

Abstract
Bendamustine, a hybrid molecule of purine analog and alkylator, induces cell death by activation of apoptosis, DNA damage response, and mitotic catastrophe. Entinostat, a selective class I inhibitor of histone deacetylase (HDAC), exerts anti-tumor activity in various cancer types, including multiple myeloma (MM). We sought to determine the combinatorial effects of bendamustine and entinostat on MM cells. Cell growth assays showed that bendamustine or entinostat inhibited proliferation in a dose-dependent manner, and their combinations synergistically induced growth inhibition in all MM cells tested. An apoptotic-ELISA and western blot assays on PARP cleavage and caspase-8 and caspase-3 revealed that bendamustine in combination with entinostat exhibited a much more potent activity than either agent alone to promote the MM cells undergoing apoptosis in a dose-dependent manner. Flow cytometric analysis found that entinostat exhibited distinct effects on cell cycle progression in different lines and bendamustine mainly arrested the cells at S phase, whereas their combinations dramatically blocked the S cells entering G2/M phase. Furthermore, studies on DNA damage response indicated that phospho-histone H2A.X (P-H2A.X), a hall marker of DNA double strand break, along with phosphorylated CHK2 (P-CHK2) was significantly enhanced by the combinations of bendamustine and entinostat as compared to either agent alone. These molecular changes were correlated with the increases in mitotic catastrophe. Collectively, our data demonstrate that bendamustine in combination with entinostat exhibit potent anti-proliferative/anti-survival activity in MM cells via induction of apoptosis and DNA damage response. Regimens consisting of bendamustine and/or entinostat may represent novel therapeutic strategies against MM.
Keywords: Bendamustine, Entinostat, Apoptosis, DNA damage, Multiple myeloma
1. Introduction
Multiple myeloma (MM) is a plasma-cell neoplasm which is characterized by clonal proliferation of malignant plasma cells and the symptoms of skeletal destruction, renal impairment, and hematological dysfunctions [1]. Despite recent progress in understanding the biology of MM and developing novel agents and strategies, the prognosis of most MM patients is still poor, and resistance to traditional chemotherapy occurs frequently. A number of investigators have focused on studying the aberrant signaling pathways in the pathogenesis of MM and identifying abnormal protein expression involved in these pathways. Such study gives promise to targeted therapy and drug combination to overcome resistance [2].
Bendamustine was first synthesized in 1960s in German. Similarly but not the same as other alkylating agents, bendamustine combines the alkylating activity of the mustard group with the anti-metabolite activity of the purine analog structure, which makes it have a unique pharmacological profile. It has been reported that bendamustine has long-lasting DNA damage action, and can induce apoptosis and mitotic catastrophe and inhibit mitotic checkpoint. In addition, it does not show cross-resistance with other cytotoxic agents [3]. In preclinical studies, bendamustine is able to overcome resistance to other alkylating agents [4], and shows synergistic inhibitory effects with cladribine or rituximab on lymphoma cells or xenograft models [5,6]. Recent clinical trials have shown that bendamustine combined with first line agents is safe and effective in the treatment of relapsed and/or refractory chronic lymphocytic leukemia (CLL) [7], lymphoma [8,9], and MM [10,11], which brings prospect to explore the therapeutic potential of bendamustine combining with other agents.
Histone acetylation is a major epigenetic modification which regulates gene expression and affects tumorigenesis in various human cancers, including MM. It is controlled by the balance between histone deacetylase (HDAC) and histone acetyltransferase (HAT) [12]. HDACs modulate many oncogenes and tumor suppressor genes instead of directly affecting oncogenesis [13]. In addition, the function of many non-histone proteins is also controlled by HDACs. In cancer cells, HDACs regulate many biological functions including proliferation, differentiation, apoptosis and survival by modulating multiple factors in signaling pathways. Furthermore, higher levels of histone acetylation are found in normal tissues as compared to tumors, and HDACs are typically overexpressed in tumor cells. Therefore, HDACs are considered as promising targets for cancer therapy, and several HDAC inhibitors (HDACi) have been designed to target different types of HDACs [14]. Entinostat (also known as SNDX-275 or MS-275) is a synthetic benzamide derivative which inhibits class I HDACs. In vitro or in vivo studies demonstrated that entinostat exhibits anti-tumor activity in multiple solid tumors or hematological malignancies [15]. Our previous studies showed that entinostat induces apoptosis via down-regulation of erbB3 expression, enhances efficacy of trastuzumab, and has potential to overcome trastuzumab resistance in erbB2-overexpressing breast cancer cells [16,17]. We have also found that entinostat in combination with melphalan synergistically enhances DNA damage response and apoptosis in MM cells [18]. In this study, we sought to determine the combinatorial effects of bendamustine and entinostat on growth inhibition, apoptosis, and DNA damage response in MM cells, with the hope to develop novel therapeutic strategies against MM.
2. Materials and methods
2.1. Reagents and antibodies
Bendamustine (Cephalon Inc., Frazer, PA) and entinostat (Syndax Pharmaceuticals, Inc., Waltham, MA) were dissolved in dimethyl sulfoxide (DMSO) to make a stock solution at 526 mmol/L and 20 mmol/L, respectively. The stock solutions were stored at −20 °C.
The sources of antibodies for western blot were as follows: caspase-3 rabbit mAb (8G10), caspase-8 mouse mAb (1C12), PARP rabbit mAb, P-Histone H2A.X (Ser139) rabbit antibody, Histone H2A rabbit polyclonal antibody II, P-CHK1 (Ser345) (133D3) rabbit mAb, CHK1 rabbit antibody, P-CHK2 (Thr68) rabbit poly-clonal antibody, and CHK2 rabbit polyclonal antibody (Cell Signaling Technology, Inc., Beverly, MA); β-actin mouse mAb (clone AC-75) (Sigma Chemical Co., St. Louis, MO). All other reagents were purchased from Sigma unless otherwise specified.
2.2. Cells and cell culture
Human MM cell line U266 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). Human MM cell line MM1.S and MM1.R [19] were kindly provided by Dr. Steven Rosen (Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL). All cell lines were maintained in RPMI1640 cell culture medium supplemented with 10% fetal bovine serum (FBS) at a 37 °C humidified atmosphere containing 95% air and 5% CO2 and were split twice a week.
2.3. Cell proliferation assays
The CellTiter96™ AQ non-radioactive cell proliferation kit (Promega Corp., Mad-ison, WI) was used to evaluate cell viability as we previously described [18]. In brief, cells were plated on 96-well plates with 0.1 ml complete medium containing 0.5% FBS as control, or 0.1 ml of the same medium with either bendamustine or entinostat alone, or their combinations, and incubated for 72 h in a cell culture incubator. After reading all wells at 490 nM with a microplate reader, the percentages of surviving cells from each group relative to controls, defined as 100% survival, were determined by reduction of MTS.
2.4. Quantification of apoptosis
An apoptosis ELISA kit (Roche Diagnositics Corp., Indianapolis, IN) was used to quantitatively measure cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) as previously reported [18].
2.5. Western blot analysis
Protein expression levels were measured as previously described [18]. In brief, cells were lysed in a buffer containing 50 mM Tris, pH 7.4, 50 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 25 μg/ml leupeptin, and 25 μg/ml aprotinin. The protein concentrations of total cell lysates were measured by the Coomassie Plus protein assay reagent (Pierce Chemical Co., Rockford, IL). Equal amounts of cell lysates were boiled in Laemmli SDS-sample buffer, resolved by SDS–PAGE, and western blot analysis with specific antibodies as described in the figure legends.
2.6. Flow cytometric analysis of cell cycle
Flow cytometric analyses were performed as described previously [18] to define the cell cycle distribution and apoptosis for treated and untreated cells. In brief, cells grown in 100-mm culture dishes were harvested and fixed with 70% ethanol. Cells were then stained for total DNA content with a solution containing 50 μg/ml propidium iodide and 100 μg/ml RNase I in PBS for 30 min at 37 °C. Cell cycle distribution was analyzed at the Flow Cytometry Core Facility of University of Colorado Cancer Center with a FACScan flow cytometer (BD Biosciences, San Jose, CA).
2.7. Morphologic evaluation of mitotic catastrophe
Cultured MM cells were harvested, resuspended with RPMI1640 medium, and cytocentrifuged for 1 min at 1000 rpm. Cells were fixed in methanol for 5 min, and then stained in Jenner solution for 5 min. Samples were transferred into Giemsa solution for at least 45 min, and then rinsed in distilled water. Slides were examined under a photomicroscope (Olympus). Pathologists were blinded on each slide set regarding the treatment group. Cells that showed abnormal mitotic figures, chromatin condensation and fragmentation were counted against normal cells, and reported in percentage.
2.8. Statistical analysis
Statistical analyses of the experimental data were performed using a two-sided Student’s t test. Significance was set at a P < 0.05. Calculation of IC50, combination index (CI) and evaluation of synergy vs antagonism between bendamustine and entinostat were performed using the Calcusyn software (Biosoft, Ferguson, MO), which was designed based on Chou–Talalay method [19,20]. CI values less than, equal to and more than 1 represent synergistic, additive and antagonistic effects, respectively.
3. Results
3.1. Bendamustine in combination with entinostat enhances growth inhibition of MM cells, and is synergistic over a wide range of effects
To explore whether bendamustine or entinostat might have therapeutic potential against MM, we first performed cell growth assays using U266, dexamethasone-sensitive (MM1.S) and dexamethasone-resistant (MM1.R) cell lines. Upon treatment with a serious dose of bendamustine or entinostat for 72 h, the proliferation of all three cell lines was significantly inhibited, although U266 cells were less sensitive to both agents than the other two cell lines (Fig. 1A and B). The response of MM cells to entinostat was in accordance with our previous findings [18]. It appeared that MM1.R cells were more sensitive to the agents, especially entinostat, than MM1.S cells (Fig. 1A and B). Thus, both bendamustine and entinostat were able to inhibit proliferation of dexamethasone-sensitive and -resistant MM cells in a dose-dependent manner.
Fig. 1.

Bendamustine or entinostat alone inhibits proliferation of MM cells in a dose-dependent manner. Human MM cells were plated onto 96-well plates with fresh RPMI1640 medium (0.5% FBS) or same medium containing indicated concentrations of bendamustine (Benda) or entinostat (Ent) for 72 h. The percentages of surviving cells as compared to controls, defined as 100% survival, were determined by reduction of MTS. Data shows the representative of three independent experiments. Bars, SD. (A) bendamustine; (B) entinostat.
Next, we sought to determine whether the combination of bendamustine and entinostat may further enhance their inhibitory effects on MM cells. After treating cells with single agent or their combinations in a fixed ratio for 72 h, we observed a significant growth inhibition upon combinatorial treatment as compared with either agent alone (Fig. 2A). The IC50s of bendamustine when used in combination with entinostat for U266, MM1.S and MM1.R cells were approximately 132.8, 13.7, and 34.5 μmol/L, respectively. In contrast, The IC50s of bendamustine when used alone for U266, MM1.S and MM1.R cells were approximately 375, 86.9, and 83.8 μmol/L, respectively. The combinatorial anti-proliferation activity was much more potent in MM1.S and MM1.R cells than that in U266 cells, which is consistent with single agent treatment. It should be emphasized that the combination enhanced inhibition dramatically at the concentration of 50 μmol/L (bendamustine) and 0.2 μmol/L (entinostat) in MM1.S cells, even though no inhibition was observed with entinostat (0.2 μmol/L) alone (Fig. 2A). This result promoted us to further explore whether the two agents may have synergistic effect. We performed combination index (CI) analysis according to the Chou–Talalay equation [19,20]. The curves showed that bendamustine and entinostat exhibit a synergistic activity over a wide range of effects with CI = 0.531 ± 0.1339 at IC50s (fraction of cells affected = 0.5) in U266 cells. Similar results were obtained with MM1.S and MM1.R (Fig. 2B). In conclusion, the combination of bendamustine and entinostat synergistically induced growth inhibition in MM cells.
Fig. 2.

Combination of bendamustine and entinostat significantly induces growth inhibition of MM cells, and is synergistic over a wide range of effects. (A) Human MM cells were plated onto 96-well plates with fresh RPMI1640 medium (0.5% FBS) or same medium containing indicated concentrations of bendamustine (Benda) or entinostat (Ent) or their combinations with a fixed ratio for 72 h. The percentages of surviving cells as compared to controls, defined as 100% survival, were determined by reduction of MTS. Data shows the representative of three independent experiments. Bars, SD. P values vs bendamustine single agent. (B) The combination index (CI) curves were calculated using Calcusyn software according to the Chou–Talalay equation.
3.2. Combination of bendamustine and entinostat significantly promotes MM cells undergoing apoptosis and induces cell cycle S phase arrest
To elucidate the molecular mechanism of bendamustine and entinostat-mediated anti-proliferation/anti-survival effects, we first tested whether bendamustine and/or entinostat may induce apoptosis in MM cells. A specific apoptotic ELISA showed that entinostat alone (0.1 μmol/L) induced minor apoptotic effect in U266, MM1.S and MM1.R cells (Fig. 3A). However, bendamustine alone induced apoptosis in a dose-dependent manner, and this effect was significantly enhanced after entinostat (0.1 μmol/L) was added into bendamustine (Fig. 3A). Furthermore, western blot analysis revealed that the combination of bendamustine and entinostat as compared to either agent alone more potently induced PARP cleavage, the hall mark of apoptosis, and activation of caspase-8 and -3 evidenced by the increases of cleaved caspase-8 and -3 in all three cell lines (Fig. 3B–D). These data indicate that entinostat significantly accelerates bendamustine-induced apoptosis in MM cells via caspase-dependent signaling pathways. Next, we examined the effects of bendamustine and/or entinostat on cell cycle progression in MM cells. In all three lines tested, treatment with bendamustine alone clearly increased the percentage of cells at S phase, which was correlated with the reduction of G1 cells (Fig. 4). However, entinostat exhibited distinct effects on different MM cells. It appeared entinostat increased G1 percentage and reduced G2/M phase in U266 cells, whereas it had a minor effect on cell cycle progression in MM1.S cells (Fig. 4A and B). In contrast, entinostat dramatically increased the MM.1R cells at S phase which was associated with a significant reduction of G1 cells (Fig. 4C). Entinostat’s effects on MM.1R cells were similarly observed in RPMI8226 cells [18]. More importantly, the combinations of entinostat and bendamustine induced a profound S phase arrest with no or few G2/M cells left in all three MM lines (Fig. 4). These data suggest that the combinatorial effects mainly blocked the MM cells at S phase to enter G2/M phase.
Fig. 3.

Combination of bendamustine and entinostat significantly promotes MM cells undergoing apoptosis. MM cells were cultured with RPMI1640 (0.5% FBS) in the absence or presence of either entinostat (Ent) or bendamustine (Benda) alone, or the combinations of entinostat and bendamustine for 24 h. Cells were collected and subjected to apoptotic ELISA (A) or western blot analyses with specific antibody directed against PARP, caspase-8 (Casp-8), caspase-3 (Casp-3), or β-actin (B, C, D). Bars, SD. P values vs bendamustine single agent.
Fig. 4.
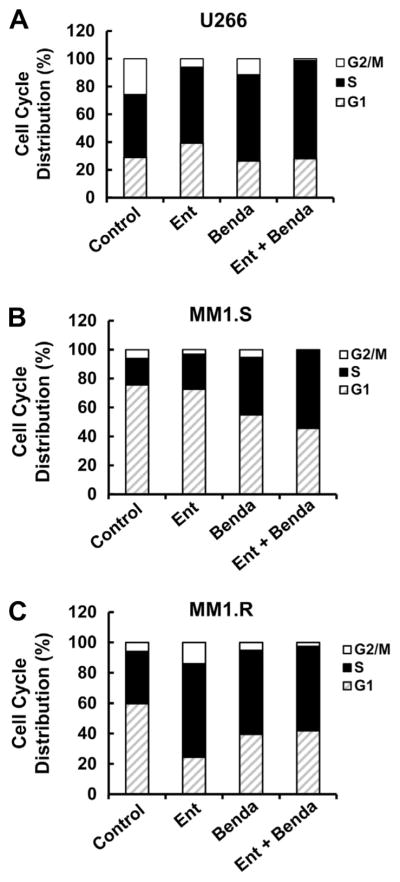
Entinostat and bendamustine block cell cycle progression. MM cells were cultured with RPMI1640 (0.5% FBS) in the absence or presence of entinostat (Ent, 0.1 μmol/L), bendamustine (Benda, 200 μmol/L for U266 cells, 100 μmol/L for MM1.S and MM1.R cells) alone or the combinations of entinostat and bendamustine for 24 h. Cells were harvested and subjected to flow cytometric analysis of cell cycle distribution. The bar graph reflects the percentage of cells in G1, S, or G2/M phase of the cell cycle. Data shows the representative of three independent experiments. (A) U266; (B) MM1.S; (C) MM1.R.
3.3. Combination of bendamustine and entinostat significantly enhances DNA damage response associated with enhanced mitotic catastrophe
Since bendamustine induces DNA damage response and mitotic catastrophe of cancer cells, which are major mechanisms for treatment, we next focused studies on investigating whether entinostat amplifies bendamustine-induced DNA damage response and mitotic catastrophe. We examined the expression of DNA damage checkpoint proteins after treating MM cells with single agent or their combinations for 24 h. Treatment with entinostat alone (0.1 μmol/L) did not increase P-H2A.X, P-CHK1, or P-CHK2 in either cell lines except MM1.S with minor increase of H2A.X phosphorylation (Fig. 4). However, the levels of P-H2A.X were dramatically increased following adding entinostat (0.1 μmol/L) to bendamustine treatment in all three cell lines, while the potent induction of P-CHK2 by combinatorial treatment was only observed in MM1.S cells (Fig. 4). In addition, bendamustine mainly upregulated P-CHK2 in U266 and MM1.S cells, whereas it enhanced CHK1 phosphorylation in all three cell lines. Moreover, morphologic observations revealed a significant increase in aberrant cells with mitotic catastrophe when treated with both bendamustine and entinostat as compared to either agent alone (Fig. 5), which was consistent with the results of our studies on apoptosis (Fig. 3) and DNA damage response (Fig. 4). Taken together, these data indicate that entinostat significantly enhances bendamustine-induced DNA damage response via induction of P-H2A.X and/or P-CHK2 in MM cells, and subsequently promotes the cells undergoing morphologic abnormalities and apoptosis.
Fig. 5.

Combination of bendamustine and entinostat enhances DNA damage response. MM cells were cultured with RPMI1640 (0.5% FBS) in the absence or presence of entinostat (Ent), bendamustine (Benda) alone or the combinations of entinostat and bendamustine for 24 h. Cells were collected and subjected to western blot analyses with specific antibody directed against P-H2AX, H2AX, P-CHK1, CHK1, P-CHK2, CHK2 or β-actin. (A) U266; (B) MM1.S; (C) MM1.R.
4. Discussion
Recent advances in identifying novel therapeutic agents and combination strategies for MM treatment have provided promising results. As an old drug, bendamustine has being given new perspective in treating hematologic malignancies such as CLL, lymphoma, and MM [21–23]. On the other side, HDACis are widely investigated for cancer treatment. Several clinical trials are ongoing to evaluate the efficacy of vorinostat, panobinostat, ITF2357 and belinostat in treating relapse/refractory MM patients [24–27]. We and others show that HDACi presents synergistic effects with conventional alkylator melphalan [18,28]. Here we provide strong evidence indicating that bendamustine shows anti-MM activity when used as single agent, and its capability to induce apoptosis of MM cells is synergistically potentiated by the specific HDACi entinostat via enhanced DNA damage response and mitotic catastrophe. Furthermore, either bendamustine alone or its combination with entinostat exhibits therapeutic potential to overcome dexamethasone resistance.
Among the three MM cell lines we tested, U266 cells appeared to be less sensitive to either bendamustine or entinostat than the other two cell lines in the presence of effective drug concentration nearly out of the range of peak plasma concentrations (approximately 129 μmol/L for bendamustine and 0.34 μmol/L for entinostat) [29,30]. In contrast, similar inhibitory activity of either agent or their combinations was found in dexamethasone-sensitive and -resistant cells (Fig. 1). The concentrations of bendamustine and entinostat we evaluated for apoptosis, cell cycle progression, and mitotic catastrophe were all within their clinically tolerated concentrations (Figs. 3–6). Thus, it is feasible to explore their efficacy in managing dexamethasone-resistant MM in an in vivo animal model, and subsequently in clinical trials of MM patients.
Fig. 6.

Combination of bendamustine and entinostat induces mitotic catastrophe. (A) MM1.S cells were cultured with RPMI1640 (0.5% FBS) in the absence or presence of entinostat, bendamustine alone or the combinations of entinostat (Ent) and bendamustine (Benda) for 24 h. Cells were collected and subjected to cytospin onto cell slides followed by Giemsa staining and examination under a photomicroscope (×100 magnification). Arrows indicate the cells with mitotic catastrophe. (B) Cells that showed atypical mitotic figures, multi-nucleation, atypical chromosome clusters, and/or apoptosis were counted against normal cells, and reported in percentage. Data shows the representative of three independent experiments. Bars, SD. P values vs bendamustine single agent.
Most MM patients are treated with multi-drug chemotherapy regimens. The clinical trials using HDACi as monotherapy for MM appeared not to get much benefit as compared to those traditional chemotherapeutics [25,31,32]. Thus, although either bendamustine or entinostat alone showed potent inhibitory effects on MM cells, it is more practical to find their therapeutic potential in the context of combinations. It has been reported that HDACis, such as romidepsin, vorinostat and panobinostat exhibit synergistic effects with bortezomib on growth inhibition of MM cells [33–35]. HDACis combined with DNA-damaging agents or other epigenetic therapies were also investigated [36]. While majority of the studies focused on synergistic effects to induce apoptosis and cell cycle distribution, we sought to determine the alterations of DNA damage response and mitotic catastrophe as well. Our data showed that the levels of P-H2A.X and/or P-CHK2 were significantly upregulated upon the combinatorial treatment of entinostat and bendamustine (Fig. 5). These data provide strong evidence supporting that entinostat potentiates bendamustine-induced apoptosis through enhancing DNA damage responses. Since chromatin structure is critical for cells to sense and repair double strand breaks [37], we hypothesize that treatment with low dose entinostat may loosen the chromatin conformation making DNA-damaging agents easier to destroy DNA structure. Further studies are warranted to test this hypothesis.
It is possible that apoptotic/survival-related cell signaling may be altered with the combinatorial treatment of entinostat and bendamustine. Our data showed that entinostat did enhance bendamustine-induced apoptosis via activation of caspase-8 and -3 (Fig. 3), but the significant decrease of phosphorylated Stat3 was only detected in U266 cells in our explorations for PI-3K/Akt, Ras/MAPK, and JAK/Stat3 signaling pathways (data not shown). This phenomenon suggests that these three pathways may not be important in MM1.S and MM1.R cell survival in the context of entinostat and bendamustine treatment. Advances in unveiling MM pathogenesis identified several key signaling which control cell biology [38]. We are currently trying to find out if other signaling pathways, such as NF-κB and IGF-1/IGF-1R may play a critical role in promoting proliferation and survival of MM cells.
In summary, we demonstrate that entinostat, a specific class I HDACi, synergistically enhances bendamustine-induced growth inhibition and apoptosis in MM cells mainly through induction of DNA damage response and mitotic catastrophe. This combinatorial activity is equally observed in dexamethasone-sensitive and -resistant MM cells. Our studies suggest that bendamustine in combination with epigenetic therapy, such as entinostat may be promising for managing MM patients and overcoming dexamethasone resistance.
Acknowledgments
The authors are grateful to Dr. Stephen P. Trusko (Cephalon, Inc., Frazer, PA) for providing bendamustine, to Dr. Peter Ordentlich (Syndax Pharmaceuticals, Inc., Waltham, MA) for providing entinostat, and to Ms. Lisa Litzenberger for her excellent assistance in arts preparation. This work was supported in part by a Research Fund from Cephalon, Inc. (to CKL & BL). BC was sponsored by China Scholarship Council (2011006001).
Abbreviations
- MM
Multiple myeloma
- CLL
chronic lymphocytic leukemia
- HDAC
histone deacetylase
- HDACi
Inhibitor of HDAC
- HAT
Histone acetyltransferase
- PARP
poly(ADP-ribose) polymerase
- ELISA
enzyme-linked immunosorbent assay
- PAGE
polyacrylamide gel electrophoresis
- IC50
inhibitory concentration 50
- CI
combination index
- PI-3K
phosphoinositide 3-kinase
- MAPK
Mitogen-activated protein kinase
- IGF-1
insulin-like growth factor-1
- IGF-1R
IGF-1 receptor
- JAK
c-Jun N-terminal kinase
- STAT
signal transducers and activators of transcription
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
Footnotes
Conflicts of interest statement
The authors declare that they have no conflicts of interest.
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KC. New insights into therapeutic targets in myeloma. Hemat Am Soc Hemat Educ Program. 2011;2011:184–190. doi: 10.1182/asheducation-2011.1.184. [DOI] [PubMed] [Google Scholar]
- 3.Leoni LM, Bailey B, Reifert J, Bendall HH, Zeller RW, Corbeil J, Elliott G, Niemeyer CC. Bendamustine (Treanda) displays a distinct pattern of cytotoxicity and unique mechanistic features compared with other alkylating agents. Clin Cancer Res. 2008;14:309–317. doi: 10.1158/1078-0432.CCR-07-1061. [DOI] [PubMed] [Google Scholar]
- 4.Kalaycio M. Bendamustine: a new look at an old drug. Cancer. 2009;115:473–479. doi: 10.1002/cncr.24057. [DOI] [PubMed] [Google Scholar]
- 5.Chow KU, Boehrer S, Geduldig K, Krapohl A, Hoelzer D, Mitrou PS, Weidmann E. In vitro induction of apoptosis of neoplastic cells in low-grade non-Hodgkin’s lymphomas using combinations of established cytotoxic drugs with bendamustine. Haematologica. 2001;86:485–493. [PubMed] [Google Scholar]
- 6.Kanekal S, Crain B, Elliott G, Multani PS. SDX-105 (Treanda (TM)) enhances the tumor growth inhibitory effect of rituximab in Daudi lymphoma xenografts. Blood. 2004;104:229b. [Google Scholar]
- 7.Fischer K, Cramer P, Busch R, Stilgenbauer S, Bahlo J, Schweighofer CD, Bottcher S, Staib P, Kiehl M, Eckart MJ, Kranz G, Goede V, Elter T, Buhler A, Winkler D, Kneba M, Dohner H, Eichhorst BF, Hallek M, Wendtner CM. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29:3559–3566. doi: 10.1200/JCO.2010.33.8061. [DOI] [PubMed] [Google Scholar]
- 8.Visani G, Malerba L, Stefani PM, Capria S, Galieni P, Gaudio F, Specchia G, Meloni G, Gherlinzoni F, Giardini C, Falcioni S, Cuberli F, Gobbi M, Sarina B, Santoro A, Ferrara F, Rocchi M, Ocio EM, Caballero MD, Isidori A. BeEAM (bendamustine, etoposide, cytarabine, melphalan) before autologous stem cell transplantation is safe and effective for resistant/relapsed lymphoma patients. Blood. 2011;118:3419–3425. doi: 10.1182/blood-2011-04-351924. [DOI] [PubMed] [Google Scholar]
- 9.Fowler N, Kahl BS, Lee P, Matous JV, Cashen AF, Jacobs SA, Letzer J, Amin B, Williams ME, Smith S, Saleh A, Rosen P, Shi H, Parasuraman S, Cheson BD. Bortezomib, bendamustine, and rituximab in patients with relapsed or refractory follicular lymphoma: the phase II VERTICAL study. J Clin Oncol. 2011;29:3389–3395. doi: 10.1200/JCO.2010.32.1844. [DOI] [PubMed] [Google Scholar]
- 10.Lentzsch S, O’Sullivan A, Kennedy RC, Abbas M, Dai L, Pregja SL, Burt S, Boyiadzis M, Roodman GD, Mapara MY, Agha M, Waas J, Shuai Y, Normolle D, Zonder JA. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: results of phase 1/2 open-label, dose escalation study. Blood. 2012;119:4608–4613. doi: 10.1182/blood-2011-12-395715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramasamy K, Hazel B, Mahmood S, Corderoy S, Schey S. Bendamustine in combination with thalidomide and dexamethasone is an effective therapy for myeloma patients with end stage renal disease. Br J Haematol. 2011;155:632–634. doi: 10.1111/j.1365-2141.2011.08754.x. [DOI] [PubMed] [Google Scholar]
- 12.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 14.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 15.Knipstein J, Gore L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin Inv Drug. 2011;20:1455–1467. doi: 10.1517/13543784.2011.613822. [DOI] [PubMed] [Google Scholar]
- 16.Huang X, Gao L, Wang S, Lee CK, Ordentlich P, Liu B. HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res. 2009;69:8403–8411. doi: 10.1158/0008-5472.CAN-09-2146. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Wang S, Lee CK, Yang X, Liu B. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 2011;307:72–79. doi: 10.1016/j.canlet.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Lee CK, Wang S, Huang X, Ryder J, Liu B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010;296:233–240. doi: 10.1016/j.canlet.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 20.Chou TC, Talaly P. A simple generalized equation for the analysis of multiple inhibitions of Michaelis-Menten kinetic systems. J Biol Chem. 1977;252:6438–6442. [PubMed] [Google Scholar]
- 21.Cheson BD, Rummel MJ. Bendamustine: rebirth of an old drug. J Clin Oncol. 2009;27:1492–1501. doi: 10.1200/JCO.2008.18.7252. [DOI] [PubMed] [Google Scholar]
- 22.Montillo M, Ricci F, Tedeschi A, Vismara E, Morra E. Bendamustine: new perspective for an old drug in lymphoproliferative disorders. Expert Rev Hematol. 2010;3:131–148. doi: 10.1586/ehm.10.7. [DOI] [PubMed] [Google Scholar]
- 23.Stewart AK. Novel therapies for relapsed myeloma. Hematol Am Soc Hematol Educ Program. 2009:578–586. doi: 10.1182/asheducation-2009.1.578. [DOI] [PubMed] [Google Scholar]
- 24.Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, Harris C, Zwiebel J, Wright JJ, Espinoza-Delgado I, Baer MR, Holleran JL, Egorin MJ, Grant S. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15:5250–5257. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N, Rambaldi A. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89:185–190. doi: 10.1007/s00277-009-0793-8. [DOI] [PubMed] [Google Scholar]
- 26.Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81:170–176. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 27.Wolf JL, Siegel D, Goldschmidt H, Hazell K, Bourquelot PM, Bengoudifa BR, Matous J, Vij R, de Magalhaes-Silverman M, Abonour R, Anderson KC, Lonial S. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma. 2012;53:1820–1823. doi: 10.3109/10428194.2012.661175. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez E, Shen J, Steinberg J, Li M, Wang C, Bonavida B, Chen H, Li ZW, Berenson JR. The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk Res. 2011;35:373–379. doi: 10.1016/j.leukres.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 29.Rasschaert M, Schrijvers D, Van den Brande J, Dyck J, Bosmans J, Merkle K, Vermorken JB. A phase I study of bendamustine hydrochloride administered once every 3 weeks in patients with solid tumors. Anticancer Drug. 2007;18:587–595. doi: 10.1097/CAD.0b013e3280149eb1. [DOI] [PubMed] [Google Scholar]
- 30.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–3922. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 31.Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, Chen-Kiang S, Sparano JA. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117:336–342. doi: 10.1002/cncr.25584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, Sun L, Ricker J, Rizvi S, Oerth C, Atkins B, Fearen I, Anderson K, Siegel D. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008;49:502–507. doi: 10.1080/10428190701817258. [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K, Noborio-Hatano K, Nobuyoshi M, Ozawa K, Kano Y, Furukawa Y. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406–417. doi: 10.1182/blood-2009-07-235663. [DOI] [PubMed] [Google Scholar]
- 34.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–3852. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 35.Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, Tassone P, Atadja P, Chauhan D, Munshi NC, Anderson KC. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108:3441–3449. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller CP, Singh MM, Rivera-Del Valle N, Manton CA, Chandra J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J Biomed Biotechnol. 2011;2011:514261. doi: 10.1155/2011/514261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkitaraman AR. Modifying chromatin architecture during the response to DNA breakage. Crit Rev Biochem Mol Biol. 2010;45:2–13. doi: 10.3109/10409230903325446. [DOI] [PubMed] [Google Scholar]
- 38.Mahindra A, Cirstea D, Raje N. Novel therapeutic targets for multiple myeloma. Future Oncol. 2010;6:407–418. doi: 10.2217/fon.10.2. [DOI] [PubMed] [Google Scholar]