

Abstract
Background
A lymphotropic hepatitis C virus strain (HCV, SB strain, hereafter “SB-HCV”) has been shown to infect established T cell lines (Molt-4 and Jurkat) and primary human naive CD4+ T cells. During T cell development and activation, transient expression of CD44 splicing variant 6 (CD44v6) plays a significant role.
Methods
SB-HCV was used to infect Molt-4 cells, and their cellular proliferation and CD44 expression was examined.
Results
SB-HCV–infected Molt-4 cells expressed a significantly lower level of the CD44v6 isoform. The infected cells could be divided into 2 carboxyfluorescein succinimidyl ester (CFSE) groups, CFSE–high (indicating low proliferation activity; 34.2% of the cells) and CFSE-low (indicating high proliferation activity; 62.5% of the cells), whereas uninfected cells consisted of only a CFSE-low population. Of the CFSE-high cells, 82.4% were positive for the HCV protein NS5A, whereas only 1.2% of the CFSE-low cells were positive for this protein. Among the HCV proteins, NS5A alone caused the down-regulation of CD44v6 expression. After cells were stimulated with phorbol myristate acetate, the amount of phosphorylated mitogen-activated protein (MAP) kinase was significantly reduced in CFSE-high, SB-HCV–infected Molt-4 cells. After Fas ligand stimulation, SB-HCV–infected Molt-4 cells had increased cleavage of caspase 8 and 3 and enhanced apoptosis, compared with the rates of cleavage and apoptosis in control groups, indicating that SB-HCV infection increased Fas-mediated apoptosis.
Conclusion
HCV replication in T cells suppresses cellular proliferation and enhances susceptibility to Fas signaling by inhibiting CD44v6 signaling and expression.
Hepatitis C virus (HCV) infects about 170 million people worldwide and is a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) [1]. HCV infection is often persistent and cellular immune response to HCV plays an important role in the pathogenesis of chronic hepatitis, cirrhosis, and HCC [2]. Several mechanisms have been proposed for the failure of HCV-specific CD4+ and CD8+ T cell response during HCV infection; these include deletion, anergy, cytotoxic T lymphocyte exhaustion, and suppression via regulatory CD4+CD25+ T cells and interleukin-10 (IL-10)– secreting regulatory CD8+ T cells [3, 4].
Previously we have shown that a strain of HCV (strain SB, hereafter “SB-HCV”) can infect and replicate in not only B cells but also T cells as well, and that HCV replication in T cells can inhibit signaling by interferon-gγ, signal transducer and activator of transcription factor 1 (STAT-1), and T-bet signaling of T cells [5]. However, the replication activity of SB-HCV in T cells is weaker than that in B cells, suggesting that the lymphotropic SB-HCV might preferentially replicate in B cells but nevertheless affect T cell development in local lymph nodes. In particular, among CD4+ cells, CD45RA+-CD45RO−CD4+ cells are the most susceptible to HCV infection [5]. It is known that during the activation that shifts T cells from naive to effector cells, T cells have to survive activation-induced cell death (AICD), which may contribute to the maintenance of an appropriate level of immune response. Fas–Fas ligand signaling is thought to be one of the mechanisms of AICD [6, 7].
There are several splicing variants of the immune molecules in T cells that contribute to proper immune response [8]. We have reported that SB-HCV could replicate in T cells during different temporal periods [5]. Therefore, we focused on CD44 splicing variants because the temporal regulation of alternative CD44 splicing in T lymphocytes plays a significant role in the maintenance of appropriate T cell development [8]. The CD44 family of transmembrane glycoproteins is present on a wide variety of cell types, including lymphocytes, neutrophils, endothelial cells, and fibroblasts [9, 10]. CD44 molecules display a multitude of functions involving a large array of signal transduction pathways, and CD44 is expressed into multiple splicing variants [11, 12]. During T cell development and activation, transient expression of CD44 splicing variant 6 (CD44v6) plays a significant role [13, 14]. The expression of CD44v6 provides a proliferative stimulus for T cells independent of the T cell receptor (TCR)–CD3 complex. Proliferative activity is accompanied by activation of the mitogen-activated protein kinase (MAPK) pathway [15]. Moreover, CD44v6 expression could interfere with Fas signaling [16].
Recently, the roles of individual HCV proteins have been studied extensively by using HCV-producing hepatocyte cell lines and the replicon system [17–21]. The processing of the HCV precursor polyprotein requires both host and viral proteases to produce structural and nonstructural proteins. Among the nonstructural proteins, NS5A interacts with a number of cellular proteins and may interfere with host cell signaling pathways [22, 23]. Several authors have reported that NS5A can inhibit the extracellular signal-regulated kinase (ERK)–MAPK pathway; however, several other HCV proteins, such as HCV core protein, have been reported to enhance this pathway [24, 25]. The mechanism of CD44v6 inclusion is still not clear, although it is known that the events upstream of this inclusion mechanism may include Ras-ERK activation [8, 26, 27]. Thus, HCV replication in T cells may affect proliferation and Fas-mediated apoptosis of T cells by altering CD44v6 expression or MAPK activation. During T cell development, T cells undergo vigorous proliferative activity which might facilitate HCV replication in T cells. However, extensive proliferation of HCV in T cells might interfere with proper T cell development.
Previously, our reports have indicated that SB-HCV, which was produced from an HCV-positive B-cell lymphoma cell line (SB cells), can infect and replicate in established B cell lines (Raji and Daudi), established T cell lines (Molt-4 and Jurkat), and primary lymphocytes [5, 28, 29]. HCV infection of these immune cells conceivably can perturb their functions. This study was conducted to analyze whether HCV replication in T lymphocytes might affect the proliferative activity and apoptosis of T cells as a result of signaling changes and aberrant expression of CD44 variants.
MATERIALS AND METHODS
Culture of cell lines
SB cells, which continuously produce infectious HCV particles, were originally cultured from splenocytes obtained from an HCV-infected patient with type 2 mixed cryoglobulinemia and monocytoid B-cell lymphoma [29]. The cells were maintained in standard RPMI 1640 (Invitrogen) medium that contained 20% fetal bovine serum (FBS) without any supplement. Huh7.5 cells, which were kindly provided by C.M. Rice (Laboratory of Virology and Infectious Disease, Center for the Study of Hepatitis C, The Rockefeller University), were cultured at 37° C in Dulbecco’s modified Eagle medium containing 10% FBS.
In vitro infection of Molt-4 and primary naive (CD45RA+-CD45RO−) CD4+ cells
SB cell culture supernatant (5 mL), which contained 2.2 × 104 copies/mL of HCV RNA, was used to infect Molt-4 and human naive CD4+ cells (1 × 105 cells). Control cells infected with UV-irradiated SB cell culture supernatant (hereafter, “UV–SB-HCV–infected cells”) were included in every experiment. Supernatant from Huh7.5 cells transfected with JFH-1 strains [19, 20, 30] at 10 days after transfection were used for several control experiments. Cells were washed 3 times at 5 days after infection. A portion of the cells (3 × 105–5 × 105 cells) was then harvested for analysis; the remaining cells (1 × 105 cells) were kept and incubated further.
Strand-specific intracellular SB-HCV RNA detection and quantitative SB-HCV RNA detection in the culture supernatant
Strand-specific intracellular SB-HCV RNA was detected by using a recently established procedure that combined methods published elsewhere [31, 32], with minor modifications [5]. Positive strand–specific and negative strand–specific SB-HCV RNA were detected by use of a nested polymerase chain reaction (PCR) method.
To quantify SB-HCV RNA in the culture supernatant, SB-HCV RNA extraction from culture supernatant (140 μL) was carried out with QIAmp Viral RNA Minikit (Qiagen). Amplification was performed in 20-μL reaction mixtures that contained AmpliTaq Gold DNA polymerase and optimized buffer (Taq-Man Universal PCR Master Mix; Applied Biosystems) with the ABI prism 7900 sequence detector system. Forward primer (300 nmol/L), nucleotide 215–197, (5′-TCCCGGGAGAGCCATAGTG-3′); reverse primer (600 nmol/L), nucleotide 158–140, (5′-TCCAAGAAAGGACCCRGT-3′); and 250 nmol/L TaqMan minor-groove-binding (MGB) probe labeled with 6-carboxy-fluorescein, nucleotide 195–181, (5′-FAM-TCTGCGGAACCGGTG-MGB-3′) were also included in the PCR mix reaction. In vitro–transcribed full-length HCV RNA was used as a standard.
Analysis of standard CD44 (CD44s) mRNA and variant mRNA
Cells were collected sequentially at various time points after addition of phorbol myristate acetate (PMA) or anti-CD3 antibody. After extraction of total RNA and a reverse-transcriptase procedure, real-time PCR and semiquantitative PCR were carried out. Real-time PCR analysis was performed in accordance with methods described elsewhere, with minor modifications [33, 34]. Forward primer for CD44s (5′-CAACTCCATCTGTGCAGCAAA-3′), CD44v3 (5′-GCAGGCTGGGAGCCAAAT-3′), and CD44v6 (5′-GCAACTCCTAGTAGTAC AACGGAAGA-3′); reverse primer for CD44s (5′-CAACTCCATCTGTGCAGCAAA-3′), CD44v3 (5′-TCATCAATGCCTGATCCAGAAA-3′), and CD44v6 (5′-CGATATCCCTCATGCCATCTGA-3′); and probe for CD44s (5′-FAM-CATATTGCTTCAATGCTTCAGCTCCACCTC-TAMRA-3′), CD44v3 (5′-FAM-AGGTGTCTGTCTCTTTCATCTTCAT-TAMRA-3′), and CD44v6 (5′-FAM-ACAGCTACCCAGAAGGAACAGTGGTTTGG-TAMRA-3′) were used for analysis. Semiquantified analysis was carried out by use of primers and cycles described elsewhere [27].
Staining of surface CD44s and variants
The surface expression of CD44s and the variants was analyzed by immunofluorescence labeling. Nonspecific binding was blocked by using PBS containing 1% bovine serum albumin. Cells were incubated for 15 min with the primary antibody. Monoclonal anti-CD44s, anti-CD44v3, and anti-CD44v6 antibodies were all obtained from R&D Systems. Murine IgG2b, IgG2a, and IgG1 were applied as isotype controls. Cells were washed 3 times in PBS, then incubated for 15 min with fluorescein isothiocyanate (FITC)– or allophycocyanin (APC)–conjugated goat anti–mouse IgG, followed by washing 3 times in PBS. Fluorescence was measured by use of a flow cytometer (FACSCalibur; Becton Dickinson).
Carboxyfluorescein succinimidyl ester (CFSE) staining and sorting
Cells were analyzed with the CellTrace CFSE Cell Proliferation Kit (Molecular Probes). CFSE staining was carried out at 5 days after infection; the cell staining methods used were in accordance with the manufacturer’s protocol. Stained cells were washed 3 times and incubated for an additional 10 days because our previous study showed that active HCV replication in T cells occurred during this period [5]. Cells were analyzed by use of flow cytometry with 488-nm excitation and emission filters.
Immunoblotting analysis: MAPK signaling, Fas signaling, and immunoprecipitation (Western blot)
Proteins were resolved by electrophoresis in sodium dodecyl sulfate–polyacrylamide gels and electrophoretically transferred onto nitrocellulose membranes (Amersham Biosciences). The membrane was incubated with anti–P44/42 MAP kinase, anti–phospho–P44/42 MAP kinase, anti–MAPK/ERK kinase (MEK) 1/2, anti–phospho–MEK 1/2, anti–phospho–cRaf, anti–caspase 8, or anti–caspase 3 antibodies (Cell Signaling) and then subjected to reaction with peroxidase-conjugated secondary antibody. Immunoreactivity was visualized with enhanced-chemiluminescence detection (Amersham Biosciences).
Annexin V and propidium iodide (PI) staining
Cells were stained with annexin V antibody and PI by use of an apoptosis detection kit (R&D systems). The staining methods used were in accordance with the manufacturer’s protocol.
Transfection of HCV individual protein expression plasmids
The various expression plasmids were constructed by inserting HCV core protein, E1, E2, NS3, NS4B, NS5A, and NS5B cDNA of genotype 1a [35] behind the cytomegalovirus virus immediate-early promoter in pCDNA3.1 (Invitrogen). Molt-4 cells were transfected by use of the Gene Pulser II (Bio-Rad), and various plasmids were purified by using the EndFree plasmid kit (QIAGEN). Viable transfected cells were isolated by Ficoll-Paque centrifugation (Amersham Biosciences) at 24 h after transfection.
Confocal laser microscopy
Molt-4 cells (3 × 106 cells/mL) in suspension were fixed and permeabilized with Fixation/Permeabilization solution (BD Biosciences) at 4°C for 25 min. The cells were then washed 2 times in BD Perm/Wash buffer (BD Biosciences) and resuspended in 50 μL of BD Perm/Wash buffer containing preconjugated polyclonal anti-NS5A antibody (Biodesign International) with an FITC-conjugated anti-mouse antibody.
Isolation of naive CD4+ T cells and culture conditions
Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by Ficoll-Paque centrifugation (Amersham Biosciences). Anti-CD3 (phycoerythrin [PE] labeled), anti-CD4 (PE-Cy3 labeled), anti-CD45RO (PE labeled), and anti-CD45RA (FITC labeled) antibodies (BD Pharmingen) were used in separating different kinds of mononuclear cells with a FACSV antage flow cytometry cell sorter (BD).
Statistical analysis
Statistical analysis of the data for figures 1A, 2A, and 3A was performed by independent-sample t test in SPSS (version 10.0; SPSS). P values <.05 were considered to be statistically significant.
Figure 1.

Quantification of standard CD44 (CD44s) mRNA as well as mRNA from splicing variants 3 and 6 (CD44v3 and CD44v6) in Molt-4 cells (A) and human CD45RA+CD45RO− naive T cells (B) (samples obtained from 1 healthy subject). Quantification of CD44s, CD44v3, and CD44v6 mRNA with or without stimulation (phorbol myristate acetate [PMA] or anti-CD3 antibody [CD3]) was carried out by real-time polymerase chain reaction with comparative cycle threshold methods. Five independent experiments yielded similar results. Mock, mock-infected cells; HCV, cells infected with hepatitis C virus, strain SB; UV, cells infected with UV-irradiated hepatitis C virus, strain SB.
Figure 2.
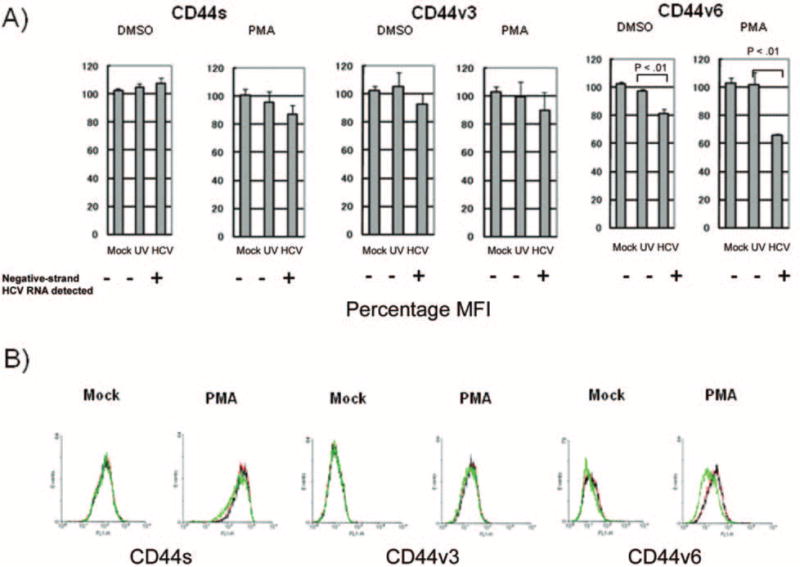
Comparison of surface expression of standard CD44 (CD44s) and splicing variants 3 and 6 (CD44v3 and CD44v6) in Molt-4 cells. A, Comparison of mean fluorescence intensity (MFI) among the 3 groups with or without phorbol myristate acetate (PMA) stimulation was carried out with flow cytometry. The percentage MFI was calculated as follows: target mean fluorescence intensity divided by mean fluorescence intensity of mock-infected cells without PMA stimulation times 100. Three independent experiments yielded similar results. B, Fluorescence-activated cell sorter analyses of surface expression of different CD44 isoforms. Red lines, mock-infected Molt-4 cells; black lines, Molt-4 cells infected with UV-irradiated hepatitis C virus, strain SB (UV–SB-HCV); green lines, Molt-4 cells infected with SB-HCV.
Figure 3.
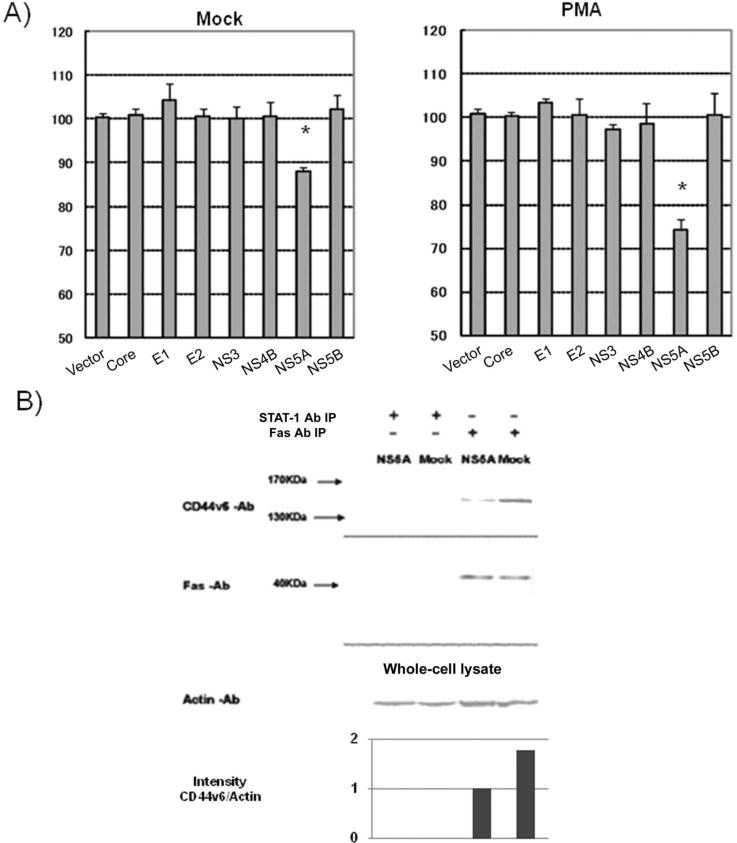
NS5A protein and suppression of CD44v6 in T cells. A, Effects of individual hepatitis C virus (HCV) proteins on surface expression of CD44 splicing variant 6 (CD44v6). Mean fluorescence intensity (MFI) of CD44v6 on Molt-4 cells transfected with individual HCV proteins was normalized by the MFI of CD44v6 on mock-infected Molt-4 cells. Five independent experiments yielded similar results. B, Coimmunoprecipitation of CD44v6 with Fas. Cell lysates were precipitated with anti-Fas antibody (Fas Ab) and detected by immunoblotting with different antibodies. Band intensity was analyzed with Image J (National Institutes of Health). The bar graph shows the relative intensity of CD44v6 and actin. Three independent experiments yielded similar results. IP, immunoprecipitation; STAT-1 Ab, anti–signal transducer and activator of transcription factor 1 antibody.
RESULTS
Suppression of CD44v6 mRNA, but not CD44s or CD44v3-mRNA, and its surface protein expression in SB-HCV–infected Molt-4 cells
Our group has previously reported that SB-HCV [29] can infect and replicate in established B cell lines (Raji and Daudi), T cell lines (Molt-4 and Jurkat), and primary lymphocytes [5]. We also reported that SB-HCV infection can affect the signaling and biological functions of B cells [28, 36, 37]. We then proceeded to study whether SB-HCV infection could affect the functions of T cells. We first examined the expression of splicing variants CD44v3 and CD44v6, the latter of which is regarded as an important factor for T cell development and activation since Molt-4 cells can be used for the analysis of CD44v3 and CD44v6 after PMA stimulation. All the splicing variants of CD44 have previously been shown to be up-regulated by PMA stimulation, which mimics TCR stimulation [38].
We first analyzed the time course of CD44s, CD44v3, and CD44v6 mRNA up-regulation in Molt-4 cells after PMA stimulation. We found that the expression of mRNA by these isoforms peaked at 8–12 h after stimulation, whereas the maximum up-regulation of surface expression of these CD44 variants was observed at 24 h after stimulation (data not shown). Therefore, to determine whether HCV could affect the expression of different splicing variants of CD44, we compared the expression level of these variants in cells that had been mock-infected, infected with UV–SB-HCV, or infected with SB-HCV; this comparison was performed at 8 h after activation for mRNA analysis and 24 h after activation for surface protein analysis. The status of HCV infection and replication was confirmed by detecting negative-strand HCV RNA by semiquantitative reverse-transcriptase PCR (figure 4, which appears only in the electronic version of the Journal, and summarized in figure 1B).
Figure 4.
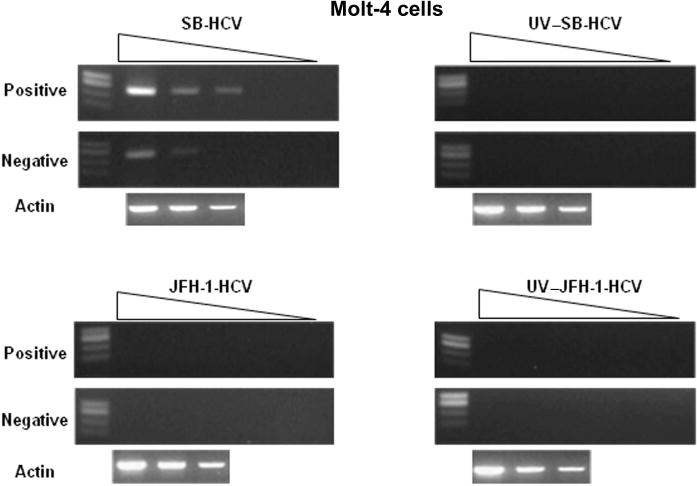
Detection of positive- and negative-strand hepatitis C virus, SB strain (SB-HCV), RNA by semiquantitative reverse-transcriptase polymerase chain reaction. JFH-1-HCV, JFH-1 HCV strain; UV–JFH-1-HCV, UV irradiated JFH-1-HCV; UV–SB-HCV, UV irradiated SB-HCV.
All the CD44 variants examined showed a low level of mRNA expression, which was significantly increased after PMA stimulation (figure 1). We noted that CD44v6 mRNA expression in SB-HCV–infected Molt-4 cells was slightly but significantly lower than that observed in control groups, even without PMA stimulation (P < .05), and this difference became more prominent after PMA stimulation (P < .001) (figure 1). In contrast, the mRNA expression of CD44s and CD44v3 was not affected by HCV infection status. We further confirmed the suppression of CD44v6 mRNA by HCV infection in primary naive T cells (figure 1B). In this case, the primary CD4+ cells were stimulated with anti-CD3 antibody, and the suppression of CD44v6 mRNA became more apparent after stimulation. Taken together, these data indicate that HCV replication in CD4+ cells specifically suppressed CD44v6 mRNA inclusion.
To corroborate this finding, we then analyzed surface expression of CD44s, CD44v3, and CD44v6 among the 3 groups of cells (mock infected, UV–SB-HCV infected, and SB-HCV infected) (figure 2). The mean fluorescence intensity (MFI) of CD44v6 with or without PMA stimulation was significantly suppressed in SB-HCV–infected Molt-4 cells, compared with Molt-4 cells that had been mock infected or infected with UV–SB-HCV (P < .01) (figure 2A). CD44s and CD44v3 also showed slightly lower levels of expression after PMA stimulation in HCV-infected cells; however, these differences were not statistically significant. The lower surface expression of CD44v6 in SB-HCV–infected cells was further confirmed by fluorescence-activated cell sorter analysis (figure 2B). These data suggested that SB-HCV infection of T cells caused down-regulation of CD44v6 and that HCV replication in T cells, not merely the binding of HCV to T cells, is required for this effect.
To further demonstrate that HCV replication was required for the down-regulation of CD44v6 expression in T cells, we used JFH-1 HCV [20] to infect Molt-4 cells. We have previously shown that JFH-1-HCV cannot infect or replicate in lymphocytes (Machida et al., unpublished data). Molt-4 cells infected with JFH-1 HCV and Molt-4 cells infected with UV-irradiated JFH-1 HCV showed no difference in mRNA or surface protein expression for CD44s, CD44v3, or CD44v6 (data not shown). Therefore, HCV replication might be necessary for the down-regulation of CD44v6 inclusion in T cells. However, we could not rule out the possibility that SB-HCV has a unique ability to down-regulate CD44v6 inclusion because we have so far only been able to use the SB strain to infect T cells.
SB-HCV infection of T cells and decreased proliferation
Since CD44v6 expression is associated with cellular proliferation, we analyzed the proliferative activity of SB-HCV–infected Molt-4 cells [15]. The results showed that SB-HCV–infected Molt-4 cells could be clearly separated into 2 groups (CFSE high and CFSE low), which were sorted by use of a flow cytometry cell sorter (FACSVantage; Becton Dickinson), whereas mock-infected cells or cells infected with UV–SB-HCV formed only 1 group (figure 5A). The CFSE-high and CFSE-low groups were separated and analyzed for NS5A expression. The data showed that 82.4% of the CFSE-high group expressed NS5A protein, whereas only 1.2% of the CFSE-low group were positive for NS5A (figure 5B). These data indicated that most cells in the CFSE-high group (i.e., the slow-growing group) were infected with SB-HCV, whereas the uninfected cells were mostly fast growing. Therefore, SB-HCV infection suppressed the proliferation activity of Molt-4 cells.
Figure 5.
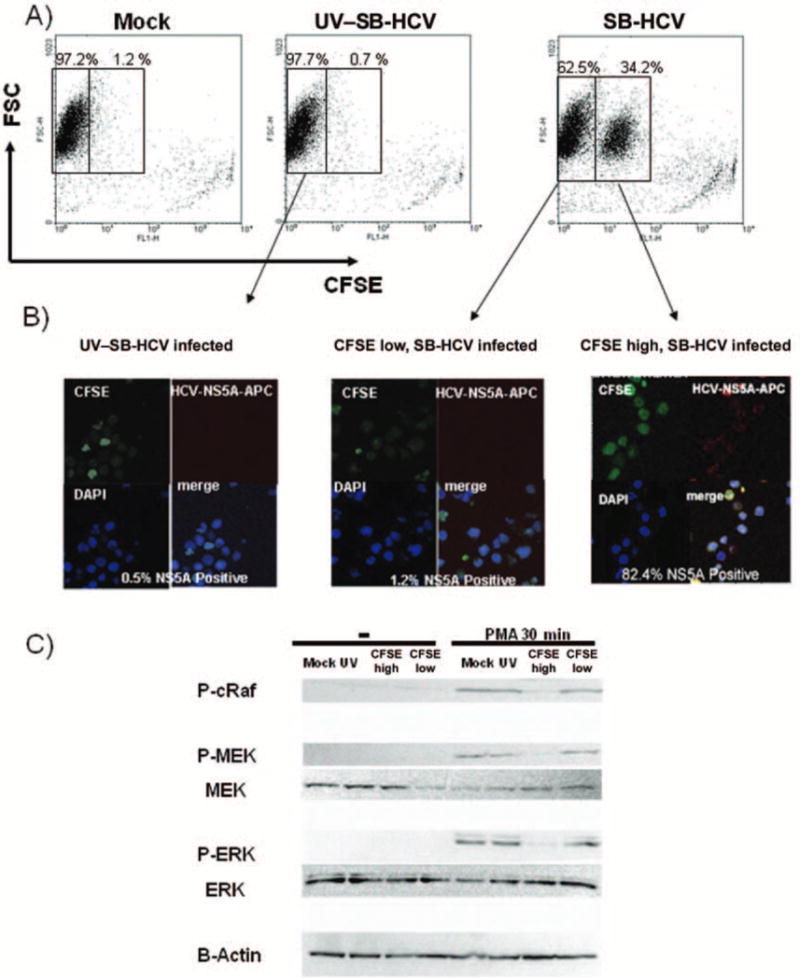
Effects of hepatitis C virus, strain SB (SB-HCV), infection on cell proliferation in Molt-4 cells. A, Representative fluorescence-activated cell sorter (FACS) analysis of cell proliferation. B, Carboxyfluorescein succinimidyl ester (CFSE)–high and CFSE-low groups were separated by FACS analysis and analyzed for NS5A expression by immunofluoresence analysis. Green stain, CFSE; red stain, NS5A; blue stain, 4′,6–diamidino–phenylindole dihydrochloride (DAPI). The percentage of infectivity was determined by counting numbers of positively stained cells per 400 cells. For the negative control, cells treated with UV-irradiated HCV and detection antibody alone were used. C, Analysis of Ras/mitogen-activated protein (MAP) kinase/extracellular signal-regulated kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling. For details about immunoblotting analysis, see Methods. Three independent experiments yielded similar results. FSC, forward scatter characteristics; P-cRaf, phospho-cRaf; P-ERK, phospho-ERK; P-MEK, phospho-MEK.
SB-HCV infection and suppression of Ras-MEK-ERK activation and CD44v6 up-regulation in T cells
We next analyzed the Ras-MEK-ERK signaling that may induce proliferation-related genes, since it has been reported that this signaling is the upstream mechanism of CD44 variant inclusion and that CD44v6 can enhance this signaling, acting like a positive loop [27]. The CFSE-high group (i.e., the enriched SB-HCV–infected Molt-4 cells) showed significantly lower levels of phospho-cRaf, phospho-MEK, and phospho-ERK after PMA stimulation than did cells in the control groups (mock-infected cells, cells infected with UV–SB-HCV, and cells in the CFSE-low group) (figure 5C). These data indicated that HCV replication in T cells suppressed the PMA-induced activation of Ras-MEK-ERK signaling.
We next carried out double staining analysis of CD44 variants and CFSE to further confirm the correlation between low CD44v6 expression and high CFSE staining. The result showed that the CFSE-high group (the cells infected with SB-HCV) had a significantly lower level of CD44v6 expression than did the cells in the CFSE-low group (P < .01) (figure 6A). CD44s and CD44v3 were only weakly suppressed in the SB-HCV–infected CFSE-high cells, compared with the CFSE-low group (figure 6B).
Figure 6.
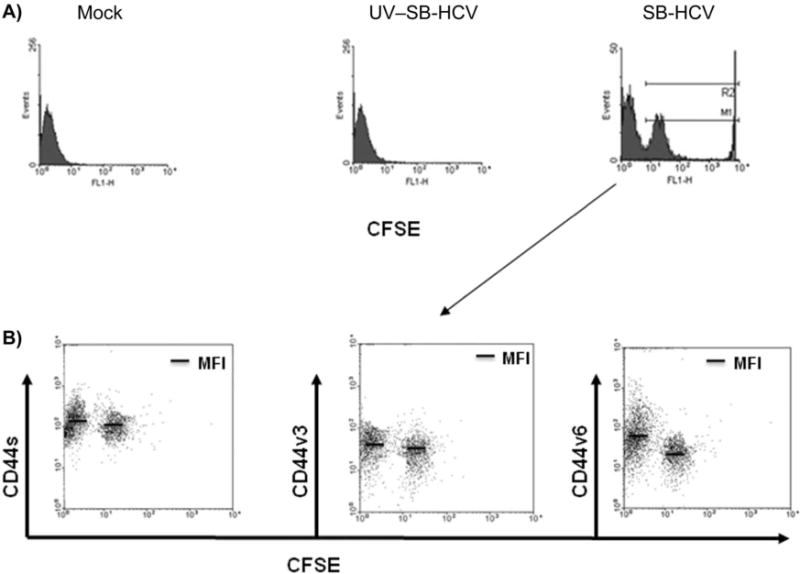
Comparison of carboxyfluorescein succinimidyl ester (CFSE)-low cells and CFSE-high cells (i.e., cells highly infected with hepatitis C virus, strain SB [SB-HCV]) with respect to expression of standard CD44 (CD44s) and the splicing variants 3 and 6 (CD44v3 and CD44v6). A, Representative graphs show CFSE intensity among mock-infected cells, cells infected with UV-irradiated SB-HCV (UV–SB-HCV), and cells infected with SB-HCV. B, CD44s and CD44v3 were only weakly suppressed in the SB-HCV–infected CFSE-high cells, compared with the CFSE-low group. Three independent experiments yielded similar results. MFI, mean fluorescence intensity.
SB-HCV infection and enhanced Fas-dependent apoptosis by suppression of CD44v6
We next analyzed Fas-mediated apoptosis because CD44v6 has been reported to cause suppression of Fas-mediated apoptosis [16]. Immunoblotting analysis showed that, after treatment with Fas ligand (FasL), Molt-4 cells infected with SB-HCV had a higher extent of cleavage of caspases 8 and 3 than did cells that had been mock infected or infected with UV–SB-HCV (figure 7A), whereas the surface expression levels of Fas and FasL were almost the same among the 3 groups (figure 7B). Moreover, pretreatment of cells with anti-CD44v6 antibody that could block the interaction of CD44v6 with Fas [15] eliminated the differences in caspase activation among the 3 groups (figure 7A). The number of early apoptotic cells, which were represented as annexin V–positive, PI-negative cells, was significantly increased among the SB-HCV–infected Molt-4 cells after Fas stimulation (the percentage of early apoptotic cells in each group was as follows: mock infected, 29.8%; UV–SB-HCV infected cells, 29.6%; and SB-HCV infected cells, 41.3%) (figure 7C). These results indicated that SB-HCV infection caused suppression of the CD44v6 isoform, leading to the enhancement of Fas-mediated apoptosis.
Figure 7.
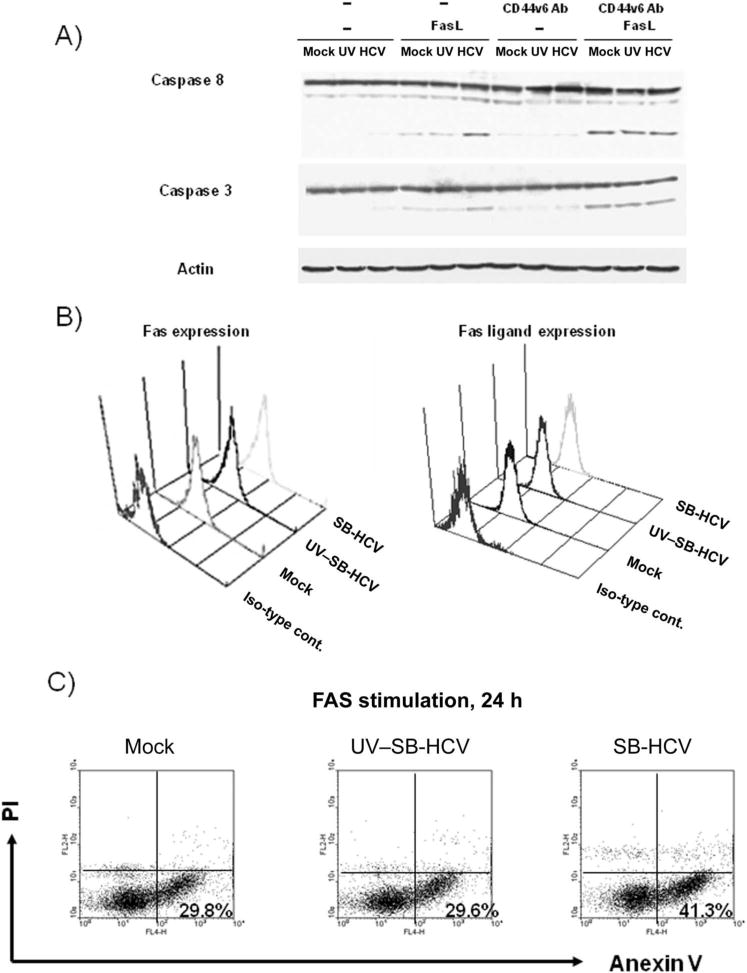
Susceptibility of different cells to Fas stimulation. A, Immunoblotting analysis was carried out by using anti-caspase 8, anti-caspase 3, and anti-actin antibodies, and the membrane was then subjected to reaction with peroxidase-conjugated secondary antibody. Immunoreactivity was visualized by use of enhanced chemiluminescence detection. Loading proteins were obtained from the following cell groups: mock-infected Molt-4 cells; Molt-4 cells infected with UV-irradiated hepatitis C virus, strain B (UV–SB-HCV); SB-HCV–infected Molt-4 cells from the carboxyfluorescein succinimidyl ester (CFSE)–high group; and SB-HCV–infected Molt-4 cells from the CFSE-low group, with or without Fas-ligand (FasL) stimulation. Pretreatment with CD44 splicing variant 6–blocking antibody (CD44v6 Ab) was carried out for some samples, as indicated. B, Surface expression of Fas and FasL as measured by flow cytometry. C, Fluorescence-activated cell sorter analysis of annexin V and propidium iodide (PI) staining. Mock-infected cells, Molt-4 cells infected with UV-SB-HCV, and Molt-4 cells infected with SB-HCV were stimulated with FasL for 24 h and analyzed to detect annexin V–positive, PI–negative early apoptotic cells. The percentages of early apoptotic cells in each group are indicated in the dot plots. Iso-type cont., isotype control antibodies.
NS5A protein and suppression of CD44v6 in T cells
To determine which viral protein was responsible for CD44v6 suppression, we expressed each HCV protein in Molt-4 cells and then studied its effects on CD44v6 expression. The transfection efficiency of each plasmid was ~50%, and cell viability after transfection was >80% after purification by Ficoll-Paque centrifugation. The results showed that, among all the viral proteins, only NS5A could suppress CD44v6 significantly (P < .01) (figure 3A). The NS5A-induced CD44v6 suppression was particularly evident after PMA stimulation. Immunoprecipitation by Western blot analysis showed that CD44v6 could coprecipitate with Fas, and the amount of CD44v6-associated Fas was reduced in NS5A-expressing Molt-4 cells, compared with Molt-4 cells without NS5A (figure 3B). As a control, STAT-1 antibody did not coprecipitate CD44v6.
DISCUSSION
The SB cell line, which was derived from an HCV-positive B-cell lymphoma, produces lymphotropic HCV particles that can infect and replicate in B cell lines, such as Raji and Daudi cells, as well as PBMCs [28, 29]. Most recently, we also demonstrated that SB-HCV could infect T cell lines, such as Molt-4 cells, and that this system could be used for signaling analysis of T cells [5]. Although there has been accumulating evidence indicating that HCV could replicate in both established T cell lines and primary T lymphocytes, so far little is known about the extent and biological significance of T cell infection [39–41]. The site of infection, as well as proliferative activity and the subset of CD4+ cells involved may be important factors for HCV replication in vivo [42]. Some authors have suggested that coinfection with human T cell lymphotropic virus type 1 or HIV might induce HCV replication in T lymphocytes [43–45]. Previously, our data indicated that with CD3 and IL-2 stimulation, naive CD4+ T cells could be one of the target cells for HCV [5]. As a result of HCV infection, changes in the development and activation of apoptosis-related molecules in T cells may contribute partially to T cell hyporesponsiveness in some patients with hepatitis C.
The temporal expression of CD44v6 is important for proliferation, activation, and apoptosis in T cells [13, 16]. The suppression of CD44v6 expression or Ras-MEK-ERK signaling by HCV replication disrupted the positive loop of proliferation in T cells [27]. We could not conclude that CD44v6 suppression was either the result or the cause of Ras-MEK-ERK suppression, since Ras-MEK-ERK have been reported as upstream regulatory molecules for CD44v6 expression and CD44v6 could enhance Ras-MEK-ERK suppression [27]. However, unexpectedly, the other CD44 splicing variants, for example CD44v3, could not be suppressed by Ras-MEK-ERK signaling in this Molt-4 cell infection system [13, 14]. Our studies further showed that the protein NS5A is responsible for CD44v6 suppression. One of the possible mechanisms for this suppression is the stimulation of phosphatase 2A activity that can suppress MAPK signaling [24]. However, many individual HCV proteins have been reported to affect MAPK signaling and apoptotic signaling in diverse ways [25]; our Molt-4 cell HCV replication system showed that HCV replication, and NS5A protein alone, could suppress CD44v6 expression and MAPK signaling in T cells. Moreover, the results of Fas signaling experiments showed that suppression of CD44v6 might contribute to the apoptosis of T cells. However, some authors have reported that NS5A can inhibit apoptosis in hepatoma cell lines [46, 47]. One of the explanations for these contradictory results probably lies in the developmental stages and characteristics of the naive T cells. During T cell activation, apoptosis is easily induced, in order to maintain an appropriate immune response. During this stage, suppression of CD44v6 might strongly affect apoptosis signaling in T cells.
We have found that our results apply not only to T cell lines, but also to primary naive T cells. We could also detect significant suppression of proliferation after SB-HCV infection (data not shown). Furthermore, in our ongoing clinical study some clinical samples (PBMCs) from patients with chronic hepatitis C showed a significant, albeit small, degree of CD44v6 down-regulation with CD3 stimulation (data not shown).
We conclude that HCV replication in T cells may play a role in the regulation of proliferation and apoptosis during T cell activation. The results suggest that NS5A expression induces the suppression of MAPK signaling and CD44v6 expression in T cells. Suppression of CD44v6 could enhance susceptibility to Fas signaling by reducing the binding of Fas and CD44v6. These biological effects may contribute to the disturbance of T cell proliferation and activation in individuals with persistent lymphotropic HCV infection (figure 8). A key issue for future research will be determining what percentage of T cells are infected with HCV during natural T cell infection.
Figure 8.

A proposed mechanism for T cell hyporesponsiveness induced by hepatitis C virus, strain SB (SB-HCV). T cell proliferation activity is strongly suppressed in SB-HCV infected T cells (thick arrows). A second effect is the induction of apoptosis through Fas. Both pathways lead to T cell hyporesponsiveness. The relative effects of these 2 pathways is not clear. CFSE, carboxyfluorescein succinimidyl ester.
Acknowledgments
Financial support: National Institutes of Health (research grants AI40038 to M.M.C.L. and CA108302 to M.M.C.L.).
Footnotes
Potential conflicts of interest: none reported.
References
- 1.Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556–62. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- 2.Chang KM, Rehermann B, Chisari FV. Immunopathology of hepatitis C. Springer Semin Immunopathol. 1997;19:57–68. doi: 10.1007/BF00945025. [DOI] [PubMed] [Google Scholar]
- 3.Manigold T, Racanelli V. T-cell regulation by CD4 regulatory T cells during hepatitis B and C virus infections: facts and controversies. Lancet Infect Dis. 2007;7:804–13. doi: 10.1016/S1473-3099(07)70289-X. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007;15:143–6. doi: 10.1016/j.tim.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Kondo Y, Sung VM, Machida K, Liu M, Lai MM. Hepatitis C virus infects T cells and affects interferon-gamma signaling in T cell lines. Virology. 2007;361:161–73. doi: 10.1016/j.virol.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407:789–95. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 7.Holmstrom TH, Schmitz I, Soderstrom TS, et al. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. Embo J. 2000;19:5418–28. doi: 10.1093/emboj/19.20.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch KW. Consequences of regulated pre-mRNA splicing in the immune system. Nat Rev Immunol. 2004;4:931–40. doi: 10.1038/nri1497. [DOI] [PubMed] [Google Scholar]
- 9.Flanagan BF, Dalchau R, Allen AK, Daar AS, Fabre JW. Chemical composition and tissue distribution of the human CDw44 glycoprotein. Immunology. 1989;67:167–75. [PMC free article] [PubMed] [Google Scholar]
- 10.Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 11.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda M, Nakano K, Yasumoto K, Tanaka Y. CD44: functional relevance to inflammation and malignancy. Histol Histopathol. 2002;17:945–50. doi: 10.14670/HH-17.945. [DOI] [PubMed] [Google Scholar]
- 13.Forster-Horvath C, Bocsi J, Raso E, et al. Constitutive intracellular expression and activation-induced cell surface up-regulation of CD44v3 in human T lymphocytes. Eur J Immunol. 2001;31:600–8. doi: 10.1002/1521-4141(200102)31:2<600::aid-immu600>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Seiter S, Schmidt DS, Zoller M. The CD44 variant isoforms CD44v6 and CD44v7 are expressed by distinct leukocyte subpopulations and exert non-overlapping functional activities. Int Immunol. 2000;12:37–49. doi: 10.1093/intimm/12.1.37. [DOI] [PubMed] [Google Scholar]
- 15.Marhaba R, Bourouba M, Zoller M. CD44v6 promotes proliferation by persisting activation of MAP kinases. Cell Signal. 2005;17:961–73. doi: 10.1016/j.cellsig.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 16.Mielgo A, van Driel M, Bloem A, Landmann L, Gunthert U. A novel antiapoptotic mechanism based on interference of Fas signaling by CD44 variant isoforms. Cell Death Differ. 2006;13:465–77. doi: 10.1038/sj.cdd.4401763. [DOI] [PubMed] [Google Scholar]
- 17.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 18.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–4. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 19.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 20.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 23.Szabo G. Hepatitis C virus NS5A protein–a master regulator? Gastroenterology. 2006;130:995–9. doi: 10.1053/j.gastro.2006.01.072. [DOI] [PubMed] [Google Scholar]
- 24.Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J Biol Chem. 2003;278:17775–84. doi: 10.1074/jbc.M210900200. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsumi T, Suzuki T, Moriya K, et al. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology. 2003;38:820–8. doi: 10.1053/jhep.2003.50399. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J, Shendure J, Mitra RD, Church GM. Single molecule profiling of alternative pre-mRNA splicing. Science. 2003;301:836–8. doi: 10.1126/science.1085792. [DOI] [PubMed] [Google Scholar]
- 27.Cheng C, Yaffe MB, Sharp PA. A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. 2006;20:1715–20. doi: 10.1101/gad.1430906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machida K, Cheng KT, Sung VM, et al. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci U S A. 2004;101:4262–7. doi: 10.1073/pnas.0303971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung VM, Shimodaira S, Doughty AL, et al. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J Virol. 2003;77:2134–46. doi: 10.1128/JVI.77.3.2134-2146.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. Cell culture and infection system for hepatitis C virus. Nat Protoc. 2006;1:2334–9. doi: 10.1038/nprot.2006.395. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Shahidi A, Park S, Guilfoyle D, Hirshfield I. Detection of extrahepatic hepatitis C virus replication by a novel, highly sensitive, single-tube nested polymerase chain reaction. Am J Clin Pathol. 2003;119:95–100. doi: 10.1309/33TA-JLB7-48KL-MXVG. [DOI] [PubMed] [Google Scholar]
- 32.Negro F, Krawczynski K, Quadri R, et al. Detection of genomic- and minus-strand of hepatitis C virus RNA in the liver of chronic hepatitis C patients by strand-specific semiquantitative reverse-transcriptase polymerase chain reaction. Hepatology. 1999;29:536–42. doi: 10.1002/hep.510290223. [DOI] [PubMed] [Google Scholar]
- 33.Yamada Y, Itano N, Narimatsu H, et al. Receptor for hyaluronan-mediated motility and CD44 expressions in colon cancer assessed by quantitative analysis using real-time reverse transcriptase-polymerase chain reaction. Jpn J Cancer Res. 1999;90:987–92. doi: 10.1111/j.1349-7006.1999.tb00846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni HM, Leong AF, Cheong D, Hooi SC. Expression of CD44 variants in colorectal carcinoma quantified by real-time reverse transcriptase-polymerase chain reaction. J Lab Clin Med. 2002;139:59–65. doi: 10.1067/mlc.2002.120425. [DOI] [PubMed] [Google Scholar]
- 35.Machida K, Cheng KT, Sung VM, Lee KJ, Levine AM, Lai MM. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J Virol. 2004;78:8835–43. doi: 10.1128/JVI.78.16.8835-8843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machida K, Kondo Y, Huang J, et al. HCV-induced immunoglobulin hypermutation reduces the affinity and neutralizing activities of antibodies against HCV envelope protein. J Virol. 2008;82:6711–20. doi: 10.1128/JVI.02582-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces Toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80:866–74. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franklin RA, Tordai A, Patel H, Gardner AM, Johnson GL, Gelfand EW. Ligation of the T cell receptor complex results in activation of the Ras/Raf-1/MEK/MAPK cascade in human T lymphocytes. J Clin Invest. 1994;93:2134–40. doi: 10.1172/JCI117209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pham TN, King D, Macparland SA, et al. Hepatitis C virus replicates in the same immune cell subsets in chronic hepatitis C and occult infection. Gastroenterology. 2008;134:812–22. doi: 10.1053/j.gastro.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 40.MacParland SA, Pham TN, Gujar SA, Michalak TI. De novo infection and propagation of wild-type Hepatitis C virus in human T lymphocytes in vitro. J Gen Virol. 2006;87:3577–86. doi: 10.1099/vir.0.81868-0. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Wang X, Douglas SD, et al. CD8+ T cell depletion amplifies hepatitis C virus replication in peripheral blood mononuclear cells. J Infect Dis. 2005;192:1093–101. doi: 10.1086/432957. [DOI] [PubMed] [Google Scholar]
- 42.Bare P, Massud I, Parodi C, et al. Continuous release of hepatitis C virus (HCV) by peripheral blood mononuclear cells and B-lymphoblastoid cell-line cultures derived from HCV-infected patients. J Gen Virol. 2005;86:1717–27. doi: 10.1099/vir.0.80882-0. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Yamada O, Kawagishi K, et al. Up-regulation of hepatitis C virus replication by human T cell leukemia virus type I-encoded Tax protein. Virology. 2007;369:198–205. doi: 10.1016/j.virol.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 44.Toro C, Bassani S, Rios P, Jimenez V, Camino N, Soriano V. Influence of HTLV-2 infection on hepatitis C virus replication in HIV-positive patients. J Clin Virol. 2005;32:338–9. doi: 10.1016/j.jcv.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Mizutani T, Kato N, Saito S, Ikeda M, Sugiyama K, Shimotohno K. Characterization of hepatitis C virus replication in cloned cells obtained from a human T-cell leukemia virus type 1-infected cell line, MT-2. J Virol. 1996;70:7219–23. doi: 10.1128/jvi.70.10.7219-7223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nanda SK, Herion D, Liang TJ. The SH3 binding motif of HCV [corrected] NS5A protein interacts with Bin1 and is important for apoptosis and infectivity. Gastroenterology. 2006;130:794–809. doi: 10.1053/j.gastro.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 47.Miyasaka Y, Enomoto N, Kurosaki M, et al. Hepatitis C virus nonstructural protein 5A inhibits tumor necrosis factor-α–mediated apoptosis in Huh7 cells. J Infect Dis. 2003;188:1537–44. doi: 10.1086/379253. [DOI] [PubMed] [Google Scholar]