

Abstract
MicroRNA-128 (miR-128) is reduced in prostate cancer (PCa) relative to normal/benign prostate tissues but causal roles are obscure. Here we show that exogenously introduced miR-128 suppresses tumor regeneration in multiple PCa xenograft models. Cancer stem-like cell (CSC) associated properties were blocked, including holoclone and sphere formation as well as clonogenic survival. Using a miR-128 sensor to distinguish cells on the basis of miR-128 expression, we found that miR-128-lo cells possessed higher clonal, clonogenic and tumorigenic activities than miR-128-hi cells. miR-128 targets the stem cell regulatory factors BMI-1, NANOG, and TGFBR1, the expression of which we found to vary inversely with miR-128 expression in PCa stem/progenitor cell populations. In particular, we defined BMI-1 as a direct and functionally relevant target of miR-128 in PCa cells, where these genes were reciprocally expressed and exhibited opposing biological functions. Our results define a tumor suppressor function for miR-128 in PCa by limiting CSC properties mediated by BMI-1 and other central stem cell regulators, with potential implications for PCa gene therapy.
Keywords: microRNA, prostate cancer, miR-128, BMI-1, cancer stem cells, tumor suppressor
Introduction
Prostate cancer (PCa) is the most common malignancy afflicting American men with estimated 238,590 new cases and ~29,720 deaths in 2013, accounting for 10% of all male cancer-related deaths (1). Development of castration-resistant disease and distant metastasis are the most common causes of PCa patient death (2). At the cellular level, most patient tumors are heterogeneous containing phenotypically differentiated cancer cells as well as a population of self-renewing tumorigenic cells termed cancer stem cells (CSC), which are implicated in tumor initiation, metastasis, recurrence and resistance to conventional therapies (3–5). Deciphering novel molecular mechanisms associated with PCa progression, especially those regulating the ‘stemness’ of CSC, is of great importance and may provide insight into developing novel therapeutics.
MicroRNAs (miRNAs) are small noncoding RNAs, roughly 19–22 nucleotides in their mature form that mediate the instability and degradation of target mRNAs in a sequence complementarity-dependent fashion (6, 7). As endogenous regulators of gene expression, miRNAs play vital roles in diverse physiological processes including proliferation (8, 9), apoptosis (10, 11), senescence (12), cell identity (13) and stem cell maintenance (14). Dysregulation of miRNA expression or function is involved in various malignancies (15). Systematic miRNA profiling and characterization studies have reported differentially regulated miRNAs in human cancers. For example, >50 miRNAs are aberrantly expressed in PCa with tumor-suppressive miRNAs (e.g., miR-34a and Let-7)repressed or deleted (16,17) and oncogenic miRNAs (e.g., miR-21, miR-125b, and miR-221) overexpressed or amplified (18–20). Our group has reported miR-34a and let-7 as PCa-suppressive miRNAs that can inhibit tumor regeneration and metastasis (21, 22). Recently, a proteomics-based study found that miR-128 expression was progressively lost from normal/benign prostate tissues to adenocarcinoma to metastatic PCa (23). Another microarray study revealed that miR-128 mRNA was reduced by 40% in PCa samples compared with the normal prostate tissues (24). These observations would suggest tumor-suppressive functions of miR-128 in PCa but up to now this suggestion has not been rigorously tested.
The goal for our current study is to investigate the biological functions of miR-128 in PCa cells and to explore the underlying mechanisms of action. By performing a spectrum of gain-of-function, miR-128 reporting sensor, and loss-of-function studies in both bulk and fractionated stem/progenitor cell-enriched PCa cells, we provide direct evidence for PCa-suppressive functions of miR-128 in vitro and in vivo. We further show that the tumor-suppressing effects of miR-128 are associated with its inhibition of CSC via targeting critical molecules such as BMI-1.
Materials and Methods
Cells, animals, and reagents
PC3, Du145, PPC-1, C4-2, and LNCaP cells were cultured in RPMI-1640 plus 7% heat-inactivated fetal bovine serum (FBS), 100 mg/ml streptomycin, and 200 U/ml penicillin (Life Technologies). VCaP cells were cultured in DMEM supplemented with 10% FBS and antibiotics. The non-tumorigenic human prostate epithelial cell line 9 (NHP9) was maintained in serum-free PrEBM medium (Clonetics) supplemented with insulin, epidermal growth factor, hydrocortisone, bovine pituitary extract, and cholera toxin. LAPC9 and LAPC4 were xenograft tumors (21,22). NOD/SCID mice were purchased from the Jackson Laboratory and breeding colonies maintained in our animal facility under standard conditions. All animal experiments were approved by Institutional Animal Care and Use Committee. Antibodies were active caspase-3 (polyclonal antibody (pAb), R&D), Ki-67 (monoclonal antibody (mAb), DAKO), BMI-1 (mAb, Cell Signaling), TGFBR1 (pAb, Abcam), EGFR (mAb, Cell Signaling), GAPDH (pAb, Santa Cruz), BrdU (mAb, Sigma), CD44 (mAb, BD Pharmingen), CD133 (mAb, Miltenyi), α2β1 (mAb, Chemicon). Isotype control antibodies and FITC- or PE-conjugated secondary antibodies were from Chemicon. Other secondary antibodies were purchased from GE Healthcare.
Oligonucleotides (oligos), plasmids, and transfection
Lipofectamine RNAiMAX (Invitrogen) was employed to transfect PCa cells with 30 nmol/L (nM) miR-128 mirVana mimic or non-targeting negative control (miR-NC) oligonucleotides (Life Technologies) according to the manufacturer’s instructions. The mirVana mimics are double-stranded oligonucleotides mimicking mature microRNA, which also have novel chemical modifications that allow for higher potency and specificity over the early-generation pre-miR precursor products (Ambion). mirVana miR-128 inhibitor (anti-miR-128) or non-targeting negative control miRNA inhibitor (anti-NC) was used in some experiments to inhibit miR-128 expression, under the same transfection conditions. Lipofectamine 2000 (Invitrogen) was used to transfect cells with vectors in medium without antibiotics. The miR-128 sensor, which contains 4 copies of miR-128 complementary binding sites inserted into pEGFP-N1 downstream of GFP, was kindly provided by Dr. F. G. Wulczyn (25). For BMI-1 functional assays, BMI-1 shRNA (shBMI-1) and its control shRNA (shCtrl), pBABE-puro BMI-1 (pBABE-BMI-1) containing full-length BMI-1 cDNA and the empty vector (pBABE), were used to knock down and overexpress BMI-1, respectively. pcDNA-CW-CAT BMI-1 (BMI-1) lacking BMI-1 3′-UTR and its parent pcDNA-CW-CAT (Ctrl), were cotransfected with miR-128 mimic for rescue experiments. These BMI-1 related vectors were courtesy of Dr. Rajeev Vibhakar (26).
Quantitative RT-PCR and Western blot
Total RNA was extracted using the mirVana miRNA isolation kit (Ambion). Levels of mature miR-128 were measured using TaqMan MicroRNA Assay (Applied Biosystems) by normalizing to the levels of RNU48. SYBR Green PCR kit (TAKARA) was used to quantify the mRNA levels of several miR-128 targets by normalizing to GAPDH. The PCR reactions were performed and analyzed using ABI 7900 system. Western blots were performed as described previously (21). Briefly, total protein was separated on a precast 4–15% polyacrylamide gel and blotted with antibodies for BMI-1, EGFR, TGFBR1 and GAPDH. Densitometric analysis of protein bands was performed via Image J software.
Clonal, clonogenic, and sphere-formation assays
Basic procedures have been described (21). For clonal experiments, cells were seeded at low density (100 cells/well) in a 6-well plate and allowed to grow until visible colonies appeared. Clones were counted within 2 weeks. For clonogenic assays, 100 μl of cells (300 cells/well) was mixed with 100 μl of cold Matrigel and then plated around the rim of a 24-well dish. After solidification at 37°C for 15 min, 200 μl warm PrEBM was added in the center of the dish. Colonies were enumerated in 1–2 weeks. For sphere formation assay, 500–800 single cells/well are seeded in serum-free PrEBM supplemented with 1X B27 (Life Technologies), 20 ng/ml epidermal growth factor and 20 ng/ml basic fibroblast growth factor in ultralow attachment plate. Medium was replenished every 4 d and spheres counted within 2 weeks. For secondary (2°) sphere formation assay, the 1° spheres were trypsinized into single cells and re-seeded (500 cells/well) in the ultralow attachment plate. The 2° spheres were counted in ~10 days.
Dual-luciferase assays
For BMI-1 and NANOG, fragments containing the predicted binding sites for miR-128 at the 3′-untranslated regions (UTR) were amplified from Du145 genomic DNA by PCR. PCR products were cloned downstream of the firefly luciferase gene in pMIR-REPORT (Ambion) to obtain wild-type pMIR-REPORT-BMI-1 3′-UTR or pMIR-REPORT-NANOG 3′-UTR. To construct mutant vectors, putative miR-128 binding sites in BMI-1 and NANOG 3′-UTR were mutated using QuickChange Site-Direct Mutagenesis Kit (Stratagene). All inserts were sequenced to verify the mutations. Primers used for PCR and sequencing arepresented in Supplementary Table 1. For luciferase assays, Du145 cells were plated in 24-well plates and, 24 h later, cotransfected with 30 nM miR-128 or NC mimic, 1 μg pMIR-REPORTER or vectors containing wild-type or mutant BMI-1 or NANOG-3′UTR, together with 0.5 μg pMIR-Renilla expressing vector (transfection control). 48 h later, luciferase activities were measured using Dual Luciferase Reporter assay kit (Promega) on a Gen-Probe chemiluminometer.
MTT and invasion assays
For MTT assays, 5,000 cells were seeded in 96-well plates and transfected with various vectors for 72 h using Lipofectamine 2000. Then, cells were stained with 100 μl MTT dye (0.5 mg/ml) for 2 h at 37°C, followed by adding 50 μl dimethyl sulphoxide (DMSO). The optical density was measured at 590 nm with a microplate reader (Bio-Rad). For invasion assays, PCa cells were transfected with miR-128 or NC mimic for 48 h, after which 50,000 cells in serum-free medium were seeded in the top chamber of 24-well transwell units (BD Pharmingen) with RPMI-1640 containing 15% FBS added to the bottom chambers. Cells were allowed to migrate for 20 h at 37°C, and then cells in the top chambers were removed and cells that invaded into the bottom chambers were fixed, stained, and quantified.
Cell cycle analysis, BrdU incorporation assay, immunofluorescence, and immunohistochemistry
These procedures were previously detailed (21). For cell cycle analysis, cells were transfected with miR-128 or NC mimics for 72 h and fixed with cold 70% ethanol overnight followed by staining in PPR buffer (0.02 mg/ml RNase A, 0.5%BSA, 0.5%Tween-20 and 0.05 mg/ml propidium iodide) at 37°C in the dark for 40 min. DNA contents were determined on a BDAria FACS machine and data were analyzed by FlowJo software. For bromodeoxyuridine (BrdU) incorporation assays, cells were transfected with mimics or vectors for 72 h and incubated in 10 μmol/L of BrdU for 4 h. At the end, cells were fixed with 4% paraformaldehyde (PFA), denatured with 6 M HCI, and then incubated with anti-BrdU antibody followed by Alexa Fluor® 594 labeled secondary antibody. A total of 500–1,000 cells per coverslip were counted to determine the percentage of BrdU+ cells with two coverslips counted for each condition. Immunofluorescence (IF) staining of BMI-1 required permeabillization and denaturation (0.5% Triton) pretreatment. For immunohistochemistry (IHC), tumor tissue sections were cut and stained for hematoxylin and eosin (H&E), Ki-67 and active caspase-3. The IF images were captured using an Olympus IX 50 inverted epifluorescence microscope.
Tumor transplantation experiments
The lab routinely maintains LNCaP, Du145, PC3, PPC-1, LAPC9, and other human PCa xenografts in NOD/SCID or NOD/SCIDγ mice (21,27,28). Human PCa cells in these xenografts were purified out using a Histopaque-1077 gradient followed by removing murine cells with a lineage-depletion kit (Miltenyi) (21,27,28). The purity of the HPCa cells from xenografts was generally ~100%. The 1° tumor transplantation experiments were performed by subcutaneously injecting purified human PCa cells in 60-μl medium-Matrigel mixture (1:1) into NOD/SCID mice. For 2° PC3 tumor transplantation, 1° tumors were dissociated into single cells, human tumor cells were purified out and then re-transfected with the NC or miR-128 mimics. The 2° tumor transplantation was performed using strategies similar to the primary implantation.
Statistical analysis
In general, unpaired two-tailed Student t-test was used to compare significance between groups for cell numbers, tumor weights, the percentages of Ki-67+, caspase-3+, BrdU+ and S-phase cells, cloning and sphere-forming efficiencies, and RNA levels of miR-128 and its targets. The Fisher exact test and χ2 test were used to compare tumor incidence. All results were presented as mean ± S.D or mean ± SEM with a P value < 0.05 considered statistically significant.
Results
Overexpression of miR-128 by oligo transfection inhibits xenograft tumor growth in vivo
Mature miR-128 can derive from either pre-miR-128a (i.e., miR-128-1) or pre-miR-128b (miR-128-2) locus located at two different chromosomal regions (Supplementary Fig. S1). miR-128 levels were recently reported to be downregulated in PCa and their metastases (23,24). Consistent with these reports, when we quantified miR-128 mRNA levels in 8 PCa cell lines including PPC-1, LAPC4, LNCaP, VCaP, PC3, DU145, LAPC9 and LNCaP-C4-2 (a ‘castration-resistant’ LNCaP subline), we found that the miR-128 levels were lower in these PCa cells compared to an immortalized non-tumorigenic prostate epithelial cell line, NHP9 (Supplementary Fig. S2), which was established in our lab (21). These observations (23,24; Supplementary Fig. S2) prompted us to investigate the functions of miR-128 in PCa cells by first performing gain-of-function studies. To this end, we transfected synthetic miR-128 mimic or the negative control (NC) oligos (30 nM, 48 h) into PC3, PPC-1, Du145 or LNCaP cells. As expected, cells transfected with miR-128 mimic showed miR-128 mRNA levels several orders of magnitude higher than cells transfected with miR-NC (Supplementary Fig. S3A). Strikingly, when miR-128 transfected PC3, PPC-1, and Du145 cells were implanted subcutaneously (s.c) into NOD/SCID mice, tumor regeneration was significantly inhibited in every case (Fig. 1A; Supplementary Fig. S3B). Specifically, miR-128 mimics significantly inhibited tumor growth as well as tumor incidence in PPC-1 cells (Fig. 1A). miR-128 overexpression inhibited both primary (1°) and secondary (2°) PC3 tumor growth with a trend of reduced tumor incidence (Fig. 1A). In 3 independent experiments, miR-128 overexpression inhibited Du145 tumor growth with a trend of reduced tumor incidence (Fig. 1A; Supplementary Fig. 3B). Taken together, these tumor regeneration experiments provide the first in vivo evidence that miR-128 possesses PCa-suppressive effects.
Figure 1. miR-128 overexpression attenuates PCa tumor growth in vivo.
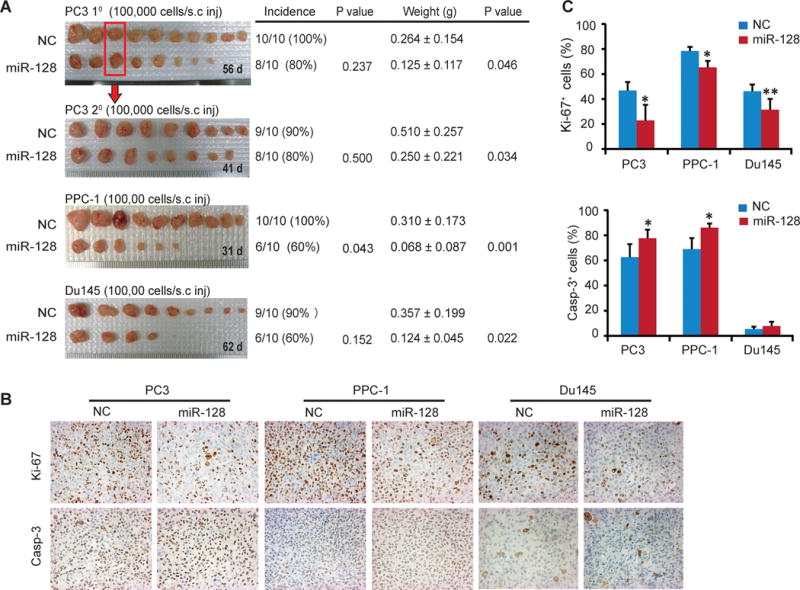
A. miR-128 overexpression inhibits PCa xenograft growth in 3 tumor models. Shown on the left are images of prostate tumors from the indicated cell types that were transfected with miR-128 mimics or miR-NC oligos with cell numbers injected and time in days (d)when tumors were harvested shown for each panel. Tumor incidences (#tumors/#injections), endpoint tumor weights (mean ± S.D) and the corresponding P values were indicated on the right. For 2° PC3 tumor transplantations, one 1° NC and miR-128 tumor each (indicated by a red box)was dissociated into single cells and purified human PCa cells were re-transfected with the miR-128 or NC mimics, respectively, and transplanted into the NOD/SCID mice.
B. Representative IHC micrographs of Ki-67 and active caspase-3 in endpoint tumor tissues. Original magnification, x200.
C. Qualification of Ki-67+ (top) and active caspase-3+ (bottom) cells in endpoint tumors. Bars represent the mean ± S.D. *P<0.05; **P<0.01.
We performed immunohistochemical (IHC) staining of Ki-67 and activate caspase-3 in the endpoint tumors. The results revealed reduced Ki-67+ cells in all three miR-128-transfected tumor systems and increased caspase-3+ cells in miR-128-overexpressing PC3 and PPC-1 tumors (Fig. 1B–C). Du145 tumors had a low percentage of caspase-3+ cells, which did not differ between miR-128 and miR-NC transfected tumors (Fig. 1B–C). These results suggest that the tumor-inhibitory effects of miR-128 may be associated with suppression of proliferation and promotion of apoptosis. Preliminary results revealed that miR-128 might also regulate PCa cell differentiation as miR-128 mimic increased both the mRNA and protein levels of differentiation markers androgen receptor (AR) and prostate-specific antigen (PSA) (data not shown).
miR-128 overexpression suppresses cell proliferation and invasion in vitro
To further help elucidate the mechanism of action of miR-128, we assessed the impact of miR-128 on several biological properties of PCa cells in vitro. miR-128 overexpression inhibited PCa cell growth (Fig. 2A) and proliferation as determined by both BrdU incorporation assays (Fig. 2B) and cell cycle analysis (Fig. 2C). miR-128 mimic decreased the percentage of S-phase cells from 30.3% to 18.6% in PC3, and from 23.1% to 9.5% in Du145 cells, compared with the corresponding NC mimic-transfected cells (Fig. 2C). miR-128 also significantly inhibited the Matrigel invasion in 3 of the 4 PCa cell lines tested except PPC-1 (Fig. 2D).
Figure 2. miR-128 overexpression suppresses PCa cell proliferation and invasion in vitro.
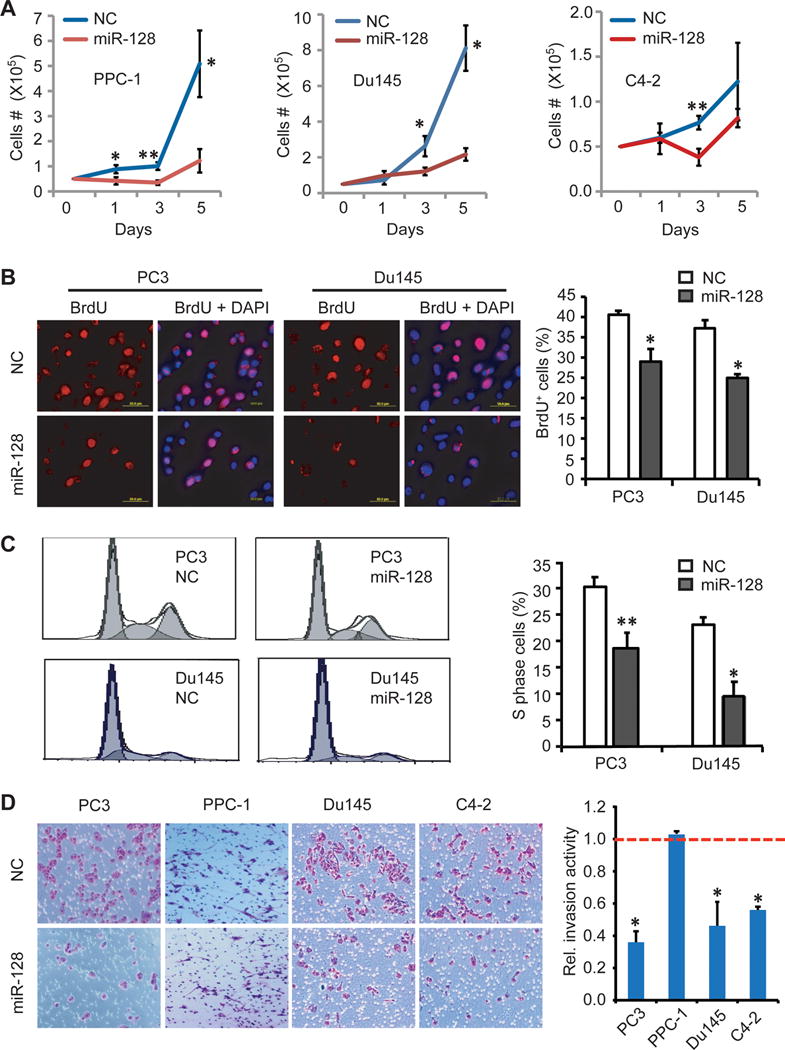
A. Effect of miR-128 on PCa cell growth. Cells as indicated and transfected with NC or miR-128 mimic (30 nM) were plated in triplicate in 6-well plate (50,000 cells/well) on day 0. Cells were harvested and counted on day 1, 3, and 5. Shown are the mean ± S.D.
B. Effect of miR-128 on BrdU incorporation. Shown are representative micrographs (left) and quantification (right) of BrdU staining in PC3 and Du145 cells transfected with miR-NC or miR-128 (30 nM; 72 h).
C. Effect of miR-128 on PCa cell cycle. PC3 and Du145 cells transfected with miR-128 or NC mimic for 72 h were used in cell-cycle analysis upon propidium iodide staining. Shown are representative histograms (left) and quantification of the S-phase cells (right; mean ± S.D).
D. Effect of miR-128 on PCa cell invasion. Shown are representative images (left) and quantification (right) of cell invasion in PC3, PPC-1, DU145 and LNCaP C4-2 cells as determined by Transwell assay. The invasive activity detected in NC-transfected cells was used as the calibrator (assigned a value of “1”, indicated by red dotted line). Error bars represent the mean ± SEM of three independent experiments with each conducted in duplicate.
In all experiments, *P<0.05. **P<0.01.
miR-128 overexpression inhibits PCa cell clonal, clonogenic and sphere-forming activities in vitro
Our previous studies indicate that a fraction of PCa cells in culture, xenografts, and patient tumors possesses certain stem cell properties such as high capacity to found holoclones in 2-D cultures, 3-D colonies in matrices such as Matrigel or methycellulose, or anchorage-independent floating spheres in ultralow attachment (ULA) plates (21,22,27–29). Here we analyzed the effects of miR-128 on these properties of PCa cells. As shown in Fig. 3 (A–F), miR-128 overexpression, compared to miR-NC transfection, greatly inhibited the formation of holoclones, which are enriched in self-renewing CSCs (29). A differential enumeration in Du145 cells indicated that miR-128 shifted holoclone to meroclone formation (Fig. 3F). Similarly, miR-128 mimic inhibited clonogenic (in Matrigel) and sphere-forming capacities of both PC3 and Du145 cells (Fig. 3, G–J). Serial sphere propagating assays revealed that miR-128 also impaired the stem cell trait of self-renewal as assessed by secondary prostasphere establishment in Du145 cells (Fig. 3J).
Figure 3. miR-128 overexpression suppresses clonal, clonogenic and sphere-forming capacities of PCa cells.
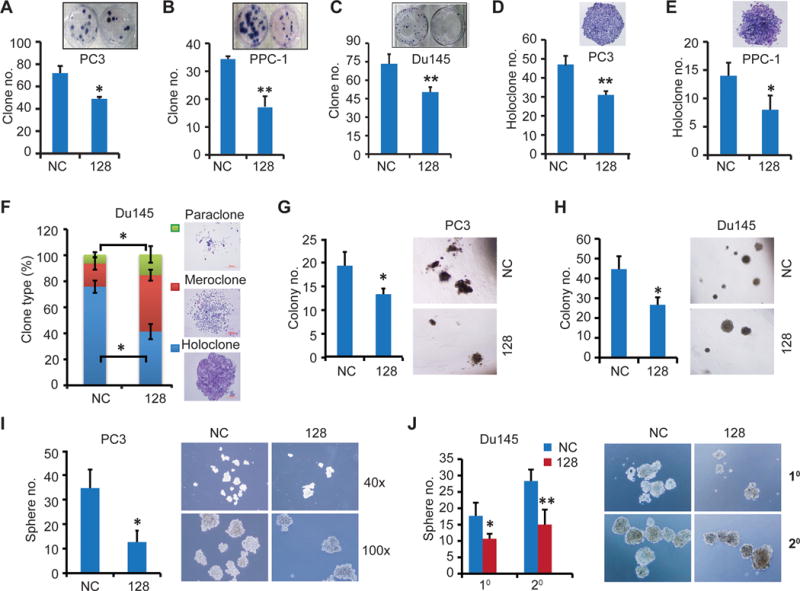
A–F. Clonal assays in PC3 (A and D), PPC-1 (B and E) and Du145 (C and F) cells. Cells transfected with miR-128 or NC mimic (30 nM, 48 h) were seeded in 6-well plates at clonal density and cultured for two weeks followed by staining with Giemsa and photography. Total number of all clones (A–C)or the number of holoclones (D–E) was enumerated and representative micrographs were shown in the insets. (F) Summary of three types of clones in Du145 cells.
G–H. Clonogenic assays in PC3 (G) and Du145 (H) cells. Cells (300) transfected as above were mixed with Matrigel and plated in 24-well plates and colonies counted in two weeks. Presented are quantification (left) and representative images (right).
I–J. Sphere assays in PC3 (I) and Du145 (J) cells. Cells (500) transfected as above were plated in 6-well ultralow attachment plate and cultured in serum-free medium. The 1° Du145 spheres were passaged and re-transfected with corresponding miR-128 or NC mimic and then equal number of cells were plated for 2° sphere assays (J). Shown are quantification (left) and representative images (right).
In all experiments, data represent the mean ± S.D from three independent experiments with triplicate in each condition.*P<0.05; **P<0.01.
Differential clonogenic and tumorigenic capacities of PCa cells expressing low and high levels of endogenous miR-128
All the preceding experiments studied the impact of exogenously overexpressed miR-128 on the biological and tumorigenic properties of PCa cells. Next, we investigated whether PCa cells that express different levels of endogenous miR-128 may possess intrinsically different biological and tumorigenic potential. To this end, we utilized a miR-128 sensor construct (25), which contains four copies of miR-128 perfect complementary binding sequences in the 3-′UTR of GFP cDNA (Supplementary Fig. S4A). The rationale was that the cells that express high endogenous levels of miR-128 (i.e., miR-128-hi) will be identified as GFP− as miR-128 will target the binding sites leading to extinguishment of GFP signal. By contrast, PCa cells that express low levels of endogenous miR-128 (i.e., miR-128-lo) will be identified as GFP+. When the control plasmid pEGFP-N1 was transfected into PC3 and Du145 cells, an average of ~80% of the cells were GFP+ and this percentage did not change in the presence of increasing amount of exogenous miR-128a mimic (Fig. 4A–B; Supplementary Fig. S4A). These results suggest that our transfection efficiency was ~80%. In contrast, when PC3 and Du145 cells were transfected with the miR-128 sensor construct, we observed that ~40% cells were GFP+ in the absence of exogenous mimic (Fig. 4A–B; Supplementary Fig. S4B), suggesting that both cell types contain a fraction of miR-128-hi cells leading to increased GFP− cells. Two pieces of evidence support that the miR-128 sensor faithfully reports endogenous levels of miR-128. First, when increasing amounts of exogenous miR-128 mimics were co-transfected, together with the sensor construct, into PC3 and Du145 cells, we observed decreasing numbers of GFP+ cells (Fig. 4B; Supplementary Fig. S4C). Second, qPCR analysis in 4 sensor-transfected PCa cell types demonstrated that GFP+ cells expressed lower levels of miR-128 compared to the corresponding GFP− PCa cells (Fig. 4C).
Figure 4. Endogenous miR-128 levels in PCa cells inversely correlate with their clonogenic and tumorigenic potential.
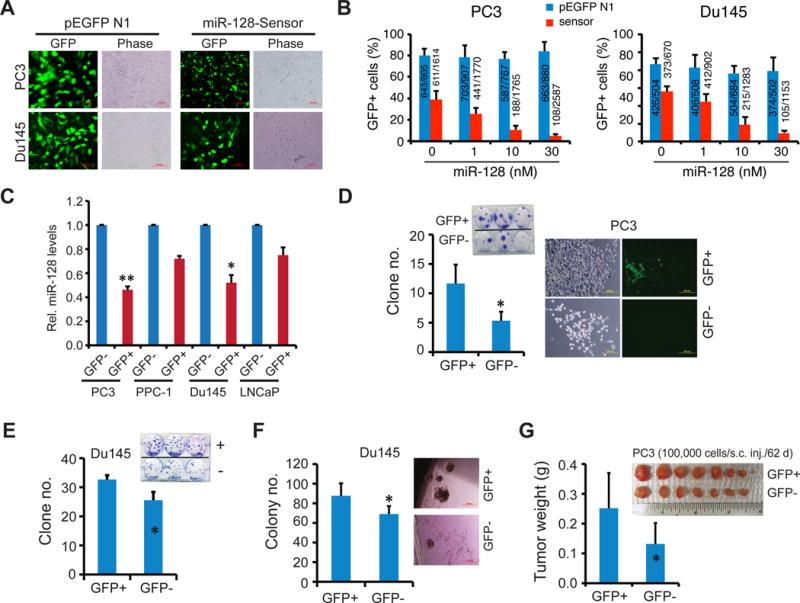
A–C. miR-128 sensor reports endogenous miR-128 mRNA levels in PCa cells. Shown in A are images of PC3 and Du145 cells transfected with either parent pEGFP-N1 or miR-128 sensor constructs in the absence of exogenous miR-128 mimic (72 h; original magnification, x400). The corresponding images in the presence of 1–30 nM of exogenous miR-128 mimic were presented in Supplementary Fig. S4C. Shown in B is quantification of GFP+ percentages in PC3 and Du145 cells transfected with the control (pEGFP N1) or sensor construct in the presence of 0–30 nM of exogenous miR-128 mimic. Data represent the results from two independent experiments. The GFP+ cells/total number of cell counted were presented in bar graphs. In C, levels of endogenous miR-128 were determined by qRT-PCR in purified GFP+ and GFP− PCa cells 48 h after transfection. Data were normalized to RNU48 and presented as values relative to corresponding GFP− cells. *p<0.05; **P<0.01.
D–F. Differential clonal (C–D) and clonogenic (E) potential of miR-128-hi (GFP−) vs. miR-128-lo (GFP+) PC3 (C) and Du145 (D–E) cells. Log-phase PC3 and Du145 cells were transfected with the miR-128 sensor plasmids and GFP+/GFP− cells purified out and used in clonal and clonogenic assays. *P<0.05.
G. miR-128-lo PC3 cells are more tumorigenic than the corresponding miR-128-hi cells. Data on the left represents the mean tumor weight (± S.D). *P<0.05.
Subsequently, GFP+ (miR-128-lo) and GFP− (miR-128-hi) cells were sorted out by fluorescence-activated cell sorting (FACS) and used in clonal, clonogenic, and tumor regeneration assays. The results demonstrated that miR-128-lo PCa cells possessed higher clonal (Fig. 4, D–E), clonogenic (Fig. 4F), and tumorigenic (Fig. 4G) potentials. Since the miR-128 sensor construct contains 4 copies of the binding sequences that perfectly match the miR-128 seed sequence, we utilized the miR-128-sensor construct as a “decoy” for loss-of-function studies (i.e., to sequester endogenous miR-128). As shown in Supplementary Fig. S5, compared to the bulk PC3 and Du145 cells transfected with pEGFP-N1, cells transfected with the miR-128 sensor constructs displayed increased clonal and sphere-forming capacities.
miR-128 targets a cohort of stem cell regulatory factors
To investigate the molecular mechanisms through which miR-128 exerts its PCa-inhibitory effects, we screened its potential targets using several bioinformatics tools including TargetScan, Microcosm V5 and miRanda. Based on the sequence complementarity to miR-128 seed sequence, we chose 5 genes as our interest of research, including BMI-1, NANOG, TGFBR1, EGFR and E2F3 (Fig. 5A), all of which are known oncogenic and stem cell regulators implicated in PCa initiation and progression. We found that miR-128 overexpression reduced the mRNA levels of different molecules in a cell type dependent manner (Fig. 5B). In PC3 cells, miR-128 significantly decreased NANOG and TGFBR1 levels whereas in PPC-1 cells miR-128 additionally reduced E2F3 mRNA levels (Fig. 5B). In Du145 cells, miR-128 attenuated mRNA levels of all molecules except EGFR (Fig. 5B). In sharp contrast, in LNCaP cells, miR-128 only reduced the mRNA levels of BMI-1 and EGFR (Fig. 5B).
Figure 5. miR-128 regulates a cohort of self-renewal genes in PCa cells.
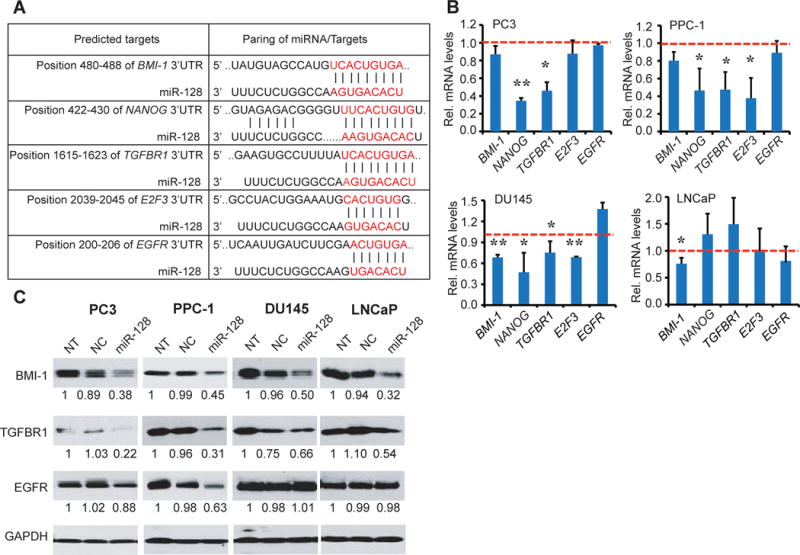
A. Predicted binding of miR-128 to the 3′-UTRs of BMI-1, NANOG, TGFβR1, EGFR, and E2F3.
B. qRT-PCR analysis of the effect of miR-128 overexpression on mRNA levels of the indicated genes in PC3, PPC-1, DU145 and LNCaP cells. Cells were transfected with either miR-128 or NC mimic (30 nM) for 48 h. RNA levels were normalized to GAPDH and data shown are relative to the corresponding NC control. Red lines were used as calibrators. Data represent the mean ± S.D from several independent measures. *P<0.05; **P<0.01.
C. Western blot analysis of the effect of miR-128 overexpression on protein levels of BMI-1, TGFBR1 and EGFR in indicated cells. Densitometric values relative to non-transfected (NT) cells were given below. GAPDH was used as a loading control.
Western blotting analysis of BMI-1, TGFBR1, and EGFR (Fig. 5C) overall corroborated the qPCR results. Specifically, miR-128 overexpression reduced BMI-1 proteins levels, to ~30–50% of the NC-transfected cells, in all 4 cell types (Fig. 5C). TGFBR1 protein levels were also reduced by miR-128 (Fig. 5C), even in LNCaP cells in which its mRNA levels were not altered by miR-128 (Fig. 5B), suggesting that miR-128 might regulate TGFBR1 in LNCaP cells through translational mechanisms. miR-128 reduced EGFR protein levels only in PPC-1 cells (Fig. 5C).
BMI-1 is a direct and functional target of miR-128 in PCa cells
In all 4 PCa lines, miR-128 reduced both mRNA and protein levels of BMI-1 (Fig. 5B–C), and the reduction of BMI-1 protein by miR-128 was also confirmed by immunofluorescence staining of nuclear BMI-1 (Fig. 6A). These results, together with recent reports on BMI-1 being an essential regulator of mouse prostate epithelial and other stem cells (30), prompted us to focus on BMI-1 in subsequent studies. In luciferase reporter assays in which we cloned the BMI-1 3′-UTR harboring the miR-128 binding site downstream of the firefly luciferase gene (Fig. 6B, left), co-transfection of Du145 cells with the luciferase construct and miR-128 mimic led to reduced luciferase activity (Fig. 6B, right). Mutation of the miR-128 binding site in the BMI-1 3′-UTR abrogated the miR-128 effects (Fig. 6B), testifying BMI-1 as a direct target of miR-128. Similar luciferase assays also confirmed NANOG as a direct miR-128 target (Fig. 6C).
Figure 6. BMI-1 is a direct target of miR-128 and its reexpression partly rescued the decreased proliferation phenotype of miR-128 overexpression.
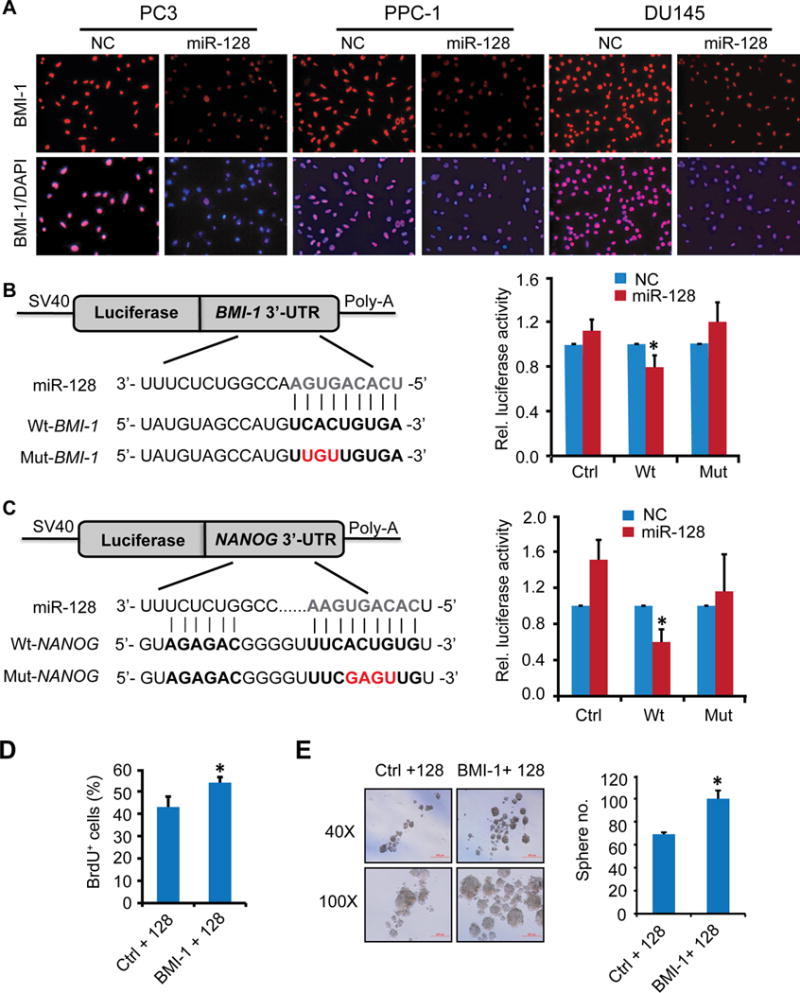
A. Immunofluorescence staining of BMI-1 in 3 PCa cell types. Cells transfected with miR-NC or miR-128 mimic (30 nM, 48 h) were cultured on coverslips overnight followed by immunostaining. Original magnifications, x100.
B–C. Dual Luciferase assays in DU145 cells. Schematic diagram of the BMI-1 and NANOG 3′-UTR pMIR-REPORT constructs (left). Sequences were compared between mature miR-128 and the wild-type (Wt) or mutant (Mut) putative target sites in the 3′-UTR of BMI-1 (top) or NANOG (below). Cells were co-transfected with pMIR-REPORT containing the empty (Ctrl), wild-type (Wt) or mutant (Mut) target site of the BMI-1 or NANOG 3′-UTR plus miR-128 or NC mimic for 48 h. The luciferase activity was normalized to the Renilla activity and presented as relative activity to the corresponding NC (assigned as value “1”). Values denote the mean ± SEM of three independent assays. *P < 0.05.
D–E. Effects of restoring BMI-1 expression on proliferation or sphere-formation ability in DU145 cells as determined by BrdU incorporation (D) and sphere formation (E) assays 72 h after co-transfection with BMI-1 cDNA lacking the 3′-UTR and miR-128 mimic. Error bars represent mean ±S.D obtained from two independent experiments. *P<0.05.
To determine whether BMI-1 is a functional target of miR-128, we performed ‘rescue’ experiments by co-transfecting Du145 cells with miR-128 mimic and BMI-1 overexpressing construct containing the BMI-1 cDNA lacking its 3′-UTR (26), which could not be inhibited by miR-128. BMI-1 overexpression was able to promote both proliferation (Fig. 6D) and sphere formation (Fig. 6E) over miR-128 and control vector co-transfected cells. Altogether, these data support the notion that BMI-1 is a direct and functional target mediating the anti-proliferative and anti-tumorigenic effects of miR-128 in PCa cells.
Inverse correlation between miR-128 and its targets in prostate CSC (PCSC) populations
We and others have characterized and reported on several tumor-initiating PCa cell populations from both cultured cell lines and xenograft tumors (21,22,27,28,31,32), which include CD44+, CD44+α2β1+, CD133+, and PSA−/lo populations. We first determined the mRNA expression patterns of miR-128 and its 5 targets (above) in some of these PCa stem/progenitor cell populations. miR-128 mRNA levels were lower in CD44+ and CD133+ Du145 cells (Fig. 7A, left) and in CD133+ LAPC9 cells (Fig. 7A, middle) compared to the corresponding marker-negative populations. Interestingly, in androgen-independent (AI) LAPC9 xenograft tumors which harbor significantly increased CD44+ and PSA−/lo PCSCs compared to the androgen-dependent (AD) xenografts (28; data not shown), the miR-128 mRNA levels were also much lower (Fig. 7A, right). Overall, these data suggests that several PCSC populations in different PCa models show reduced miR-128 expression relative to non-CSC populations.
Figure 7. Inverse correlation between miR-128 and BMI-1 in prostate CSC populations.
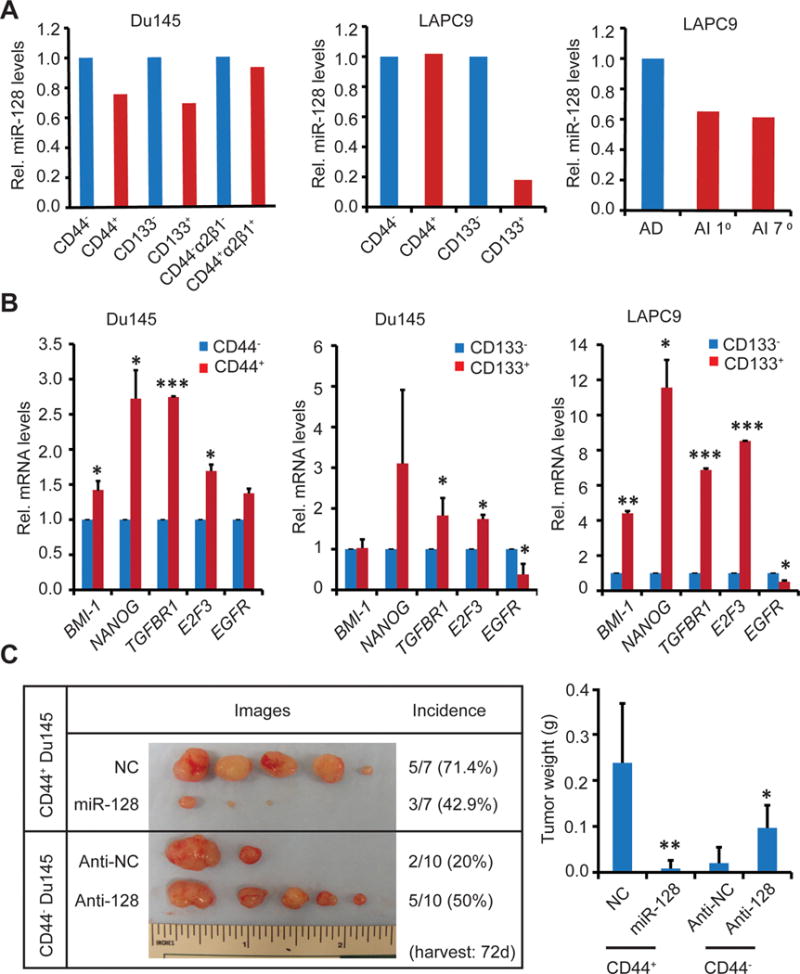
A. miR-128 mRNA levels in the indicated CSC populations or AD and AI LAPC9 tumors. Shown are the mean values of the miR-128 mRNA levels measured by qRT-PCR in the indicated marker-positive populations (or AD LAPC9) relative to the corresponding marker-negative (or AI LAPC9) populations. Due to the rarity of these populations, only duplicate measures from one batch of sorted samples were performed.
B. Assessment of miR-128 target mRNA levels in purified CD44+ or CD133+ populations. Shown are the relative expression levels of the indicated genes in CD44+ DU145 (left), CD133+ DU145 (middle), and CD133+ LAPC9 (right) cells relative to the corresponding marker-negative populations. *P<0.05; **P<0.01; ***P<0.001.
C. Effects of miR-128 and anti-miR-128 on the tumorigenecity of CD44+ and CD44− Du145 cells, respectively. CD44+ and CD44− Du145 cells were purified out by FACS, transfected with the mimics (30 nM), and 48 h later, and transplanted subcutaneously into NOD/SCID mice (100,000 cells/injection). Mean tumor weights are plotted on the right (*P<0.05; **P<0.01). Note that the mean tumor latency for NC, miR-128, anti-NC, and anti-128 groups was 27, 42, 40, and 34 d, respectively.
Consistent with miR-128 expression patterns, the mRNA levels of its target molecules, i.e., BMI-1, NANOG, TGFBR1, E2F3, and EGFR, showed upregulation in some of the PCSC populations (Fig. 7B). For example, in CD44+ Du145 cells, all 5 molecules were overexpressed (Fig. 7B, left). In CD133+ Du145 (Fig. 7B, middle) and LAPC9 (Fig. 7B, right) cells, 3 or 4 of these target molecules were over-represented. These results (Fig. 7, A–B), together, reveal an inverse correlation between miR-128 and its targets in some PCSC populations.
Negative regulation of PCSCs by miR-128
Because miR-128 was expressed at lower levels in some PCa stem/progenitor cell populations, we asked whether miR-128 might negatively regulate the tumor-regenerating capacity of these cells. To address this question, we manipulated miR-128 expression in CD44+ and CD44− Du145 cells followed by tumor regeneration assays. As shown in Fig. 7C, overexpression of miR-128 in purified CD44+ Du145 cells significantly inhibited tumor development in that fewer Du145 tumors were regenerated and the developed tumors were much smaller. In contrast, overexpression of anti-miR-128 (i.e., anti-128) in CD44− Du145 cells promoted tumor development (Fig. 7C).
Next, we manipulated the BMI-1 expression levels in purified CD44+ and CD44− Du145 cells to determine how that would affect their respective biological behaviors in vitro. As expected, the lentiviral shBMI-1 downregulated BMI-1 mRNA and protein in CD44+ Du145 cells (Fig. 8A) and resulted in reduced proliferation (Supplementary Fig. S6A), holoclone establishment (Fig. 8B), and sphere formation (Fig. 8C). BMI-1 downregulation also inhibited proliferation of bulk PC3 and PPC-1 cells (Supplementary Fig. S6A). In contrast, BMI-1 overexpression (Fig. 8D) promoted proliferation (Supplementary Fig. S6B) and holoclone (Fig. 8E) and sphere (Fig. 8F) formation in CD44− Du145 cells, as well as the proliferation of bulk PC3 and PPC-1 cells (Supplementary Fig. S6B).
Figure 8. Effects of manipulating BMI-1 expression on the CSC properties of PCa cells in vitro.
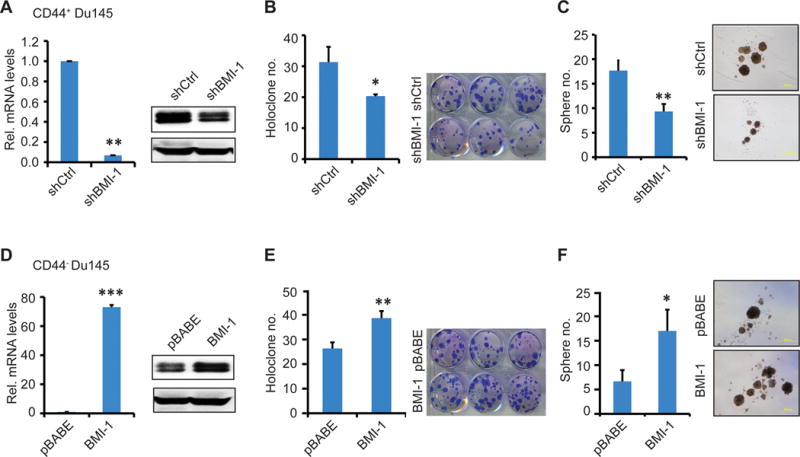
A–C. In vitro assays in CD44+ Du145 cells with BMI-1 knocked down using shBMI-1. Validation of BMI-1 knockdown effects by qRT-PCR (left) and Western blotting (right) analyses. Holoclone (B) and sphere formation (C) assays in shCtrl/shBMI-1 infected CD44+Du145 cells.
D–F. In vitro assays in CD44− Du145 cells with BMI-1 overexpression via infecting with pBABE-Bmi1 vector encoding full-length BMI-1. pBABE was used as control. Validation of BMI-1 overexpression in CD44− Du145 cells by qRT-PCR (left) and Western blot (right) analyses (D). Holoclone (E) and sphere formation (F) assays in pBABE and pBABE-BMI-1 infected CD44− cells.
In all above experiments, shown are data from 3 independent experiments carried out in triplicate per condition. *P<0.05; **P<0.01; ***P<0.001.
Considering the reciprocal expression patterns of miR-128 and BMI-1 in CD44+ Du145 cells, the above results suggest that the PCSC-inhibitory effects of miR-128 are mediated, at least in part, via downregulating BMI-1.
Discussion
Our present study has revealed the following novel findings: 1) exogenously overexpressed miR-128 suppresses tumor regeneration in 3 PCa xenograft models and inhibits cell proliferation in vitro and in vivo; 2)miR-128 overexpression inhibits CSC-associated properties including holoclone and sphere formation; 3) PCa cells that express low levels of endogenous miR-128 possess high clonal, clonogenic, and tumorigenic properties; 4) miR-128 targets several stem cell regulatory factors in PCa cells including BMI-1, NANOG, and TGFBR1; 5)miR-128 and its targets exhibit reciprocal expression patterns in several PCSC populations; 6) BMI-1 represents a direct and functionally relevant target of miR-128 in PCa cells; and 7) miR-128 and BMI-1 are reciprocally expressed and also exhibit opposite biological functions in the CD44+ Du145 PCa cells.
miR-128 was originally identified as a ‘brain-specific’ miRNA whose expression profile is associated with brain development:it is present mainly in mature, terminally differentiated neurons but absent in neural stem cells (25, 33) implicating miR-128 in neuronal differentiation. Aberrant expression of miR-128 has been observed in some malignancies and several studies have lent credence to the notion that miR-128 functions as a tumor suppressor in glioblastoma multiform (34) and medulloblastoma (26). On the other hand, upregulation of miR-128 has also been reported in acute lymphoblastic leukemia (35) and in letrozole-resistant breast cancer cell lines (36). These findings imply miR-128 as either a tumor-suppressive or oncogenic miRNA, probably depending on specific cancer types. Recently, miR-128 was found to be downregulated in PCa compared to the normal/benign prostate tissues, and even more so in metastases in comparison to primary tumors (23,24). However, a causal role of miR-128 in PCa has not been reported. Here, for the first time, we thoroughly characterize the biological activities of miR-128 and provide direct evidence for its tumor-suppressive functions in PCa.
Exhaustive overexpression studies by introducing exogenous miR-128 mimic reveal its tumor-inhibitory effects in 3 PCa cell models. Complementary studies using the miR-128-reporting sensor demonstrate that the miR-128-lo PCa cells possess higher clonal, clonogenic, and tumorigenic activities than the isogenic miR-128-hi PCa cells. These latter observations highlight the cellular heterogeneity of PCa cells (3,37) in that the cells expressing different levels of a single miRNA can manifest intrinsic differences in many fundamental tumor-associated biological traits. Along the same line, the miR-128 sensor construct, when used as a decoy to neutralize the endogenous miR-128 in bulk PCa cells, behaves opposite to miR-128 mimic and promotes PCa cell clonal and clonogenic growth.
We explored the potential mechanisms underlying the PCa-inhibitory effects of miR-128 by focusing on molecular and cellular targets. At the molecular level, miR-128 appears to target, in a cell type-dependent manner, several stem cell related genes including BMI-1, NANOG, and TGFBR1. BMI-1, a component of the PRC2 polycomb repressor complex, is crucial for self-renewal and malignant transformation of prostate stem cells (38). BMI-1 is frequently overexpressed in cancer including PCa and is a potential biomarker for the diagnosis and prognosis of PCa (39,40). In PCa cells, we find that miR-128 directly regulates BMI-1 through its 3′-UTR. BMI-1 represents a bona fide as well as a functional target of miR-128, as expression of a BMI-1 cDNA lacking the miR-128 binding site at the 3′-UTR partly rescues the proliferation and sphere-forming defects in miR-128 over-expressing PCa cells. These observations suggest that BMI-1 is an important downstream target for miR-128 to exert its tumor inhibitory functions. miR-128 likely also targets other important stem cell regulators such as NANOG, which has previously been shown by our lab to be enriched in cancer stem/progenitor cells and to play important positive roles in promoting tumorigenicity (41). Inducible expression of NANOG in PCa cells upregulates molecules such as c-Myc, CXCR4, CD133 and ALDH1 and reprograms bulk PCa cells to CSCs (42). In this study, we also find that miR-128 can directly target the NANOG mRNA in PCa cells. Whether this will lead to reduced NANOG protein levels and whether NANOG (protein and/or RNA) represents a functionally relevant target of miR-128 in PCa cells await further investigations.
At the cellular level, miR-128 seems to negatively regulate PCa stem/progenitor cells. miR-128 overexpression inhibits the holoclone and sphere-forming activities of bulk PCa cells. Vice versa, miR-128-lo PCa cells possess high clonal and sphere-forming capacity. Furthermore, miR-128 is under-expressed whereas its targets are overexpressed in several PCa stem/progenitor cell populations. Importantly, miR-128 overexpression in CD44+ PCa cells suppresses whereas anti-miR-128 expression in CD44− PCa cells promotes tumor regeneration. The inhibitory effects of miR-128 on PCa-initiating cells are likely mediated through its targeting critical stem cell molecules such as BMI-1 and NANOG. In support, silencing BMI-1 in CD44+ Du145 cells significantly inhibits their CSC properties whereas BMI-1 overexpression in CD44− cells confers an oncogenic effect in vitro similar to the phenotype of miR-128 inhibition. Consistent with our results, BMI-1 has been shown to be a relevant target of miR-128 in regulating glioblastoma (43) and breast (44) CSCs. Future work will determine whether NANOG also represents a functionally relevant target of miR-128 in PCa stem/progenitor cells. Our present results on miR-128, together with our recent data on miR-34a (21) and let-7 (22), reinforce the concept that multiple key tumor-suppressive miRNAs function, concertedly and coordinately, to negatively regulate tumor-initiating cells (45). In principle, these tumor-suppressive miRNAs can be developed into CSC-targeting ‘replacement’ therapeutics (21,45).
Supplementary Material
Acknowledgments
We thank Dr. F. Gregory Wulczyn for providing the miR-128 sensor vectors and Dr. Rajeev Vibhakar for sharing the BMI-1 plasmids. We also thank Ms. P. Whitney for assistance in flow sorting, Dr. J.J. Shen and his group for qRT-PCR analysis, and the other lab members for advice and discussions. This project was supported, in part, by grants from NIH (R01-CA155693), Department of Defense (W81XWH-13-1-0352 and PC130483), CPRIT (RP120380), and the MDACC RNA Center-Laura & John Arnold Foundation grant (D.G.T). C. Jeter was supported, in part, by CPRIT RP120394. C. Liu was supported, in part, by DOD post-doctoral fellowship (PC121553) and NSFC grant 81302208. R. Liu was supported, in part, by NSFC grant 81302209, Shanghai Municipal Natural Science Fund 13ZR1459900, and the Fundamental Research Funds for Central Universities 2012KJ041. M. Jin was supported by the China Scholar Council (CSC) as a joint-training Ph.D student from Tongji Medical College of Huazhong University of Science and Technology, China, and the Department of Molecular Carcinogenesis of the University of Texas M.D Anderson Cancer Center, USA.
Footnotes
Disclosure of Potential Conflicts of Interest:
The authors claim no potential conflicts of interest.
Authors’ Contributions:
Conception and design:M. Jin, C. Liu, D.G. Tang
Development of methodology: M. Jin, C. Liu, D.G. Tang
Acquisition of data: M. Jin, D.G. Tang
Analysis and interpretation of data: M. Jin, C. Liu, D.G. Tang
Writing, review, and/or revision of the manuscript: M. Jin, C. Liu, R. Liu, D.G. Tang
Administrative, technical, or material support:D.G. Tang
Study supervision: T. Zhang, D.G. Tang
Coordinating collaborating groups: D.G. Tang
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Egan A, Dong Y, Zhang H, Qi Y, Balk SP, Sartor O. Castration-resistant prostate cancer: Adaptive responses in the androgen axis. Cancer Treat Rev. 2014;40:426–33. doi: 10.1016/j.ctrv.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22:457–72. doi: 10.1038/cr.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, Tang DG. Prostate cancer stem cells and their potential roles in metastasis. J Surg Oncol. 2011;103:558–62. doi: 10.1002/jso.21806. [DOI] [PubMed] [Google Scholar]
- 5.Liu T, Xu F, Du X, Lai D, Zhao Y, Huang Q, et al. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Mol Cell Biochem. 2010;340:265–73. doi: 10.1007/s11010-010-0426-5. [DOI] [PubMed] [Google Scholar]
- 6.Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–63. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 7.Makeyev EV, Maniatis T. Multilevel regulation of gene expression by microRNAs. Science. 2008;319:1789–90. doi: 10.1126/science.1152326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirotkin AV, Laukova M, Ovcharenko D, Brenaut P, Mlyncek M. Identification of microRNAs controlling human ovarian cell proliferation and apoptosis. J Cell Physiol. 2010;223:49–56. doi: 10.1002/jcp.21999. [DOI] [PubMed] [Google Scholar]
- 9.Lee YS, Kim HK, Chung S, Kim KS, Dutta A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J Biol Chem. 2005;280:16635–41. doi: 10.1074/jbc.M412247200. [DOI] [PubMed] [Google Scholar]
- 10.Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- 11.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez I, Almstead LL, DiMaio D. MicroRNAs and senescence. Aging. 2011;3:77–8. doi: 10.18632/aging.100282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivey KN, Srivastava D. MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cells. 2010;7:36–41. doi: 10.1016/j.stem.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the microRNA pathway. Nature. 2005;435:974–8. doi: 10.1038/nature03816. [DOI] [PubMed] [Google Scholar]
- 15.Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett. 2009;285:116–26. doi: 10.1016/j.canlet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 16.Kashat M, Azzouz L, Sarkar SH, Kong D, Li Y, Sarkar FH. Inactivation of AR and Notch-1 signaling by miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res. 2012;4:432–42. [PMC free article] [PubMed] [Google Scholar]
- 17.Schubert M, Spahn M, Kneitz S, Scholz CJ, Joniau S, Stroebel P, et al. Distinct microRNA expression profile in prostate cancer patients with early clinical failure and the impact of let-7 as prognostic marker in high-risk prostate cancer. PLoS ONE. 2013;8:e65064. doi: 10.1371/journal.pone.0065064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reis ST, Pontes-Junior J, Antunes AA, Dall’Oglio MF, Dip N, Passerotti CC, et al. miR-21 may acts as an oncomir by targeting RECK, a matrix metalloproteinase regulator, in prostate cancer. BMC Urol. 2012;12:14. doi: 10.1186/1471-2490-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi XB, Xue L, Ma AH, Tepper CG, Kung HJ, White RW. miR-125b promotes growth of prostate cancer xenograft tumor through targeting pro-apoptotic genes. Prostate. 2011;71:538–49. doi: 10.1002/pros.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun T, Wang X, He HH, Sweeney CJ, Liu SX, Brown M, et al. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene. 2013 Jun 17; doi: 10.1038/onc.2013.230. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nature Med. 2011;17:211–5. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu C, Kelnar K, Vlassov AV, Brown D, Wang J, Tang DG. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res. 2012;72:3393–404. doi: 10.1158/0008-5472.CAN-11-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan AP, Poisson LM, Bhat VB, Fermin D, Zhao R, Kalyana-Sundaram S, et al. Quantitative proteomic profiling of prostate cancer reveals a role for miR-128 in prostate cancer. Mol Cell Proteomics. 2010;9:298–312. doi: 10.1074/mcp.M900159-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–70. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smirnova L, Grafe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005;21:1469–77. doi: 10.1111/j.1460-9568.2005.03978.x. [DOI] [PubMed] [Google Scholar]
- 26.Venkataraman S, Alimova I, Fan R, Harris P, Foreman N, Vibhakar R. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS ONE. 2010;5:e10748. doi: 10.1371/journal.pone.0010748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 28.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. The PSA−/lo prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–69. doi: 10.1016/j.stem.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Chen X, Calhoun-Davis T, Claypool K, Tang DG. PC3 human prostate carcinoma cell holoclones contain self-renewing tumor-initiating cells. Cancer Res. 2008;68:1820–5. doi: 10.1158/0008-5472.CAN-07-5878. [DOI] [PubMed] [Google Scholar]
- 30.Laffin B, Tang DG. An old player on a new playground: Bmi-1 as a regulator of prostate stem cells. Cell Stem Cell. 2010;7:639–40. doi: 10.1016/j.stem.2010.11.019. [DOI] [PubMed] [Google Scholar]
- 31.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+α2β1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67:6796–805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 32.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 33.Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, et al. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2007;21:415–26. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 34.Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–8. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 35.Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB, et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci USA. 2007;104:19971–6. doi: 10.1073/pnas.0709313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masri S, Liu Z, Phung S, Wang E, Yuan YC, Chen S. The role of microRNA-128 in regulating TGFbeta signaling in letrozole-resistant breast cancer cells. Breast Cancer Res Treat. 2010;124:89–99. doi: 10.1007/s10549-009-0716-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyd LK, Mao X, Lu YJ. The complexity of prostate cancer: genomic alterations and heterogeneity. Nat Rev Urol. 2012;9:652–64. doi: 10.1038/nrurol.2012.185. [DOI] [PubMed] [Google Scholar]
- 38.Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell. 2010;7:682–93. doi: 10.1016/j.stem.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan C, He L, Kapoor A, Gillis A, Rybak AP, Cutz JC, et al. Bmi1 promotes prostate tumorigenesis via inhibiting p16(INK4A) and p14(ARF) expression. Biochim Biophys Acta. 2008;1782:642–8. doi: 10.1016/j.bbadis.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Siddique HR, Parray A, Zhong W, Karnes RJ, Bergstralh EJ, Koochekpour S, et al. BMI1, stem cell factor acting as novel serum-biomarker for Caucasian and African-American prostate cancer. PLoS ONE. 2013;8:e52993. doi: 10.1371/journal.pone.0052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeter C, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993–1005. doi: 10.1002/stem.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–45. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–30. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 44.Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, et al. Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res. 2011;17:7105–15. doi: 10.1158/1078-0432.CCR-11-0071. [DOI] [PubMed] [Google Scholar]
- 45.Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res. 2011;71:5950–4. doi: 10.1158/0008-5472.CAN-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.