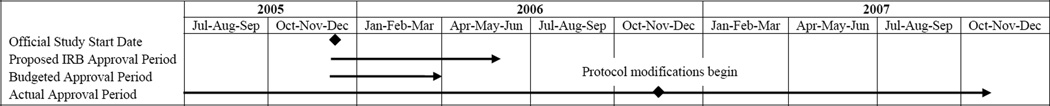
Impact of IRB and R&D Approval Process on Study Timeline
IRB = Institutional Review Board; R&D = VA Research & Development Committee; Jan = January; Feb = February; Mar = March; Apr = April; Jun = June; Jul = July; Aug = August; Sep = September; Oct = October; Nov = November; Dec = December
We began participant recruitment at a site as soon as their regulatory boards had provided full approval of the protocol modification. By the time the study was approved at all sites, seven physician participants had withdrawn due to a position change, transfer, maternity leave, or retirement. In December 2007, while randomizing sites to study arms, the IRB for one of the sites where we had exceeded our physician recruitment goal shut down, preventing the study from continuing at that site. We had to replace that site with another where we had not met our recruitment goal; after four more months of recruiting, we met the goal at the replacement site.
Of the 25 sites initially eligible for inclusion in this study, only 12 ultimately were included. The average patient complexity index at included facilities was significantly greater than that at the 13 excluded facilities (p=0.017; Table 2). Included sites also had a significantly greater mean ratio of medical resident slots to 10,000 unique patients (p=0.004) and a significantly greater average number of hospital beds in their community (p=0.005) than sites that could not be included, suggesting that included facilities were more urban.
Table 2.
Characteristics of Facilities That Were and Were Not Included in the Trial*
| Facility Characteristics | Included (N = 12) |
Excluded (N = 13) |
P Value |
|---|---|---|---|
| Patient complexity index† | 1584 (1191) | 541 (821) | .017 |
| Ratio of medical resident slots to 10,000 unique patients | 23.0 (10.2) | 8.0 (10.1) | .004 |
| Number of hospital operating beds | 174 (118) | 103 (70) | .115 |
| Number of hospital beds in community | 6814 (5594) | 2626 (2909) | .005 |
Data are presented as mean (standard deviation).
The patient complexity index is a measure of patient complexity based on the relative weight and frequency of each Diagnosis Related Group.
DISCUSSION
The Department of Health and Human Services (DHHS) recently called for public comment on human subjects research protections (12). Our experience suggests that this request is timely. For a multisite trial of a health services intervention to improve the delivery of evidence-based care, the human subjects review process at 17 sites involved 115 submissions, consumed over 6,700 staff hours, and lasted almost two years longer than planned. The time to initial approval was shortest for the first submission, and the longest times occurred at the midpoint of the submission process, suggesting that the protracted approval process was not due to a “learning curve” on the part of the study team. This process greatly impacted the trial. First, changes required at one site necessitated a protocol modification to all sites to preserve the study’s internal validity. Second, the approval process had a profound financial impact on the project, costing close to $170,000 in staff salaries. Third, delays in approval affected participant recruitment and retention; seven physician participants had left their primary care setting before all approvals were received. Finally, requirements for local site PIs and for IRB and R&D committee approvals effectively resulted in the inclusion of more highly-affiliated, urban sites that were treating more complex patients, potentially affecting the external validity (generalizability) of the study findings.
All 14 IRBs that approved the study and provided risk determinations classified the study as “minimal risk”, making the time and costs involved in the review process seem quite incongruous, especially when compared to those involved within other research disciplines. For example, genomewide association studies routinely use more than 100,000 single-nucleotide polymorphisms to genotype individuals, yet an individual can be uniquely identified with access to fewer than 100 single-nucleotide polymorphisms (17). Surprisingly, according to the Office for Human Research Protections, these data are not considered identifiable, and no IRB oversight or informed consent is mandated, nor does the Health Insurance Portability and Accountability Act necessarily provide protection for participants (17).
To our knowledge, this is the first health services research study to examine the IRB process for a randomized controlled trial of an intervention to improve the provision of evidence-based care, and the first to quantitatively evaluate the impact of human subjects requirements on the external validity of study findings. We conducted a PubMed search of empirical studies of the IRB process in the implementation of multisite studies in the US. Other studies also have documented marked site-to-site variation in the time from submission to approval (2, 3, 6, 8, 18–20), in the number of resubmissions required (3, 6, 19), and in IRB review decisions (2, 3, 6, 8, 11, 18–21). Two studies also estimated the costs involved; one cited $17,000 spent on coordinating center personnel, space and supplies for an 8-site study (21), and a 14-site study estimated that staff salary spent on the IRB process cost over $53,000 within the first year (8). We found that staff salaries involved in securing IRB approvals for this study, a process that took more than 27 months, amounted to almost $170,000. Several prior studies also have cited concerns about the generalizability of their research due to changes mandated by the IRB. In one survey of patients about how to improve health care quality, opt-in and opt-out procedures imposed by several IRBs resulted in a loss of up to 37% of potential patients (2). Authors noted that such hurdles to participation may have affected minority and low-income patients disproportionately (2). In another study, changes imposed by the IRB resulted in a protocol that was not translatable into clinical practice; asked the authors, “Is it ethical to involve humans in research if the research is not likely to yield valid answers to the proposed research questions?” (22). They noted how the implied social contract between researchers and society is to ensure that research has the greatest impact possible by making the study as generalizable to clinical practice as possible (22). In our study, the 12 sites that ultimately participated in the study were more likely to be urban, academically affiliated, and care for more complex patients than excluded sites. External validity is of concern because of the expectation that health services and comparative effectiveness research will yield findings that are directly implementable and translatable into improvements in patient care (23). While some sites were excluded due to their inability to fulfill requirements for IRB review; others were excluded because the IRB and R&D committee either disapproved the study or provided conflicting rulings. When multisite studies receive very different IRB determinations as we experienced here, regulations do not provide clear guidance about how to resolve conflicts (10).
Much of the variations in IRB processes are due to the system of local review whereby a multisite study has to be reviewed by local IRBs to ensure that the protocol addresses any problems that might arise from local contexts (24). Some variation in review may be appropriate due to local values in assessing human subjects’ risks and benefits. However, many of the revisions requested by local IRBs, when compared to what was approved by the IRB of a multisite study’s coordinating center, have been shown to add little in the way of essential local subject protections and usually make little if any substantive changes to the study protocol (21, 24). Our experience confirms this finding. One underlying issue responsible for the type of local variation we experienced is that IRBs do not appear to agree upon the limits of their sphere of, and do not confine themselves to reviewing the ethical issues relating to, human research protections (25). For example, one IRB required that we provide documentation of union approval and then asked whether we were providing any incentives to the institution itself.
Several authors have made recommendations for easing the burden of IRB review in multisite studies. These suggestions include increasing standardization of the review process across IRBs (1, 26); centralized IRB review in which the coordinating center’s local IRB or an independent IRB reviews the protocol and takes responsibility for human subjects protections for all sites (24, 27); and the use of a single, central IRB (3, 4, 7, 8, 21, 24). While a single, central IRB may appear to be a logical answer to standardizing reviews, one study estimated that the cost of running the National Cancer Institute’s central IRB was greater than the amount of money it saved (28). Additionally, one study suggested several methods for streamlining the IRB process, including creating model IRB applications, starting the process and communication with the IRB early, maintaining that communication throughout the study, and being prepared for changes during the IRB application process (29). Although the authors of this study met their timeline, they noted that several of their procedures may have placed undue burden on the sites in their study and the sites may have unduly influenced their local IRBs (29).
Several limitations of our study need to be addressed. First, the VA health care system employs a two-stage review process. Research must be approved by both the local IRB and the local VA R&D committee prior to implementation. Second, the trial tested a novel intervention, providing financial incentives for high quality care. Although non-invasive, the lack of a precedent may have prompted regulatory boards to err on the side of caution in granting approval. Finally, the data from this study are derived from a single multicenter trial involving regulatory submissions to only 17 different sites.
Our study shows that obstacles presented by the IRB review process exist even for a minimal risk health services research study employing a randomized controlled design. IRB rulings that affect study design can threaten the internal validity of a study, and the barriers to obtaining IRB approval may favor studies taking place at highly selected sites that do not necessarily reflect health care delivery in the majority of the US. Furthermore, the Office for Human Research Protections regulations do not appear consistent with the nature of minimal risk studies (10). An overall review of the standards for research as planned by DHHS appears welcome.
ACKNOWLEDGEMENT
The authors thank Eric J. Thomas, MD, MPH, for his comments on an earlier version of the paper.
Funding/Support
The Veterans Affairs Health Services Research & Development (HSR&D) Investigator-Initiated Research (IIR) 04-349 (PI Laura A. Petersen, MD, MPH), National Institutes of Health (NIH) R01 HL079173-01 (PI Laura A. Petersen, MD, MPH), and Houston VA HSR&D Center of Excellence HFP90-020 (PI Laura A. Petersen, MD, MPH), funded the design and conduct of the study; the collection, management, analysis, and interpretation of the data; and the preparation of this manuscript.
Footnotes
Significant Author Contributions
Laura A. Petersen, MD, MPH, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Conception and design: Petersen
Acquisition of data: Petersen, Simpson, SoRelle, Urech
Analysis and interpretation of data: Chitwood, Petersen, Simpson
Drafting of the manuscript: Chitwood, Petersen, Simpson, SoRelle
Critical revision of the manuscript for important intellectual content: Chitwood, Petersen, Simpson, Urech
Statistical analysis: Simpson
Obtaining funding: Petersen
Administrative, technical, or material support: Chitwood, Petersen, Simpson, Urech
Supervision: Petersen
Potential Conflicts of Interest
There are no conflicts of interest to disclose.
References
- 1.Hirshon JM, Krugman SD, Witting MD, Furuno JP, Limcangco R, Perisse AR, et al. Variability in institutional review board assessment of minimal-risk research. Acad Emerg Med. 2002;9(12):1417–1420. doi: 10.1111/j.1553-2712.2002.tb01612.x. [DOI] [PubMed] [Google Scholar]
- 2.Dziak K, Anderson R, Sevick MA, Weisman CS, Levine DW, Scholle SH. Variations among institutional review board reviews in a multisite health services research study. Health Serv Res. 2005;40(1):279–290. doi: 10.1111/j.1475-6773.2005.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green LA, Lowery JC, Kowalski CP, Wyszewianski L. Impact of institutional review board practice variation on observational health services research. Health Serv Res. 2006;41(1):214–230. doi: 10.1111/j.1475-6773.2005.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stark AR, Tyson JE, Hibberd PL. Variation among institutional review boards in evaluating the design of a multicenter randomized trial. J Perinatol. 2010;30(3):163–169. doi: 10.1038/jp.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman H, Hull SC, Sugarman J. Variability among institutional review boards’ decisions within the context of a multicenter trial. Crit Care Med. 2001;29(3):235–241. doi: 10.1097/00003246-200102000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stair TO, Reed CR, Radeos MS, Koski G, Camargo CA MARC (Multicenter Airway Research Collaboration) Investigators. Variation in institutional review board responses to a standard protocol for a multicenter clinical trial. Acad Emerg Med. 2001;8(6):636–641. doi: 10.1111/j.1553-2712.2001.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 7.Helfand BT, Mongiu AK, Roehrborn CG, Donnell RF, Bruskewitz R, Kaplan SA, et al. MIST Investigators. Variation in institutional review board responses to a standard protocol for a multicenter randomized controlled surgical trial. J Urol. 2009;181(6):2674–2679. doi: 10.1016/j.juro.2009.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vick CC, Finan KR, Kiefe C, Neumayer L, Hawm MT. Variation in institutional review processes for a multisite observational study. Am J Surg. 2005;190(5):805–809. doi: 10.1016/j.amjsurg.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Pronovost P, Needham D, Berenholtz S, Sinopoli D, Chu H, Cosgrove S, et al. An intervention to decrease catheter-related bloodstream infections in the ICU. N Engl J Med. 2006;355(26):2725–2732. doi: 10.1056/NEJMoa061115. [DOI] [PubMed] [Google Scholar]
- 10.Baily MA. Harming through protection? N Engl J Med. 2008;358(8):768–769. doi: 10.1056/NEJMp0800372. [DOI] [PubMed] [Google Scholar]
- 11.Silberman G, Kahn KL. Burdens on research imposed by institutional review boards: the state of the evidence and its implications for regulatory reform. Milbank Q. 2011;89(4):599–627. doi: 10.1111/j.1468-0009.2011.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emanuel EJ, Menikoff J. Reforming the regulations governing research with human subjects. N Engl J Med. 2011;365(12):1145–1150. doi: 10.1056/NEJMsb1106942. [DOI] [PubMed] [Google Scholar]
- 13.Petersen LA, Woodard LD, Urech T, Daw C, Sookanan S. Does pay-for-performance improve the quality of health care? Ann Intern Med. 2006;145(4):265–272. doi: 10.7326/0003-4819-145-4-200608150-00006. [DOI] [PubMed] [Google Scholar]
- 14.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr, et al. National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 15.Petersen LA, Urech T, Simpson K, Pietz K, Hysong SJ, Profit J, et al. Design, rationale, and baseline characteristics of a cluster randomized controlled trial of pay for performance for hypertension treatment: study protocol. ImplementSci. 2011;6:114. doi: 10.1186/1748-5908-6-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrne MM, Daw CN, Nelson HA, Urech TH, Pietz K, Petersen LA. Method to develop health care peer groups for quality and financial comparisons across hospitals. Health Serv Res. 2009;44(2 Pt 1):577–592. doi: 10.1111/j.1475-6773.2008.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuire AL, Gibbs RA. Genetics. No longer de-identified. Science. 2006;312(5772):370–371. doi: 10.1126/science.1125339. [DOI] [PubMed] [Google Scholar]
- 18.Greene SM, Geiger AM. A review finds that multicenter studies face substantial challenges but strategies exist to achieve Institutional Review Board approval. J Clin Epidemiol. 2006;59(8):784–790. doi: 10.1016/j.jclinepi.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 19.Greene SM, Geiger AM, Harris EL, Altschuler A, Nekhlyudov L, Barton MB, et al. Impact of IRB requirements on a multicenter survey of prophylactic mastectomy outcomes. Ann Epidemiol. 2006;16(4):275–278. doi: 10.1016/j.annepidem.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Thompson DA, Kass N, Holzmueller C, Marsteller JA, Martinez EA, Gurses AP, et al. Variation in local Institutional Review Board Evaluations of a Multicenter Patient Safety Study. J Healthc Qual. 2011 May 25; doi: 10.1111/j.1945-1474.2011.00150.x. Epub. [DOI] [PubMed] [Google Scholar]
- 21.Humphreys K, Trafton J, Wagner TH. The cost of institutional review board procedures in multicenter observational research. Ann Intern Med. 2003;139(1):77. doi: 10.7326/0003-4819-139-1-200307010-00021. [DOI] [PubMed] [Google Scholar]
- 22.Gittner LS, et al. Health service research: the square peg in human subjects protection regulations. J Med Ethics. 2011;37:118–122. doi: 10.1136/jme.2010.037226. [DOI] [PubMed] [Google Scholar]
- 23.Naik AD, Petersen LA. The neglected purpose of comparative effectiveness research. N Engl J Med. 2009 May 7;360(19):1929–1931. doi: 10.1056/NEJMp0902195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravina B, Deuel L, Siderowf A, Dorsey ER. Local institutional review board (IRB) review of a multicenter trial: local costs without local context. Ann Neurol. 2010;67(2):258–260. doi: 10.1002/ana.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angell EL, Bryman A, Ashcroft RE, Dixon-Woods M. An analysis of decision letters by research ethics committees: the ethics/scientific quality boundary examined. Qual Saf Health Care. 2008;17(2):131–136. doi: 10.1136/qshc.2007.022756. [DOI] [PubMed] [Google Scholar]
- 26.Larson E, Bratts T, Zwanziger J, Stone P. A survey of IRB process in 68 U.S. hospitals. J Nurs Scholarsh. 2004;36(3):260–264. doi: 10.1111/j.1547-5069.2004.04047.x. [DOI] [PubMed] [Google Scholar]
- 27.Pogorzelska M, Stone PW, Cohn EG, Larson E. Changes in the institutional review board submission process for multicenter research over 6 years. Nurs Outlook. 2010;58(4):181–187. doi: 10.1016/j.outlook.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner TH, Murray C, Goldberg J, Adler JM, Abrams J. Costs and benefits of the national cancer institute central institutional review board. J Clin Oncol. 2010;28(4):662–666. doi: 10.1200/JCO.2009.23.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blustein J, Regenstein M, Siegel B, Billings J. Notes from the field: jumpstarting the IRB approval process in multicenter studies. Health Serv Res. 2007 Aug;42(4):1773–1782. doi: 10.1111/j.1475-6773.2006.00687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]