

Abstract
Inconsistent results obtained with published methods for the elution of antibodies from tissue sections prompted the assessment of both old and new methods in combination with monoclonal rabbit antibodies of known, increased affinity (above 1×10-9 KD). We tested an acidic (pH 2) glycine buffer, a 6 M urea hot buffer and a 2-Mercaptoethanol, SDS buffer (2-ME/SDS). Some antibodies were not removed by the glycine pH 2 or 6 M urea hot buffers, indicating that antibodies survive much harsher conditions than previously believed. We found that the elution is dependent upon the antibody affinity and is reduced by species-specific crosslinking via a dimeric or Fab fragments of a secondary antibody. The high affinity bond of exogenous streptavidin with the endogenous biotin can be removed by 6 M urea but not by the other buffers. 2-ME/SDS buffer is superior to glycine pH 2 and 6 M urea hot elution buffers for all antibodies because of its irreversible effect on the structure of the antibodies. It also has a mild retrieving effect on some antigens present on routinely treated sections and no detrimental effect on the immunoreactivity of the tissue. Therefore, 2-ME/SDS buffer is the method of choice to perform multiple rounds of immunostaining on a single routine section.
Keywords: affinity, antibody, multiple immunostaining, elution, stripping
Introduction
To demonstrate two or more antigens on the very same tissue section simultaneously or with sequential rounds of immunostaining, several conditions must first be met. The pigments used to visualize each individual antigen must not block the visualization of others (Sternberger and Joseph 1979) and this is easily accomplished with light-emitting fluorochromes, selectively visualized by optical filter combinations (Buscone et al. 2013). In addition, each primary-secondary antibody pair may not crossreact with another, particularly if two or more antigens are to be immunostained simultaneously (Mason et al. 2000), but also if they are added to the section in sequence. An exception to this is when the blocking effect of the pigment used to visualize the first antigen is sought on purpose to prevent the crosstalk of a potentially interacting second layer of antibodies (Sternberger and Joseph 1979). A second method to allow for the simultaneous visualization of two or more antibodies of the same species is the exploitation of the differential representation in the tissue, the differential signal amplification of the detection systems, or a combination of both (Tóth and Mezey 2007). A third method relies on the use of monomeric Fab forms of secondary antibodies such that the passive capture of a second primary antibody by the previous layer is prevented (Brouns et al. 2002).
Alternatively, when the first layer(s) is physically removed from the section (Kolodziejczyk and Baertschi 1986; Tramu et al. 1978; Wählby et al. 2002) and its reporter, a fluorochrome, is inactivated (Gerdes et al. 2013), the section is ready for a second or subsequent deposition of differently labelled, directly conjugated antibodies (Gerdes et al. 2013; Gerner et al. 2012) or a fresh round of indirect immunofluorescence. Combinations of these methods allow for the simultaneous identification of up to 61 different antigens on the very same tissue (Gerdes et al. 2013), leading to the development of complex digital analysis tools collectively called histo-cytometry or image cytometry.
Published methods to erase a previously deposited layer of indirect immunofluorescence or immunohistochemistry consist of a combination of physical agents, namely heat and solutions of high ionic strength. Gentle (moderate changes in pH and ionic strength of the buffer) or harsh buffers (extreme pH, <2 or >10) aim to denature the bound antibody, which detaches from the antigen and can then be removed.
These methods are not native to histopathology, but adapted from methods extensively used in biochemistry and antibody production and characterization. The closest examples are methods describing the sequential detection of antigens immobilized on a solid artificial substrate, usually a nitrocellulose sheet, which is probed and re-probed with antibodies, alternating with the elution (also known as stripping) with acidic (pH 2.0) glycine-containing buffers, followed by blocking of unwanted background with a protein solution and re-probing with the second round of antibodies.
Other methods are borrowed from antibody purification and elution techniques, in which antibodies are eluted from an antigen attached to a solid support (agarose beads) with a combination of denaturing agents; e.g., urea, guanidine hydrochloride, among others (Goding 1996).
The vast experience gathered from these fields tells that, unlike other proteins, antibodies survive heating above 60C (Wang et al. 2007); as we have previously shown, not only are antibodies long-lasting (Argentieri et al. 2013), but they also withstand temperatures much greater than 60C (this manuscript). In addition, the affinity for the antigens of antibodies, such as the ones used in immunohistochemistry, varies: the affinity constant of polyclonal and monoclonal mouse antibodies ranges between dissociation constants of 10-6 and 10-9 (Goding 1996). As a reference, the bond between avidin and biotin, one of the strongest noncovalent bonds in nature (Merkel et al. 1999), is in the 10-14–10-15 dissociation constant range. A high antibody affinity is not always desired because of the difficulties in separating the antibody from the antigen (Hillman et al. 2001). As a result, elution from an affinity column may require harsher and potentially denaturing methods (Goding 1996).
Unlike nitrocellulose filters and agarose beads, a tissue section is a hodgepodge of thousands of proteins, sugars and nucleic acids randomly and haphazardly crosslinked in a time- and temperature-dependent fashion with methylene bridges (Fox et al. 1985). The fixed tissue is dehydrated with a graded alcohol series, partially extracted by terpenic solvents and embedded in paraffin. This process causes many epitopes to be masked, an effect that can be reversed by enzymatic etching of the surface or a temperature- and calcium chelator-dependent process (otherwise known as antigen retrieval or AR) (Shi et al. 1991; Shin et al. 1991). An epitope within the fixed and embedded tissue, whose physical status is largely unknown, must be available to a specific antibody, for which the affinity constant is largely unknown as well.
It is thus understandable why there are different (Kolodziejczyk and Baertschi 1986; Tramu et al. 1978; Wählby et al. 2002) and occasionally contradictory (Bauer et al. 2001; Tornehave et al. 2000) reports concerning the efficiency of the various methods of elution of antibodies from formalin-fixed, paraffin embedded (FFPE) tissue sections. It should not be discounted the fact that an antibody elution method will also interact with the tissue itself and may affect either the detectability of antigens and/or the stability of the section on the glass slide.
The introduction of rabbit monoclonal antibodies (Spieker-Polet et al. 1995)—characterized, on average, by a higher affinity for the antigen as compared with the existing poly- or monoclonal reagents (Pope et al. 2009; Rossi et al. 2005)—has prompted us to revisit the existing methods of antibody elution from FFPE sections with known parameters and in a rational fashion.
Materials & Methods
Tissues
FFPE fully anonymous human leftover material was used as samples, and thus, these samples were exempt from the San Gerardo Institutional Review Board (IRB) approval as per Hospital regulations (ASG-DA-050 Donazione di materiale biologico a scopo di ricerca e/o sperimentazione, May 2012).
Three-µm sections were cut and placed on polylysine-coated glass slides, baked in a 60C oven for 1 hr, deparaffinized in a graded alcohol series, and brought to water until further use.
Hematoxylin and Eosin (H&E), Periodic Acid Shiff (PAS) and reticulin staining were performed as per standard routine laboratory stains.
Antigen Retrieval
Mounted sections were dewaxed using D-limonene, rinsed in a graded alcohol series, rehydrated in distilled water and inserted into radio-transparent slide holders (Model #S2029, Dako, Glostrup, Denmark). The latter were transferred to an 800 ml glass container filled with the retrieval solutions (10 mM EDTA in Tris-buffer pH 8 or 6 M urea) (Cattoretti et al. 1993) and irradiated in a household microwave oven at full speed for 8 min, followed by 20 min of intermittent electromagnetic radiation to maintain constant boiling.
Immunohistochemistry (IHC) / Immunofluorescence (IF)
Antigen-retrieved sections were rinsed in Tris-buffered saline (TBS, pH 7.5) containing 0.01% Tween-20 (TBS-T) and blocked for non-specific binding with a 5% w/v defatted (<1.25% fat) powdered milk solution in TBS (Latte Scremato Spray, Reire s.r.l., Reggio Emilia, Italy) for 1 hr. Subsequently, the slides were incubated with the appropriately diluted primary antibodies (Table 1 and 2) overnight in a humid chamber at room temperature (Kartell, Milano, Italy). Primary and secondary antibodies were diluted in TBS containing 5% bovine serum albumin (Sigma-Aldrich, Milan, Italy). The sections were then washed four times for 15 min each in TBS-T and incubated for an additional 30 min with the appropriate second or third immunoreagent (Table 3), as detailed in the subsequent sections.
Table 1.
Primary Antibodies of Defined Affinity (KD).
| Antibody | Clone | KD | Working Dilution | Source |
|---|---|---|---|---|
| CD45 Rabbit Monoclonal | EP322Y | 3.60×10-11 | 1:50 | Epitomics Abcam, Cambridge, MA |
| Anti Cytocheratin 7 Rabbit Monoclonal | EPR1619Y | 2.10×10-10 | 1:200 | Epitomics Abcam |
| CD34 Rabbit Monoclonal | EP373Y | 1.15×10-10 | 1:200 | Epitomics Abcam |
The KD for the rabbit monoclonal antibodies was provided by the supplier.
Table 2.
Primary Antibodies Used and Effect of Prolonged 2-Mercaptoethanol/SDS Exposure.
| Protein Class | Antigen/Antibody | Type | Clone | Isotype | Working Dilution | Source | 2-ME Cycles§ | Effect |
|---|---|---|---|---|---|---|---|---|
| Peptides, Small proteins | ||||||||
| Calcitonin | Rb poly | 1:400 | Dako, Glostrup, Denmark | 5 | = | |||
| Tireoglobulin | Rb poly | 1:10000 | Dako | 5 | = | |||
| Transmembrane proteins | ||||||||
| IgM (FITC conjugated) | Rb poly | 1:500 | Dako | 5 | = | |||
| Kappa light chain | Rb poly | 1:500 | Dako | 5 | = | |||
| CD3 | Rb poly | 1:2000 | Sigma- Aldrich, Milano, Italy | 2 | ↑ | |||
| CD20 | Mo Mab | L26 | IgG2a | 2 µg/ml | Santa Cruz Biotechnology, Dallas, TX | 2 | ↑ | |
| CD31 | Mo Mab | JC/70A | IgG1 | 1:25 | NeoMarkers, Thermo Scientific, Wilmington, DE | 5 | ↑ | |
| CD34 | Mo Mab | Qbend/10 | IgG1 | 1:50 | Dako | 2 | = | |
| CD45 (C-term) | Rb Mab | EP322Y | IgG | 1:50 | Epitomics Abcam, Cambridge, MA | 5 | = | |
| CD45 (extracellular) | Mo Mab | Bra55 | IgG1 | 1:250 | Thermo Scientific, Wilmington, DE | 5 | ↓ | |
| CD45RB (Exon B) | Mo Mab | PD7/26 | IgG1 | 1:1000 | Thermo Scientific | 5 | ↑ | |
| CD68R | Mo Mab | PGM1 | IgG3 | 1:200 | Dako | 2 | = | |
| ALK/CD246 (AA 419-520) | Mo Mab | 5A4 | IgG1 | 1 µg/ml | Santa Cruz Biotechnology | 5 | = | |
| ALK/CD246 (C-term) | Rb Mab | SP8 | IgG | 1:300 | Thermo Scientific | 5 | = | |
| ALK/CD246 (AA 1359-1460) | Mo Mab | ALK1 | IgG3 | 1:300 | Dako | 5 | = | |
| HER-2/neu (intracellular) | Mo Mab | e2-4001 | IgG1 | 1 µg/ml | Thermo Scientific | 5 | = | |
| HER-2/neu (extracellular) | Rb Mab | SP3 | IgG | 1:200 | Thermo Scientific | 5 | = | |
| HER-2/neu | Rb poly | 1:500 | Dako | 5 | = | |||
| Cytoplasmic/Cytoskeletal proteins | ||||||||
| Cytokeratins (acidic 56.5, 50, 48, 40 kD) | Mo Mab | AE-1 | IgG1 | 1:50 | Argentieri et al., 2013 | 5 | ↑ | |
| Cytokeratins (basic 65-67, 64, 59, 58,56, 52 kD) | Mo Mab | AE-3 | IgG1 | 1:100 | Argentieri et al., 2013 | 5 | ↑ | |
| CK7 | Rb Mab | EPR1619Y | IgG | 1:200 | Epitomics Abcam | 3 | = | |
| S100 | Rb poly | 1:4000 | Dako | 2 | = | |||
| Vimentin | Mo Mab | V9 | IgG1 | 1:1000 | Dako | 5 | = | |
| Transcription Factors and Nuclear Antigens | ||||||||
| CDKN2A (p16) | Mo Mab | JC8 | IgG2a | 0.5 µg/ml | Santa Cruz Biotechnology | 1 | = | |
| IRF4 | Rb Mab | EP5699 | IgG | 1:500 | Epitomics Abcam | 2 | = | |
| Ki-67 | Mo Mab | MIB 1 | IgG1 | 1:200 | Dako | 5 | ↓ | |
| MCM5 | Mo Mab | E10 | IgG2b | 1 µg/ml | Santa Cruz Biotechnology | 5 | = | |
| P63 protein | Mo Mab | 4A4 | IgG2a | 1:100 | Imgenex, San Diego, CA | 5 | ↓ | |
| NKX2-1 (a.k.a. TTF1) | Mo Mab | 8G7G3/1 | IgG1 | 1:50 | Dako | 5 | = | |
| Phosphoproteins | ||||||||
| Phospho-MEK1/2 (Ser217/221) | Rb poly | 1:200 | Cell Signaling Technology, Beverly, MA | 5 | ↑ | |||
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Rb Mab | D13.14.4E | IgG | 1:200 | Cell Signaling | 5 | = | |
| Carbohydrates | ||||||||
| CD15 (Lewis X) | Mo Mab | C3D-1 | IgM | 1:50 | Dako | 5 | = | |
| EMA | Mo Mab | E29 | IgG2a | 1:50 | Dako | 5 | = | |
indicates 2.5 hr continuous incubation in 2-Mercaptoethanol, SDS buffer (2-ME/SDS), which has been used as a proxy to five cycles of re-staining, each including a 30’ elution step.
Abbreviations: Mo= mouse; Rb = rabbit; Mab = monoclonal antibody.
Table 3.
Secondary Antibodies.
| Antibody | Target | Working Dilution | Source |
|---|---|---|---|
| Goat Anti Rabbit, FITC | Ig H+L | 1:1000 | Sigma-Aldrich, Milano, Italy |
| Sheep anti Fluorescein Fab, Alkaline Phosphatase | FITC | 1:700 | Roche Applied Science; Indianapolis, IN |
| EnVision+ System - HRP Labelled Polymer Anti Mouse | Ig H+L | neat | Dako, Glostrup, Denmark |
| EnVision+ System - HRP Labelled Polymer Anti Rabbit | Ig H+L | neat | Dako |
| EnVision+ System - HRP Labelled Polymer Anti Mouse + Rabbit | Ig H+L | neat | Dako |
| Donkey Anti Rabbit Alkaline Phosphatase | Ig H+L | 1:1000 | Jackson ImmunoResearch Laboratories, West Grove, PA |
| Donkey Anti Rabbit Fab | Ig H+L | 1:100 | Rockland |
| Goat Anti Mouse, Biotin-SP | IgH γ1 | 1:500 | Jackson ImmunoResearch |
| Goat Anti-Mouse IgG3, Rhodamine Red-X | IgH γ3 | 1:500 | Jackson ImmunoResearch |
| Goat Anti-Mouse IgG2a, Rhodamine Red-X | IgH γ2a | 1:500 | Jackson ImmunoResearch |
| Goat Anti Mouse IgG2a, ATTO 488 | IgH γ2a | 1:200 | Rockland Immunochemicals; Gilbertsville, PA |
| Donkey Anti Rabbit, DyLight 549 | Ig H+L | 1:500 | Jackson ImmunoResearch |
| Goat Anti Mouse IgG1, ATTO 647N | IgH γ1 | 1:200 | Rockland |
| Donkey Anti Rabbit, Alexa Fluor 680 | Ig H+L | 1:100 | Invitrogen, Carlsbad, CA |
| Streptavidin, DyLight 488 | biotin | 1:500 | Jackson ImmunoResearch |
Peroxidase IHC: Endogenous peroxidase was quenched with one 30-min incubation in 1% NaN3 solution, to which 3% H2O2 was added. After the primary antibody incubation rinse, the slides were incubated for 30 min with a species-specific peroxidase-conjugated polymer (Dako, Glostrup, Denmark). After a final rinse, the peroxidase was developed with 3-3’-diaminobenzidine (DAB) or aminoethylcarbazole (AEC) as a chromogen.
Immunoalkaline IHC: Endogenous phosphatases were inactivated by heat (Cattoretti et al. 1993). After the primary antibody incubation rinse, the slides were incubated for 30 min with species-specific, calf Alkaline Phosphatase (AP)-conjugated secondary antibodies (Table 3) for 30 min, rinsed for 60 min in a solution of 0.5 M NaCl and 0.005 M TBS, and then developed with the nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) chromogen (Sigma-Aldrich) at 37C in a humidity chamber with visual inspection for 20 min–2 hr (min–max in these experiments). The developing time was recorded for each experiment.
Counterstaining and coverslipping: Light hematoxylin counterstain was applied when appropriate and the sections coverslipped with Eukitt mounting medium (Sigma-Aldrich) or Glycergel (Dako), with the latter used in the cases of water soluble dyes such as AEC or NBT-BCIP. In order to minimize the shearing force applied by the solidifying gelatin during mounting, the sections and the molten gelatin were placed on a heat plate with a waterbath (model VPPS 200050, Lanzoni S.r.l., Bologna, Italy).
Indirect immunofluorescence: Nuclear dyes and species- and/or isotype-specific fluorochrome-conjugated secondary antibodies were used (Table 3) as published previously (Buscone et al. 2013; Cattoretti et al. 2006). Fluorescence slides were coverslipped with optically clear mounting medium (Fluoromount G With DAPI, Electron Microscopy Sciences, Hatfield, PA).
Endogenous biotin detection and elution: Abundant endogenous biotin in kidney tubules was stained with fluorescent Steptavidins (Table 3).
Alternatively, kidney sections stained with tetrameric streptavidin were reacted with an irrelevant biotin-conjugated goat secondary antibody, which would be captured by the free streptavidin arms; the goat antibody was stained using immunoalkaline IHC with an AP-conjugated anti-goat antibody.
Antibody Crosslinking
Antibodies on sections were crosslinked either via species-specific FITC-conjugated secondary antibody whole molecules or Fab fragments. Subsequently, the sections underwent immunostaining, with or without a preceding elution step. Detection of the eluted reagent(s) was performed by staining the first antibody or the FITC hapten. In the case of Fab secondary antibodies, the same primary antibody was re-applied on the presumably vacated antigen and immunostained: a negative stain would signal the presence of a blocking layer of monomeric fragments attached to the primary antibody layer, which prevents binding of a second identical primary Ab or staining by a different secondary reagent of the already covered primary Ab.
Chemical crosslinks were introduced by incubatinga FITC-conjugated rabbit-anti human IgM antibody (1:500)-stained section with 10% buffered formalin (3.4% formaldehyde) for 30 min (Carey et al. 2009), followed by a TBS-T wash and a milk block. The slides were then eluted as appropriate.
Antibody Elution
AR in 6 M urea as an elution method was performed as published (Cattoretti et al. 1993). Three grams of pure urea (Sigma-Aldrich) were dissolved in 50 ml of distilled water. The sections were placed in the buffer in a temperature-controlled waterbath (Dako), pre-heated to 95C and incubated for 15 min, washed for 1 hr in TBS-T and then processed for the next immunodetection procedure.
For the Gycine-SDS elution buffer, 0.94 g of glycine (Sigma-Aldrich) was dissolved in 25 ml 20% w/v SDS in water (Sigma-Aldrich), the volume brought to 500 ml with distilled water, and the solution was adjusted to pH 2 with HCl under a hood, as published (Pirici et al. 2009, with minor modifications). The slides were incubated for 30 min in the buffer, pre-heated to 50C in a dry oven (Heraterm, Thermo Scientific, Langenselbold, Germany), washed for 1 hr in TBS-T, and then processed for the next immunodetection procedure.
Elution with 2-Mercaptoethanol/SDS (2-ME/SDS) was performed by mixing 20 ml 10% w/v SDS with 12.5 ml 0.5 M Tris-HCl, pH 6.8, and 67.5 ml ultra-pure water. Under a fume hood, 0.8 ml 2-ME (Sigma) was added to the solution. Buffer incubation was performed in a shaking waterbath (Model SW23, JULABO Italia SRL, Milano) pre-heated at 56C for 30 min. Subsequently, the sections were washed for 1 hr in distilled water, with water changes every 15 min. Finally, the sections were rinsed in TBS-T for 5 min and non-specific binding was blocked with a 5% w/v defatted milk solution in TBS-T for 1 hr before subsequent immunostaining.
Elution experiments were repeated at least twice on at least two different tissues/specimens.
Sequential Immunostaining
Immunofluorescent sequential immunostaining was performed by applying a 2-ME/SDS elution, a TBS-T wash and a milk block to a previously stained slide, of which a digital virtual slide was secured. Subsequently, another IF staining was performed and another whole slide was acquired.
Multiple rounds of light microscopy staining were obtained using the alcohol-soluble AEC chromogen. After a whole slide image of the stained section was secured and the coverslip removed from the section, the chromogen was removed in a 70% ethanol bath (Glass et al. 2009), the antibody layers removed with the 2-ME/SDS buffer, and a second immunostain applied.
Virtual Whole Slide Imaging
Stained slide images were acquired with an Aperio FL and an Aperio CS whole slide scanners (Leica Microsystems, Italy), as published (Buscone et al. 2013). Individual single stain images in light and fluorescent microscopy were acquired with the ImageScope software (Aperio), optimized for contrast with Adobe Photoshop CS3 (Adobe Systems Incorporated, San Jose, CA) and mounted with Adobe Illustrator.
Results
Elution Is Dependent on the Antibody Affinity and Is Reduced by Crosslinking
Physical elution from sections of a small panel of antibodies with defined affinity (KD) (Table 1) was attempted with two different methods, one based on low pH (Pirici et al. 2009), the other on high temperature and a chaotropic agent, urea (Cattoretti et al. 1993). These two methods are expected to reversibly denature the antibodies, and indeed, the glycine buffer cleared all antibodies. The 6 M urea buffer was unable to strip the sections of the antibody with the highest affinity, CD45 (Table 4 and Fig. 1). This suggests that the equilibrium dissociation constant between the antibody and its antigen (that is, its affinity) may determine the successful elution by a method employing pH, temperature or a chaotropic agent.
Table 4.
Effect of the Elution Buffers on Bound Antibodies.
| Primary Antibody | Control (untreated) | 6 M UREA | Glycine-SDS, pH 2 | 2-Mercaptoethanol, SDS buffer | ||
|---|---|---|---|---|---|---|
| Primary Ab only | with 2nd Antibody | Primary Ab only | with 2nd Antibody | With primary only or with 2nd antibody | ||
| CD45 Rabbit Monoclonal KD= 3.60×10-11 | +++ | ++ | ++ | − | + | − |
| Anti Cytocheratin 7 Rabbit Monoclonal KD=2.10×10-10 | +++ | − | ++ | − | − | − |
| CD34 Rabbit Monoclonal KD=1.15×10-10 | +++ | − | +/- | − | − | − |
| CD20 Mouse Monoclonal KD unknown | +++ | − | traces | − | − | − |
The amount of staining after the elution is semi-quantitatively rated from none (-) to the equivalent of that of the untreated control section (+++).
Figure 1.

Elution from tissue sections of antibodies as single layers or counterstained with secondary antibodies by three elution buffers. Serial sections of human colonic mucosa (top two rows) or tonsil (bottom two rows) were stained with the primary antibody alone (1st Ab) or primary and secondary antibodies (1st+2nd Ab), eluted with the buffers listed across the top, and then counterstained with an appropriate secondary or tertiary anti-hapten antibody sequence. (A–H) CD45 (KD 3.6×10-11); (I–P) Cytokeratin 7 (CK7) (KD 2.1×10-10). Note that Glycine (Gly) pH2 buffer fails to elute a counterstained CD45 antibody (G) and 6 M Urea (6MU) fails to elute a counterstained CK7 antibody (N). The faint background staining is due to prolonged NBT-BCIP development to ensure optimal sensitivity. Scale = 100 µm.
In a real-life scenario, primary antibodies are counterlabeled in indirect immunostaining by a secondary antibody unless they are directly carrying a reporter molecule. We thus tested the panel of primary antibodies indirectly labeled with a second step and we chose a diluted, FITC-conjugated secondary antibody, whereby FITC was used as a small-size hapten. The results showed that the acidic glycine buffer was no longer able to consistently strip the topmost avid antibody, CD45, from the sections. 6 M urea also no longer could remove the second most avid antibody, CK7, from the sections and left stainable residues for other antibodies (Table 4 and Fig. 1). Appropriate controls showed that a successful elution comprised both the primary and the secondary antibody. These findings may explain the inconsistencies we have noticed with at least one of these two methods in eluting a range of primary antibodies of varying or unknown affinity from the section (data not shown).
The retention in the tissue induced by the binding of the secondary antibody may be the effect of one or multiple separate causes: the simple binding of the secondary antibody to the primary, the additional stiffening effect caused by the binding of one molecule of secondary antibody to the neighboring primary antibodies (Oda 2004), particularly when highly diluted (Mason et al. 1969), or the mere local macromolecular crowding effect (Ellis 2001) of bystander Ig molecules. Because we used the secondary antibody at a high dilution, we could not distinguish between the first or second scenario. Thus, we tested the effect of a Fab secondary Ab monomer, which is unable to crosslink neighboring molecules, on the elution of CD45 and CK7 antibodies. The results of this experiment show that monomeric Fab fragments of secondary antibodies also prevent the elution (Fig. 2 and data not shown).
Figure 2.

Effect of Fab monomers vs whole Igs on the elution of high affinity antibodies. Tonsil sections were stained with CD45 rabbit monoclonal antibodies, secondary reagents and control-treated or eluted with 6 M Urea (6MU) or Glycine (Gly) pH2. CD45 was then re-applied in the case of Fab staining, followed by counterstaining. The staining sequence for each experiment is described in the referenced square. The absence of staining in (A), (B) and (D) is expected when the CD45 antigen in the tissue is masked by an existing primary antibody-Fab complex. Bar = 100 µm.
2-ME/SDS Buffer Is Superior to Glycine pH 2 and 6 M Urea Elution Buffers for All Antibodies
High molar urea + heat and the acidic pH-glycine buffer reversibly act on the physical structure of the bound primary antibody such that a transient deformation of the structure may allow the removal of the bound antibody. However, the sets of experiments performed here showed that enough antibody or antibody complexes remains in the tissue such that, upon removal of the buffer, they quickly reconstitute the antigen-primary-secondary complex in situ.
To overcome this effect, we chose a chemical and permanent inactivation of the bound antibodies via 2-ME-mediated destruction of the intramolecular disulfide bridges. The results show that, in each and every test tried, the 2-ME/SDS buffer completely and permanently erased the primary antibodies and secondary reagents (FITC- or biotin- conjugated antibodies, immunoenzymatic polymers) from the sections (Table 4). In addition, it cleaned the non-specific binding of excess antibodies on sections (data not shown), where the other buffers could not. Thus, the 2-ME/SDS buffer was able to entirely remove a chemically crosslinked antibody from the section, whereas 6 M urea only partially did.
2-ME/SDS Buffer Has a Mild AR Effect and Positively Enhances Immunoreactivity
The effect of 2-ME/SDS on the basic tissue constituents was tested on paired serial tissue sections that were deparaffinized, one of which underwent the elution buffer treatment for 2 hr and 30 min. No differences were detected by two blinded pathologists in H&E- and PAS-stained sections (Fig. 3). A very mild reduction of reticulin staining was noticed on treated sections. We found, however, that the 2ME/SDS buffer is a potent decolorizer for H&E as well, and the time to complete the decolorization matches the time needed for a complete erasure of the bound antibody layers.
Figure 3.

Effect of the exposure to a 2-Mercaptoethanol, SDS buffer (2-ME/SDS) on the detection of tissue antigens. Paired serial sections were routinely stained for H&E and Periodic Acid Shiff, subjected to antigen retrieval, and then immunostained for a specific antigen/epitope (indicated on the left). Before staining, one (right column) of the two sections was exposed to the 2-ME/SDS buffer for 2 hr and 30 min, which is equivalent to five 30 min cycles. The tissues shown are kidney (top two rows) and tonsil (the remaining rows). Bar = 100 µm.
To test whether 2-ME/SDS buffer would be beneficial or detrimental to the immunodetection of antigens in FFPE tissue, we tested its effect on plain, dewaxed sections followed by immunodetection of molecules for which AR is mandatory (Ki-67, MCM5, TP63 and CK7). We found that the exposure of plain, dewaxed sections (i.e., without any AR) to 2-ME/SDS for 2 hr and 30 min was unable to unmask the epitopes.
In order to test the effect of 2-ME on the tissue antigens, we tested a small panel of antibodies directed against nuclear, cytoplasmic and transmembrane proteins, with glycosylated and phosphorylated modifications, as well as small peptides and immunoglobulins. The sections were exposed to repeated 30 min elutions or a prolonged (up to 2.5 hr) exposure to 2-ME/SDS; this latter exposure time equates to five elution cycles (Table 2). Transmembrane, cytoskeletal, cytoplasmic, nuclear and secreted antigens were not affected or enhanced by the long elution treatment (Table 2) with the exception of Ki-67 and TP63 (Table 2 and Fig. 3). Keratins, rich in disulfide bonds, were considerably enhanced by the 2-ME treatment (Table 2 and Fig. 3). Proteins with post-transcriptional modifications, such as glycosylation or phosphorylation preserved the antigenicity after the treatment (Table 2).
Because the removal of intramolecular bridges by 2-ME may render an epitope unavailable for binding or remove it from the tissue, whereas the remaining portion of the protein may remain anchored, we tested multiple epitopes for Her2, ALK, CD45 proteins after a 2.5 hr treatment with 2-ME/SDS. None of the proteins we tested showed a loss of epitopes after prolonged treatment. Interestingly, two epitopes of the disulfide bond-rich extracellular portion of CD45 had an opposite effect upon 2-ME treatment (Table 2 and Fig. 3): one was enhanced, the other reduced. The standard 30 min treatment did not remove FITC-tyramide deposits (data not shown).
Avidin-biotin Bond is Selectively Affected by 6 M Urea
Endogenous biotin, a vitamin ubiquitously present in mammalian cells, is bound by avidin or streptavidin with one of the highest affinities known. We thus tested the effect of 6 M urea and 2-ME/SDS elution methods on this bond. A biotin-rich tissue (kidney) was stained with streptavidin tetramers and eluted with one of each buffers. The results (Fig. 4) show that 2-ME/SDS did not elute the bound streptavidin, whereas 6 M urea did.
Figure 4.
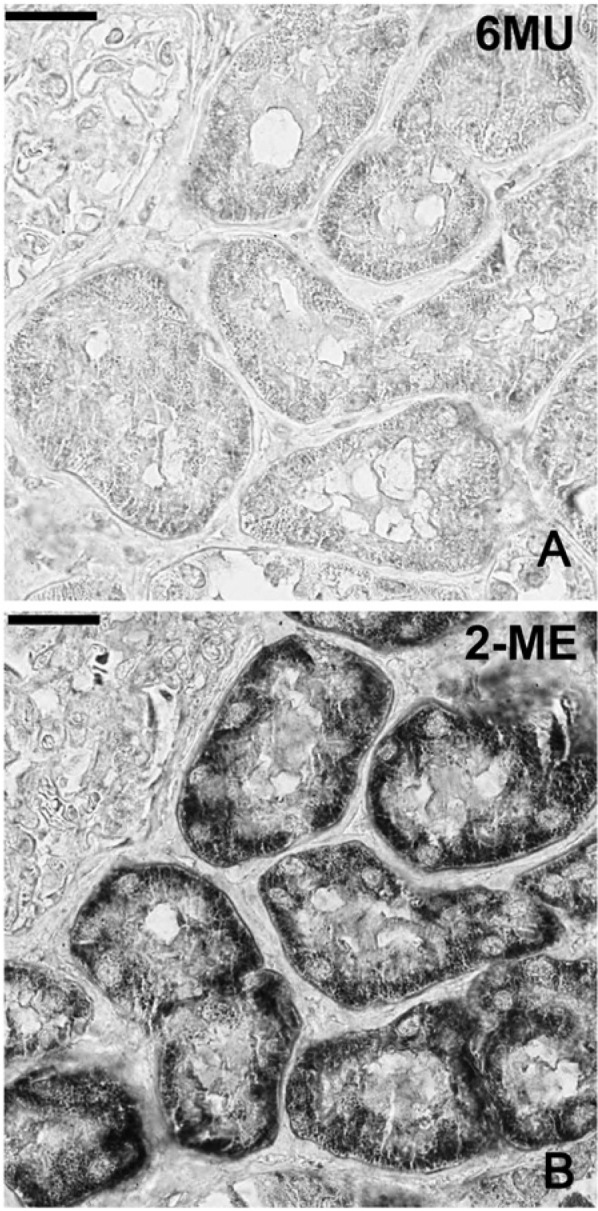
Elution of streptavidin bound to endogenous biotin by two elution buffers. 6 M Urea (6MU) elution (A) removes the granular endogenous staining in the kidney tubules, which remains evident (B) after 2-ME/SDS elution. Bar = 100 µm
Unlike avidin, which contains a single intramolecular disulfide bond (Airenne et al. 1999), streptavidin does not; thus, 2-ME/SDS was expected to not be able to remove streptavidin from the sections. We could not assess the effect of the buffers on the antibody separately from the biotin-streptavidin bond, in case of secondary, biotin-conjugated antibodies.
2-ME/SDS Elution Allows Sequential Multiple Immunostaining for Research and Diagnosis
The practical use of sequential rounds of light microscopy or fluorescent immunostaining after 2-ME/SDS elution was shown by co-localizing lymphoid and non-lymphoid subsets in the human tissue (Figs. 5 and 6).
Figure 5.

Routinely processed sections sequentially immunostained after elution and re-staining with diagnostic antibodies. One section of normal colon (left two images) and one section from a cutaneous nevus (right) are stained sequentially for the antigens named above each image. The primary antibodies have been counterstained with a multilink (mouse + rabbit) HRP-conjugated polymer. Note the clean background and no crossover staining left by the earlier staining. Bar = 100 µm.
Figure 6.

Multiple immunofluorescent staining of a human tonsil. The same tonsil section was stained three times, each with a three-combination set of primary antibodies, and counterstained with species-specific, fluorochrome-conjugated secondary antibodies. (A) Keratin (blue), Ki-67 (red) and IRF4 (green). (B) S100 (green), CD68 (red), DAPI (blue). (C) CD3 (red), CD20 (green), Ki-67 (blue). (D) CD68 (red), CD34 (green), Keratin (blue). The thumbnails on the right depict each of the nine stains obtained. Bar = 100 µm.
Elution of a previous immunostain and the application of another or multiple rounds of additional stains may be a crucial aid in diagnostics whenever there is a limited amount of tissue or slides. To test our system, we successfully applied five rounds of conventional IHC staining to sections of skin, GI tract and kidney biopsies. As shown in Figure 5, the same section can be successfully re-stained for diagnostic markers using IHC. Furthermore, a section can be stained with nine or more antibodies using four-color immunofluorescence (Fig. 6). The number of sequential elutions that can be applied is limited by the wear and tear on the section due to coverslipping and coverslip removal.
Discussion
The availability of rabbit monoclonal antibodies of known affinity for the antigen allowed us to test modifications of the bond between an antibody and an antigen on a poorly understood, largely neglected substrate, the histological tissue section. We found that primary antibodies survive much harsher conditions (pH 2, temperatures above 60C, chaotropic agents) than previously published. In addition, our results show that, despite extensive blocking and washing in excess of what is normally used (e.g., with the automated immunostainers), the primary and secondary antibodies linger on the sections and cannot be displaced except with drastic measures such as chemical breaks of the antibody disulfide bonds.
The binding of a divalent secondary antibody is sufficient to retain the primary antibody on the section and prevent its removal by a buffer of defined molar strength or pH; this effect may be due to an increase in the affinity of the primary antibody or an increased stability of the antibody chain complexed with the antigen. An analogous effect has been demonstrated for avidin upon biotin binding in an acidic environment (Green 1963), resulting in an increased thermal stability (Gonzalez et al. 1999). Bauer et al. (2001) reported a similar effect for CD18, which failed to be completely removed by microwaving after being crosslinked by a secondary antibody.
Experiments with antibody Fab monomers suggest that the mere binding prevents the removal from the section; it is not necessary to crosslink neighboring primary antibody molecules via a dimeric secondary reagent. Fab monomers are even more efficient at preventing the elution. However, we cannot exclude that the additional effect of macromolecular crowding (Ellis 2001) may take place, due to the higher protein density attainable with smaller molecules.
Not all antibodies require 2-ME/SDS for elution, but the increasing use of high affinity monoclonal antibodies in immunostaining and on western blotting may require a re-assessment of the requirements for multiple elution steps.
Endogenous biotin represents a nuisance for avidin-based immunostaining systems; 6 M urea efficiently removes the streptavidin bound to tissue biotin, in keeping with the known competing and dissociating effect of this chaotropic molecule (Merkel et al. 1999; Sano and Cantor 1990). We found that a 6 M urea step can be combined with the 2-ME/SDS buffer in order to remove the bound streptavidin, without much damage to the section (data not shown).
The use of 2-ME/SDS buffer seems more reproducible than any other buffer we tested and gentle enough with the sections such that multiple rounds of immunostaining can be performed on routine material. As published by others with different elution buffers (Glass et al. 2009; Pirici et al. 2009), we successfully repeatedly re-used the very same section in up to five sequential staining steps and up to nine markers in the case of multiple immunofluorescent immunostaining.
2-ME, an antioxidant, has a selective action on disulfide bonds. This allowed us to investigate its effect on tissue proteins for which the biochemical structure is known. The extracellular portion of transmembrane proteins, such as CD3, CD31, CD45, is rich in labile disulfide bonds (Metcalfe et al. 2011), yet all survived an extended exposure to the chemical, most were even better exposed. IgM, which is more sensitive to 2-ME than other Ig subclasses (Deutsch and Morton 1957), was unmodified in the tissue after the 2-ME treatment and so were other cytoplasmic, cytoskeletal, secreted and nuclear proteins; albeit, we could not demonstrate loss of individual epitopes within a retained protein within the small panel of proteins tested.
Among the nuclear proteins, Ki-67 and TP63 were not enhanced; to the contrary, a prolonged exposure to 2-ME reduced, but not abolished the staining. The bond between Ki-67 and the nuclear matrix is enhanced in conditions favoring the preservation or de novo formation of disulfide bonds (Kreitz et al. 2000) and reversed in the presence of oxidizing compounds; this effect may account for the partial loss from the nuclei upon exposure to 2-ME.
In general, the effect of the 2-ME/SDS treatment on tissues seems to be unrelated to the content in disulfide bonds of the target (with exceptions) and it may act as an additional unmasking agent in combination with AR, a property to be fully explored.
We tested a limited selection of antibodies; therefore, any investigator should test upfront the performance of their chosen antibody after 2-ME/SDS exposure. For the few antigens which were reduced upon prolonged treatment (Ki-67 or TP63), it is advisable to stain those first on sections to be serially de-stained and re-stained; however, one may switch to other proliferation-associated antigens, such as MCM5.
In conclusion, 2-ME/SDS buffer action is limited to the exogenous antibody or antibodies and leaves largely untouched the antigens embedded in the section. The 2-ME/SDS buffer is an important tool at the disposal of the pathologist for an extensive exploitation of often unique material for diagnosis and research.
Acknowledgments
We wish to thank Ms. Lorella Riva, Loredana Tusa, Martina Aliquó for technical help, Dr. Franco Ferrario for continuous support, Reire s.r.l. (Reggio Emilia, Italy) for providing powdered milk samples, Andrew James Smith for useful critique.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article. SB is supported by a grant from the Fondazione per la Ricerca Scientifica Termale (FoRST). RG and CRS are funded by a GlaxoSmithKline clinical research contract with the Azienda Ospedaliera San Gerardo (HGS1006-C1121). The Aperio Scanscope FL was provided through a grant from the Regione Lombardia (Call for Independent Research, DDG 6716 del 1/7/2009). This project has been supported by: Fondazione per la Ricerca Scientifica Termale (FoRST), IV call grants (Project “Lymphopoiesis In Secondary Lymphoid Tissue”) and by Departmental Hospital funds.
References
- Airenne KJ, Marjomäki VS, Kulomaa MS. (1999). Recombinant avidin and avidin-fusion proteins. Biomol Eng 16:87-92 [DOI] [PubMed] [Google Scholar]
- Argentieri MC, Pilla D, Vanzati A, Lonardi S, Facchetti F, Doglioni C, Parravicini C, Cattoretti G. (2013). Antibodies are forever: a study using 12-26-year-old expired antibodies. Histopathology 63:869-876 [DOI] [PubMed] [Google Scholar]
- Bauer M, Schilling N, Spanel-Borowski K. (2001). Limitation of microwave treatment for double immunolabelling with antibodies of the same species and isotype. Histochem Cell Biol 116:227-232 [DOI] [PubMed] [Google Scholar]
- Brouns I, Van Nassauw L, Van Genechten J, Majewski M, Scheuermann DW, Timmermans J-P, Adriaensen D. (2002). Triple immunofluorescence staining with antibodies raised in the same species to study the complex innervation pattern of intrapulmonary chemoreceptors. J Histochem Cytochem 50:575-582 [DOI] [PubMed] [Google Scholar]
- Buscone S, Argentieri MC, Pilla D, Cattoretti G. (2014) Whole-slide, Quadruple Immunofluorescence Labeling of Routinely Processed Paraffin Sections. Appl immunohistochem Mol Morphol 22:e1-7 [DOI] [PubMed] [Google Scholar]
- Carey MF, Peterson CL, Smale ST. (2009). Chromatin immunoprecipitation (ChIP). Cold Spring Harbor protocols 2009:pdb.prot5279. [DOI] [PubMed] [Google Scholar]
- Cattoretti G, Büttner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. (2006). Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood 107:3967-3975 [DOI] [PubMed] [Google Scholar]
- Cattoretti G, Pileri S, Parravicini C, Becker MH, Poggi S, Bifulco C, Key G, D’Amato L, Sabattini E, Feudale E, Reynolds F, Gerdes J, Rilke F. (1993). Antigen unmasking on formalin-fixed, paraffin-embedded tissue sections. J Pathol 171:83-98 [DOI] [PubMed] [Google Scholar]
- Deutsch HF, Morton JI. (1957). Dissociation of human serum macroglobulins. Science 125:600-601 [DOI] [PubMed] [Google Scholar]
- Ellis RJ. (2001). Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 26:597-604 [DOI] [PubMed] [Google Scholar]
- Fox CH, Johnson FB, Whiting J, Roller PP. (1985). Formaldehyde fixation. J Histochem Cytochem 33:845-853 [DOI] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A 110:11982-11987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. (2012). Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37:364-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass G, Papin JA, Mandell JW. (2009). SIMPLE: a sequential immunoperoxidase labeling and erasing method. J Histochem Cytochem 57:899-905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding JW. (1996). Monoclonal Antibodies. Principles and Practice, 3rd edition, Waltham, Massachusetts: Academic Press [Google Scholar]
- Gonzalez M, Argaraña CE, Fidelio GD. (1999). Extremely high thermal stability of streptavidin and avidin upon biotin binding. Biomol Eng 16:67-72 [DOI] [PubMed] [Google Scholar]
- Green NM. (1963). Avidin 3. The Nature of the biotin-binding site. Biochem J 89:599-609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman MC, Yang LS, Sun S, Duke JL, O’Neil KT, Kochie JE, Karjoo A, Nath P, Breth LA, Murphy K, Ross OH, Burn TC, Hollis GF, Wynn R. (2001). A comprehensive system for protein purification and biochemical analysis based on antibodies to c-myc peptide. Protein Expr Purif 23:359-368 [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk E, Baertschi AJ. (1986). Multiple immunolabeling in histology: a new method using thermo-inactivation of immunoglobulins. J Histochem Cytochem 34:1725-1729 [DOI] [PubMed] [Google Scholar]
- Kreitz S, Fackelmayer FO, Gerdes J, Knippers R. (2000). The proliferation-specific human Ki-67 protein is a constituent of compact chromatin. Exp Cell Res 261:284-292 [DOI] [PubMed] [Google Scholar]
- Mason DY, Micklem K, Jones M. (2000). Double immunofluorescence labelling of routinely processed paraffin sections. J Pathol 191:452-461 [DOI] [PubMed] [Google Scholar]
- Mason TE, Phifer RF, Spicer SS, Swallow RA, Dreskin RB. (1969). An immunoglobulin-enzyme bridge method for localizing tissue antigens. J Histochem Cytochem 17:563-569 [DOI] [PubMed] [Google Scholar]
- Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. (1999). Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature 397:50-53 [DOI] [PubMed] [Google Scholar]
- Metcalfe C, Cresswell P, Ciaccia L, Thomas B, Barclay AN. (2011). Labile disulfide bonds are common at the leucocyte cell surface. Open Biol 1:110010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda M. (2004). Antibody flexibility observed in antigen binding and its subsequent signaling. J Biol Macromol 2:45-56 [Google Scholar]
- Pirici D, Mogoanta L, Kumar-Singh S, Pirici I, Margaritescu C, Simionescu C, Stanescu R. (2009). Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J Histochem Cytochem 57:567-575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope ME, Soste MV, Eyford BA, Anderson NL, Pearson TW. (2009). Anti-peptide antibody screening: selection of high affinity monoclonal reagents by a refined surface plasmon resonance technique. J Immunol Methods 341:86-96 [DOI] [PubMed] [Google Scholar]
- Rossi S, Laurino L, Furlanetto A, Chinellato S, Orvieto E, Canal F, Facchetti F, Dei Tos AP. (2005). Rabbit monoclonal antibodies: a comparative study between a novel category of immunoreagents and the corresponding mouse monoclonal antibodies. Am J Clin Pathol 124:295-302 [DOI] [PubMed] [Google Scholar]
- Sano T, Cantor CR. (1990). Cooperative biotin binding by streptavidin. Electrophoretic behavior and subunit association of streptavidin in the presence of 6 M urea. J Biol Chem 265:3369-3373 [PubMed] [Google Scholar]
- Shi SR, Key ME, Kalra KL. (1991). Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem 39:741-748 [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J. (1991). Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab Inv 64:693-702 [PubMed] [Google Scholar]
- Spieker-Polet H, Sethupathi P, Yam PC, Knight KL. (1995). Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit-rabbit hybridomas. Proc Natl Acad Sci U S A 92:9348-9352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberger LA, Joseph SA. (1979). The unlabeled antibody method. Contrasting color staining of paired pituitary hormones without antibody removal. J Histochem Cytochem 27:1424-1429 [DOI] [PubMed] [Google Scholar]
- Tornehave D, Hougaard DM, Larsson L. (2000). Microwaving for double indirect immunofluorescence with primary antibodies from the same species and for staining of mouse tissues with mouse monoclonal antibodies. Histochem Cell Biol 113:19-23 [DOI] [PubMed] [Google Scholar]
- Tóth ZE, Mezey E. (2007). Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J Histochem Cytochem 55:545-554 [DOI] [PubMed] [Google Scholar]
- Tramu G, Pillez A, Leonardelli J. (1978). An efficient method of antibody elution for the successive or simultaneous localization of two antigens by immunocytochemistry. J Histochem Cytochem 26:322-324 [DOI] [PubMed] [Google Scholar]
- Wählby C, Erlandsson F, Bengtsson E, Zetterberg A. (2002). Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry 47:32-41 [PubMed] [Google Scholar]
- Wang W, Singh S, Zeng DL, King K, Nema S. (2007). Antibody structure, instability, and formulation. J Pharm Sci 96:1-26 [DOI] [PubMed] [Google Scholar]