

SUMMARY
Photoswitchable fluorescent proteins (PSFPs) that change their color in response to light have led to breakthroughs in studying static cells. However, using PSFPs to study cells in dynamic conditions is challenging. Here we introduce a method for in vivo ultrafast photoswitching of PSFPs that provides labeling and tracking of single circulating cells. Using in vivo multicolor flow cytometry, this method demonstrated the capability for studying recirculation, migration, and distribution of circulating tumor cells (CTCs) during metastasis progression. In tumor-bearing mice, it enabled monitoring of real-time dynamics of CTCs released from primary tumor, identifying dormant cells, and imaging of CTCs colonizing a primary tumor (self-seeding) or existing metastasis (reseeding). Integration of genetically encoded PSFPs, fast photoswitching, flow cytometry, and imaging makes in vivo single cell analysis in the circulation feasible to provide insights into the behavior of CTCs and potentially immune-related and bacterial cells in circulation.
INTRODUCTION
Most cancer deaths are related to metastases in distant organs due to disease dissemination by circulating tumor cells (CTCs) shed from the primary tumor (Chaffer and Weinberg, 2011; Christofori, 2006; Lazebnik, 2010; Fidler, 2003; Talmadge and Fidler, 2010). Detection of CTCs appears to be a marker of metastasis development, cancer recurrence, and therapy efficacy (Alix-Panabières et al., 2012; Hayes and Smerage, 2010; Attard and de Bono, 2011; Balic et al., 2013). Although substantial efforts have been made to develop new methods for studying CTCs in vitro and recently in vivo (Alix-Panabières et al., 2012; Hayes and Smerage, 2010; Attard and de Bono, 2011; Balic et al., 2013; Georgakoudi et al., 2004; He et al., 2007; Galanzha et al., 2009; Hwu et al., 2011; Yu et al., 2011), many aspects of CTC dissemination, recirculation, migration, and final destination (e.g., dormancy and self-seeding) remain poorly known (Alix-Panabières et al., 2012; Attard and de Bono, 2011; Wicha and Hayes, 2011). For example, it is not clear how long spontaneous CTCs (i.e., naturally shed from a primary tumor or metastasis) linger in circulation (referred to as CTC lifespan); how their lifespan depends on their biochemical, molecular, and genetic properties; or how their lifespan correlates with metastasis progression. Answers to these and many other questions require labeling single cells in the circulation to track their fate over a long period. Despite its importance, this task cannot be accomplished by means of existing imaging techniques.
In particular, the use of genetically encoded fluorescent proteins, such as green fluorescent protein (GFP), depicts all cells expressing this protein, in particular bulk CTCs (Georgakoudi et al., 2004). More specific molecular targeting, involving exogenous labels bioconjugated with antibodies against a cell-surface marker, can identify a specific subpopulation among bulk CTCs (e.g., stem CTCs), but once inside the bloodstream, the bioconjugated labels can target many cells with the same marker (He et al., 2007; Galanzha et al., 2009; Pitsillides et al., 2011). To label and track individual cells and ultimately a single cell in vivo, attention needs to be paid to new imaging and labeling strategies.
Among many imaging agents, genetically encoded, photoswitchable (called also photoconvertible) fluorescent proteins (PSFPs) with controllable spectral shifts in excitation and emission in response to light offer a solution to this problem because PSFPs are able to create unique cellular spectral signatures (Kedrin et al., 2008; McKinney et al., 2009; Subach et al., 2011, 2012; Lombardo et al., 2012).
Applications of PSFPs such as green-to-red Dendra2 (Kedrin et al., 2008), green-to-red mEos2 (McKinney et al., 2009), orange-to-far-red PSmOrange (Subach et al., 2011), and orange-to-far-red PSmOrange2 (Subach et al., 2012) have already led to breakthroughs in the study of cell biology in vitro. In addition, we have demonstrated the promise of PSFPs for monitoring primary tumors in vivo (Kedrin et al., 2008). However, to our knowledge, PSFPs have not been used to detect CTCs because fast moving cells in vivo represent the most challenging target for labeling and photoswitching. In particular, the high velocity of CTCs prevents conventional photoswitching of PSFPs (i.e., changing of their color), which typically takes 50- to 1,000-fold more time (e.g., 0.5–10 s) than the lifetime (e.g., 10–30 ms) of CTCs in the detection volume (Tuchin et al., 2011; Novak et al., 2004; Boutrus et al., 2007; Galanzha and Zharov, 2012; Markovic et al., 2013). Because photoswitching time clearly depends on laser power and laser exposure time (Subach et al., 2012), we suggest that photoswitching time can be reduced by increasing the laser power level, with the total energy deposition for the fast moving cells still remaining at a safe level because of their short lifetime in the irradiated volume. To minimize changes in surrounding static cells, the laser beam can be strongly focused into blood vessels (i.e., moving cells in blood flow) as we have previously reported (Galanzha and Zharov, 2012).
Whereas a cell is labeled in circulation, it should be tracked with a technique capable of monitoring millisecond-scale dynamic events in fast blood flow at the single cell level in vivo. Among a wide range of in vivo imaging and detection techniques, such as intravital microscopy, MRI, and positron emission tomography, in vivo flow cytometry (FC) offers an impressive example in which the blood vessels are used as natural tubes for detection, among the native cells in flow, of rare CTCs, bacteria, clots, and other abnormal objects (Georgakoudi et al., 2004; He et al., 2007; Galanzha et al., 2009; Pitsillides et al., 2011; Tuchin et al., 2011; Novak et al., 2004; Boutrus et al., 2007; Galanzha and Zharov, 2012; Nedosekin et al., 2013). Two basic detection platforms of FC—fluorescence (Georgakoudi et al., 2004; He et al., 2007; Hwu et al., 2011; Pitsillides et al., 2011; Novak et al., 2004; Boutrus et al., 2007) and photoacoustics (Galanzha et al., 2009; Tuchin et al., 2011; Galanzha and Zharov, 2012)— have been explored separately and, recently, together (Nedosekin et al., 2013) with the use of intrinsic (e.g., melanin, hemoglobin), genetically encoded (e.g., GFP), or exogenous (e.g., dyes or nanoparticles) labels. However, in vivo FC has never been used to detect and photoswitch PSFPs in circulating cells.
In view of the above considerations, the problem of ultrafast photoswitching could be solved by integrating in vivo FC with a laser to photoswitch PSFPs, a technique referred to below as a “photoswitchable” flow cytometry (PFC).
In this report, we introduce ultrafast photoswitching of genetically encoded fluorescent proteins in individual circulating cells and tracking of these cells in a whole organism in vivo. Using CTCs, we demonstrate the capability of PFC for labeling a controlled number of CTCs, potentially one cell, in blood flow and tracking them over time, as well as for studying the lifespan, recirculation, migration, and final destination of individual CTCs.
RESULTS
In Vivo Time-Resolved Multicolor PFC Platform
In PFC (Figure 1A), three continuous-wave lasers emit parallel linear (~10 μm × 80 μm) beams that are placed across the blood flow, completely covering a vessel's cross-section and not overlapping with each other (~30 μm gaps). This setup allows sequentially implementing four diagnostic procedures: (1) identifying each nonphotoswitched CTC moving through the vessel cross-section, (2) photoswitching (i.e., photolabeling) of this cell, (3) detecting the photolabeled cell to control photoswitching efficacy, and (4) tracking the photolabeled cell over time. Specifically for CTCs genetically encoded with the Dendra2 PSFP, the first laser at a wavelength of 488 nm excites the green fluorescence of Dendra2 (excitation, 488 nm; emission, 507 nm; Kedrin et al., 2008; Figure 1B, top); the second laser at 405 nm induces an irreversible shift in Dendra2′s excitation and emission spectra >150 nm (i.e., photoswitching) so that the PSFP emits a red fluorescence upon excitation (Figure 1B, middle); and the third laser at 540 nm excites the photoswitched PSFP's red fluorescence (excitation, 540 nm; emission, 560 nm, Kedrin et al., 2008; Figure 1B, bottom). To monitor and control these events, the fluorescence from each cell is collected, detected, and reproduced on a computer monitor as real-time signal traces in two channels: green and red (Figure 1A, right, top). Thus, once the cell is identified in the first (green) channel by a high-amplitude signal (Figure 1C, top), it is immediately photolabeled, and the change in its fluorescent color is detected when the signal appears in the second (red) channel (Figure 1C, bottom).
Figure 1. Principles of Multicolor PFC.

(A) Schematic of the setup, together with signal traces in the green and red channels before, during, and after photoswitching (upper right inset) and a diagram of the alignment of the laser beams on the sample (lower right inset). CL, cylindrical lens; DM, dichroic mirror; F, bandpass filter; AL, achromatic lens; S, mechanical slit; PMT, photo-multiplier tube, and PC, a computer.
(B and C) The underlying principle (B) and PFC traces (C) of photoswitching and detection are depicted.
The monitoring of multicolor signals also allows us to estimate photolabeling efficacy and identify individual spectral signatures (color fingerprints) of CTCs. Specifically, photoswitching (i.e., labeling) efficacy is estimated as the ratio of signal amplitudes in the green and red channels before and after photoswitching. If almost the entire amount of intracellular PSFP is photoswitched (effective photolabeling) in a cell, the signal amplitudes before (green channel) and after (red channel) photoswitching can have similar values after the proper green and red channel's sensitivities are adjusted. Furthermore, the signal amplitude from a PSFP-labeled cell reflects the cell's fluorescence intensity attributable to the amount of intracellular PSFP that depends on an individual feature of a cell to express PSFP and, thus, can be used to distinguish one CTC from another. Taken together, the specific values of green signal amplitude before photoswitching, red signal amplitude after photoswitching, and their ratio at specific setup parameters represent an individual spectral/color signature (fingerprint) for each CTC labeled by PSFP and can be used to recognize individual CTCs in vivo. In addition, the schematic with three nonoverlapping linear laser beams provides the opportunity to measure the velocity (ν) and size of labeled objects (e.g., CTCs). Specifically, the temporal width of the signal, which depends on the time (Δt1 Figure 1C) it takes an object with certain diameter to pass the laser beam (Georgakoudi et al., 2004; Galanzha and Zharov, 2012), was typically used to measure its size and thus distinguish single CTCs (14–30 μm; Table S1 available online) from their aggregates (>30 μm) and microparticles (<5 μm). To estimate velocity, we measured the delay between signals in the green and red channels (Δt; Figure 1C) related to the time that it took a cell to travel the distance Δd (Figures 1B and 1C) from the centerline of the first laser (488 nm) to the centerline of the third laser (540 nm). In the last case, the velocity is calculated as ν = Δd/Δt. In our experiments, most labeled CTCs had a velocity in the range of 0.7–3 mm/s, which is in line with data in the literature for circulation in the mouse ear (Georgakoudi et al., 2004).
Selection of the PSFP for CTC Labeling
We compared three PSFPs, Dendra2, PSmOrange, and PSmOrange2, using laser and mercury lamp light at wavelengths of 405 nm and 488 nm to photoswitch Dendra2 and PSmOranges, respectively. We determined that Dendra2 had the fastest, most efficient photoswitching (Figure S1), which is in line with previously reported results in vitro (Subach et al., 2011). Because the kinetics and the degree of photoswitching of both PSmOranges are highly dependent on the concentration of oxidants in its environment (in the cuvette if in vitro or in the cytoplasm if in cells) and the total concentration of intracellular oxidants typically does not exceed 250 μM, which is suboptimal for PSmOrange photoconversion, the PSmOrange photoswitching in cells is rather inefficient (Subach et al., 2011). Thus, Dendra2 was chosen for further studies.
Characterization and Photoswitching of Tumor Cells Expressing Dendra2
Using highly metastatic carcinoma cells (MTLn3 adenocarcinoma) expressing Dendra2 (Dendra2-MTLn3 cells), we demonstrated that (1) culturing of Dendra2-MTLn3 cells do not cause loss of fluorescence after many passages, and disappearance of fluorescence is observed only in dead cells (Figure S2A); (2) Dendra2-MTLn3 cells growing in an incubator (Figure S2B, left) and CTCs extracted from a mouse (Figure S2B, right) 21 days after inoculation with Dendra2-MTLn3 cells have similar brightness that confirms stable expression of Dendra2 by the cancer cells during proliferation in vivo; and (3) photoswitched (i.e., red) Dendra2-MTLn3 cells growing in an incubator changed their color to green after 48–72 hr proliferation (Figures S2C and S2D).
Taking into account these features, as well as the well-known inability of moving cells to proliferate in the blood circulation, we assume that all photoswitched CTCs will retain their switched color (i.e., red for Dendra2) and can be distinguished from nonphotoswitched CTCs (i.e., green for Dendra2) until they are dead or extravasate and proliferate in tissues.
At the next step, the 405-nm laser parameters were optimized for photoswitching static Dendra2-MTLn3 cells in PBS (i.e., the almost ideal condition for photoswitching). Fluorescence imaging was used to quantify the changes in fluorescence. After photoswitching, we obtained up to 182-fold increase in red fluorescence, resulting in up to a 1,050-fold increase in the red/green ratio of fluorescence intensities without essential photobleaching or an effect on cell viability (Figure S3A and Table S2). Notably, the total fluorescence from the same cell measured in different focal planes slightly varied perhaps due to uneven intracellular distribution of Dendra2 (Figure S3B). From this, the repeatable detection of red fluorescence from the circulating cells that change their orientation and position in flow can be slightly different.
As expected, the presence of blood decreased both red and green fluorescence. Nevertheless, photoswitching of Dendra2-MTLn3 cells in the 120-μm thick sample of whole blood in static conditions (i.e., approximately two times thicker than the diameter of the blood vessels examined in the mouse ear) resulted in an ~230-fold increase in the red/green fluorescence ratio (Figures S3C and S3D).
To optimize dynamic photoswitching of moving cells (mimic CTCs), a slide with cells suspended in PBS and whole blood (sample thickness 120 μm) was scanned with a 405-nm linear laser beam at different velocities and laser power levels (Figure 2 and Figure S4). At beam velocities in the range of 1–3 mm/s (expected cell velocity in mouse ear vessels), optimal photoswitching was achieved at a laser power of 10–30 mW. In particular, the photoswitching of cells in PBS, moving at 1 mm/s at a laser power of 30 mW (power intensity at the sample 3 × 103 W/cm2), led to a 70-fold increase in the red/green fluorescence ratio (Figures S4A and S4B). The chosen parameters (e.g., laser power of ≤ 30mW) did not cause photobleaching because of the short cell-exposure time (a few milliseconds; Figure S4A). Photoswitching of moving cancer cells in a 120-μm thick layer of whole blood at the indicated above parameters increased the red/green fluorescence ratio 40-fold (Figure 2). Despite the predictable loss of photoswitching contrast against the background signals of blood, fluorescence microscopy clearly distinguished photoswitched from nonphotoswitched cells in the blood (Figure 2B). Based on these results, we can conclude that effective photoswitching can be achieved in selected animal models in blood vessels with a diameter ≤ 100–120 μm at a flow velocity up to 1–3 mm/s.
Figure 2. Photoswitching of Cancer Cells in Whole Blood In Vitro.
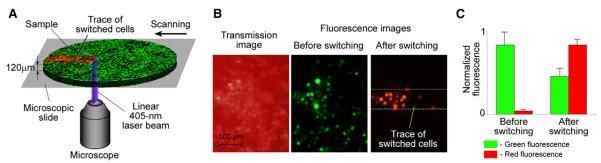
Schematic (A), images (B), and normalized fluorescence (C) in cancer cells moving with a velocity of 1 mm/s across a linear laser beam having a power of 30 mW. Error bars represent SEM; data were collected from 15 cells (C).
See also Figures S2, S3, and S4.
In Vivo Photoswitching Optimization
In healthy mice, we explored whether irradiation of 405-nm laser affects the functions of ear blood vessels. Based on the results above and previous data (Lombardo et al., 2012), we tested power level in the range of 20–40 mW, which is appropriate for photoswiching of Dendra2. From our observations, all vessels had good blood flow before and after photoswitching, and no mice exhibited blood flow stasis (Movie S1). Only one mouse showed a temporary ~50%–60% constriction of an irradiated vessel after a 10 min exposure to the 405-nm laser at a power of 38 mW. To reduce the risk of such effects in the experimental mice, we kept the laser power at ≤30 mW and carefully controlled blood flow stability with video recording. Then, we estimated background autofluorescence from blood irradiated by 488-nm and 540-nm lasers. In analogy to Lin's study (Georgakoudi et al., 2004; Boutrus et al., 2007), a signal-amplitude threshold in each channel was determined as the mean and an increment of the SD (typically five SDs) of the background signal. The intact ear skin without visible vessels provided a background autofluorescence-to-noise ratio ≤ 3. Variations in the autofluorescence background from mouse to mouse and day to day were not significant for vessels of the similar size, at least within the established signal threshold. Signals having a higher amplitude than this threshold were considered to be associated with CTCs.
Injection of nonphotoswitched (green) and photoswitched (red) Dendra2-MTLn3 cells (~2 × 105 cells per injection) showed that PFC detected CTCs with a signal-to-background noise ratio up to 15 (Figure S5). In addition, both, green and red fluorescent cells exhibited similar dynamic rates after injection, which allowed an estimation of PFC's sensitivity. Although the maximal number of CTCs per minute varied from mouse to mouse because of individual injection conditions and distribution of cells in the body, we could detect an average of up to 18 CTCs/min in vessels with a mean diameter of 52 ± 5 μm. This was close to the theoretical estimation of up to 24 CTCs/min in a 50-μm diameter vessel at a velocity of 2 mm/s after the introduction of 2 × 105 cells.
Labeling and Tracking of Single CTCs In Vivo
For proof-of-principle implementation, nonphotoswitched (green) cells were injected into mice and monitored with PFC (Figure 3A). When the rate of cells became rare and relatively stable at a level of 1–3 CTCs per minute (25–30 min after injection), we photoswitched single CTCs by turning on a 405-nm laser at the power of 30 mW (power intensity on the vessel 3 × 103 W/cm2). During photoswitching, blood flow and vessel diameter were monitored by video-recording (Movie S1), and CTC velocity was controlled as described above. All photoswitched CTCs had velocity ranging from 1 to 2 mm/s.
Figure 3. In Vivo Photolabeling and Tracking of Individual CTCs.

(A and B) Diagram of the experiment (A) and traces of three CTCs with different fingerprints (B).
(C) Effective photoswitching and fingerprint creation for CTC 3 (velocity, 1.7 mm/s).
(D) Recognition of CTC 3 based on its fingerprint.
Using a PFC platform, we were able to photolabel the desired number of CTCs, even just one cell. For example, Figure 3 (25 min after cell injection; 1.5 ± 0.7 green CTCs/5 min) shows the photoswitching of three CTCs. Each CTC created an individual fingerprint (purple rectangles in Figures 3B and 3C) that can be defined as the ratio of green signal amplitude before photoswitching to red signal amplitude after photoswitching. The value of red signal amplitude was used to later identify these cells in circulation (Figures 3B and 3D). Specifically, CTC 1 had a signal amplitude of 2.7 arbitrary units (a.u.) in the green channel before photoswitching and a signal amplitude of 2.5 a.u. in the red channel after photoswitching. This means that the green-to-red fluorescence signature (fingerprint) for CTC 1 was 2.7/2.5. Similar amplitude values before and after photoswitching (difference of ≤10%) allowed us to conclude that labeling was effective. CTCs 2 and 3 were also effectively labeled with fingerprints of 5.0/4.8 and 4.7/4.4, respectively. Continuous 90 min monitoring demonstrated that the CTC with a red amplitude of ~2.5 a.u. (i.e., CTC 1) did not repeat its appearance at the detection point, while CTCs 2 and 3 were monitored again at the point of detection within 30–40 min. Unexpectedly, the CTC with a red amplitude of 4.4 a.u. (i.e., likely CTC 3) appeared before the CTC with a red amplitude of 4.8 a.u. (i.e., likely CTC 2), suggesting that CTCs, like normal white blood cells, may use different trafficking pathways (Gowans and Steer, 1980; Halin et al., 2005).
Thus, we demonstrated the potential to fingerprint CTCs. However, in some cases, the value of the CTC signal amplitude in the red channel was not exactly the same at the repeated crossing of the detection laser as it was when measured after photoswitching. This presented a challenge. Based on our in vitro tests (Figure S3B) and previous results (Novak et al., 2004), this finding may be due to a change in the axial location and/or orientation of the moving cell (Novak et al., 2004). Although the differences in amplitude in our experiments did not exceed 5%–10%, this issue requires additional study before fingerprinting can be routinely used in experimental studies. Additionally, although this pilot study demonstrated the labeling of single CTCs after intracardiac injection, we did not see any difficulties in using PFC for tumor-bearing mice. Thus, this method would help to answer the question of how the lifespan of spontaneous CTCs correlates with their metastatic potential and the progression of metastatic disease.
Real-Time Shedding of CTCs from Primary Tumor
To test the capability of fast photoswitching and PFC for advanced study of metastatic disease and naturally shedding (i.e., spontaneous) CTCs, we first developed a mouse ear model of metastatic carcinoma (Figure 4 and Figure S6). This model offers a few advantages. First, the relatively isolated location of the primary tumor from other parts of the body allows successful laser photoswitching without undesirable irradiation of other organs with possible metastasis (e.g., lymph nodes, brain, liver, lungs). Second, the thin (~250 μm), relatively transparent ear structure allows noninvasive optical imaging (Figures 4A and 4B) to control tumor development and photoswitching. Third, CTCs appear early, starting from day 5 after tumor inoculation, disseminate by both blood and lymphatic pathways, and the development of multiple metastases follows (Figure 4C, Figures S6A–S6F, and Supplemental Experimental Procedures). With this model in hand, we define the in vivo process by which tumor cells are released from a primary tumor into blood circulation. We photoswitched a primary tumor (Figure 5A) in four mice and performed real-time monitoring of CTCs in the blood circulation before, during, and after photoswitching. The efficiency of ear-tumor switching was easily controlled with fluorescence imaging (Figure 5A, bottom). As expected, before photoswitching of the primary tumor, all CTCs were green (i.e., only green signals were detected with PFC; Figure 5B). The green-to-red photoswitching of the primary tumor caused the appearance of red signals in the PFC traces (Figure 5B, bottom) that were associated with “new” CTCs just shed from the primary tumor.
Figure 4. In Vivo Monitoring of Primary Tumor and CTCs in Mice with Metastatic Carcinoma.

(A) Photo of the ear tumor and combined fluorescence-transmission microscopic image of the tumor area of interest (inset).
(B) Imaging of intratumor vessels and intravascular rolling of a CTC (inset).
(C) CTC kinetics in mice bearing ear tumors (n = 7). In vivo measurements at the final point of tumor development were verified by fluorescent imaging of blood samples in vitro (inset). Error bars represent SEM.
See also Figure S6.
Figure 5. Tracking CTCs Spontaneously Shedding from the Primary Tumor.
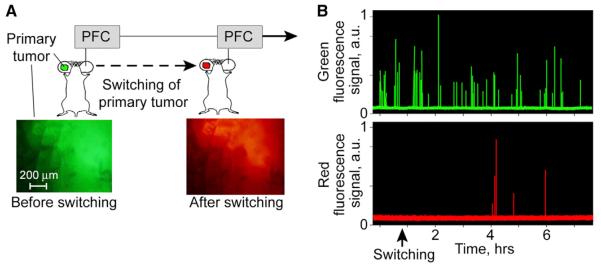
(A) Workflow diagram for studying the real-time dynamics of the release of CTCs from a primary tumor after it is photoswitched.
(B) Green and red fluorescence traces recorded in a 56-μm diameter vein shows dynamic of CTC shedding from a primary tumor. Green signals correspond to nonswitched (“old”) CTCs, while the red signals indicate photoswitched (“new”) CTCs.
We found that an advanced carcinoma was able to release 3–7 CTCs per 4–6 hr and the process of CTC shedding was characterized by temporal irregularity. As shown in Figure 5B, for example, the first “new” CTCs (red) were detected only at ~180 min after photoswitching, and three of them appeared almost simultaneously. Notably, this triplet of “new” CTCs was recognized by PFC as single cells not as aggregates. Later, at ~50 and ~120 min after the first group, we monitored the next two red CTCs. Thus, we monitored in real time the dynamic process of the appearance of CTCs in circulation after their spontaneous shedding from primary tumor.
Dormancy, Self-Seeding, and Reseeding
Finally, we explored whether PFC can study dormancy (colonization of metastatic sites by CTCs without proliferation) and self-seeding (return of CTCs to the primary tumor; Kim et al., 2009; Langley and Fidler, 2007; Townson and Chambers, 2006; Klein, 2011). In mice with ear tumors (Figure 6A), we photoswitched CTCs in a tail vein with a 405-nm laser. The reason for using a tail vein is to exclude irradiation (i.e., undesired photoswitching) of both the primary tumor and metastases throughout the body. Despite stronger scattering of laser light in the tail skin and higher blood velocity in tail vessels, as compared to the ear model, the PFC, which followed the photoswitching procedure, revealed 44% ± 5% of signals in the red channel (Figure 6B), indicating that approximately half of bulk CTCs were photolabeled. In some cases, photoswitching was incomplete, and CTCs emitted signals in the red channels (white arrows in Figure 6B) with two to three times lower amplitude than in the green channel before photoswitching. Nevertheless, the number of labeled CTCs was sufficient to distinguish these cells after they were extravasated into intact tissues, primary tumor, or existing metastases (Figures 6C–6E). For example, the cells that had 15–70 times higher red fluorescence than autofluorescence of liver, brain, and lymph nodes and did not proliferate for up to 9 days were associated with dormant cells (Figure 6C and Figures S6G and S6H). We also imaged rare red fluorescent cells within green tissue of a primary tumor that were associated with self-seeding of the primary tumor (Figure 6D). Self-seeding CTCs exhibited up to 52-fold increased red fluorescence compared to the surrounding tumor mass. The 15–30 μm diameter of red cells additionally confirmed their relation to cancer cells (Table S1). Using the same methodology, we unexpectedly identified red fluorescent cells inside a green metastasis (Figure 6E), providing direct evidence of the colonization of the existing metastatic sites by CTCs (reseeding phenomenon).
Figure 6. Dormacy, Self-Seeding, and Reseeding.

(A and B) Workflow of photoswitching of CTCs in a tail vein (A) and monitoring CTCs in the green and red channels (B); the white arrows indicate the signals from a partly switched CTC.
(C) Combined, transmission and fluorescence, image of the dormant cell in liver (day 9 after CTC photoswitching); the red fluorescence of the dormant cell is 18-fold higher than the autofluorescence of an intact liver.
(D and E) Combined images from the green and red fluorescence channels of self-seeding (D) and reseeding (E) cells with a 48-fold and 77-fold, respectively, increase of red fluorescence compared to a green primary tumor or existing metastasis; visible vessels indicated by white dotted lines.
See also Figures S2 and S6.
DISCUSSION
We developed a method for labeling a controlled number of circulating cells (e.g., CTCs) and, potentially even one cell, through ultrafast photoswitching (10–50 ms [PFC] versus 1–10 s [existing methods]) directly in the bloodstream in vivo for the purpose of tracking these labeled cells within whole animals; this capability is beyond the scope of current labeling and detection methods.
Our method is based on a technological platform termed PFC and in vivo ultrafast photoswitching of circulating cells genetically encoded with PSFPs. Notably, circulating cells in vivo represent the most challenging target for photoswitching because multiple undesirable factors cannot be excluded, such as light scattering in skin, autofluorescence of the blood background, and the high velocity of moving cells. Nevertheless, in studies in vivo, we demonstrated sufficient sensitivity and specificity of our method to effectively label a single CTC and successfully recognized the labeled cell from nonphotoswitched PSFP-expressing cells, as well as from other cells in circulation.
Among different PSFPs and various biomedical applications of PFC, we used carcinoma cells with Dendra2 PSFP to demonstrate the capability of this method to enhance CTC-related research. Subsequent in vitro and in vivo experiments allowed us to estimate the optimal parameters for lasers and vessels (e.g., laser energy, range of vessel diameters, and flow velocity) for effective photoswitching of Dendra2 in cancer cells against the background of blood. To minimize possible photodamage of surrounding tissues, the laser beams were strongly focused into blood flow to ensure a short thermal relaxation time and hence more effective cooling of the heated localized zone.
As a result, we developed a protocol for implementing four procedures: (1) identifying each nonphotoswitched Dendra2-CTC crossing the first laser beam, (2) photoswitching (i.e., photolabeling) it while it moves through the second laser beam, (3) detecting the photolabeled CTC to control photoswitching efficacy when it crosses the third laser beam, and (4) tracking the labeled (red fluorescent) CTC(s) over time. Thus, the key advantage of PFC is its ability to photolabel a single cell to create a unique light-activated fluorescent signature for that cell while it circulates within a whole organism in vivo. This allows CTC behavior to be continuously tracked in its natural environment under the complex regulation of a whole living organism. In addition, PFC can yield information on the velocity and number of CTCs, as well as information on the size of labeled objects that allows us to distinguish single CTCs from their aggregates and fragments (e.g., microparticles).
In tests in vitro, we ascertained that (1) CTCs with Dendra2 retain bright fluorescence for many days in vivo if they are not dead, and (2) photoswitched (red) CTCs can become green only when they proliferate. From these results, we concluded that (1) individual viable CTCs switched in flow can be distinguished from bulk CTCs, until they leave the circulation; and (2) photoswitched CTCs that extravasate into tissue and do not proliferate will retain their switched color (i.e., red for Dendra2). These features of the cells with PSFPs represent an advanced way to track CTCs and study their shedding, recirculation, migration, and destination.
For the first time, we showed that some CTCs (not all) may recirculate quickly and repeatedly appeared at the detection point within 30 min. Such a scenario is possible, given that CTCs move relatively slowly at a velocity of a few millimeters per second in small blood vessels, and, then, their motion is dramatically accelerated in central circulation to achieve velocity of ~23 cm/s in the mouse aorta at a cardiac output of 14–15 ml/min (Hartley et al., 1995; Hedrich, 2004). We cannot exclude that CTCs, like lymphocytes (Gowans and Steer, 1980; Halin et al., 2005), may recirculate locally using the bridges between the arterial and venous systems (arteriolovenular anastomoses; Schmidt and Carmeliet, 2010) and/or a network of lymphatic vessels. The latter pathway is quite possible because we demonstrated the ability of carcinoma CTCs to disseminate via peripheral lymph vessels (Figure S6F). Our future studies will be directed to understand the process and purpose of CTC recirculation for the development of metastatic disease.
PFC has considerable potential to measure the lifespan of spontaneous CTCs and define correlations between CTC lifespan and metastatic progression. This, for example, would help clarify whether a decrease in the number of CTCs observed during disease progression is the result of a shortened lifespan with accelerated extravasation into target organs or shedding from the primary tumor at a lower rate. Such knowledge would be important for judging clinical prognosis because these mechanisms are related to alternative scenarios of disease development: the first by possibly facilitating the metastasis progression, and the second by possibly indicating regression of this process. If CTC lifespan strongly correlates with metastatic progression, then the lifespan of CTCs can be used as a biomarker of disease prognosis. Hypothetically, in line with the concept of parallel progression of primary tumor and metastasis (Klein, 2009), we expect to find shorter lifespans for CTCs during the initial stages of disease when CTCs actively colonize new metastatic sites.
Our results demonstrated that PSFPs and PFC are useful for uncovering new knowledge on the real-time dynamics of CTC shedding from primary tumors. Using PFC with the photoswitching of a primary tumor, we observed that an advanced carcinoma sheds CTCs at the relatively low rate of a few cells per 4–6 hr. Although it is currently too early to make conclusions about the generality of this phenomenon, our results demonstrated a methodological solution to studying the dynamics of CTC renewal that might be applied to various tumors at different stages. We also found that the rate of shedding CTCs varied over time. These temporal fluctuations seem to be significant for CTC diagnosis in patients because, for instance, they can explain false-negative results of a one-time snapshot test. Our recent clinical studies indirectly confirmed the dynamic fluctuation of CTCs in circulation (Juratli et al., 2014). In three blood samples taken at different times from the same patient with metastatic melanoma, we detected different numbers of CTCs. Moreover, these results are in line with previous data obtained in animals: different numbers of CTCs were found in blood samples taken at different times from the same mouse (Glaves et al., 1988).
Labeling of CTCs with PSFPs also offers a relatively simple approach to studying the phenomena of self-seeding at the single cell level. Moreover, our findings showed that some CTCs were able to colonize existing metastases. The role of such re-seeding is yet unclear and requires clarification in future studies.
PFC improves the quantification of bulk CTCs. Indeed, when researchers previously detected CTCs with in vivo flow cytometry (Georgakoudi et al., 2004; He et al., 2007; Galanzha et al., 2009; Hwu et al., 2011; Pitsillides et al., 2011), they could never be certain whether they had detected the same CTCs returning to the detection point or newly shed cells. PFC with PSFP-labeled cells can prevent repeated counting of CTCs by the switching of previously detected cells, thus providing more accurate quantification of CTCs.
The flexibility of the technical platform (e.g., easy replacement or addition of lasers) makes this approach universally applicable for detecting various PSFPs. We also expect a further leap forward when photolabeling of CTCs is combined with molecular labeling. Technically, this can be achieved by injection of nanoparticles and/or fluorescent dyes bioconjugated with CTC-specific biomarkers (e.g., antibodies) into the circulation and then detection of specific molecular profiles of photo-switched and nonphotoswitched CTCs with the use of combining fluorescence and photoacoustic FC in vivo that we recently developed (Nedosekin et al., 2013). Integrated labeling of CTCs with PSFPs and bioconjugated nanoparticles would clarify the in vivo interrelationships between CTC lifespan, molecular profiles, and metastasis progression. The molecular signatures of CTCs with different lifespans could be estimated and expected to be different. The molecular patterns of gene expression characterizing tumor-initiating properties, epithelialmesenchymal transition, and high migration activity will likely be found in mice models, in which the lifespan of CTCs is shorter. This approach will also define how the in vivo behavior of CTCs correlates with biochemical processes in them, including oxygenation and processes responsible for cell migration. For example, labeling of calcium and/or transient receptor potential channels that modulate intracellular calcium concentration and contribute to cancer cell migration (Fiorio Pla and Gkika, 2013) would instantly facilitate exploring the role of transient receptor potential channels in CTC shedding into the circulation.
A limitation of the current setup related to the incomplete photoswitching of CTCs in large vessels could be overcome by replacing the continuous-wave laser with a pulsed laser. Optimized pulse exposure triggering by signals from the first (green) channel would significantly improve photolabeling efficacy and decrease tissue photodamage. As shown in the three-beam schematic (Figure 1B), immediately after photoswitching cells can be detected using the red channel only, whereas their detection simultaneously in two (green-red) channels can be performed after they come back to detection point.
In general, PFC can be broadly applied for studying any solid metastatic tumor (e.g., breast, ovarian, prostate, or melanoma) and for determining tumor cell dissemination in hematologic malignancies (e.g., leukemia or multiple myeloma). Moreover, single cell photolabeling can uniquely trace the fate of any circulating cell or group of cells of interest in different animal models to discover and assess various physiologic and pathologic processes related to health and disease, including immune function, bacteremia, sepsis, and clotting. In the future, this knowledge could help in developing advanced diagnostic techniques and individualized therapy.
SIGNIFICANCE
Our work presents a technical approach for the analysis of single circulating cells in vivo that integrates photolabeling of individual cells directly in the bloodstream and tracking of the labeled cells over time. Specifically, our technology provides a noninvasive, ultrafast photolabeling of single circulating cells in vivo using genetically encoded photo-switchable fluorescent proteins (PSFPs), which change their fluorescent color (e.g., from green to red for Dendra2) in response to light irradiation. This approach has been shown to be capable of the efficient labeling and tracking single circulating tumor cells (CTCs) in tumor-bearing mice to study dynamic processes of the CTC recirculation, migration, distribution, and final localization.
EXPERIMENTAL PROCEDURES
Schematics of the Multicolor PFC Platform
The system (Figure 1A) is based on an Olympus IX81 inverted microscope (Olympus America) with incorporated three continuous-wave lasers: (1) wavelength, 488 nm; power, 1 mW at the sample (IQ1C45[488–60]G26, Power Technology); (2) 540 nm (He-Ne; power, 0.17 mW at the sample, Research Electro-Optics); and (3) 405 nm (continuous-wave diode); power, up to 40 mW at the sample (IQu6C225[405–500S]/8726, Power Technology).
As shown in Figure 1A, a cylindrical lens created a linear beam configuration measuring ~10 μm × 80 μm for each laser. A 40× objective (Olympus America) with a numerical aperture (NA) of 0.65 was used to focus light from each of the three lasers onto a mouse ear blood vessel with a depth of focus of ~60 μm, matching the vessel's diameter (50–60 μm). The same micro-objective collected the emitted fluorescence from each cell. To operate the three lasers (focusing, collecting fluorescence) with one objective, the laser beams were filled full NA of the objective and were displaced from the main microscope axis as indicated in Figure 1A (lower right inset). Collected fluorescence passed through bandpass filters (520 ± 15 nm for the 488-nm laser and 580 ± 15 nm for the 540-nm laser), achromatic lenses, and slits (200 μm × 3 mm) and was then detected with two photomultiplier tubes (PMTs; model R928, socket HC123-01; Hamamatsu) and recorded with a computer equipped with a high-speed (200 MHz) analog-to-digital converter board (PCI-5152, 12-bit card, 128-MB memory; National Instruments). PMT signals were continuously sampled at a rate of 4 MHz and down-sampled to a 10-kHz rate, with 400 points on average being sampled. The resultant traces were displayed on the computer monitor in real time (Figure 1A, upper right inset). Custom software (LabVIEW; National Instruments) was used for off-line processing of saved traces and calculating amplitudes, locations, and peak widths exceeding the background level.
For video-recording of blood flow in vivo, as well as imaging of tissue samples or cell suspensions ex vivo/in vitro and primary tumor in vivo, the microscope was equipped with a cooled color CCD camera (DP72, Olympus) and a black-and-white, high-speed, highly sensitive CCD camera (Cascade:512; Photometrics/Roper Scientific). Images were obtained in either transmission mode or fluorescence mode with two emission filters (530 ± 30 nm and 590 ± 30 nm) and a variety of objectives with magnifications ranging from 4× to 40× in vivo and from 10× to 100× (oil immersion) ex vivo/in vitro. The same exposure time was applied for the same sample before and after photoswitching for green and red fluorescence. Images were acquired and combined (if necessary) at a workstation and processed with the use of Adobe Photoshop 7.0.1 software (Adobe Systems) and ImageJ 1.46 for Windows (available at http://rsb.info.nih.gov/ij/).
In Vitro Photoswitching
The PSFPs Dendra2 and the PSmOranges were photoswitched in solution and in cells with, respectively, 405-nm and 488-nm wavelength diode lasers. A linear laser beam configuration was used to photoswitch the cells in a 120-μm thick, 8-mm diameter well (S-24737; Molecular Probes/Life Technologies) attached to a microscope slide. To photoswitch a large number of cells in suspension (e.g., for in vivo injection) in a cuvette or cells proliferated in a dish, the laser spot was defocused by a ground-glass diffuser, which resulted in a 15-mm diameter beam that was comparable to the size of the cuvette/dish. In some comparative tests, PSFP suspensions were photoswitched with a mercury lamp and the use of excitation filters of 387 ± 6 nm and 520 ± 15 nm for Dendra2 and PSmOranges, respectively.
Fast photoswitching of moving cells in vitro was modeled by scanning Dendra2-MTLn3 cells in a well with a two-dimensional (X-Y) translation stage (H117 ProScan II; Prior Scientific) with a positioning accuracy of 50 nm.
Photoswitching Efficacy
The efficacy of photoswitching CTCs in the blood circulation in vivo was estimated by comparison of signal amplitudes from the CTCs before photo-switching (green channel) and after photoswitching (red channel). Photo-switching cells ex vivo/in vitro were quantified by fluorescent imaging with the use of ImageJ 1.46 software. In particular, we measured both green and red fluorescence as the total fluorescence in the same cell(s) before and after photoswitching after subtracting background signal from surrounding solution, blood, or tissue. The value of green fluorescence was normalized to the initial fluorescence level (before photoswitching), whereas the value of red fluorescence was normalized to the highest fluorescence level after photoswitching. We also calculated the non-normalized ratio of redto-green fluorescence intensity (red/green ratio).
PSFPs and Mammalian Cells
Recombinant Dendra2, PSmOrange, and PSmOrange2 cells were expressed in bacteria and purified as previously described (Kedrin et al., 2008; Subach et al., 2011, 2012). The PSFP stock solution was diluted with PBS to 3 μM, which approximately corresponded to its concentration in the cytoplasm of stably expressing cells. The solution of each protein was mixed with inert optical transparent oil and rigorously shaken to prepare small drops of PSFPs, which were placed on slides for further study.
The MTLn3 adenocarcinoma cells, originally isolated by Neri and Nicolson (Institute for Molecular Medicine, Huntington Beach, CA), were maintained in α-minimum essential medium (Invitrogen/Life Technologies) with 5% fetal bovine serum and penicillin-streptomycin (Invitrogen/Life Technologies). A preclonal mixture of MTLn3 cells expressing Dendra2 was used in all experiments. Viable cells were resuspended in PBS to the desired concentration.
Animal Models
Animals were used in accordance with a protocol approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee. Nude mice (nu/nu), 8–10 weeks old, weighing 20–30 g, were procured from a commercial source for use in the experiments. The animals were anesthetized by isoflurane and placed on a heated microscope stage (at 38°C [body temperature]).
Control measurements were performed (1) on intact vessels (mean diameter, 51 ± 6.5 μm) in eight healthy mice and (2) on the vessels before any injection or inoculation in experimental groups of mice. For a valid comparison of the data, PFC was performed in accordance with the same parameters (laser power, monitoring time, etc.) on vessels of similar diameters to those in the experimental groups. In addition, tissue samples from healthy mice were used to estimate autofluorescence level in microscope images.
To photoswitch individual cells, 2 × 105 Dendra2-MTLn3 cells resuspended in 50 μl of PBS were introduced into the mouse circulation through intracardiac injection into the left ventricle.
To create a primary tumor with metastatic dissemination (see Supplemental Experimental Procedures), mice (n = 8) were inoculated with 1 × 106 Dendra2-MTLn3 cells in an ear. The mice were examined weekly with (1) PFC and (2) intravital fluorescence imaging of the inoculated ear to estimate the development of primary tumors (e.g., size, vascularity), and (3) whole-body imaging with an IVIS Spectrum imaging system (Caliper Life Sciences) to define the appearance of overt metastases. In one subgroup of tumor-bearing mice (n = 4), primary ear tumors (30 days after inoculation) were switched with an unfocused 405-nm laser (spot size, ~20 mm; laser power, 30 mW; exposure, 3–5 min). During photoswitching, the ear was isolated from other parts of the mouse body by a foil. Fluorescence imaging was used to control the efficacy of switching and avoid overexposure. In a second subgroup (n = 4), the mice underwent photoswitching of CTCs in tail veins (exposure, ~5 min). To prevent possible overheating, the irradiated skin area was cooled with a customized, optically transparent cooler.
Blood Sampling and Tissue Isolation for Ex Vivo Tests
At the end of the in vivo studies, blood and multiple organs (e.g., liver, lymph nodes, lungs, and brain) were extracted for examination. Samples of whole blood were placed in 120-μm thick wells and tissue samples in 1-mm thick wells (both from Molecular Probes) attached to slides for ex vivo/in vitro imaging.
To optimize photoswitching, fresh stabilized whole blood was obtained from healthy mice and spiked with of Dendra2-MTLn3 cells at a concentration of 2 × 106 cells per 1 ml of blood, which is close to the real situation in blood circulation in vivo because carcinoma can shed up to 3–4 × 106 CTCs (Butler and Gullino, 1975) and the total volume of mouse blood is ~2 ml.
Statistical Analysis
MATLAB 7.0.1 software (The MathWorks) was used for statistical analyses. Results were expressed as means ± SEM of at least three independent experiments. A p value < 0.05 indicated a significant difference.
Supplementary Material
ACKNOWLEDGMENTS
We thank L.J. Hennings for her histologic contributions and S. Foster and J. Ye for assistance with cell culturing and imaging. This work was supported by grants CA131164 and EB009230 (both to V.P.Z.), GM073913 and CA164468 (both to V.V.V.) from NIH, grant W81XWH-11-1-0129 from the Department of Defense, and grant UL1TR000039 from the Arkansas Breast Cancer Research Program (both to E.I.G.), and grants from the Arkansas Biosciences Institute and the Translational Research Institute at UAMS (both to V.P.Z.). We also thank the Office of Grants and Scientific Publications at UAMS for editorial assistance.
Footnotes
SUPPLEMENTAL INFORMATION Supplemental Information includes Supplemental Experimental Procedures, six figures, two tables, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2014.03.012.
REFERENCES
- Alix-Panabières C, Schwarzenbach H, Pantel K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012;63:199–215. doi: 10.1146/annurev-med-062310-094219. [DOI] [PubMed] [Google Scholar]
- Attard G, de Bono JS. Utilizing circulating tumor cells: challenges and pitfalls. Curr. Opin. Genet. Dev. 2011;21:50–58. doi: 10.1016/j.gde.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Balic M, Williams A, Lin H, Datar R, Cote RJ. Circulating tumor cells: from bench to bedside. Annu. Rev. Med. 2013;64:31–44. doi: 10.1146/annurev-med-050311-163404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutrus S, Greiner C, Hwu D, Chan M, Kuperwasser C, Lin CP, Georgakoudi I. Portable two-color in vivo flow cytometer for real-time detection of fluorescently-labeled circulating cells. J. Biomed. Opt. 2007;12:020507. doi: 10.1117/1.2722733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512–516. [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the `seed and soil' hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Fiorio Pla A, Gkika D. Emerging role of TRP channels in cell migration: from tumor vascularization to metastasis. Front Physiol. 2013;4:311. doi: 10.3389/fphys.2013.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanzha EI, Zharov VP. Photoacoustic flow cytometry. Methods. 2012;57:280–296. doi: 10.1016/j.ymeth.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanzha EI, Shashkov EV, Kelly T, Kim JW, Yang L, Zharov VP. In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells. Nat. Nanotechnol. 2009;4:855–860. doi: 10.1038/nnano.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakoudi I, Solban N, Novak J, Rice WL, Wei X, Hasan T, Lin CP. In vivo flow cytometry: a new method for enumerating circulating cancer cells. Cancer Res. 2004;64:5044–5047. doi: 10.1158/0008-5472.CAN-04-1058. [DOI] [PubMed] [Google Scholar]
- Glaves D, Huben RP, Weiss L. Haematogenous dissemination of cells from human renal adenocarcinomas. Br. J. Cancer. 1988;57:32–35. doi: 10.1038/bjc.1988.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans JL, Steer HW. The function and pathways of lymphocyte recirculation. Ciba Found. Symp. 1980;71:113–126. doi: 10.1002/9780470720547.ch7. [DOI] [PubMed] [Google Scholar]
- Halin C, Mora JR, Sumen C, von Andrian UH. In vivo imaging of lymphocyte trafficking. Annu. Rev. Cell Dev. Biol. 2005;21:581–603. doi: 10.1146/annurev.cellbio.21.122303.133159. [DOI] [PubMed] [Google Scholar]
- Hartley CJ, Michael LH, Entman ML. Noninvasive measurement of ascending aortic blood velocity in mice. Am. J. Physiol. 1995;268:H499–H505. doi: 10.1152/ajpheart.1995.268.1.H499. [DOI] [PubMed] [Google Scholar]
- Hayes DF, Smerage JB. Circulating tumor cells. Prog. Mol. Biol. Transl. Sci. 2010;95:95–112. doi: 10.1016/B978-0-12-385071-3.00005-8. [DOI] [PubMed] [Google Scholar]
- He W, Wang H, Hartmann LC, Cheng JX, Low PS. In vivo quantitation of rare circulating tumor cells by multiphoton intravital flow cytometry. Proc. Natl. Acad. Sci. USA. 2007;104:11760–11765. doi: 10.1073/pnas.0703875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich H. The Laboratory Mouse. Elsevier Academic Press; New York: 2004. [Google Scholar]
- Hwu D, Boutrus S, Greiner C, DiMeo T, Kuperwasser C, Georgakoudi I. Assessment of the role of circulating breast cancer cells in tumor formation and metastatic potential using in vivo flow cytometry. J. Biomed. Opt. 2011;16:040501. doi: 10.1117/1.3560624. [DOI] [PubMed] [Google Scholar]
- Juratli MA, Sarimollaoglu M, Nedosekin DA, Melerzanov AV, Zharov VP, Galanzha EI. Dynamic Fluctuation of Circulating Tumor Cells during Cancer Progression. Cancers (Basel) 2014;6:128–142. doi: 10.3390/cancers6010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, van Rheenen J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Methods. 2008;5:1019–1021. doi: 10.1038/nmeth.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massagué J. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CA. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr. Opin. Genet. Dev. 2011;21:42–49. doi: 10.1016/j.gde.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Langley RR, Fidler IJ. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr. Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- Lazebnik Y. What are the hallmarks of cancer? Nat. Rev. Cancer. 2010;10:232–233. doi: 10.1038/nrc2827. [DOI] [PubMed] [Google Scholar]
- Lombardo VA, Sporbert A, Abdelilah-Seyfried S. Cell tracking using photoconvertible proteins during zebrafish development. J. Vis. Exp. 2012;67 doi: 10.3791/4350. http://dx.doi.org/10.3791/4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic S, Li B, Pera V, Sznaier M, Camps O, Niedre M. A computer vision approach to rare cell in vivo fluorescence flow cytometry. Cytometry A. 2013;83:1113–1123. doi: 10.1002/cyto.a.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat. Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedosekin DA, Sarimollaoglu M, Galanzha EI, Sawant R, Torchilin VP, Verkhusha VV, Ma J, Frank MH, Biris AS, Zharov VP. Synergy of photoacoustic and fluorescence flow cytometry of circulating cells with negative and positive contrasts. J Biophotonics. 2013;6:425–434. doi: 10.1002/jbio.201200047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak J, Georgakoudi I, Wei X, Prossin A, Lin CP. In vivo flow cytometer for real-time detection and quantification of circulating cells. Opt. Lett. 2004;29:77–79. doi: 10.1364/ol.29.000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsillides CM, Runnels JM, Spencer JA, Zhi L, Wu MX, Lin CP. Cell labeling approaches for fluorescence-based in vivo flow cytometry. Cytometry A. 2011;79:758–765. doi: 10.1002/cyto.a.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Carmeliet P. Blood-vessel formation: Bridges that guide and unite. Nature. 2010;465:697–699. doi: 10.1038/465697a. [DOI] [PubMed] [Google Scholar]
- Subach OM, Patterson GH, Ting LM, Wang Y, Condeelis JS, Verkhusha VV. A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat. Methods. 2011;8:771–777. doi: 10.1038/nmeth.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Entenberg D, Condeelis JS, Verkhusha VV. A FRET-facilitated photoswitching using an orange fluorescent protein with the fast photoconversion kinetics. J. Am. Chem. Soc. 2012;134:14789–14799. doi: 10.1021/ja3034137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5:1744–1750. doi: 10.4161/cc.5.16.2864. [DOI] [PubMed] [Google Scholar]
- Tuchin VV, Tárnok A, Zharov VP. In vivo flow cytometry: a horizon of opportunities. Cytometry A. 2011;79:737–745. doi: 10.1002/cyto.a.21143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicha MS, Hayes DF. Circulating tumor cells: not all detected cells are bad and not all bad cells are detected. J. Clin. Oncol. 2011;29:1508–1511. doi: 10.1200/JCO.2010.34.0026. [DOI] [PubMed] [Google Scholar]
- Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J. Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.