

Abstract
The AKT signaling pathway is activated in soft tissue sarcoma (STS). However, AKT blockade has not yet been studied as a potential targeted therapeutic approach. Here we examined the in vitro and in vivo effects of AKT inhibition in STS cells. Western blot analysis was utilized to evaluate the expression of AKT pathway components and the effect of AKT stimulation and inhibition on their phosphorylation. Cell culture assays were used to assess the impact of AKT blockade (using a PI3 kinase inhibitor, and a specific AKT inhibitor) on STS cell growth, cell cycle and apoptosis. Oligoarrays were used to determine gene expression changes in response to AKT inhibition. RT-PCR was used for array validation. Specific siRNA was used to knockdown GADD45α. Human STS xenografts in nude mice were used for in vivo studies and immunohistochemistry was utilized to assess the effect of treatment on GADD45α expression, proliferation and apoptosis. Multiple STS cell lines expressed activated AKT. AKT inhibition decreased STS downstream target phosphorylation and growth in vitro; G2 cell cycle arrest and apoptosis were also observed. AKT inhibition induced GADD45α mRNA and protein expression in all STS cells treated independent of p53 mutational status. GADD45α knockdown attenuated the G2 arrest induced by AKT inhibition. In vivo, AKT inhibition led to decreased STS xenograft growth. AKT plays a critical role in survival and proliferation of STS cells. Modulation of AKT kinase activity may provide a novel molecularly based strategy for STS targeted therapies.
Keywords: Soft tissue sarcoma, AKT, GADD45 α, G2 arrest, p53, Apoptosis
Introduction
Use of standard chemotherapy (even in drug combinations) for soft tissue sarcoma (STS) therapy remains problematic due to toxicity, expense, and the markedly chemoresistant nature of these malignancies; the search for better systemic agents is therefore crucial(1). Molecularly targeted therapy has recently emerged as a new treatment paradigm that seeks to improve conventional systemic therapy by specifically and selectively targeting cancers while minimizing treatment-related morbidities. This approach has led to successful advances in several diseases, including GIST(2, 3), a STS subtype. To utilize targeted therapy for STS, an increased knowledge of potential targets and their roles in STS progression and metastasis is needed(4, 5).
One potential molecular target is AKT kinase and its signaling pathways. AKT, also referred to as protein kinase B (PKB), is a serine-threonine kinase activated by phosphorylation of two critical residues: threonine 308 (T308) located in the activation loop and serine 473 (S473) at the C-terminal portion of the protein(6, 7). AKT activation is mediated by PI3 kinase which in turn is activated by a multitude of cell surface receptors and other related molecules(8–10); the negative regulation of AKT activation is achieved via tumor suppressor genes such as PTEN(11) and Src homology 2 domain-containing inositol 5-phosphatase1/ 2 (SHIP1/2)(12). The role of AKT has been investigated in a variety of epithelial origin tumors; upon phosphorylation, AKT activates downstream pathways that promote pro-tumorigenic, pro-metastatic processes (13). Activation of Akt has been found in brain(14), prostate(15), breast(16), lung(17), liver(18), gastric(19), colon(20), ovarian(21), and endometrial cancers(22) in association with cancer progression and chemoresistance. These findings have led to identification of several specific AKT inhibitors that are currently in clinical trials for a variety of epithelial malignancies.
While not extensively explored, evidence points to potential involvement of the AKT pathway in STS development and progression. Recently Hernando et al reported increased expression of activated AKT in a large panel of human leiomyosarcoma, malignant fibrous histocytoma, and de-differentiated liposarcoma (23); using a conditional PTEN knockout mouse model, they demonstrated a critical role for the AKT pathway in smooth muscle transformation and leiomyosarcoma development. Tomita et al identified a correlation between phospho-AKT (pAKT) expression in human STS specimens and subsequent tumor recurrence and patient survival (24). These findings suggest that determining the impact of AKT inhibition on STS in vitro and in vivo may facilitate inclusion of specific AKT targeted therapy in the anti-STS treatment armamentarium.
We report that AKT activity blockade induces STS cell growth inhibition, G2 cell cycle arrest, and apoptosis both in vitro and in vivo using human STS xenograft murine models. Relevant to STS, which harbor a high rate of p53 mutations contributory to the STS chemoresistance phenotype(25), is the finding that anti-tumor effects induced by AKT inhibition were observable in both wtp53 as well as mutated p53 STS cell lines. In addition, we identified a p53 independent increase in GADD45α, which is at least partially responsible for AKT-induced STS growth inhibition.
Materials and Methods
Cell culture and reagents
Human SKLMS1 (leiomyosarcoma), HT1080 (fibrosarcoma), RD (rhabdomyosarcoma), A204 (unclassified sarcoma), SW872 (liposarcoma), SW684 (fibrosarcoma), MES-SA and its multi-drug resistant derived MES-SA/DX (uterine sarcoma) STS cell lines were obtained from the American Type Culture Collection (ATCC). Cells were cultured in DMEM medium (A204 in McCoy's 5A) supplemented with 10% FCS (Life Technologies, Inc). p53 mutational status of these cells was previously determined by sequencing*. The specific AKT kinase inhibitor A674563 (A563) was a kind gift from Abbott laboratories (Abbott Park, IL); the PI3-kinase inhibitor Ly294002 was purchased from Cayman Chemical (Ann Arbor, MI). Doxorubicin (Ben Venue Lab, Bedford, OH) was obtained from the UTMDACC Pharmacy. Recombinant human EGF (R&D Systems, Minneapolis, MN) was used for EGFR stimulation.
Commercially available antibodies were used to detect Akt, pAkt (S473), pGSK3 (S21/9), pMDM2 (S166), activated-Caspase-3, PTEN, SHIP2, EGFR, c-MET, HER2 and IGF-IRα (Cell Signaling, Beverly, MA); GADD45α, p53, p21/WAF1, MDM2, GSK3, β-actin (Santa Cruz Biotechnology, Santa Cruz, CA); PCNA (Dako Cytomation, Carpinteria, CA). The Dead End Fluorometric TUNEL System (Promega, Madison, WI) was used for TUNEL staining. Secondary antibodies included HRP-conjugated (Universal kit HRP; Biocare Medical, Concord, CA) and fluorescent secondary antibodies (anti-rabbit Alexa488 and anti-mouse Alexa 594; Jackson Immuno Research, West Grove, PA). Other reagents included CytoQ FC Receptor block (Innovex Bioscience, Richmond, CA), Hoechst 33342 (Polysciences, Inc., Warrington, PA) and propyl gallate (ACROS Organics, Morris Plains, NJ).
Western blot analysis (WB)
WB was performed by standard methods. Briefly, 25–50 µg of proteins extracted from cultured cells were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked and blotted with relevant antibodies. Horseradish peroxidase–conjugated secondary antibodies were detected by ECL chemiluminesence (Amersham Biosciences, Plc., UK). IRdye680- and IRdye800-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were detected using Odyssey Imaging (LICOR Biosciences, Lincoln, NE).
Measurement of cell proliferation
Cell growth assays were done utilizing CellTiter96 Cell Prolifetation Assay kit (Promega, Madison, WI), per manufacturer’s instructions. STS cell lines were plated at concentrations of 1.5×103 to 4×103 cells/well (depending on cell doubling time) in 96-well plates. The next day, cells were treated with either 0.1% DMSO as control, or different concentrations of LY294002 or A563 (for 24, 48 and 72hr).. Absorbance was measured at a wavelength of 490 nm; absorbance values of treated cells are presented as a percentage of the absorbance of untreated cells. Drug concentrations required to inhibit cell growth by 50% (IC50) were determined by interpolation of dose-response curves.
Cell cycle analysis
STS cell monolayers were treated with relevant agents for varying time periods. Cells were harvested, washed and fixed. Fixed cells were treated with 50 µg/ml RNase and stained with 50 ug/ml propidium iodide for 30 min. Cells were analyzed in a FACSCalibur, and data were analyzed with Cell Quest and Flowjo software (Philadelphia, PA) or ModFitLT v3.1 software (Verity Software House).
Apoptosis assay
Apoptosis was measured using the Apoptosis Detection Kit I (BD Biosciences, Mountain View, CA). As a standard, 1 × 106/ml cells per treatment condition were fixed and stained with 5ul Annexin-V-FITC (BD PharMingen) and 5µl propidium iodide (Sigma). Flow-cytometric analysis was performed for 1 × 104 cells and analyzed by FACScan (Becton Dickinson, NJ) using a single laser emitting excitation light at 488 nm. Data was analyzed by CellQuest software (Becton Dickson, San Jose, CA).
Caspase-3 apoptosis assay
DEVD-NucView™ 488 caspase-3 assay kit for live cells (cat #30029) was purchased from Biotium, INC (Hayward, CA). Apoptotic cells were detected per manufacturer’s instructions. Briefly, STS cells grown on chamber slides were treated with A563 (1µM) or DMSO for 24 hours. NucView™ substrate stock solution (5 µL from 0.2 mM stock) was added to culture medium and Incubated at room temperature for 30 minutes. Fluorescence was determined via a florescent microscope using FITC filters, and images captured.
Microarray hybridization
Total RNA isolated from STS cells treated with relevant agents was used to synthesize cDNA as template for generating Biotin-16-UTP (non-radioactive) labeled cRNA target using TrueLabeling-AMP linear RNA amplification kit (SuperArray Bioscience Corporation, Frederick, MD). Labeled cRNA was purified using SuperArray ArrayGrade cRNA cleanup kit and quantified by spectrophotometry (1 O.D. unit = 40 ug/ml). Gene expression profiling was performed using OligoGEArray Human Cell Cycle OHS-020 (SuperArray Bioscience Corporation). This microarray is designed to profile the expression of 112 key genes in cell cycle regulation#. Prehybridization (2h) and hybridization (overnight) was performed in a hybridization oven (60°C) using 4 µg of labeled cRNA target. High stringency washing at 60°C (0.1xSSC, 0.5% SDS) was followed by chemiluminescent detection. The array image was recorded using X-ray film and a flatbed desktop scanner to create grayscale (16 bit) files in TIFF format that were analyzed by GEArray Expression Analysis Suite online software† (http://geasuite.superarray.com).
Reverse transcription-PCR
RT-PCR was done as previously described ((26)). Briefly, total RNA was isolated from cultured STS cells using TRIzol reagent (Invitrogen Corp) per manufacturer instructions. Total RNA was reverse-transcribed using superscript II reverse transcriptase (Invitrogen) and 2µL of the product were used as templates for multiplex PCR containing both target GADD45α and GAPDH primers for normalization. PCR primers were designed using primer 3 software. GADD45α: 5’-GGAGAGCAGAAGACCGAAA-3’ and 5’-TCACTGGAACCCATTGATC-3’; GAPDH: 5’-GAGCCACATCGCTCAGAC-3’ and 5’-CTTCTCATGGTTCACACCC-3’. The PCR reaction solution contained 25ul of Taq PCR Master Mix (Qiagen), 0.2µM/L of forward and reverse primers respectively, 2µl of cDNA and ddH2O to final volume of 50µl. PCR consisted of denaturation for 3 min at 94°C, 26 cycles of denaturation for 30 seconds at 94°C, annealing for 40 seconds at 56°C, and an extension for 50 seconds at 72 °C. PCR cycles were terminated by an extension at 72°C for 7min and products were resolved on a 2% agarose gel.
Small inhibitory RNA (siRNA) knock-down of GADD45α
5×104 RD cells were plated per well of 6-well plate and incubated overnight at 37°C. The following morning SmartPool GADD45α siRNA or non-targeting siRNAs constructs (Dharmacon Inc., IL) were transfected using lipofectamine 2000 (Invitrogen, NY) reagents according to manufacturer instructions. Mock-transfected cells were treated with Lipofectamine 2000 only. Incubation time for transfection reagents was 24 hours, at which time media was replaced with fresh regular media containing appropriate inhibitors.(A563 or DMSO). The next day cells were harvested for RT-PCR and cell cycle analysis by flow cytometry.
In vivo therapeutic animal model
All animal procedures and care was approved by the Institutional Animal Care and Usage Committee of UTMDACC. Animals received humane care as per the Animal Welfare Act and the NIH "Guide for the Care and Use of Laboratory Animals." Trypan blue staining–confirmed viable HT1080 STS cells (1 × 106/ 0.1ml HBSS/mouse) were injected subcutaneously into the flank of 6 week-old female nude/nude mice (NCI/NIH; n=20). Subcutaneous tumors were measured twice weekly by digital caliper; tumor volume was calculated as V = L × W2 × π/6, where V = volume, L = length, and W = width. When average subcutaneous tumor volume reached about 100 mm3, mice were assigned into two treatment groups (n=10): 1) control (vehicles only); 2) A563 (20mg/kg/bid, gavage.). Mice were followed for tumor size and body weight, and were sacrificed when control group tumors reached an average of 1.5cm in largest dimension. Tumor was resected, weighed, and frozen or fixed in formalin and paraffin embedded for further immunohistochemical studies.
Immunohistochemical analysis
Immunohistochemistry was performed as previously described (27). Briefly, paraffin sections were dewaxed and rehydrated prior to antigen retrieval. Endogenous peroxidase activity was quenched with 0.6% hydrogen peroxide before blocking with horse serum. Staining with primary antibodies (described above) was done at concentrations based on manufacturer’s recommendation. Biotinylated secondary antibodies were applied at 1:200 prior to ABC peroxidase system application (Vectastain ABComplex; Vector Laboratories, Inc., CA), DAB color development (Sigma Chemical Co., St. Louis, MO), and Mayer’s hematoxylin counterstaining. For immunofluorencence staining, fluorescence-conjugated secondary antibodies were used, followed by nuclear staining with Hoechst. In Situ Cell Death Detection Kit (Roche Applied science) was used per manufacturer’s instructions for TUNEL assay. Staining distribution and intensity was evaluated and scored by two independent reviewers (BK and QZ). Photographs were obtained using a Leica DM4000B microscope (Leica Microsystems, Gmb, HErnst-Leitz-Strasse) and a Leica HCxPL-S-APO 40×/0.75 numeric aperture objective lens. Images were captured using SPOT digital camera and were processed using SPOT advanced Acquisition software (Diagnostic Instruments Inc. Sterling Heights, MI).
Statistical analysis
Cell culture assays were repeated at least three times and mean± SD was calculated. Cell lines were examined separately. For outcomes that were measured at a single time point, two sample t-tests were used to assess the differences. Differences in xenograft growth in vivo were assessed using a two-tailed Student’s t test. Significance was set at p≤0.05.
Results
STS cell lines express activated AKT
Previous reports have suggested a possible role for AKT activation in STS (24). To study the impact of AKT inhibition on STS cell growth, we initially evaluated the expression of pAKT in a panel of human STS cell lines of diverse histology (Fig 1A). AKT activation to varying levels was demonstrable in all STS cells examined by immunobloting for the pAKT serine 473 epitope. Moreover, increased AKT phosphorylation was observed when STS cells were under serum starvation conditions (Supp Fig 1). Serum independent pAKT expression is possibly an even better discriminator of STS cell AKT-dependence.
Figure 1.
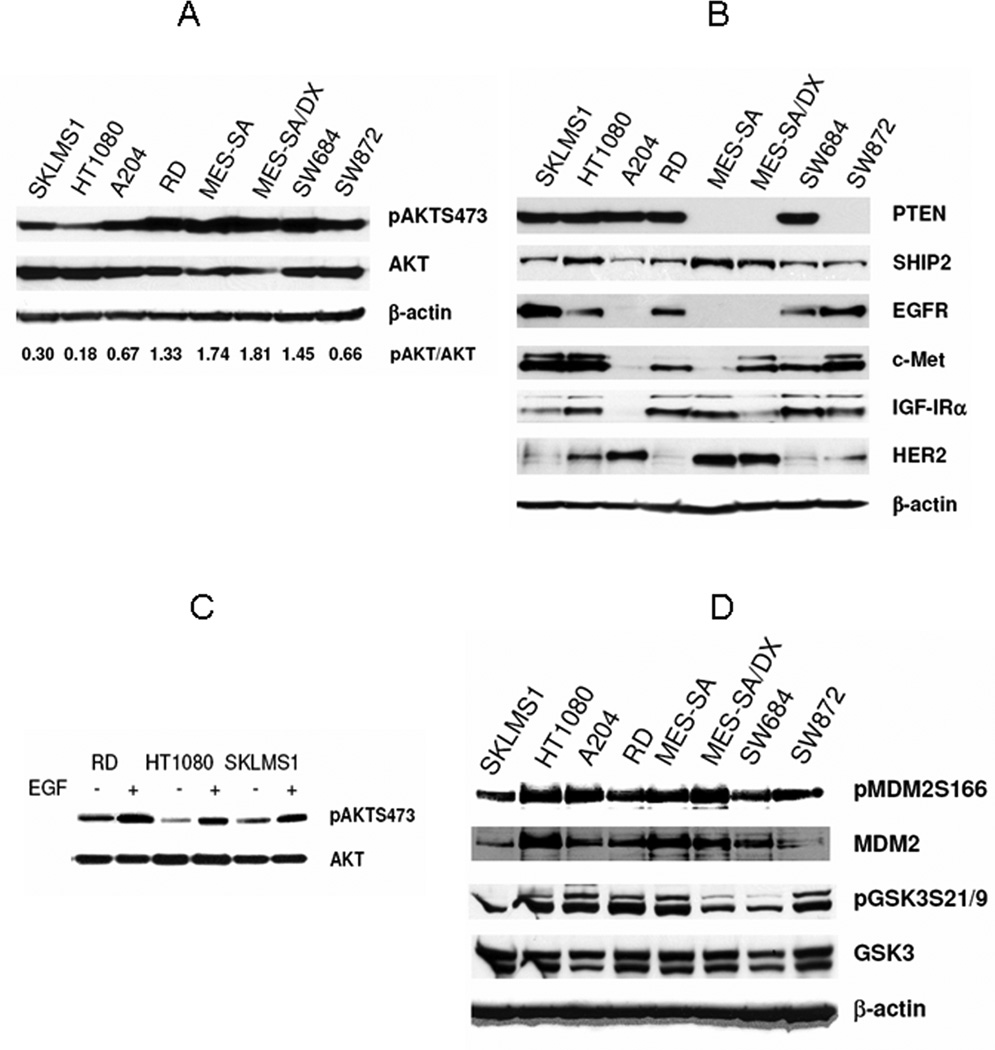
STS cells express high levels of activated and functional AKT: A, WB demonstrating increased pAKT (S473) in a panel of STS cell lines. pAKT: total AKT ratio (calculated based on densitometry analysis) is recorded at the bottom. B, WB demonstrating the STS expression of several possible upstream AKT modifiers. C, EGF stimulation (80ng/ml for 15min) increases AKT phosphorylation in STS; D) AKT downstream targets (MDM2 and GSK3) are phosphorylated in STS cells.
There are multiple molecular mechanisms which could possibly result in the activation of AKT. Dysregulation in intrinsic components of the PI3K-AKT pathway, e.g., PTEN mutation resulting in the loss/dysfunction of the PI3 kinase negative regulator PTEN protein, has been identified in many cancers. While not well described in STS, some evidence suggests that PTEN is dysregulated in STS, especially leiomyosarcomas(23). Screening human STS cells‡ for PTEN expression (Fig 1B), we observed that two cell lines (MES-SA - uterine sarcoma and its derivative MES-SA-Dx, and SW872 – liposarcoma) do not exhibit PTEN protein. It is possible that the loss of PTEN in these cells is at least partially responsible for their pAKT expression. Similarly, we screened cells for the expression of another phosphatase that negatively regulates PI3K, SHIP2 (Fig 1B). All STS cells were found to express SHIP2. As with STS cells expressing PTEN, we can not exclude the possibility that loss of function, rather than loss of expression of these phosphatases per se, contributes to AKT activation.
The AKT pathway is a common point of convergence for a multitude of upstream activators such as tyrosine kinase receptors, several of which have been previously shown to be dysregulated in STS(28, 29). For example, WB analysis (Fig 1B) demonstrated that all STS cells evaluated in the current study express at least one of the growth factor receptors examined, and that most express multiple such receptors. Thus, it is possible that upstream modulators are at least partially responsible for the AKT phosphorylation observed in the STS cell lines. Moreover, stimulation of upstream receptors with an appropriate ligand (i.e. EGF/EGFR; Fig 1C) can induce further AKT phosphorylation. These findings are of special importance in that the tumor microenvironment is rich in cytokines secreted by a variety of cells composing the tumor stroma, suggesting that AKT could possibly be even more activated in vivo.
To further evaluate whether AKT activation is of significance in STS cells, we examined the phosphorylation status of downstream AKT targets such as MDM2 and GSK3. As shown in Figure 1D, phosphorylation of MDM2 at Ser 166 and phosphorylation of GSK3 at Ser 21(α subunit) and Ser 9 (β subunit) were detectable at varying levels in all STS cell lines. These data suggest that AKT kinase is activated and functional in STS cells, mediating intracellular signaling and downstream target phosphorylation.
AKT blockade results in decreased STS cell downstream target phosphorylation and tumor cell growth inhibition
Next, we wanted to evaluate the effect of AKT inhibition on STS cells. Utilizing a commercially available inhibitor of the AKT activator PI3 kinase (LY294002), we were able to demonstrate a dose dependent reduction in AKT phosphorylation in all STS cell lines tested (Fig 2A). As suggested above, these data indicate that phosphorylation of AKT in STS is dependent upon PI3 kinase activity. Treatment of the different STS cell lines with increasing doses (0.1–10µM) of LY294002 resulted in a dose dependent decrease in cell proliferation as was identified by MTS assay, with IC50s (at 48hr) ranging from 3.5 to 7.5µM (data not shown)
Figure 2.
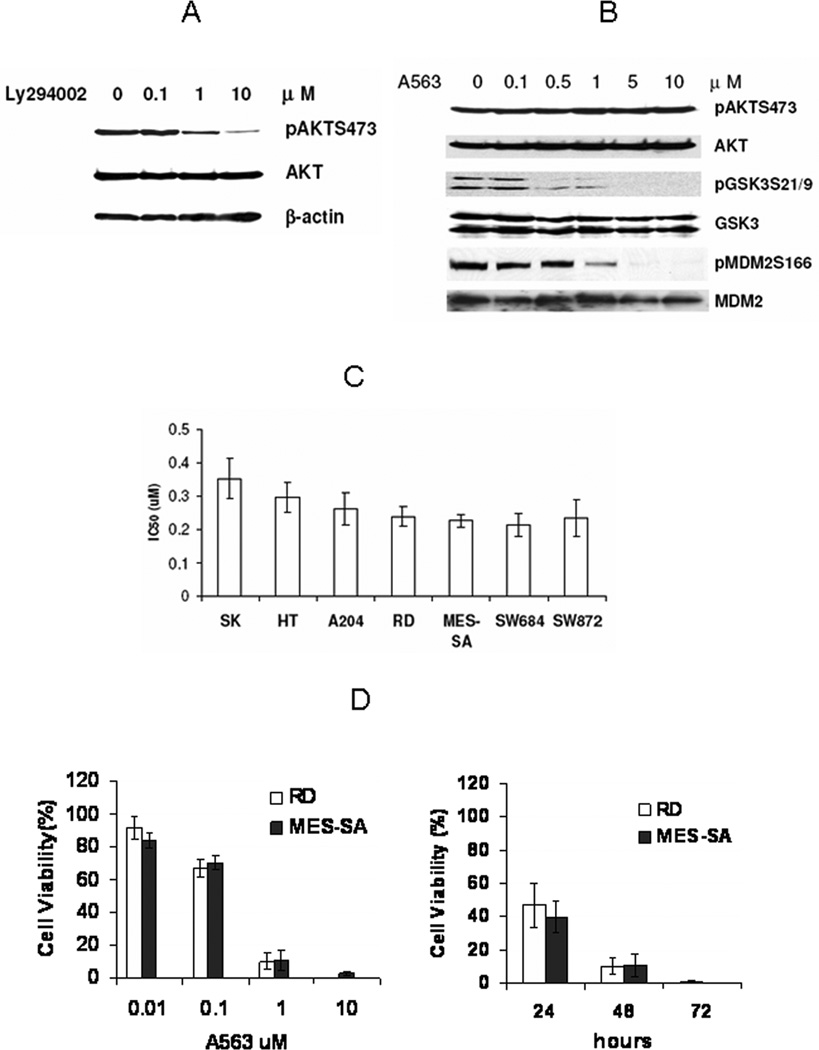
AKT blockade inhibits AKT downstream target phosphorylation and induces STS growth inhibition. A, PI3 kinase inhibitor LY294002 (4hr treatment) induced a dose dependent reduction in AKT phosphorylation in RD cells (similar responses were observed in other STS cells). B, A563 does not affect AKT phosphorylation in STS cells (RD is shown), but directly inhibits its kinase activity. WB demonstrating A563 (4hr treatment) dose dependent inhibition of AKT downstream target phosphorylation (GSK3 and MDM2). C, IC50 values for different STS cell lines (48hr; MTS assays). Drug concentration required to inhibit cell growth by 50% (IC50) for each cell line was determined by interpolation from dose-response curve (each bar represents results of four independent experiments). D &E, A563 inhibits STS cell growth in a dose and time dependent manner (MTS assays; results were expressed as percent of cell viability compared to control DMSO-treated cells, each bar represents results of four independent experiments).
While PI3K blockade results in effective inhibition of AKT phosphorylation, it is not specific and possibly affects other signaling pathways(30, 31). Therefore, we next analyzed the efficacy of a specific AKT inhibitor, A-674563 (A563)(32). This compound is an ATP competitor that binds to the ATP site of the AKT kinase domain, inhibiting AKT-catalyzed phosphorylation activity. As depicted in Fig 2B, A563 does not inhibit AKT phosphorylation per se, but blocks the phosphorylation of AKT downstream targets in a dose dependent manner. Exposure of STS cells to increasing doses of A563 significantly decreased GSK3 and MDM2 phosphorylation (Fig 2B). No effect on MAPK phosphorylation could be observed in STS cells after A563 treatment (data not shown).
Next, we determined the effect AKT activity blockade induced by A563 on STS cell proliferation. We examined the sensitivity of the different cell lines exposed to increasing concentrations of A563 for varying lengths of time (24, 48, and 72 hrs). Our results indicate that all STS cell lines were sensitive to A563, with the IC50 values at 48 hours ranging from 0.22 ±0.034µM (SW684) to 0.35 ±0.06 µM (SKLMS1; Fig 2C). AKT inhibition induced growth inhibition in a dose- and time-dependent manner (Fig 2D). Cells expressing the highest level of pAKT (RD, MES-SA, and SW684; see densitometry Fig 1A) were found to be most sensitive to this inhibition. Additionally, because p53 mutations are the most common genetic alteration in STS and p53 mutated STS are more therapeutically resistant, it is important that no significant differences in response to A563 could be found when comparing STS cells bearing wtp53 (HT1080, A204, MES-SA) versus mutated p53 genes (SKLMS1, RD, SW684, SW872). This observation suggests the possible importance of further investigating the AKT inhibition-induced p53 independent pathway in STS
AKT activity inhibition induces G2 cell cycle arrest and apoptosis in STS cells
Based on the observed effect of A563 on STS cell growth, we next evaluated the effect of A563 on STS cell cycle progression and apoptosis. Cell cycle analysis after treatment of STS cell lines with A563 using PI staining/FACS induced G2 cell cycle arrest in all STS cells tested (p<0.05; Fig 3A). Additionally, an increase in apoptotic cells (cells in G0/sub-G1) could be also observed. When treating STS cells with A563, we observed clear morphological changes, i.e. loss of elongated shape and cytoplasmic and nuclear condensation, confirming A563- induced apoptosis (Supp. Fig 2). To further quantitatively evaluate the impact of A563 on STS cell apoptosis, we utilized an Annexin V apoptosis detection assay (Figure 3B). AKT inhibition induced significant early and late apoptosis in all STS cell lines tested (p<0.01; Fig 3B). WB demonstrated increase in caspase-3 cleavage (Fig 3C), and using a NucView 488 caspase-3 assay kit, we detected apoptosis in living cells when treated with AKT kinase inhibitor as shown in Fig 3D. Similar to the cell growth assays, the impact of A563 on the cell cycle and apoptosis observed in the STS cells was independent of p53 mutational status, suggesting that these findings can not solely be explained by wtp53 stabilization found to occur secondary to AKT inhibition (33). Taken together, these data indicate that AKT activity is highly co-related to proliferation and survival of STS cells. AKT blockade is effective in significantly inhibiting STS cell proliferation, as well as induction of G2 cell cycle arrest and apoptosis.
Figure 3.
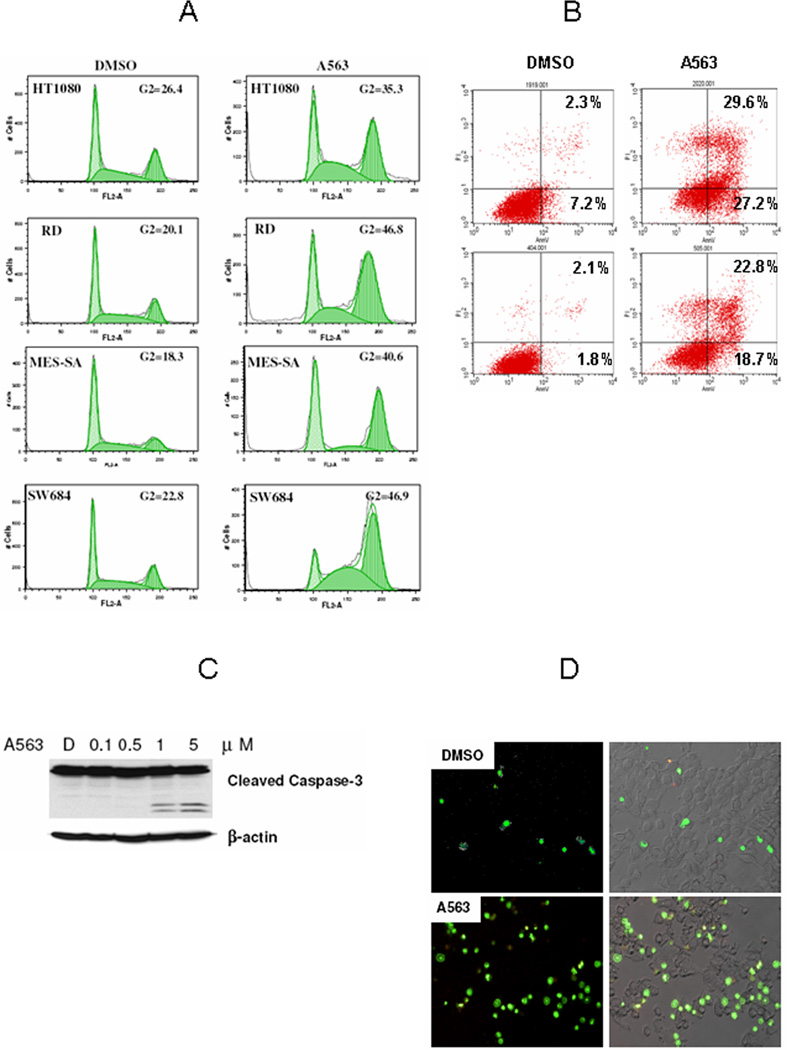
AKT blockade induced G2 cell cycle arrest and apoptosis in STS cells. A, PI staining/FACS analysis demonstrates A563 (1µM/24hr) induced G2 cell cycle arrest in STS cells. B, Annexin-V staining demonstrating increase in early and late apoptosis in STS cell treated with A563 (1µM/24hr). C A563 (24hr) induces a dose dependent increase in cleaved caspase-3 in RD cells (WB). D, Caspase 3 apoptosis assay demonstrates increased apoptosis in MES-SA cells treated with A563 (1µM/24hr). Nuclear DNA of apoptotic cells were stained green by the enzymatically released DNA dye (all figure panels are representative of three independent experiments).
AKT inhibition upregulated the expression of GADD45A independent of p53
To identify possible AKT downstream modulators responsible for the effect of AKT blockade on STS G2 cell cycle arrest in both wtp53 and mutp53 cells, we utilized the commercial Human Cell Cycle OligoGEArray to determine the expression profile of a panel of genes involved in cell cycle progression. Human RD cells (mutp53) and MES-SA cells (wtp53) were treated with A563 (1µM/12hrs), and total RNA from treated and untreated cells was used to identify changes in gene expression (Supp Fig 3). Our results demonstrated that GADD45α, a gene encoding for the Growth Arrest and DNA Damage 45α protein, was the only gene upregulated in both cell lines (5.69± 0.09 fold in MES-SA and 2.37±0.62 fold in RD); therefore, we elected to study its possible role in AKT blockade-induced effects. Initially we wanted to confirm the Oligoarray results in RD and MES-SA cell lines as well as evaluate whether the GADD45α increase could also be seen in other human STS cells. Increase in GADD45α mRNA expression level after A563 treatment was identified in all STS cell lines tested (MES-SA, HT1080, A204 – wtp53, and RD, SW684, SKLMS1 – mutp53; Fig 4A). Furthermore, WB demonstrated that the changes observed in mRNA expression were translated to an increase in GADD45α protein levels in all STS cell lines treated with A563 (Fig 4B). Our data also indicate that GADD45α induction can be observed after treatment with low doses of A563 (0.1µM) in both wtp53 and mutp53 cells (Fig 4C), while p21 increase occurs only after exposure to a higher A563 dose, and is dependent on p53 mutational status.
Figure 4.
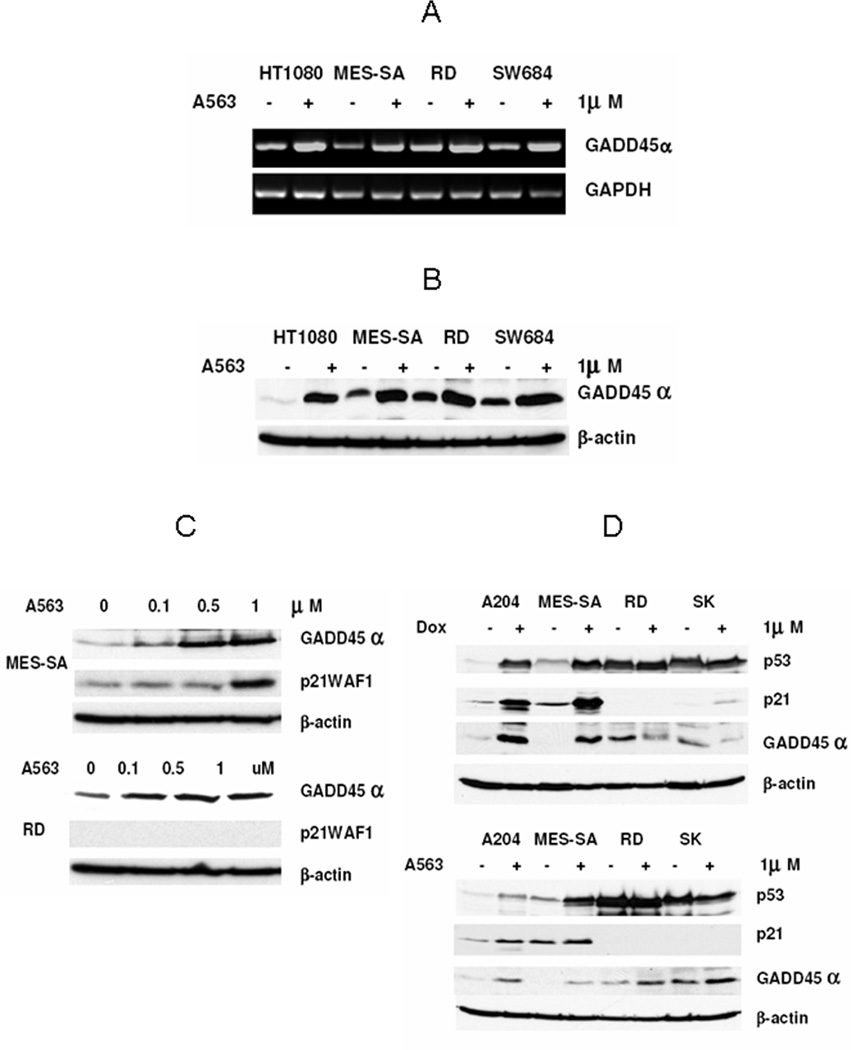
AKT inhibition induces expression of GADD45α. A, Increase in GADD45α mRNA in STS cells is demonstrated (RT-PCR) after treatment with A563 (1µM/12hr). B, similarly, an increase in GADD45α protein is observed after A563 treatment (1µM/24hr; WB). C, A563 (24hr) induces GADD45α expression in both wtp53 (MES-SA) and mutp53 (RD) STS cell lines, while A563-induced expression of p21 occurs only with a higher A563 dose and in cells harboring wtp53 (WB). D, Doxorubicin induces GADD45α as well as p53 and p21 expression only in wtp53 STS cells (A204 and MES-SA). A563-induced GADD45α is p53 status independent, while A563-induced p21 expression is observed only in wtp53 cells (WB) (all figure panels are representative of three independent experiments).
As its name implies, GADD45α has a role in cell cycle arrest and apoptosis in response to DNA damage in eukaryotic cells(34). The GADD45α gene was the first ever described p53 response gene; it was found to be transcriptionally upregulated by p53 via a p53 consensus-binding site located in the third intron of the GADD45α gene (35). It has also been reported to be regulated in a p53-independent manner(34) To further evaluate whether the increase in GADD45α observed in STS cells after AKT inhibition is a general stress response or whether it is a specific response to AKT inhibition in these cells, we compared the effect of doxorubicin (the most commonly utilized chemotherapeutic agent for the treatment of STS) on GADD45α expression to that of A563 (Fig 4D). Doxorubicin treatment resulted in an increase in GADD45α only in wtp53 STS cell lines (A204 and MES-SA). The chemotherapy induced stabilization and increase in p53 levels accompanied by increased p21 levels in these cells suggests that the wtp53 pathway is at least partially intact. While not the focus of this manuscript, the lack of doxorubicin-mediated GADD45α induction in mutp53 cells (RD and SKLMS1) might possibly offer an additional explanation of our previous observation of increased doxorubicin resistance in STS harboring mutp53(36, 37). In contrast, treatment with the AKT inhibitor increased the expression of GADD45α in both wt and mutp53 cell lines, while p21 was only induced in wtp53 cells. These data suggest that AKT inhibition-induced upregulation of GADD45α in STS cells is specific and independent of wtp53 protein function.
GADD45α knockdown attenuates G2 arrest induced by AKT inhibition
To further study whether AKT inhibition-induced GADD45α upregulation is of functional significance in STS cells, we used smartpool siRNA directed against human GADD45α mRNA to knock down its endogenous and inducible expression. RD cells were mock-transfected, transfected with non-targeting siRNA or with GADD45α smart pool siRNA for 24 hours, at which point cells were treated with 0.1% DMSO or A563 (1µM) for an additional 24 hours; cells were then harvested for total RNA isolation and for cell cycle analysis. As shown in figure 5A, RT-PCR demonstrated that endogenous and AKT inhibition-induced GADD45α mRNA was knocked down effectively (>86% as measured by densitometry) after siRNAGADD45α transfection. Furthermore, when the cells were subjected to cell cycle analysis by FACS (Fig 5B), GADD45α knock down attenuated the AKT inhibition-induced G2 arrest. These data suggest that AKT inhibition-induced GADD45α is at least partially responsible for the observed G2 cell cycle arrest in response to A563 in STS cells.
Figure 5.
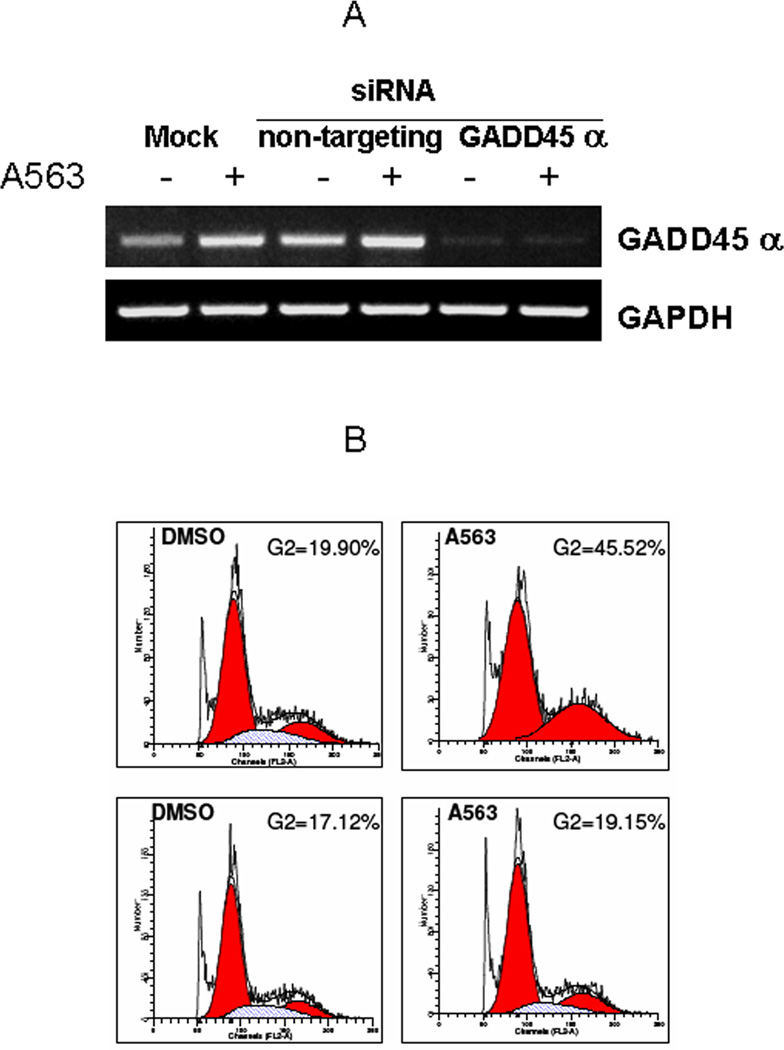
GADD45α knockdown attenuates G2 arrest induces by AKT inhibition. A, GADD45α siRNA transfection to RD cells decreased constitutive and A563-induced GADD45α expression compared to mock (lipofectamine only) or non targeting siRNA transfection (RT-PCR). B, AKT inhibition-induced G2 arrest was attenuated by GADD45α knockdown. RD cells transfected with non-targeting siRNA (top panel) exhibit increased G2 arrest after A563 treatment (1µM/24hr). No significant G2 arrest was observed in GADD45α siRNA transfected cells (bottom panel) after treatment with A563 (PI/FACS analysis; all figure panels are representative of three independent experiments).
AKT blockade decreases STS growth in vivo
To investigate if the effect of A563 on STS cells in culture can also be recapitulated in vivo, therapeutic animal studies were conducted using human fibrosarcoma (HT1080) subcutaneous xenografts. Treatment with A563 (20mg/kg/bid; p.o.) was initiated when tumors reached 5mm, tumor growth was compared to those of mice treated with vehicle only (5% dextrose). A563-treated mice exhibited slower tumor growth (Fig 6A) and a significant difference in tumor volume at the termination of the study (320.76±86.8) compared to that in control group (667.92±97.41; p<0.01). No significant weight loss was observed in A563 treated mice compared to control groups. Death occurred in one of the A563 treated mice.
Figure 6.
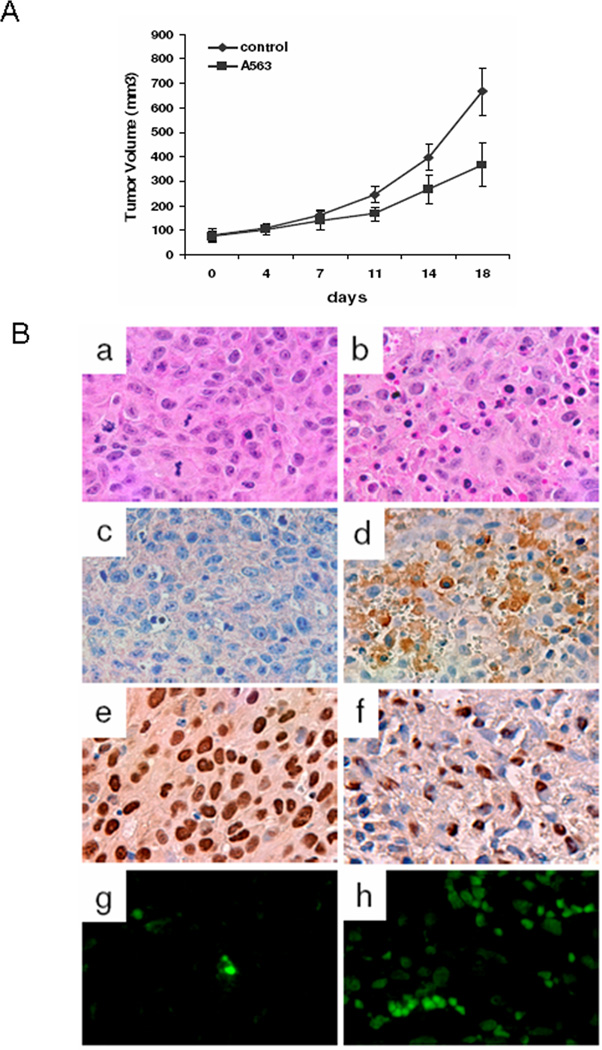
AKT blockade decreases STS growth in vivo. A, A563 (per gavage, 20mg/kg/BID) significantly inhibited HT1080 xenograft growth in nude mice (P<0.01). B, H&E staining demonstrated A563-induced necrosis in treated tumors (a,b). Immunohistochemical staining showed increased GADD45α (c,d) and decreased PCNA (e,f) expression in A563 treated tumors. TUNEL staining demonstrated increased apoptotic cells in A563 treated tumors (g.h). All original images were captured at × 400 magnification.
H&E staining of tumor specimens revealed pronounced tumor necrosis in A563-treated groups (Fig 6B a & b). Sections containing viable tumor cells were then selected for further immunohistochemical studies. A563-treated tumors were found to express increased levels of GADD45α (Fig 6B, c & d) and decreased levels of PCNA (a nuclear marker for proliferation; Fig 6B, e & f). Additionally, an increase in TUNEL assay staining levels (marker for apoptosis) in the A563- treated specimens was observed (Fig 6B, g & h). Taken together, these data suggest that AKT blockade results in significant STS tumor growth inhibition, an observation of potential clinical utility.
Discussion
Overall STS patient survival is approximately 50% at five years depending upon tumor size, histology, grade, location and presence of regional or distant disease. Development of lung metastasis is particularly ominous, accounting for approximately 80% of sarcoma-specific deaths with the remainder attributable to aggressive local STS effects(38). The need to successfully control STS growth and dissemination via systemic approaches remains compelling but is hampered by the availability of few drugs available with meaningful STS efficacy: toxicity ratios; new and effective systemic agents are crucially needed. Several reports suggest a possible role for activated AKT in STS development and progression(39–41), rendering it a potentially attractive therapeutic target. It is therefore encouraging that our studies demonstrate that AKT signaling pathway blockade results in STS cell G2 arrest and apoptosis in vitro, as well as inhibition of STS growth in vivo. AKT blockade combined with conventional chemotherapy should be explored in STS; such combinations have shown effectiveness in other tumors(42, 43).
STS include more than 50 distinct histological subtypes which are grouped together due to their shared putative mesenchymal origin and unique clinical behaviors which distinguish them from the more common epithelial tumors. Due to their relative rarity (less than 1% of adult solid tumors), it is difficult to accrue sufficient numbers of individual STS histiotype patients needed to study the efficacy of new therapies. We have shown high expression of pAKT in STS cells of different histological origin, perhaps due to an array of upstream modulators which could vary between different STS histiotypes. Functioning as a common convergence point of multiple dysregulated pathways operative in various STS, AKT per se may be more useful than its various upstream modulators as a target for STS therapy.
The effect of AKT inhibition on STS cells was reproducible using either Ly294002, a PI3 kinase inhibitor, or A563, a potent direct inhibitor of AKT kinase activity. While the effect of Ly294002 could be due to the inhibition of other PI3K downstream pathways, A563 is highly selective for the AKT pathway, suggesting its observed effects are directly related to AKT inhibition. However encouraging these results are in supporting our hypothesis, A563 cannot be utilized in the clinical arena. Others have shown that mice could not tolerate A563 for longer than 25 days, thereafter becoming moribund. While a significant A563 impact on tumor growth was observed, tumors re-grew rapidly upon A563 cessation (32). The dose limiting toxicities of A563 and other pan-AKT inhibitors are mainly due to the physiological role of AKT in insulin signaling and glucose metabolism(32, 44). Efforts to develop specific high affinity AKT inhibitors with less toxic side effects are currently underway. It may be a possible approach to utilize specific AKT1 inhibitors in lieu of pan-AKT inhibitors such as A563. AKT2 inhibition is perhaps the major cause of A563 toxic effects and predominates in insulin signaling; while AKT null mice develop typical type II diabetes, AKT1 null mice do not (32).
Another approach to reducing severe metabolic side effects is via inhibiting individual AKT down-stream substrates. There are caveats to such approaches: critical AKT downstream effectors may vary between different STS histiotypes or even within the same subtype, and inhibiting individual downstream components of the AKT pathway may miss key entities that are involved in the AKT-induced STS-promoting effects. However, encouraging preliminary results using mTOR inhibitors (a down stream target of AKT) in phase I/II clinical trials for advanced STS(45) suggests the feasibility of this approach, and supports further study of potentially significant AKT down stream substrates in STS.
In the current study we have identified GADD45α as a common AKT down stream target upregulated after AKT inhibition in STS cells of different histological background and p53 mutational status. The exact function of GADD45α, one of several growth arrest– and DNA damage–inducible proteins, and its expression and role in STS is not well elucidated. It has been implicated in G2 cell cycle arrest and apoptosis, thereby serving as a tumor suppressor(46). GADD45α interacts with several important intracellular signaling molecules such as proliferating cell nuclear antigen, cyclin B/CDC2 complex, p21/WAF1, histone, and aurora-A kinase all of which participate in the regulation of DNA replication, DNA repair, and cell cycle progression (47). Our studies show that the GADD45α increase in response to AKT blockade at least partially mediates the anti-STS AKT inhibition-induced G2 cell cycle arrest. It is possible that loss of GADD45α expression and function in STS due to increased activated AKT may play a role in the dysregulated cell cycle progression of these tumors.
AKT-induced suppression of GADD45α, as well as AKT blockade-induced GADD45α has not yet been extensively explored and may be due to several molecular mechanisms. AKT blockade is known to upregulate and stabilize wtp53 expression through the inhibition of MDM2 phosphorylation (7); GADD45α is a known transcriptional downstream target of wtp53, however this can not solely explain the AKT inhibition-induced GADD45α, which also occurs in mutp53 cells. FOXO3 has also been identified as a GADD45α transcription activator(34, 48). AKT inhibition induces the translocation of cytoplasmic FOXO3 to the nucleus, thus increasing its transcriptional activity; previously FOXO3 has been shown to increase GADD45α expression(34). In our studies we have failed to demonstrate FOXO3 expression or subcellular localization in STS cells in response to AKT inhibition (data not shown); however, it is possible that other FOXO family members having similar functions might play a role in STS. Additionally, AKT inhibition-induced post transcriptional mechanisms resulting in GADD45α mRNA stability are also possible. For example, Zheng et al (49) reported that NF-κB inhibition resulted in post transcriptional stabilization of GADD45α mRNA; AKT blockade inhibited the function of NFkB (50). Current studies in our laboratory are investigating the STS-specific mechanisms resulting in AKT induced GADD45α suppression and AKT inhibition-induced GADD45α.
In summary, we demonstrate that AKT inhibition results in significant anti-tumor activity against human STS in vitro and in vivo. Our results suggest the possibility of AKT blockade as a promising therapeutic intervention for the treatment of patients burdened by this disease.
Supplementary Material
Acknowledgement
We thank Dr. Vincent L. Giranda (Abbot Laboratories, Abbott Park, IL) for kindly providing A674563 (A563).
Funding: This manuscript was supported in part by a Radiation Therapy Oncology Group (RTOG) seed grant.
Footnotes
Information is avilable online in Cosmic database (www.samger.ac.uk)
For a comprehensive list of genes on the array see (http://geasuite.superarray.com).
For information see (http://geasuite.superarray.com).
All previously found to harbor wt PTEN see (www.sanger.ac.uk/genetics/CGP/CellLines)
References
- 1.Mocellin S, Rossi CR, Brandes A, Nitti D. Adult soft tissue sarcomas: conventional therapies and molecularly targeted approaches. Cancer Treat Rev. 2006;32:9–27. doi: 10.1016/j.ctrv.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Barnes G, Bulusu VR, Hardwick RH, et al. A review of the surgical management of metastatic gastrointestinal stromal tumours (GISTs) on imatinib mesylate (Glivectrade mark) Int J Surg. 2005;3:206–212. doi: 10.1016/j.ijsu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Tarn C, Godwin AK. Molecular research directions in the management of gastrointestinal stromal tumors. Curr Treat Options Oncol. 2005;6:473–486. doi: 10.1007/s11864-005-0026-x. [DOI] [PubMed] [Google Scholar]
- 4.Yang JL, Crowe PJ. Targeted therapies in adult soft tissue sarcomas. J Surg Oncol. 2007;95:183–184. doi: 10.1002/jso.20636. [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanian L, Evens AM. Targeting angiogenesis for the treatment of sarcoma. Curr Opin Oncol. 2006;18:354–359. doi: 10.1097/01.cco.0000228741.64541.ca. [DOI] [PubMed] [Google Scholar]
- 6.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 8.Franke TF, Yang SI, Chan TO, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 9.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 10.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 11.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J Biol Chem. 2002;277:6266–6272. doi: 10.1074/jbc.M105969200. [DOI] [PubMed] [Google Scholar]
- 13.List K, Szabo R, Molinolo A, Sriuranpong V, Redeye V, Murdock T, Burke B, Nielsen BS, Gutkind JS, Bugge TH, et al. Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 2005;19:1934–1950. doi: 10.1101/gad.1300705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO. Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res. 2001;61:6674–6678. [PubMed] [Google Scholar]
- 15.Carson JP, Kulik G, Weber MJ. Antiapoptotic signaling in LNCaP prostate cancer cells: a survival signaling pathway independent of phosphatidylinositol 3'-kinase and Akt/protein kinase B. Cancer Res. 1999;59:1449–1453. [PubMed] [Google Scholar]
- 16.Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 17.Moore SM, Rintoul RC, Walker TR, Chilvers ER, Haslett C, Sethi T. The presence of a constitutively active phosphoinositide 3-kinase in small cell lung cancer cells mediates anchorage-independent proliferation via a protein kinase B and p70s6k-dependent pathway. Cancer Res. 1998;58:5239–5247. [PubMed] [Google Scholar]
- 18.Ueda S, Basaki Y, Yoshie M, et al. PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by gefitinib. Cancer Res. 2006;66:5346–5353. doi: 10.1158/0008-5472.CAN-05-3684. [DOI] [PubMed] [Google Scholar]
- 19.Bae IH, Park MJ, Yoon SH, et al. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006;66:4991–4995. doi: 10.1158/0008-5472.CAN-05-4254. [DOI] [PubMed] [Google Scholar]
- 20.Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002;94:3127–3134. doi: 10.1002/cncr.10591. [DOI] [PubMed] [Google Scholar]
- 21.Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19:2324–2330. doi: 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]
- 22.Gagnon V, Mathieu I, Sexton E, Leblanc K, Asselin E. AKT involvement in cisplatin chemoresistance of human uterine cancer cells. Gynecol Oncol. 2004;94:785–795. doi: 10.1016/j.ygyno.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 23.Hernando E, Charytonowicz E, Dudas ME, et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. 2007;13:748–753. doi: 10.1038/nm1560. [DOI] [PubMed] [Google Scholar]
- 24.Tomita Y, Morooka T, Hoshida Y, et al. Prognostic significance of activated AKT expression in soft-tissue sarcoma. Clin Cancer Res. 2006;12:3070–3077. doi: 10.1158/1078-0432.CCR-05-1732. [DOI] [PubMed] [Google Scholar]
- 25.Zhan M, Yu D, Liu J, Glazer RI, Hannay J, Pollock RE. Transcriptional repression of protein kinase Calpha via Sp1 by wild type p53 is involved in inhibition of multidrug resistance 1 P-glycoprotein phosphorylation. J Biol Chem. 2005;280:4825–4833. doi: 10.1074/jbc.M407450200. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Zhan M, Hannay JA, et al. Wild-type p53 inhibits nuclear factor-kappaB-induced matrix metalloproteinase: 9 promoter activation: implications for soft tissue sarcoma growth and metastasis. Mol Cancer Res. 2006;4:803–810. doi: 10.1158/1541-7786.MCR-06-0201. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Hannay JA, Liu J, et al. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer Res. 2006;66:8770–8778. doi: 10.1158/0008-5472.CAN-06-1217. [DOI] [PubMed] [Google Scholar]
- 28.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of growth factor receptors, the focal adhesion kinase, and other tyrosine kinases in human soft tissue tumors. Ann Surg Oncol. 1994;1:18–27. doi: 10.1007/BF02303537. [DOI] [PubMed] [Google Scholar]
- 29.Gee MF, Tsuchida R, Eichler-Jonsson C, Das B, Baruchel S, Malkin D. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene. 2005;24:8025–8037. doi: 10.1038/sj.onc.1208939. [DOI] [PubMed] [Google Scholar]
- 30.van der Heide LP, Hoekman MF, Biessels GJ, Gispen WH. Insulin inhibits extracellular regulated kinase 1/2 phosphorylation in a phosphatidylinositol 3-kinase (PI3) kinase-dependent manner in Neuro2a cells. J Neurochem. 2003;86:86–91. doi: 10.1046/j.1471-4159.2003.01828.x. [DOI] [PubMed] [Google Scholar]
- 31.Yoshizumi M, Tsuchiya K, Kirima K, Kyaw M, Suzaki Y, Tamaki T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol Pharmacol. 2001;60:656–665. [PubMed] [Google Scholar]
- 32.Luo Y, Shoemaker AR, Liu X, et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 33.Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. Phospholipase D elevates the level of MDM2 and suppresses DNA damage-induced increases in p53. Mol Cell Biol. 2004;24:5677–5686. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 35.Smith ML, Ford JM, Hollander MC, et al. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol. 2000;20:3705–3714. doi: 10.1128/mcb.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannay JA, Liu J, Zhu QS, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007;6:1650–1660. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 37.Zhan M, Yu D, Lang A, Li L, Pollock RE. Wild type p53 sensitizes soft tissue sarcoma cells to doxorubicin by down-regulating multidrug resistance-1 expression. Cancer. 2001;92:1556–1566. doi: 10.1002/1097-0142(20010915)92:6<1556::aid-cncr1482>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 38.Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg. 1999;229:602–610. doi: 10.1097/00000658-199905000-00002. discussion 10–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer. 2007;97:785–791. doi: 10.1038/sj.bjc.6603952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerrero S, Figueras A, Casanova I, et al. Codon 12 and codon 13 mutations at the K-ras gene induce different soft tissue sarcoma types in nude mice. Faseb J. 2002;16:1642–1644. doi: 10.1096/fj.02-0050fje. [DOI] [PubMed] [Google Scholar]
- 41.Dobashi Y, Suzuki S, Sugawara H, Ooi A. Involvement of epidermal growth factor receptor and downstream molecules in bone and soft tissue tumors. Hum Pathol. 2007;38:914–925. doi: 10.1016/j.humpath.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3'-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–1092. [PubMed] [Google Scholar]
- 43.Dai Y, Grant S. Small molecule inhibitors targeting cyclin-dependent kinases as anticancer agents. Curr Oncol Rep. 2004;6:123–130. doi: 10.1007/s11912-004-0024-3. [DOI] [PubMed] [Google Scholar]
- 44.Martelli AM, Nyakern M, Tabellini G, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–928. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 45.Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19:6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]
- 46.Tront JS, Hoffman B, Liebermann DA. Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res. 2006;66:8448–8454. doi: 10.1158/0008-5472.CAN-06-2013. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A. 2006;103:9976–9981. doi: 10.1073/pnas.0603728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomis RR, Alarcon C, He W, et al. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006;103:12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng X, Zhang Y, Chen YQ, Castranova V, Shi X, Chen F. Inhibition of NF-kappaB stabilizes gadd45alpha mRNA. Biochem Biophys Res Commun. 2005;329:95–99. doi: 10.1016/j.bbrc.2005.01.105. [DOI] [PubMed] [Google Scholar]
- 50.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.