

Abstract
Microbiome analysis has identified a state of microbial imbalance (dysbiosis) in patients with chronic intestinal inflammation and colorectal cancer. The bacterial phylum Proteobacteria is often overrepresented in these individuals, with Escherichia coli being the most prevalent species. It is clear that a complex interplay between the host, bacteria and bacterial genes is implicated in the development of these intestinal diseases. Understanding the basic elements of these interactions could have important implications for disease detection and management. Recent studies have revealed that E. coli utilizes a complex arsenal of virulence factors to colonize and persist in the intestine. Some of these virulence factors, such as the genotoxin colibactin, were found to promote colorectal cancer in experimental models. In this Review, we summarize key features of the dysbiotic states associated with chronic intestinal inflammation and colorectal cancer, and discuss how the dysregulated interplay between host and bacteria could favor the emergence of E. coli with pathological traits implicated in these pathologies.
KEY WORDS: Adherent-invasive E. coli, Dysbiosis, IBD, CRC, Colibactin
Introduction
Inflammatory bowel disease (IBD) is a group of inflammatory disorders of the intestine. The most common forms of IBD are ulcerative colitis (colitis of the large intestine) and Crohn’s disease (a type of IBD that can affect any part of the digestive tract), which combine to affect ~1.4 million Americans (~0.45% of the US population), with up to 70,000 new cases being diagnosed in the United States each year (CCFA, 2011). IBD is a multifactorial immune disorder: it is influenced by the genetic susceptibility of an individual and by environmental and lifestyle factors (diet, smoking, etc.) (Kaser et al., 2010). Although IBD predominantly affects people in the Western world (e.g. Europe and North America), both the incidence and the prevalence of this pathology have risen in Asia, especially in areas that have adopted an industrialized lifestyle (Ng et al., 2012; Prideaux et al., 2012). Current treatment for IBD, which mainly involves anti-inflammatory drugs (e.g. mesalazine), corticosteroids (e.g. prednisone), immunosuppressant drugs (e.g. azathioprine), biological therapy [e.g. tumor necrosis factor α (TNF-α) inhibitors such as infliximab and adalimumab] and surgery, is costly and often associated with severe adverse effects (Baumgart and Sandborn, 2012; Ordás et al., 2012). IBD is associated with severe morbidity and impaired quality of life owing to its chronic nature and high recurrence (Høivik et al., 2013; Netjes and Rijken, 2013), and represents a substantial socioeconomic burden in the United States (~$2.2 billion/year, including healthcare costs and loss of earnings) (CCFA, 2011).
Colorectal cancer (CRC) is the type of cancer that develops in the colon or rectum. Despite the increasing implementation of colonoscopy screening and advances in chemotherapeutic and biological-agent-based therapies, CRC remains one of the most common and deadliest malignancies in the United States (Siegel et al., 2014). The American Cancer Society estimates that, in 2014, there will be ~140,000 new CRC cases and over 50,000 CRC-related deaths nationwide (Siegel et al., 2014). CRC includes hereditary, sporadic and colitis-associated CRC. Colitis-associated CRC represents a severe medical complication for patients with IBD, and often shows rapid progression, poor response to treatment and high mortality (Feagins et al., 2009). Individuals with IBD are at increased risk for developing CRC (Herrinton et al., 2012).
Both IBD and CRC evolve in the context of a vast and complex gut microbial ecosystem. The human intestinal microbiota consists mainly of bacteria (~1014) and contains more than 106 bacterial genes (Human Microbiome Project Consortium, 2012). Through the action of various microbial structural components, microbial gene products and/or metabolites, this microbiota plays essential roles in intestinal homeostasis, regulating host immunity, gut barrier function and metabolic activity (Clemente et al., 2012). Changes in the richness, diversity and stability of the gut bacterial ecosystem, a state referred to as microbial dysbiosis, is commonly associated with intestinal pathologies such as IBD and CRC (discussed below). In this Review, we will summarize key characteristics of the microbial dysbiosis associated with these intestinal diseases, and discuss connections between the gut microbiota, intestinal inflammation and carcinogenesis. We will focus on potential microbial candidates – specifically Escherichia coli – that have been identified in studies of the human CRC microbiota and are implicated in the carcinogenesis. We will describe important biological events implicated in the development of microbial dysbiosis and the emergence of carcinogenic microorganisms. Finally, we will review recent studies that demonstrate the pro-tumorigenic role of E. coli in animal models, in particular of those strains that produce the genotoxin colibactin.
Microbiota and intestinal pathologies
In the 1990s, it was found that re-routing the intestine out onto the surface of the skin to divert the fecal stream away from a patient’s bowel prevented recurrence of Crohn’s disease (Rutgeerts et al., 1991). Fecal-stream diversion also induced clinical and histopathological remission of collagenous colitis, a type of IBD that specifically affects the colon (Järnerot et al., 1995). These findings suggest that some agents that are present in the luminal contents are important for IBD pathogenesis. Consistent with this concept, reinfusion of the luminal contents into bypassed colonic segments caused recurrent Crohn’s disease (D’Haens et al., 1998). Later, studies using genetically engineered animal models of IBD showed that colitis was often attenuated or absent when these animals were kept in germ-free conditions (Kamada et al., 2013), indicating that the gut microbiota is essential for triggering and/or enhancing chronic intestinal inflammation. This is supported by the observation that antibiotic treatment ameliorates clinical symptoms in certain IBD patients (Feller et al., 2010; Selby et al., 2007; Thia et al., 2009). Moreover, transplantation of the microbiota from genetically engineered IBD mice to healthy wild-type mice induced colitis in the recipients (Grivennikov, 2013), although similar experiments have not been performed with the human dysbiotic microbiota. Nevertheless, an accumulating body of evidence supports the idea that intestinal microbes play important roles in the etiology and pathology of IBD.
More recently, researchers have started to unravel the genetic factors that contribute to IBD pathogenesis. The first polymorphism associated with susceptibility to Crohn’s disease was identified in the innate immunity sensor gene NOD2 (nucleotide-binding oligomerization domain-containing protein 2) in 2001 (Hugot et al., 2001; Ogura et al., 2001). Thus far, more than 160 genomic loci have been correlated with IBD susceptibility in humans (Jostins et al., 2012). Noticeably, many of these IBD genetic loci are implicated in innate and adaptive immunity, gut barrier function, and bacterial handling (Jostins et al., 2012). However, the low concordance rates of IBD between monozygotic twins indicate that genetic predisposition only partially accounts for susceptibility to this disease (Halme et al., 2006). Moreover, evidence that IBD susceptibility genes promote microbial imbalance in the intestine is lacking. These observations clearly highlight the complexity of this intestinal pathology; as such, determining the events that lead to IBD development will require a holistic view and an integrative approach to understanding host-bacteria interactions.
In the last decade, advancements in DNA sequencing technologies and sequence analysis have enabled a comprehensive characterization of the gut microbiota at an unprecedented level. Many studies have shown that the intestinal microbial ecosystem is markedly altered in individuals with IBD as compared to healthy individuals (Manichanh et al., 2012). These microbial composition analyses, combined with functional studies, have revealed dynamic roles of the microbiota in IBD. The pathology is likely caused by the action of various groups of bacteria (polymicrobial), either through the production of noxious factors and/or antigens or through the depletion of protective mediators such as short-chain fatty acids (SCFA) (Kamada et al., 2013; Kostic et al., 2014; Strober, 2013). The dual nature of the microbiota, pathogenic or protective, highlights the complex relationship that exists between microbes and their host (Kamada et al., 2013).
To date, several enteropathogens (e.g. Mycobacterium avium subspecies paratuberculosis, Yersinia spp., Listeria monocytogenes, Salmonella spp., Campylobacter concisus) that are found to be abundantly present in some IBD cases have been speculated to contribute to the disease pathogenesis (Chassaing and Darfeuille-Michaud, 2011; Mukhopadhya et al., 2012). However, there has been no evidence that these pathogenic microorganisms cause IBD (Packey and Sartor, 2009). By contrast, some bacteria [e.g. Faecalibacterium prausnitzii (Sokol et al., 2008), Clostridium spp. (clusters IV and XIVa) (Atarashi et al., 2011), commensal Bacteroides fragilis (Round et al., 2011)] that are able to induce immunosuppressive responses are considered to be protective against IBD. Bacteria can also enhance gut epithelial barrier function by stimulating mucus secretion and producing certain metabolites. For example, Bifidobacterium spp. protect mice from the lethal action of enterohemorrhagic E. coli O157:H7 infection through the enhanced production of acetate, an effect linked to the inhibition of translocation of shiga toxin (a toxin produced by this E. coli strain) from the lumen to the gut tissue (Fukuda et al., 2011; Fukuda et al., 2012). For more information on the role of bacteria in modulating intestinal homeostasis and inflammation, we direct the reader to a series of recent Reviews (Hardy et al., 2013; Kamada et al., 2013; Kostic et al., 2014; Strober, 2013).
Cancer genome studies have linked many somatic mutations to CRC (Esteban-Jurado et al., 2014; Watson et al., 2013). But, similarly to IBD, the etiology of CRC can only be partially attributed to genetics (Rustgi, 2007), indicating the involvement of environmental factors in this disease pathogenesis. Noticeably, marked changes in the intestinal microbiota are also observed in individuals with CRC (Ahn et al., 2013; Castellarin et al., 2012; Chen et al., 2012; Kostic et al., 2012; McCoy et al., 2013; Sobhani et al., 2011; Wu et al., 2013). Accumulating evidence suggests that the microbiota influences intestinal carcinogenesis. In particular, a number of studies using germ-free animals have revealed that the microbiota has cancer-promoting effects in spontaneous and genetically induced cancer models (Schwabe and Jobin, 2013; Sears and Garrett, 2014).
The microbiota might promote colonic carcinogenesis via a variety of mechanisms, and we direct readers to several recent reviews for more information on this topic (Elinav et al., 2013; Grivennikov, 2013; Schwabe and Jobin, 2013). Some key mechanisms include the pro-tumorigenic inflammatory responses induced by the dysbiotic microbiota, the inflammation-associated tissue injury and repair process, and the expansion of ‘keystone’ bacteria – those harboring specific virulence traits to drive colon cancer development. These ‘alpha bugs’, which usually exist at very low levels at gut homeostasis, could contribute substantially to colonic carcinogenesis when overrepresented (Sears and Pardoll, 2011). Thus far, several ‘alpha bugs’ that promote intestinal carcinogenesis in animal models have been described: enterotoxigenic B. fragilis (Wu et al., 2009), Fusobacterium nucleatum (Kostic et al., 2013; Rubinstein et al., 2013) and colibactin-producing E. coli (Arthur et al., 2012; Bonnet et al., 2014; Cougnoux et al., 2014).
It is worth noting that the microbiota might also prevent carcinogenesis. Recent work by Zhan et al. demonstrates that the gut microbiota protects mice from intestinal carcinogenesis induced by epithelial injury (Zhan et al., 2013). The authors attributed the beneficial effects of the microbiota to its ability to promote epithelial restitution and injury recovery. Key mechanisms by which commensal bacteria suppress colonic carcinogenesis include the induction of immunosuppressive responses, the detoxification of carcinogens and the production of cancer-suppressing metabolites (Boleij and Tjalsma, 2012; Schwabe and Jobin, 2013).
Dysbiotic states during inflammation and carcinogenesis
Although a considerable degree of variation is found among the microbial compositions of different individuals, the gut microbiota of healthy adults is commonly dominated by four major bacterial phyla: Firmicutes and Bacteroidetes (two groups of obligate anaerobes), which constitute ~90% of the microbial ecosystem, and Proteobacteria and Actinobacteria, which contribute to a lesser degree (Human Microbiome Project Consortium, 2012).
The structure of the human gut microbiota is established early in life and remains relatively stable for decades (Costello et al., 2009; Yatsunenko et al., 2012). However, during chronic intestinal inflammation, as experienced by individuals with IBD, the gut microbial composition displays marked alterations both taxonomically and functionally (Kostic et al., 2014; Manichanh et al., 2012). In general, the IBD-associated gut microbiota exhibits lower stability and diversity as compared with the microbial community found in a healthy human gut. At the phylum level, levels of both Firmicutes and Bacteroidetes are decreased, whereas those of Proteobacteria and Actinobacteria are significantly increased. Concurrently, there is a marked drop in the abundance of protective anaerobic commensals, mostly Firmicutes (e.g. F. prausnitzii, Clostridium spp.) and Bacteroidetes (e.g. B. fragilis), and an expansion of translocating facultative aerobes, especially bacteria belonging to the Enterobacteriaceae family (phylum Proteobacteria). Overall, the gut microbiota of individuals with IBD has a higher prevalence of Gram-negative bacteria, largely owing to the reduced ratio of Firmicutes (Gram-positive) relative to Bacteroidetes (Gram-negative).
In addition to the structural imbalance, profound perturbations of the functions of gut microbiota are also observed in IBD, e.g. bacterial amino acid biosynthesis and carbohydrate metabolism are diminished, whereas nutrient uptake is enhanced (Morgan et al., 2012). Importantly, bacterial genes involved in survival and pathogenesis processes, such as redox tolerance, secretion systems, adherence and/or invasion, are overrepresented in the microbiota of individuals with ileal Crohn’s disease, indicative of a functional shift to a ‘pathogenic’ community (Morgan et al., 2012). Conversely, pathways linked to the production of bacterial SCFAs, microbial metabolites known for their immunosuppressive functions (Arpaia et al., 2013; Furusawa et al., 2013; Maslowski et al., 2009; Singh et al., 2014; Smith et al., 2013), are repressed (Morgan et al., 2012). Using the T-bet−/−×Rag2−/− ulcerative colitis (TRUC) mouse, an animal model of ulcerative colitis that features expanded Enterobacteriaceae (Garrett et al., 2010), Rooks et al. recently reported increased bacterial motility, tetrathionate respiration [a metabolic pathway promoting intestinal colonization of the pathogenic Salmonella enterica subsp. Typhimurium in the inflamed gut (Winter et al., 2010)], and benzoate degradation [a pathway linking to Enterobacteriaceae growth and virulence (Freestone et al., 2007; Lyte et al., 2011)] in active colitis (Rooks et al., 2014). It is not clear whether changes in microbial activities influence IBD pathogenesis. However, it is reasonable to speculate that improper microbial function would impact mucosal immune responses (Kamada et al., 2013; Kostic et al., 2014; Strober, 2013). Interestingly, therapeutic interventions, such as anti-TNF-α therapy, in TRUC mice impacts gut microbiota composition (Rooks et al., 2014), revealing the plasticity of the microbiota to external cues.
E. coli, a key member of the Enterobacteriaceae family, is a common colonizer of the human intestine, with an average of five commensal E. coli strains found in a human digestive tract (Apperloo-Renkema et al., 1990). In healthy individuals, Enterobacteriaceae constitute only a small fraction (less than 1%) of the gut microbiota (Eckburg et al., 2005). However, Enterobacteriaceae, in particular E. coli, become dominant in the gut microbiota of individuals with IBD and in several animal models of gut inflammation (Carvalho et al., 2012; Morgan et al., 2012; Mukhopadhya et al., 2012). E. coli strains isolated from individuals with IBD are often adherent and invasive, displaying pathogenic properties (Baumgart et al., 2007; Darfeuille-Michaud et al., 2004; Darfeuille-Michaud et al., 1998; Elliott et al., 2013).
Microbial dysbiosis has also been identified in CRC (Jobin, 2013), and E. coli seem to play an important role in the pathogenesis of this disease (Arthur and Jobin, 2013). For example, mono-association of the adherent-invasive E. coli (AIEC) mouse strain NC101, which produces colibactin, enhanced colonic tumor development in azoxymethane (AOM)-treated interleukin-10-knockout (Il-10−/−) mice (Table 1), a mouse model of colitis-associated CRC (Arthur et al., 2012). The human CRC mucosa-associated E. coli strain 11G5 promoted intestinal neoplastic changes when introduced in Apcmin/+ mice, which carry a mutation in one allele of the tumor suppressor gene adenomatous polyposis coli (Apc) and are genetically predisposed to developing intestinal tumors (Bonnet et al., 2014) (Table 1). Another human CRC E. coli isolate, CCR20, increased tumor formation in AOM–dextran-sulphate-sodium (DSS)-treated mice (Cougnoux et al., 2014) (Table 1). These E. coli strains thus constitute a novel group of ‘alpha bugs’. Importantly, a high prevalence of mucosa-associated E. coli, especially those producing cyclomodulins (bacterial toxins and effectors that interfere with the eukaryotic cell cycle), is observed in individuals with CRC (Arthur et al., 2012; Bonnet et al., 2014; Buc et al., 2013; Martin et al., 2004; Prorok-Hamon et al., 2014). Therefore, AIEC represents a group of microorganisms that is implicated in the pathogenesis of both IBD and CRC. In the following section, we will discuss important host-bacteria and bacteria-bacteria interactions that modulate E. coli colonization and fitness in the intestine, and review key mechanisms by which E. coli mediate carcinogenesis.
Table 1.
Colibactin-producing E. coli promotes tumor development in animal models

Factors affecting E. coli colonization and fitness in the intestine
Host regulation at gut homeostasis
E. coli colonize the gut of a human infant within hours of birth, largely owing to their ability to respire oxygen that is present in the newborn’s intestine (Palmer et al., 2007), and thereafter establishes a symbiotic relationship with their host.
The gut luminal surface is covered by a single layer of epithelial cells, which is composed of absorptive enterocytes, secretory enteroendocrine cells, mucus-producing goblet cells, Paneth cells and a stem cell compartment that assures renewal of all cell lineages. Underneath the gut epithelium is the lamina propria, which harbors diverse immune cell populations (neutrophils, macrophages, dendritic cells, innate lymphoid cells, B and T cells, etc.) that are involved in gut immunity (Fig. 1A). At gut homeostasis, uncontrolled interaction between E. coli, or other commensal bacteria, and intestinal epithelial cells is prevented by a layer of mucus on the apical side of the epithelium (Bergstrom et al., 2010; Johansson et al., 2008) (Fig. 1A). The organization of this protective mucus system varies along the intestine. Mucus in the small bowel is unattached to the gut epithelium and is easily removable, whereas mucus in the colon forms two layers: one inner dense and attached mucus layer and an outer loose and unattached layer (Johansson et al., 2013). Mucus in both the small and the large intestine is composed of the same mucin protein, MUC2 (mucin 2), but the distinct two-layer organization in the colon provides a stronger protective barrier against the bacteria that are abundantly present in this region of the intestine (Johansson et al., 2013). Interestingly, mucin was reported to facilitate biofilm formation by E. coli (Bollinger et al., 2003; Bollinger et al., 2006), suggesting its potential role in modulating E. coli colonization in the gut.
Fig. 1.
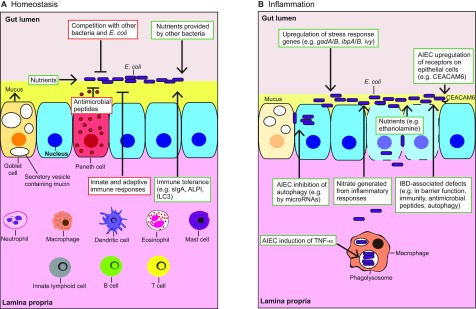
Events modulating E. coli colonization and fitness in the intestine. Events promoting E. coli fitness are illustrated in green boxes and those inhibiting E. coli growth are shown in red boxes. (A) At homeostasis, bacteria such as E. coli are prevented from attaching to epithelial cells (blue cells; for simplicity, villi are not shown) by a mucus layer present at the apical surface of the gut epithelium. The host immune system and the Paneth-cell-derived antimicrobial peptides regulate microbial growth. Major cell types that are involved in immune regulation of the gut microbes are illustrated in the lamina propria, where they are found. The host gut provides nutrients to support E. coli colonization, and immune tolerance mechanisms help to maintain a healthy E. coli community in the intestine. E. coli colonization is also modulated by bacterial competition for nutrients. Other bacteria can provide nutrients (e.g. mono- and disaccharides) to E. coli and promote its growth. (B) During intestinal inflammation, mucus depletion facilitates E. coli attachment to the epithelium. E. coli adjust their metabolism, and upregulate stress response genes to promote their survival. E. coli can take advantage of inflammation by using nutrients provided by dead cells and by respiring host-derived nitrate. Adherent invasive E. coli (AIEC) can disrupt the epithelial barrier, inhibit epithelial cell autophagy to promote intracellular survival, and upregulate surface receptors on epithelial cells to increase adherence. AIEC infection stimulates TNF-α expression in macrophages, which promotes their intracellular replication. IBD-associated deficiencies such as reduced secretion of antimicrobial peptides due to Paneth cell dysfunction (Paneth cells not shown) can further allow bacterial attachment, invasion and growth. CEACAM6, carcinoembryonic antigen-related cell adhesion molecule 6; gadA/B, glutamic acid decarboxylase A and B; ALPI, intestinal alkaline phosphatase; ibpA/B, inclusion body protein A and B; ILC3, group 3 innate lymphoid cells; ivy, inhibitor of C-type lysozyme; sIgA, secretory immunoglobulin A; TNF-α, tumor necrosis factor α.
Commensal microorganisms, such as E. coli, are constantly monitored by the host immune system (Slack et al., 2009) (Fig. 1A). The innate immune receptors expressed by intestinal epithelial cells and mucosal immune cells can respond to various E. coli-derived antigens. For example, Toll-like receptor 4 (TLR4) responds to lipopolysaccharide (LPS); Toll-like receptor 2 (TLR2) and NOD2 respond to peptidoglycan; and Toll-like receptor 5 (TLR5) responds to flagellin. These innate immune responses result in activation and recruitment of phagocytic cells, such as neutrophils and macrophages, which eliminate the microbes that breach the mucosal barrier. The adaptive immune system, which consists mainly of highly specialized B and T cells, provides additional protection against invading microbes and keeps the microbial population in check (Kamada et al., 2013). In particular, intestinal B cells produce large amounts of non-inflammatory immunoglobulin A (IgA) antibodies, which play important roles in maintaining appropriate bacterial communities within specific intestinal segments (Cerutti and Rescigno, 2008). Interestingly, similarly to mucin, IgA can also promote E. coli biofilm formation (Bollinger et al., 2003; Bollinger et al., 2006), and potentially modulates E. coli colonization and persistency in the intestine (Barbas et al., 2009). Another layer of protection is provided by Paneth cells, a type of cell found in the epithelium of the small intestine and appendix, which secrete a consortium of antimicrobial peptides. Many of these antimicrobial peptides are active against E. coli (Cunliffe and Mahida, 2004) and might restrict E. coli colonization and growth in the intestine (Fig. 1A).
Importantly, the host has also evolved immunoregulatory mechanisms that prevent excessive immune activation in response to symbiotic microbes, such as commensal E. coli (Fig. 1A). For example, the Gram-negative bacterium-derived LPS, upon activation of the host TLR4, upregulates intestinal alkaline phosphatase (ALPI), which functions to dephosphorylate LPS and thereby dampen the LPS-TLR4 innate immune response (Bates et al., 2007). In addition, secretory IgA in the gut mucosa can downregulate the expression of pro-inflammatory bacterial epitopes by commensal bacteria, and prevent microbes and microbial components from attaching to the gut epithelium (Cerutti and Rescigno, 2008). More recently, group 3 innate lymphoid cells (ILC3) have been demonstrated to inhibit inflammatory T-cell responses to commensal bacteria in the intestine (Hepworth et al., 2013). A delicate balance between immune surveillance and immune tolerance is required to maintain the mutually beneficial relationship between the host and the gut microbiota.
It should be emphasized that here we focus on commensal bacterial strains, not highly virulent strains (enteropathogenic E. coli, enterohemorrhagic E. coli, etc.) that have evolved sophisticated machineries to subvert the aforementioned regulatory mechanisms (Clements et al., 2012).
Bacterial competition
The intestine provides a nutrient-rich environment for commensal microbes such as E. coli. Upon intestinal colonization, E. coli genes involved in carbohydrate and amino acid metabolism and transport are strongly induced (Alpert et al., 2009). Similar metabolic function changes are observed in other commensal bacteria, for example, in Bifidobacterium longum (Yuan et al., 2008) and Lactococcus lactis (Roy et al., 2008). The high number of microbes that colonize the intestine implies that competition for common nutrients potentially exists among different commensal bacterial species, which could restrict the growth of E. coli in the intestine (Fig. 1A). The bioavailability of certain substrates could be critical for E. coli fitness in the intestine given that impaired carbohydrate metabolism or deficient purine and pyrimidine synthesis significantly diminishes the ability of E. coli to colonize the mouse intestine (Chang et al., 2004; Vogel-Scheel et al., 2010).
However, the relationship between commensal bacteria, in terms of nutritional needs, is not always competitive. Commensal E. coli strains colonize the mouse large intestine by growing in intestinal mucus (Miranda et al., 2004; Myhal et al., 1982; Sweeney et al., 1996a; Sweeney et al., 1996b; Wadolkowski et al., 1988) (Fig. 1A). Mucus is an important source of carbohydrates, mostly in the form of polysaccharides, which support bacterial colonization and growth (Fabich et al., 2008). However, E. coli do not secrete extracellular polysaccharide hydrolases (Henrissat and Davies, 1997; Hoskins et al., 1985) and therefore cannot directly use mucin-derived polysaccharides as a carbohydrate source. It is likely that polysaccharide-degrading anaerobes provide the mono- and disaccharides that E. coli need for growth (Conway et al., 2004), thereby promoting E. coli fitness in the intestine (Fig. 1A). The model of anaerobes feeding E. coli has been described by Leatham-Jensen et al. as the ‘restaurant’ hypothesis (Leatham-Jensen et al., 2012), which states that E. coli inhabits mixed biofilms in the intestine and grows on sugars served locally by surrounding anaerobes.
Colonization competition could also occur within the same species. For example, germ-free mice that are colonized with B. fragilis are resistant to further colonization of the same species (Lee et al., 2013). This ‘colonization resistance’ phenomenon could arise as a result of competition for a limited-space niche, as in the case of B. fragilis (Lee et al., 2013), or because of competition for nutrients, as has been proposed for E. coli (Fabich et al., 2008; Maltby et al., 2013) (Fig. 1A). Carbohydrates are fundamental nutrients for E. coli, and different E. coli strains can compete for the same kinds of sugar molecule, affecting each other’s fitness (Fabich et al., 2008; Maltby et al., 2013). It is not known whether different E. coli strains also compete for a specific colonization niche in the intestine, like B. fragilis.
Moreover, quorum sensing, a system that bacteria use to coordinate gene expression and behavior according to their local population density, could also be involved in modulating E. coli colonization and dissemination along the intestine. The quorum-sensing transcriptional regulator SdiA has been found to be necessary for E. coli O157:H7 colonization of the bovine intestine (Sharma and Bearson, 2013). Additionally, a mutant strain of E. coli O157:H7 that lacks the qseBC-encoded quorum-sensing system, which regulates the motility of E. coli O157:H7 in response to bacterial autoinducer 3 and the mammalian stress hormones epinephrine and norepinephrine, outcompetes its parental strain during colonization of the bovine intestine (Sharma and Casey, 2014). However, it remains unknown whether such mechanisms are involved in maintaining the colonization niche and growth of commensal E. coli in the human intestine.
Intestinal inflammation
Intestinal inflammation creates a markedly different environment for gut microbes. For example, the healthy intestine has a well-organized structure, and can be recognized by the lining of regular villi and the presence of columnar epithelial cells and goblet cells (Fig. 2). In comparison, the inflamed gut as seen in the Il-10−/− mouse, a spontaneous mouse model of colitis triggered by microbial colonization (Sellon et al., 1998), often shows loss of the villus structures, thickening of the mucosa, infiltration of inflammatory immune cells, depletion of goblet cells, disruption of the brush border and loss of surface epithelial cells (Fig. 2).
Fig. 2.

Intestinal inflammation creates a different environment for the gut microbes. Hematoxylin and eosin (H&E)-stained colon Swiss roll sections from a 9-month-old wild-type (WT) mouse (left; healthy) and an age-matched interleukin-10 knockout (Il-10−/−) mouse (right, inflamed). (Left, inset) The WT mouse colon has a well-organized lining of villi (gray box), with the presence of columnar epithelial cells (with microvilli; blue arrow) and goblet cells (black arrow). (Right, inset) The inflamed Il-10−/− mouse colon shows loss of the regular villus structures, thickening of the mucosa, crypt elongation (brown arrow), infiltration of inflammatory immune cells (yellow arrow), depletion of goblet cells, disruption of the brush border (red arrow) and loss of surface epithelial cells (green arrow).
As highly versatile microorganisms, E. coli can quickly adapt to the new environment. E. coli adjust their metabolic functions and upregulate stress response genes [e.g. glutamic acid decarboxylase A and B (gadA and gadB), inclusion body protein A and B (ibpA and ibpB) and inhibitor of C-type lysozyme (ivy)] during intestinal inflammation (Fig. 1B), as shown in experimental models (Patwa et al., 2011; Schumann et al., 2012). Inflammation is characterized by oxidative stress (high levels of reactive oxygen and nitrogen species), which is hazardous to bacteria. However, E. coli are equipped with a variety of responding strategies to help their survival (Imlay, 2013). It is worth noting that different E. coli strains likely rely on distinct mechanisms to survive in the inflamed environment. For example, as compared with E. coli NC101, the E. coli strain Nissle 1917 shows a much stronger upregulation of ivy expression, which promotes cell resistance to lysis by lysozyme, when colonizing the gut of germ-free Il-10−/− mice (Schumann et al., 2013).
E. coli might even exploit the environment in an inflamed gut to obtain a growth advantage. Chronic intestinal inflammation results in continuous epithelial cell death and tissue damage. The dead cells that slough off the intestinal luminal surface could provide extra nutrients, such as ethanolamine, to support E. coli growth (Bertin et al., 2011; Garsin, 2010) (Fig. 1B). The disruption of gut epithelial barrier during inflammation exposes deep gut tissue for E. coli translocation and infection (Fig. 1B). Recently, it was demonstrated that nitrate, a by-product of the host inflammatory response, can be used by E. coli for anaerobic respiration, thus conferring benefits to E. coli in the inflamed gut (Spees et al., 2013; Winter et al., 2013) (Fig. 1B). Although this represents a mechanism that potentially helps E. coli to outcompete fermenting anaerobes during intestinal inflammation, other IBD-associated factors could also promote bacterial growth. For example, individuals with IBD often have impaired innate and adaptive immune responses, with mutations in NOD2, NF-ΚB1 (nuclear factor kappa-light-chain-enhancer of activated B cells 1) and interleukin-related genes (IL2, IL21, IL23R, etc.) (Jostins et al., 2012), which might further facilitate E. coli adherence and invasion (Fig. 1B). In addition, defects in Paneth cell function in individuals with IBD (Wehkamp et al., 2005) and subsequently decreased secretion of antimicrobial peptides (Salzman et al., 2010) (Fig. 1B) could impair the control of bacterial infection by the intestinal mucosa, giving rise to a higher prevalence of E. coli.
E. coli, especially AIEC (Boudeau et al., 1999), have been extensively studied in individuals with IBD. AIEC type 1 pili and flagellae bind to the epithelial cell surface protein CEACAM6 (carcinoembryonic antigen-related cell adhesion molecule 6), which is upregulated in individuals with Crohn’s disease (Barnich et al., 2003; Barnich et al., 2007; Boudeau et al., 2001). Overexpression of CEACAM6 promotes AIEC colonization, and AIEC infection can further upregulate CEACAM6 (Fig. 1B) through the induction of the pro-inflammatory cytokines interferon γ (IFN-γ) and TNF-α (Barnich et al., 2007). The AIEC outer-membrane protein OmpA interacts with the endoplasmic reticulum (ER) stress-response glycoprotein Gp96, which is also overexpressed at the apical surface of ileal epithelial cells in individuals with Crohn’s disease (Rolhion et al., 2010). ER stress is commonly associated with inflammation (Garg et al., 2012; Kaser et al., 2013), and AIEC might take advantage of the ER stress response that occurs in individuals with IBD to increase its adherence to the intestinal epithelia. Moreover, the AIEC strain LF82, which was isolated from an IBD patient, has been shown to disrupt the polarized gut epithelial barrier and to replicate inside epithelial cells (Wine et al., 2009), implicating AIEC in the mucosal-barrier dysfunction observed in individuals with IBD (D’Incà et al., 2006; Hilsden et al., 1999; Wyatt et al., 1993).
A distinct feature of AIEC is the ability to survive and replicate in phagolysosomes within macrophages (De la Fuente et al., 2014; Glasser et al., 2001). AIEC infection stimulates expression of TNF-α in macrophages (Glasser et al., 2001). Interestingly, exogenous TNF-α treatment results in dose-dependent increases in the number of LF82 inside macrophages, whereas neutralization of TNF-α secreted by infected macrophages significantly reduces the number of intramacrophagic bacteria (Bringer et al., 2012). Therefore, AIEC-infection-induced TNF-α could promote bacterial growth inside macrophages (Fig. 1B).
Individuals with IBD who have mutations in innate response genes [e.g. NOD2, ATG16L1 (autophagy related 16-like 1), IRGM (immunity-related GTPase family M)] might also have impaired autophagic responses (Jostins et al., 2012), which could contribute to the overgrowth of AIEC, given that autophagy restricts the replication of intracellular AIEC (Lapaquette et al., 2010; Lu et al., 2014) (Fig. 1B). In fact, the AIEC strain LF82 has been shown to attenuate host-mediated autophagy by inducing microRNAs 30C and 130A, which downregulate genes required for the autophagic response (Nguyen et al., 2014) (Fig. 1B). A proper autophagy response is essential for intestinal homeostasis: defective function of ATG16L1 or XBP1 (X-box binding protein-1), which are involved in autophagy, in Paneth cells promotes inflammation in the ileum in mouse models (Adolph et al., 2013). Whether these mice are impaired in handling any microorganism or a specific one (e.g. E. coli) is currently unknown.
In summary, at gut homeostasis, E. coli are tightly controlled by a variety of host- and bacteria-derived mechanisms. During intestinal inflammation, they can quickly adjust to and even exploit the inflamed environment to gain a growth advantage. In the following section, we will review recent studies that demonstrate the cancer-promoting role of E. coli and discuss the key mechanisms by which dysregulated E. coli could mediate intestinal carcinogenesis.
E. coli cancer-promoting mechanisms
E. coli that colonize in the gut provide a source of antigens and toxins that foster host inflammatory responses, many of which have been linked to carcinogenesis (discussed earlier). For example, microbial activation of the innate immune pathways, in particular the LPS-TLR4 signaling pathway, can promote cancer development (Schwabe and Jobin, 2013). TLR4 is overexpressed in individuals with colitis-associated CRC (Fukata et al., 2007), and constitutive activation of TLR4 enhances cancer development in mouse models of this disease (Fukata et al., 2011). Therefore, microbial LPS could be involved in CRC development by activating TLR4 (Fig. 3). Although the cancer-promoting effect of LPS-TLR4 signaling is probably negligible at gut homeostasis, it could be markedly increased in IBD, where Gram-negative bacteria such as E. coli are abundantly present and the host gut barrier function and immune system are impaired.
Fig. 3.

E. coli cancer-promoting mechanisms. E. coli-induced innate immune responses (e.g. LPS-TLR4 signaling) can result in enhanced cell proliferation and survival through activating NF-κB and STAT3. Bacterial products (e.g. LPS) can promote cell proliferation by activating myeloid-cell-derived IL-23 and subsequently IL-17A. E. coli can induce epithelial-cell-derived IL-17C and subsequently Bcl-2 and Bcl-xL to enhance cell survival. Reactive oxygen and nitrogen species (ROS, RNS) produced by innate immune cells (e.g. macrophages and neutrophils) during E. coli infection, and E. coli-derived genotoxins (e.g. colibactin) can cause DNA damage and thus genomic instability. Colibactin-producing E. coli infection causes epithelial cell senescence, and senescent cells release growth factors (e.g. HGF) to promote cell proliferation. Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; HGF, hepatocyte growth factor; IL-17A, interleukin 17A; IL-17C, interleukin 17C; IL-23, interleukin 23; LPS, lipopolysaccharide; NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells; ROS, reactive oxygen species; RNS, reactive nitrogen species; STAT3, signal transducer and activator of transcription 3; TLR4, Toll-like receptor 4.
A key cancer-promoting downstream effect of innate immune signaling could be the induction of cell proliferation and survival pathways mediated by NF-κB and STAT3 (signal transducer and activator of transcription 3) (Elinav et al., 2013; Schwabe and Jobin, 2013) (Fig. 3). It should be emphasized though that innate immune responses have complex functions, and some innate immune sensors, such as NOD-like receptors NOD1, NOD2, NLRP3 and NLRP6, have been shown to play cancer-suppressing roles (Elinav et al., 2013; Schwabe and Jobin, 2013). Another model of inflammation-mediated carcinogenesis involves gut infiltration of phagocytes and their generation of reactive species, which causes DNA damage and thus genomic instability (Mangerich et al., 2012) (Fig. 3). Enhanced cell proliferation and survival combined with increased genomic instability heightens the risk of carcinogenesis.
Using the CPC-APC mouse model (mice that have Apc allelic loss specifically in the colonic epithelium and develop tumors primarily in the distal colon), Grivennikov et al. recently showed that microbial products, such as LPS, could activate interleukin 23 (IL-23) in tumor-associated myeloid cells and, subsequently, interleukin 17A (IL-17A) to drive tumor growth and progression (Grivennikov et al., 2012). This IL-23–IL-17A adaptive immune response might also contribute to the cancer-promoting effects of E. coli (Fig. 3). It is worth noting that the pro-tumorigenic function of LPS requires the presence of microbes, because LPS administration reduces colonic carcinogenesis in germ-free AOM-DSS-treated mice (Zhan et al., 2013).
More recently, Song et al. have shown that IL-17C, which is induced in intestinal epithelial cells by the gut microbiota, upregulates the anti-apoptotic factors Bcl-2 (B-cell lymphoma 2) and Bcl-xL (B-cell lymphoma-extra large), resulting in enhanced epithelial cell survival and thereby carcinogenesis (Song et al., 2014). Noticeably, E. coli colonization upregulates IL-17C in the colon of DSS-treated germ-free or antibiotic-treated mice (Song et al., 2014). Thus, the induction of IL-17C in intestinal epithelial cells could represent another mechanism by which E. coli promotes cancer development (Fig. 3).
Genomic instability can also arise from DNA damage induced by E. coli-derived genotoxins such as cytolethal distending toxin (CDT) and colibactin. CDT is the best-studied bacterial genotoxin and is secreted by many Gram-negative bacterial species, including E. coli (Guerra et al., 2011). CDT consists of two binding subunits (CdtA and CdtC), which mediate CDT internalization into target cells, and a catalytic subunit (CdtB), which cleaves DNA and induces DNA double-strand breaks (DSBs) (Guerra et al., 2011). The genotoxic property of CDT could be important for the carcinogenic potential of CDT-producing bacteria, because CdtB-mutant strains of Campylobacter jejuni (Fox et al., 2004) and of Helicobacter cinaedi (Shen et al., 2009) fail to elicit intestinal hyperplasia in NF-κB-deficient mice and intestinal dysplasia in Il-10−/− mice, respectively. However, it is not clear to what extent CDT contributes to the pro-tumorigenic potential of E. coli.
The genotoxin colibactin, which is encoded by a ~54-kb polyketide synthase (pks) pathogenicity island, is predominantly found in E. coli of the phylogroup B2 (Nougayrède et al., 2006). Approximately 75% of E. coli strains within the phylogroup B2 are pks-positive (pks+) (Johnson et al., 2008). Other bacterial species, such as Proteus mirabilis and Klebsiella pneumoniae, have also been found to produce colibactin (Putze et al., 2009). Colibactin-producing E. coli induce DSBs in vitro and in vivo, in various cells, including intestinal epithelial cells (Arthur et al., 2012; Cuevas-Ramos et al., 2010; Nougayrède et al., 2006; Secher et al., 2013). Unlike CDT, the genotoxic activity of colibactin requires cell-bacterium contact (Nougayrède et al., 2006). To date, little is known about how colibactin exerts its genotoxic effect, although the induction of DSBs is a key feature of its known activity. From the view point of microbial fitness, the presence of colibactin seems to correlate with the capacity of E. coli to maintain long-term colonization in the intestine (Nowrouzian and Oswald, 2012). The mechanism by which colibactin promotes colonization persistence in the intestine remains to be elucidated.
Several recent studies have demonstrated the cancer-promoting effect of E. coli and revealed that colibactin is a major contributing factor to E. coli-mediated intestinal carcinogenesis (Fig. 3). Arthur et al. reported that intestinal colonization of Il-10−/− mice by the human commensal Enterococcus faecalis strain OG1RF or the colibactin-producing AIEC strain NC101 caused comparable intestinal inflammation, but only NC101 was able to induce cancer in AOM-treated Il-10−/− mice (Arthur et al., 2012) (Table 1). This study also shows that colibactin is required for E. coli to mediate carcinogenesis in these mice, because a mutant strain of NC101 that lacks the pks pathogenicity island failed to elicit the carcinogenic effect even though intestinal inflammation was still severe (Arthur et al., 2012). This indicates that inflammation alone is not sufficient for CRC development, and provides the first evidence that specific bacterial activities, such as the production of colibactin by E. coli, are required for intestinal carcinogenesis. It is worth noting that this study uses Il-10−/− mice mono-associated with E. coli or E. faecalis (Arthur et al., 2012), and therefore the contribution of colibactin-producing E. coli to inflammation-related CRC pathogenesis in the presence of a complex microbiota remains to be elucidated. Considering the murine origin of E. coli NC101, it would also be interesting to test other E. coli strains, for example, clinical human CRC isolates, in this model.
Importantly, E. coli that produce colibactin show a higher prevalence in human cases of IBD and CRC than in non-IBD non-CRC controls (Arthur et al., 2012; Bonnet et al., 2014; Buc et al., 2013; Martin et al., 2004; Prorok-Hamon et al., 2014). These findings suggest that E. coli prevail during intestinal inflammation and contribute to carcinogenesis by producing colibactin.
The key role of E. coli colibactin in CRC development was subsequently confirmed in the Apcmin/+ mouse model (Bonnet et al., 2014). The human CRC-associated E. coli strain 11G5, which also produces colibactin, was shown to stimulate the development of colonic polyps in specific-pathogen-free (SPF) Apcmin/+ mice (Bonnet et al., 2014) (Table 1). In contrast, the E. coli strain K-12 MG1655, which does not produce colibactin, showed no pro-tumorigenic effect in this model (Bonnet et al., 2014). This study demonstrates the carcinogenic potential of colibactin-producing E. coli in the non-inflamed gut in the presence of a complex microbiota. This study also indicates that genetic predisposition (in this case, altered APC signaling) is required for E. coli to elicit its carcinogenic effect because wild-type mice infected with E. coli 11G5 did not show any neoplastic changes (Bonnet et al., 2014). Noticeably, Apcmin/+ mice colonized by E. coli 11G5 did not develop advanced carcinomas (Bonnet et al., 2014), suggesting that additional factors are required for the full development of cancers in this model. It is also worth noting that this E. coli/Apcmin/+ model involves pre-treating mice with streptomycin before E. coli infection (Bonnet et al., 2014). Because this broad-spectrum antibiotic can drastically perturb the commensal microbiota, this experimental procedure precludes investigation of the role for an intact commensal microbial community in regulating E. coli colonization and E. coli-mediated carcinogenesis.
Colibactin-producing E. coli have also been found to induce cell senescence in vitro, a phenomenon that might be linked to the carcinogenic effect of the bacteria (Cougnoux et al., 2014; Secher et al., 2013). Recently, Cougnoux et al. reported that senescent cells arising from a colibactin-producing E. coli infection secreted growth factors and promoted tumor growth in mouse models (Cougnoux et al., 2014) (Fig. 3). Nude mice (genetically engineered mice that have a greatly reduced number of T cells and therefore mount no rejection response to implanted materials) injected with a mixture of senescent HCT116 cells, induced by pks+ E. coli infection, and uninfected cells developed larger xenograft tumors than did mice injected with non-infected cells alone (Cougnoux et al., 2014) (Table 1). This enhanced tumor-growth phenotype was abrogated by the administration of HGF (hepatocyte growth factor) inhibitor, suggesting that the pro-tumorigenic effect of pks+ E. coli-induced senescent cells is mainly mediated by HGF (Cougnoux et al., 2014). In addition, the authors found that the human pks+ E. coli strain CCR20, but not the isogenic CCR20 pks mutant, significantly increased the number of tumors in AOM-DSS-treated mice (Cougnoux et al., 2014) (Table 1). Interestingly, CCR20 colonization did not affect inflammation, neoplastic grade or tumor size in this mouse model (Cougnoux et al., 2014). This study indicates that colibactin-producing E. coli mediate carcinogenesis through a variety of mechanisms, including senescence-induced HGF production and induction of host genomic instability (Fig. 3). However, given that, in the AOM-DSS mouse model, microbial colonization of germ-free mice attenuated colonic polyp formation (Zhan et al., 2013), this model might not be ideal for studying the contribution of pks+ E. coli to CRC. Moreover, this model requires the use of streptomycin (Cougnoux et al., 2014), as previously mentioned, which could have confounding effects on the interplay between the host and the microbiota.
In addition to genotoxin-induced DNA damage, E. coli might also encode factors, such as MutY (Khan and Cash, 2013), that could interfere with host-cell DNA-repair mechanisms to further increase genomic instability in the host and promote cancer susceptibility. Recently, Maddocks et al. reported that enteropathogenic E. coli-secreted effector protein EspF was required for the depletion of host-cell DNA mismatch repair (MMR) proteins (Maddocks et al., 2013). Future research will need to identify other E. coli-associated factors that interfere with host DNA repair, characterize their presence and working mechanisms in various E. coli strains, and evaluate their clinical relevance.
Other E. coli virulence factors, such as cytotoxic necrotizing factor 1 (CNF1) (Fabbri et al., 2013), have also been proposed to contribute to the cancer-promoting potential of E. coli. However, the in vivo relevance of these bacterial factors to human CRC remains to be determined.
Conclusions and future perspectives
E. coli is a common gut commensal, the colonization of which is tightly controlled in healthy individuals by both host- and bacteria-derived mechanisms. However, overrepresentation of E. coli is often observed in disease states, for example, in IBD and CRC. As an extremely versatile species, E. coli is able to adapt to and even take advantage of the environment present in the inflamed intestine to gain a growth advantage.
In the last decade, substantial evidence has been obtained from both clinical and basic research that the gut microbiota can profoundly affect intestinal inflammation and tumor development. In particular, certain members of the microbiota (so-called ‘alpha bugs’, which possess unique virulence traits) have been identified as direct drivers of intestinal carcinogenesis. The first example of such carcinogenic bacteria was the enterotoxigenic B. fragilis (Wu et al., 2009). However, although the microorganism has been linked to inflammatory diarrhea, its involvement in human IBD and CRC remains unclear. The recently reported colibactin-producing E. coli might be part of this novel group of ‘alpha bugs’.
Several research groups have recently demonstrated the carcinogenic role of colibactin-producing E. coli by using mouse models (Arthur et al., 2012; Bonnet et al., 2014; Cougnoux et al., 2014). However, certain limitations associated with the experimental models used in these studies should be recognized (discussed earlier). Using a model of vertical bacterial transmission (from mother to pup) of AIEC would help to determine the physiological role of early colonization by these bacteria in intestinal homeostasis and pathology. Such a model has recently been used to demonstrate that pks+ E. coli colonization results in DNA damage in neonates and genotoxic stress in adult mice (Payros et al., 2014).
Recent advances in our understanding of the regulation and function of E. coli in gut pathologies, such as IBD and CRC, also raise important new questions. It would be particularly interesting to know, for example, what factors and mechanisms lead to the dysbiotic state in the inflamed gut and especially to the high prevalence of E. coli. Because the microbiota responds to anti-inflammatory treatment (Rooks et al., 2014), it would be important to determine whether the activity of genotoxic factors, such as colibactin, is affected by the intestinal environment and therapeutic interventions. We recently observed transcriptional changes in pks-associated genes in E. coli strain NC101 during the development of colitis-associated CRC (Arthur et al., 2014), suggesting a dynamic microbial response to the intestinal environment. Colibactin modulates numerous aspects of host responses (DSB, cell senescence and transformation), and we need more information on how the microbial product is assembled, regulated and transported to mediate host responses. Purifying colibactin and resolving its structure would help to reveal the functional mechanisms underlying its genotoxic activity. This knowledge would be particularly valuable for designing drugs that could interfere with the activity of colibactin and prevent carcinogenesis mediated by colibactin-producing E. coli. Another interesting question is how other cyclomodulins (such as CDT) and toxins (such as CNF1) contribute to the pro-tumorigenic potential of E. coli.
IBD and CRC are likely polymicrobially driven, and dissecting the microbial-community actions that foster dysbiosis and CRC development is essential for understanding the disease etiology and for developing future therapies. Pathogenic traits of some bacteria might necessitate interaction with other members of the microbial community. Understanding the ‘cooperation’ between disease-associated bacteria is crucial for defining the role of the gut microbiota in IBD and CRC.
It is likely that microbial production of metabolites (e.g. SCFA), toxins (e.g. colibactin) and gas (e.g. H2S) influences the course of disease development, with some factors preventing while others promote it. The biological effect of a metabolite might depend on other environmental factors (diet, inflammation, stress, etc.) as well as host genetics. For example, a fiber-rich diet fosters the production of butyrate and attenuates CRC development through the enhanced function of regulatory T cells (Treg) (Singh et al., 2014). However, butyrate production induced by a carbohydrate-rich diet promotes neoplastic development in the intestine of Apcmin/+ mice that are also deficient for the DNA mismatch repair gene MutS homolog 2 (Msh2) (Belcheva et al., 2014). Dissecting the contribution of the microbial metabolites and their relationship with host genetic factors and other environmental components would provide a clearer picture of the events by which microbes influence intestinal pathology.
Although deciphering the function and activities of gut microbes that are associated with intestinal protection and pathology remains a daunting task, one that requires a wide range of expertise (microbiology, genomics, proteomics, metabolomics, etc.), it is evident that this field of research holds much promise for disease prevention and treatment.
Acknowledgments
We would like to thank the Jobin lab (especially Dr Xiaolun Sun, Ernesto Perez-Chanona, Sarah E. Tomkovich and Chelsea C. Jacobs) for critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing financial interests.
Funding
This work was supported by the National Institutes of Health [RO1DK047700 and RO1DK073338 to C.J.].
References
- Adolph T. E., Tomczak M. F., Niederreiter L., Ko H. J., Böck J., Martinez-Naves E., Glickman J. N., Tschurtschenthaler M., Hartwig J., Hosomi S., et al. (2013). Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J., Sinha R., Pei Z., Dominianni C., Wu J., Shi J., Goedert J. J., Hayes R. B., Yang L. (2013). Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 105, 1907–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert C., Scheel J., Engst W., Loh G., Blaut M. (2009). Adaptation of protein expression by Escherichia coli in the gastrointestinal tract of gnotobiotic mice. Environ. Microbiol. 11, 751–761 [DOI] [PubMed] [Google Scholar]
- Apperloo-Renkema H. Z., Van der Waaij B. D., Van der Waaij D. (1990). Determination of colonization resistance of the digestive tract by biotyping of Enterobacteriaceae. Epidemiol. Infect. 105, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N., Campbell C., Fan X., Dikiy S., van der Veeken J., deRoos P., Liu H., Cross J. R., Pfeffer K., Coffer P. J., et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J. C., Jobin C. (2013). The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 4, 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J. C., Perez-Chanona E., Mühlbauer M., Tomkovich S., Uronis J. M., Fan T. J., Campbell B. J., Abujamel T., Dogan B., Rogers A. B., et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J. C., Gharaibeh R. Z., Mühlbauer M., Perez-Chanona E., Uronis J. M., McCafferty J., Fordor A. A., Jobin C. (2014). Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. (in press) 10.1038/ncomms5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K., Tanoue T., Shima T., Imaoka A., Kuwahara T., Momose Y., Cheng G., Yamasaki S., Saito T., Ohba Y., et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbas A. S., Lesher A. P., Thomas A. D., Wyse A., Devalapalli A. P., Lee Y. H., Tan H. E., Orndorff P. E., Bollinger R. R., Parker W. (2009). Altering and assessing persistence of genetically modified E. coli MG1655 in the large bowel. Exp. Biol. Med. (Maywood) 234, 1174–1185 [DOI] [PubMed] [Google Scholar]
- Barnich N., Boudeau J., Claret L., Darfeuille-Michaud A. (2003). Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol. Microbiol. 48, 781–794 [DOI] [PubMed] [Google Scholar]
- Barnich N., Carvalho F. A., Glasser A. L., Darcha C., Jantscheff P., Allez M., Peeters H., Bommelaer G., Desreumaux P., Colombel J. F., et al. (2007). CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J. Clin. Invest. 117, 1566–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates J. M., Akerlund J., Mittge E., Guillemin K. (2007). Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart D. C., Sandborn W. J. (2012). Crohn’s disease. Lancet 380, 1590–1605 [DOI] [PubMed] [Google Scholar]
- Baumgart M., Dogan B., Rishniw M., Weitzman G., Bosworth B., Yantiss R., Orsi R. H., Wiedmann M., McDonough P., Kim S. G., et al. (2007). Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 1, 403–418 [DOI] [PubMed] [Google Scholar]
- Belcheva A., Irrazabal T., Robertson S. J., Streutker C., Maughan H., Rubino S., Moriyama E. H., Copeland J. K., Kumar S., Green B., et al. (2014). Gut microbial metabolism drives transformation of msh2-deficient colon epithelial cells. Cell 158, 288–299 [DOI] [PubMed] [Google Scholar]
- Bergstrom K. S., Kissoon-Singh V., Gibson D. L., Ma C., Montero M., Sham H. P., Ryz N., Huang T., Velcich A., Finlay B. B., et al. (2010). Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 6, e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin Y., Girardeau J. P., Chaucheyras-Durand F., Lyan B., Pujos-Guillot E., Harel J., Martin C. (2011). Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ. Microbiol. 13, 365–377 [DOI] [PubMed] [Google Scholar]
- Boleij A., Tjalsma H. (2012). Gut bacteria in health and disease: a survey on the interface between intestinal microbiology and colorectal cancer. Biol. Rev. Camb. Philos. Soc. 87, 701–730 [DOI] [PubMed] [Google Scholar]
- Bollinger R. R., Everett M. L., Palestrant D., Love S. D., Lin S. S., Parker W. (2003). Human secretory immunoglobulin A may contribute to biofilm formation in the gut. Immunology 109, 580–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger R. R., Everett M. L., Wahl S. D., Lee Y. H., Orndorff P. E., Parker W. (2006). Secretory IgA and mucin-mediated biofilm formation by environmental strains of Escherichia coli: role of type 1 pili. Mol. Immunol. 43, 378–387 [DOI] [PubMed] [Google Scholar]
- Bonnet M., Buc E., Sauvanet P., Darcha C., Dubois D., Pereira B., Déchelotte P., Bonnet R., Pezet D., Darfeuille-Michaud A. (2014). Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 20, 859–867 [DOI] [PubMed] [Google Scholar]
- Boudeau J., Glasser A. L., Masseret E., Joly B., Darfeuille-Michaud A. (1999). Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 67, 4499–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J., Barnich N., Darfeuille-Michaud A. (2001). Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol. Microbiol. 39, 1272–1284 [DOI] [PubMed] [Google Scholar]
- Bringer M. A., Billard E., Glasser A. L., Colombel J. F., Darfeuille-Michaud A. (2012). Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-α secretion. Lab. Invest. 92, 411–419 [DOI] [PubMed] [Google Scholar]
- Buc E., Dubois D., Sauvanet P., Raisch J., Delmas J., Darfeuille-Michaud A., Pezet D., Bonnet R. (2013). High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 8, e56964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho F. A., Koren O., Goodrich J. K., Johansson M. E., Nalbantoglu I., Aitken J. D., Su Y., Chassaing B., Walters W. A., González A., et al. (2012). Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12, 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellarin M., Warren R. L., Freeman J. D., Dreolini L., Krzywinski M., Strauss J., Barnes R., Watson P., Allen-Vercoe E., Moore R. A., et al. (2012). Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCFA (2011). Facts about Inflammatory Bowel Diseases. New York, NY: CCFA [Google Scholar]
- Cerutti A., Rescigno M. (2008). The biology of intestinal immunoglobulin A responses. Immunity 28, 740–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D. E., Smalley D. J., Tucker D. L., Leatham M. P., Norris W. E., Stevenson S. J., Anderson A. B., Grissom J. E., Laux D. C., Cohen P. S., et al. (2004). Carbon nutrition of Escherichia coli in the mouse intestine. Proc. Natl. Acad. Sci. USA 101, 7427–7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B., Darfeuille-Michaud A. (2011). The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140, 1720–1728.e3 [DOI] [PubMed] [Google Scholar]
- Chen W., Liu F., Ling Z., Tong X., Xiang C. (2012). Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 7, e39743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente J. C., Ursell L. K., Parfrey L. W., Knight R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements A., Young J. C., Constantinou N., Frankel G. (2012). Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes 3, 71–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway T., Krogfelt K. A., Cohen P. S. (2004). The life of commensal escherichia coli in the mammalian intestine. In EcoSal-Escherichia Coli and Salmonella: Cellular and Molecular Biology (ed. Curtiss R., III). Washington, DC: ASM Press; [DOI] [PubMed] [Google Scholar]
- Costello E. K., Lauber C. L., Hamady M., Fierer N., Gordon J. I., Knight R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougnoux A., Dalmasso G., Martinez R., Buc E., Delmas J., Gibold L., Sauvanet P., Darcha C., Déchelotte P., Bonnet M., et al. (2014). Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. [DOI] [PubMed] [Google Scholar]
- Cuevas-Ramos G., Petit C. R., Marcq I., Boury M., Oswald E., Nougayrède J. P. (2010). Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 107, 11537–11542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunliffe R. N., Mahida Y. R. (2004). Expression and regulation of antimicrobial peptides in the gastrointestinal tract. J. Leukoc. Biol. 75, 49–58 [DOI] [PubMed] [Google Scholar]
- D’Haens G. R., Geboes K., Peeters M., Baert F., Penninckx F., Rutgeerts P. (1998). Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 114, 262–267 [DOI] [PubMed] [Google Scholar]
- D’Incà R., Annese V., di Leo V., Latiano A., Quaino V., Abazia C., Vettorato M. G., Sturniolo G. C. (2006). Increased intestinal permeability and NOD2 variants in familial and sporadic Crohn’s disease. Aliment. Pharmacol. Ther. 23, 1455–1461 [DOI] [PubMed] [Google Scholar]
- Darfeuille-Michaud A., Neut C., Barnich N., Lederman E., Di Martino P., Desreumaux P., Gambiez L., Joly B., Cortot A., Colombel J. F. (1998). Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 115, 1405–1413 [DOI] [PubMed] [Google Scholar]
- Darfeuille-Michaud A., Boudeau J., Bulois P., Neut C., Glasser A. L., Barnich N., Bringer M. A., Swidsinski A., Beaugerie L., Colombel J. F. (2004). High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 [DOI] [PubMed] [Google Scholar]
- De la Fuente M., Franchi L., Araya D., Díaz-Jiménez D., Olivares M., Álvarez-Lobos M., Golenbock D., González M. J., López-Kostner F., Quera R., et al. (2014). Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int. J. Med. Microbiol. 304, 384–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P. B., Bik E. M., Bernstein C. N., Purdom E., Dethlefsen L., Sargent M., Gill S. R., Nelson K. E., Relman D. A. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E., Nowarski R., Thaiss C. A., Hu B., Jin C., Flavell R. A. (2013). Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771 [DOI] [PubMed] [Google Scholar]
- Elliott T. R., Hudspith B. N., Wu G., Cooley M., Parkes G., Quiñones B., Randall L., Mandrell R. E., Fagerquist C. K., Brostoff J., et al. (2013). Quantification and characterization of mucosa-associated and intracellular Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 19, 2326–2338 [DOI] [PubMed] [Google Scholar]
- Esteban-Jurado C., Garre P., Vila M., Lozano J. J., Pristoupilova A., Beltrán S., Abulí A., Muñoz J., Balaguer F., Ocaña T., et al. (2014). New genes emerging for colorectal cancer predisposition. World J. Gastroenterol. 20, 1961–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri A., Travaglione S., Ballan G., Loizzo S., Fiorentini C. (2013). The cytotoxic necrotizing factor 1 from E. coli: a janus toxin playing with cancer regulators. Toxins (Basel) 5, 1462–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabich A. J., Jones S. A., Chowdhury F. Z., Cernosek A., Anderson A., Smalley D., McHargue J. W., Hightower G. A., Smith J. T., Autieri S. M., et al. (2008). Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 76, 1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagins L. A., Souza R. F., Spechler S. J. (2009). Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol 6, 297–305 [DOI] [PubMed] [Google Scholar]
- Feller M., Huwiler K., Schoepfer A., Shang A., Furrer H., Egger M. (2010). Long-term antibiotic treatment for Crohn’s disease: systematic review and meta-analysis of placebo-controlled trials. Clin. Infect. Dis. 50, 473–480 [DOI] [PubMed] [Google Scholar]
- Fox J. G., Rogers A. B., Whary M. T., Ge Z., Taylor N. S., Xu S., Horwitz B. H., Erdman S. E. (2004). Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect. Immun. 72, 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freestone P. P., Walton N. J., Haigh R. D., Lyte M. (2007). Influence of dietary catechols on the growth of enteropathogenic bacteria. Int. J. Food Microbiol. 119, 159–169 [DOI] [PubMed] [Google Scholar]
- Fukata M., Chen A., Vamadevan A. S., Cohen J., Breglio K., Krishnareddy S., Hsu D., Xu R., Harpaz N., Dannenberg A. J., et al. (2007). Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133, 1869–1869.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M., Shang L., Santaolalla R., Sotolongo J., Pastorini C., España C., Ungaro R., Harpaz N., Cooper H. S., Elson G., et al. (2011). Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm. Bowel Dis. 17, 1464–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S., Toh H., Hase K., Oshima K., Nakanishi Y., Yoshimura K., Tobe T., Clarke J. M., Topping D. L., Suzuki T., et al. (2011). Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–547 [DOI] [PubMed] [Google Scholar]
- Fukuda S., Toh H., Taylor T. D., Ohno H., Hattori M. (2012). Acetate-producing bifidobacteria protect the host from enteropathogenic infection via carbohydrate transporters. Gut Microbes 3, 449–454 [DOI] [PubMed] [Google Scholar]
- Furusawa Y., Obata Y., Fukuda S., Endo T. A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T., et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 [DOI] [PubMed] [Google Scholar]
- Garg A. D., Kaczmarek A., Krysko O., Vandenabeele P., Krysko D. V., Agostinis P. (2012). ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med. 18, 589–598 [DOI] [PubMed] [Google Scholar]
- Garrett W. S., Gallini C. A., Yatsunenko T., Michaud M., DuBois A., Delaney M. L., Punit S., Karlsson M., Bry L., Glickman J. N., et al. (2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin D. A. (2010). Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 8, 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser A. L., Boudeau J., Barnich N., Perruchot M. H., Colombel J. F., Darfeuille-Michaud A. (2001). Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 69, 5529–5537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S. I. (2013). Inflammation and colorectal cancer: colitis-associated neoplasia. Semin. Immunopathol. 35, 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S. I., Wang K., Mucida D., Stewart C. A., Schnabl B., Jauch D., Taniguchi K., Yu G. Y., Osterreicher C. H., Hung K. E., et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L., Guidi R., Frisan T. (2011). Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 278, 4577–4588 [DOI] [PubMed] [Google Scholar]
- Halme L., Paavola-Sakki P., Turunen U., Lappalainen M., Farkkila M., Kontula K. (2006). Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 12, 3668–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy H., Harris J., Lyon E., Beal J., Foey A. D. (2013). Probiotics, prebiotics and immunomodulation of gut mucosal defences: homeostasis and immunopathology. Nutrients 5, 1869–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B., Davies G. (1997). Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 [DOI] [PubMed] [Google Scholar]
- Hepworth M. R., Monticelli L. A., Fung T. C., Ziegler C. G., Grunberg S., Sinha R., Mantegazza A. R., Ma H. L., Crawford A., Angelosanto J. M., et al. (2013). Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrinton L. J., Liu L., Levin T. R., Allison J. E., Lewis J. D., Velayos F. (2012). Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 143, 382–389 [DOI] [PubMed] [Google Scholar]
- Hilsden R. J., Meddings J. B., Hardin J., Gall D. G., Sutherland L. R. (1999). Intestinal permeability and postheparin plasma diamine oxidase activity in the prediction of Crohn’s disease relapse. Inflamm. Bowel Dis. 5, 85–91 [DOI] [PubMed] [Google Scholar]
- Høivik M. L., Moum B., Solberg I. C., Henriksen M., Cvancarova M., Bernklev T., IBSEN Group (2013). Work disability in inflammatory bowel disease patients 10 years after disease onset: results from the IBSEN Study. Gut 62, 368–375 [DOI] [PubMed] [Google Scholar]
- Hoskins L. C., Agustines M., McKee W. B., Boulding E. T., Kriaris M., Niedermeyer G. (1985). Mucin degradation in human colon ecosystems. Isolation and properties of fecal strains that degrade ABH blood group antigens and oligosaccharides from mucin glycoproteins. J. Clin. Invest. 75, 944–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot J. P., Chamaillard M., Zouali H., Lesage S., Cézard J. P., Belaiche J., Almer S., Tysk C., O’Morain C. A., Gassull M., et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay J. A. (2013). The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järnerot G., Tysk C., Bohr J., Eriksson S. (1995). Collagenous colitis and fecal stream diversion. Gastroenterology 109, 449–455 [DOI] [PubMed] [Google Scholar]
- Jobin C. (2013). Colorectal cancer: looking for answers in the microbiota. Cancer Discov 3, 384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. E., Phillipson M., Petersson J., Velcich A., Holm L., Hansson G. C. (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 105, 15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. E., Sjövall H., Hansson G. C. (2013). The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10, 352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. R., Johnston B., Kuskowski M. A., Nougayrede J. P., Oswald E. (2008). Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 46, 3906–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L., Ripke S., Weersma R. K., Duerr R. H., McGovern D. P., Hui K. Y., Lee J. C., Schumm L. P., Sharma Y., Anderson C. A., et al. International IBD Genetics Consortium (IIBDGC) (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N., Seo S. U., Chen G. Y., Núñez G. (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 13, 321–335 [DOI] [PubMed] [Google Scholar]
- Kaser A., Zeissig S., Blumberg R. S. (2010). Inflammatory bowel disease. Annu. Rev. Immunol. 28, 573–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A., Adolph T. E., Blumberg R. S. (2013). The unfolded protein response and gastrointestinal disease. Semin. Immunopathol. 35, 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. A., Cash P. (2013). E. coli and colon cancer: is mutY a culprit? Cancer Lett. 341, 127–131 [DOI] [PubMed] [Google Scholar]
- Kostic A. D., Gevers D., Pedamallu C. S., Michaud M., Duke F., Earl A. M., Ojesina A. I., Jung J., Bass A. J., Tabernero J., et al. (2012). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic A. D., Chun E., Robertson L., Glickman J. N., Gallini C. A., Michaud M., Clancy T. E., Chung D. C., Lochhead P., Hold G. L., et al. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic A. D., Xavier R. J., Gevers D. (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapaquette P., Glasser A. L., Huett A., Xavier R. J., Darfeuille-Michaud A. (2010). Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 12, 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatham-Jensen M. P., Frimodt-Møller J., Adediran J., Mokszycki M. E., Banner M. E., Caughron J. E., Krogfelt K. A., Conway T., Cohen P. S. (2012). The streptomycin-treated mouse intestine selects Escherichia coli envZ missense mutants that interact with dense and diverse intestinal microbiota. Infect. Immun. 80, 1716–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. M., Donaldson G. P., Mikulski Z., Boyajian S., Ley K., Mazmanian S. K. (2013). Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Chen J., Xu H. G., Zhou X., He Q., Li Y. L., Jiang G., Shan Y., Xue B., Zhao R. X., et al. (2014). MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 146, 188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyte M., Vulchanova L., Brown D. R. (2011). Stress at the intestinal surface: catecholamines and mucosa-bacteria interactions. Cell Tissue Res. 343, 23–32 [DOI] [PubMed] [Google Scholar]
- Maddocks O. D., Scanlon K. M., Donnenberg M. S. (2013). An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. MBio 4, e00152–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltby R., Leatham-Jensen M. P., Gibson T., Cohen P. S., Conway T. (2013). Nutritional basis for colonization resistance by human commensal Escherichia coli strains HS and Nissle 1917 against E. coli O157:H7 in the mouse intestine. PLoS ONE 8, e53957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich A., Knutson C. G., Parry N. M., Muthupalani S., Ye W., Prestwich E., Cui L., McFaline J. L., Mobley M., Ge Z., et al. (2012). Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. USA 109, E1820–E1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichanh C., Borruel N., Casellas F., Guarner F. (2012). The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9, 599–608 [DOI] [PubMed] [Google Scholar]
- Martin H. M., Campbell B. J., Hart C. A., Mpofu C., Nayar M., Singh R., Englyst H., Williams H. F., Rhodes J. M. (2004). Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127, 80–93 [DOI] [PubMed] [Google Scholar]
- Maslowski K. M., Vieira A. T., Ng A., Kranich J., Sierro F., Yu D., Schilter H. C., Rolph M. S., Mackay F., Artis D., et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. N., Araújo-Pérez F., Azcárate-Peril A., Yeh J. J., Sandler R. S., Keku T. O. (2013). Fusobacterium is associated with colorectal adenomas. PLoS ONE 8, e53653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda R. L., Conway T., Leatham M. P., Chang D. E., Norris W. E., Allen J. H., Stevenson S. J., Laux D. C., Cohen P. S. (2004). Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect. Immun. 72, 1666–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan X. C., Tickle T. L., Sokol H., Gevers D., Devaney K. L., Ward D. V., Reyes J. A., Shah S. A., LeLeiko N., Snapper S. B., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhya I., Hansen R., El-Omar E. M., Hold G. L. (2012). IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol 9, 219–230 [DOI] [PubMed] [Google Scholar]
- Myhal M. L., Laux D. C., Cohen P. S. (1982). Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1, 186–192 [DOI] [PubMed] [Google Scholar]
- Netjes J. E., Rijken M. (2013). Labor participation among patients with inflammatory bowel disease. Inflamm. Bowel Dis. 19, 81–91 [DOI] [PubMed] [Google Scholar]
- Ng S. C., Tsoi K. K., Kamm M. A., Xia B., Wu J., Chan F. K., Sung J. J. (2012). Genetics of inflammatory bowel disease in Asia: systematic review and meta-analysis. Inflamm. Bowel Dis. 18, 1164–1176 [DOI] [PubMed] [Google Scholar]
- Nguyen H. T., Dalmasso G., Müller S., Carrière J., Seibold F., Darfeuille-Michaud A. (2014). Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology 146, 508–519 [DOI] [PubMed] [Google Scholar]
- Nougayrède J. P., Homburg S., Taieb F., Boury M., Brzuszkiewicz E., Gottschalk G., Buchrieser C., Hacker J., Dobrindt U., Oswald E. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851 [DOI] [PubMed] [Google Scholar]
- Nowrouzian F. L., Oswald E. (2012). Escherichia coli strains with the capacity for long-term persistence in the bowel microbiota carry the potentially genotoxic pks island. Microb. Pathog. 53, 180–182 [DOI] [PubMed] [Google Scholar]
- Ogura Y., Bonen D. K., Inohara N., Nicolae D. L., Chen F. F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R. H., et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606 [DOI] [PubMed] [Google Scholar]
- Ordás I., Eckmann L., Talamini M., Baumgart D. C., Sandborn W. J. (2012). Ulcerative colitis. Lancet 380, 1606–1619 [DOI] [PubMed] [Google Scholar]
- Packey C. D., Sartor R. B. (2009). Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr. Opin. Infect. Dis. 22, 292–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C., Bik E. M., DiGiulio D. B., Relman D. A., Brown P. O. (2007). Development of the human infant intestinal microbiota. PLoS Biol. 5, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwa L. G., Fan T. J., Tchaptchet S., Liu Y., Lussier Y. A., Sartor R. B., Hansen J. J. (2011). Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141, 1842–51 e1–10 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payros D., Secher T., Boury M., Brehin C., Ménard S., Salvador-Cartier C., Ceevas-Ramos G., Watrin C., Marcq I., Nougayrède J. P., et al. (2014). Maternally acquired genotoxic Escherichia coli alters offspring’s intestinal homeostasis. Gut Microbes 5, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prideaux L., Kamm M. A., De Cruz P. P., Chan F. K., Ng S. C. (2012). Inflammatory bowel disease in Asia: a systematic review. J. Gastroenterol. Hepatol. 27, 1266–1280 [DOI] [PubMed] [Google Scholar]
- Prorok-Hamon M., Friswell M. K., Alswied A., Roberts C. L., Song F., Flanagan P. K., Knight P., Codling C., Marchesi J. R., Winstanley C., et al. (2014). Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 63, 761–770 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putze J., Hennequin C., Nougayrède J. P., Zhang W., Homburg S., Karch H., Bringer M. A., Fayolle C., Carniel E., Rabsch W., et al. (2009). Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 77, 4696–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion N., Barnich N., Bringer M. A., Glasser A. L., Ranc J., Hébuterne X., Hofman P., Darfeuille-Michaud A. (2010). Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut 59, 1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks M. G., Veiga P., Wardwell-Scott L. H., Tickle T., Segata N., Michaud M., Gallini C. A., Beal C., van Hylckama-Vlieg J. E., Ballal S. A., et al. (2014). Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 8, 1403–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round J. L., Lee S. M., Li J., Tran G., Jabri B., Chatila T. A., Mazmanian S. K. (2011). The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K., Anba J., Corthier G., Rigottier-Gois L., Monnet V., Mistou M. Y. (2008). Metabolic adaptation of Lactococcus lactis in the digestive tract: the example of response to lactose. J. Mol. Microbiol. Biotechnol. 14, 137–144 [DOI] [PubMed] [Google Scholar]
- Rubinstein M. R., Wang X., Liu W., Hao Y., Cai G., Han Y. W. (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustgi A. K. (2007). The genetics of hereditary colon cancer. Genes Dev. 21, 2525–2538 [DOI] [PubMed] [Google Scholar]
- Rutgeerts P., Goboes K., Peeters M., Hiele M., Penninckx F., Aerts R., Kerremans R., Vantrappen G. (1991). Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 338, 771–774 [DOI] [PubMed] [Google Scholar]
- Salzman N. H., Hung K., Haribhai D., Chu H., Karlsson-Sjöberg J., Amir E., Teggatz P., Barman M., Hayward M., Eastwood D., et al. (2010). Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann S., Alpert C., Engst W., Loh G., Blaut M. (2012). Dextran sodium sulfate-induced inflammation alters the expression of proteins by intestinal Escherichia coli strains in a gnotobiotic mouse model. Appl. Environ. Microbiol. 78, 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann S., Alpert C., Engst W., Klopfleisch R., Loh G., Bleich A., Blaut M. (2013). Mild gut inflammation modulates the proteome of intestinal Escherichia coli. Environ. Microbiol. [DOI] [PubMed] [Google Scholar]
- Schwabe R. F., Jobin C. (2013). The microbiome and cancer. Nat. Rev. Cancer 13, 800–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears C. L., Garrett W. S. (2014). Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears C. L., Pardoll D. M. (2011). Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J. Infect. Dis. 203, 306–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secher T., Samba-Louaka A., Oswald E., Nougayrède J. P. (2013). Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS ONE 8, e77157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby W., Pavli P., Crotty B., Florin T., Radford-Smith G., Gibson P., Mitchell B., Connell W., Read R., Merrett M., et al. Antibiotics in Crohn’s Disease Study Group (2007). Two-year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn’s disease. Gastroenterology 132, 2313–2319 [DOI] [PubMed] [Google Scholar]
- Sellon R. K., Tonkonogy S., Schultz M., Dieleman L. A., Grenther W., Balish E., Rennick D. M., Sartor R. B. (1998). Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V. K., Bearson S. M. (2013). Evaluation of the impact of quorum sensing transcriptional regulator SdiA on long-term persistence and fecal shedding of Escherichia coli O157:H7 in weaned calves. Microb. Pathog. 57, 21–26 [DOI] [PubMed] [Google Scholar]
- Sharma V. K., Casey T. A. (2014). Escherichia coli O157:H7 lacking the qseBC-encoded quorum-sensing system outcompetes the parental strain in colonization of cattle intestines. Appl. Environ. Microbiol. 80, 1882–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z., Feng Y., Rogers A. B., Rickman B., Whary M. T., Xu S., Clapp K. M., Boutin S. R., Fox J. G. (2009). Cytolethal distending toxin promotes Helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 77, 2508–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R., Ma J., Zou Z., Jemal A. (2014). Cancer statistics, 2014. CA Cancer J. Clin. 64, 9–29 [DOI] [PubMed] [Google Scholar]
- Singh N., Gurav A., Sivaprakasam S., Brady E., Padia R., Shi H., Thangaraju M., Prasad P. D., Manicassamy S., Munn D. H., et al. (2014). Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack E., Hapfelmeier S., Stecher B., Velykoredko Y., Stoel M., Lawson M. A., Geuking M. B., Beutler B., Tedder T. F., Hardt W. D., et al. (2009). Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325, 617–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P. M., Howitt M. R., Panikov N., Michaud M., Gallini C. A., Bohlooly-Y M., Glickman J. N., Garrett W. S. (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhani I., Tap J., Roudot-Thoraval F., Roperch J. P., Letulle S., Langella P., Corthier G., Tran Van, Nhieu J., Furet J. P. (2011). Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6, e16393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H., Pigneur B., Watterlot L., Lakhdari O., Bermúdez-Humarán L. G., Gratadoux J. J., Blugeon S., Bridonneau C., Furet J. P., Corthier G., et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 105, 16731–16736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Gao H., Lin Y., Yao Y., Zhu S., Wang J., Liu Y., Yao X., Meng G., Shen N., et al. (2014). Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40, 140–152 [DOI] [PubMed] [Google Scholar]
- Spees A. M., Wangdi T., Lopez C. A., Kingsbury D. D., Xavier M. N., Winter S. E., Tsolis R. M., Bäumler A. J. (2013). Streptomycin-induced inflammation enhances Escherichia coli gut colonization through nitrate respiration. MBio 4, e00430–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. (2013). Impact of the gut microbiome on mucosal inflammation. Trends Immunol. 34, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney N. J., Klemm P., McCormick B. A., Moller-Nielsen E., Utley M., Schembri M. A., Laux D. C., Cohen P. S. (1996a). The Escherichia coli K-12 gntP gene allows E. coli F-18 to occupy a distinct nutritional niche in the streptomycin-treated mouse large intestine. Infect. Immun. 64, 3497–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney N. J., Laux D. C., Cohen P. S. (1996b). Escherichia coli F-18 and E. coli K-12 eda mutants do not colonize the streptomycin-treated mouse large intestine. Infect. Immun. 64, 3504–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thia K. T., Mahadevan U., Feagan B. G., Wong C., Cockeram A., Bitton A., Bernstein C. N., Sandborn W. J. (2009). Ciprofloxacin or metronidazole for the treatment of perianal fistulas in patients with Crohn’s disease: a randomized, double-blind, placebo-controlled pilot study. Inflamm. Bowel Dis. 15, 17–24 [DOI] [PubMed] [Google Scholar]
- Vogel-Scheel J., Alpert C., Engst W., Loh G., Blaut M. (2010). Requirement of purine and pyrimidine synthesis for colonization of the mouse intestine by Escherichia coli. Appl. Environ. Microbiol. 76, 5181–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadolkowski E. A., Laux D. C., Cohen P. S. (1988). Colonization of the streptomycin-treated mouse large intestine by a human fecal Escherichia coli strain: role of growth in mucus. Infect. Immun. 56, 1030–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson I. R., Takahashi K., Futreal P. A., Chin L. (2013). Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 14, 703–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehkamp J., Salzman N. H., Porter E., Nuding S., Weichenthal M., Petras R. E., Shen B., Schaeffeler E., Schwab M., Linzmeier R., et al. (2005). Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 102, 18129–18134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine E., Ossa J. C., Gray-Owen S. D., Sherman P. M. (2009). Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 9, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S. E., Thiennimitr P., Winter M. G., Butler B. P., Huseby D. L., Crawford R. W., Russell J. M., Bevins C. L., Adams L. G., Tsolis R. M., et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S. E., Winter M. G., Xavier M. N., Thiennimitr P., Poon V., Keestra A. M., Laughlin R. C., Gomez G., Wu J., Lawhon S. D., et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Rhee K. J., Albesiano E., Rabizadeh S., Wu X., Yen H. R., Huso D. L., Brancati F. L., Wick E., McAllister F., et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N., Yang X., Zhang R., Li J., Xiao X., Hu Y., Chen Y., Yang F., Lu N., Wang Z., et al. (2013). Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 66, 462–470 [DOI] [PubMed] [Google Scholar]
- Wyatt J., Vogelsang H., Hübl W., Waldhöer T., Lochs H. (1993). Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 341, 1437–1439 [DOI] [PubMed] [Google Scholar]
- Yatsunenko T., Rey F. E., Manary M. J., Trehan I., Dominguez-Bello M. G., Contreras M., Magris M., Hidalgo G., Baldassano R. N., Anokhin A. P., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J., Wang B., Sun Z., Bo X., Yuan X., He X., Zhao H., Du X., Wang F., Jiang Z., et al. (2008). Analysis of host-inducing proteome changes in bifidobacterium longum NCC2705 grown in vivo. J. Proteome Res. 7, 375–385 [DOI] [PubMed] [Google Scholar]
- Zhan Y., Chen P. J., Sadler W. D., Wang F., Poe S., Núñez G., Eaton K. A., Chen G. Y. (2013). Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 73, 7199–7210 [DOI] [PMC free article] [PubMed] [Google Scholar]