

Abstract
Many cellular processes are controlled by multisubunit protein complexes. Frequently these complexes form transiently and require native environment to assemble. Therefore, to identify these functional protein complexes, it is important to stabilize them in vivo before cell lysis and subsequent purification. Here we describe a method used to isolate large bona fide protein complexes from Drosophila embryos. This method is based on embryo permeabilization and stabilization of the complexes inside the embryos by in vivo crosslinking using a low concentration of formaldehyde, which can easily cross the cell membrane. Subsequently, the protein complex of interest is immunopurified followed by gel purification and analyzed by mass spectrometry. We illustrate this method using purification of a Tudor protein complex, which is essential for germline development. Tudor is a large protein, which contains multiple Tudor domains - small modules that interact with methylated arginines or lysines of target proteins. This method can be adapted for isolation of native protein complexes from different organisms and tissues.
Keywords: Developmental Biology, Issue 86, Drosophila, Germ cells, embryonic development, RNA-protein complexes, in vivo crosslinking, Tudor domain
Introduction
Isolation of multisubunit protein assemblies and DNA- or RNA-protein complexes is performed to identify protein complexes, genomic loci recognized by DNA-binding regulatory proteins or RNA targets of RNA binding proteins. Different methods allow genome-wide identification of DNA sites recognized by transcription factors or chromatin proteins (ChIP-seq)1 and RNA targets associated with a given RNA-binding protein (CLIP-seq)2. The libraries of RNA-derived cDNAs or DNA targets are then deeply sequenced. These methods use chemical or UV-induced cross-linking to stabilize the complexes followed by immunoprecipitation (IP) with an antibody against a protein component of the studied complex.
During development of an organism, many protein complexes form transiently. Therefore, it is crucial to analyze the composition and function of these complexes in vivo to understand the molecular mechanisms that control development. Such an in vivo analysis would be superior to in vitro approach since it is virtually impossible to reproduce native concentrations of the interacting components and cellular biochemical environment in vitro. Here we demonstrate an in vivo approach that we successfully use to isolate large protein complexes from Drosophila embryos. In this method, protein complexes in the living embryos are crosslinked with a low concentration of formaldehyde and subsequently protein complexes of interest are isolated by IP with an antibody against a known component of the complexes followed by gel purification of the complexes and mass spectrometry analysis to identify unknown complex components. Since formaldehyde is able to permeate the cell membrane and has a crosslinking range of 2.3-2.7 Å3, protein complexes can be crosslinked in vivo and the complex components are likely to be close to each other. In this article we describe this method using the isolation of Tudor (Tud) protein complex as an example. Tud is a germline protein which is essential for germline development4-7. This protein contains 11 Tud domains known to interact with methylated arginines or lysines of other polypeptides8-10.
Previously, we have generated a transgenic Drosophila line which expresses HA-tagged functional Tud5 and therefore, specific anti-HA antibody is used to pull down Tud complex after crosslinking.
In addition to protein-protein crosslinks, formaldehyde can generate nucleic acid-protein crosslinks and is used in ChIP-seq experiments. Furthermore, in Drosophila, in vivo crosslinking with formaldehyde has allowed the identification of an RNA target of Vasa RNA helicase protein11.
While in this article we describe a method for in vivo crosslinking and purification of protein complexes from Drosophila embryos, this method can be adapted for other organisms and tissues.
Protocol
1. Preparing Large Apple Juice-agar Plates
To make 4 plates, add 375 ml H2O, 11.25 g fly agar and a stir bar to a 1,000 ml flask. This is Mix A. Autoclave Mix A with the flask lid loosely capped on one 30 min-sterilization cycle for liquid goods.
Add 125 ml apple juice, 12.5 g table sugar and a stir bar to a 500 ml beaker. This is Mix B. Heat Mix B on a heated platform while stirring and maintain the temperature at approximately 70 °C until the autoclaving of Mix A is finished.
Upon the completion of Mix A autoclaving, transfer Mix B to Mix A. Keep stirring the combined mixture gently and let it cool for 15 min. Avoid over-cooling and the resulted agar solidification before the addition of preservative.
Make 10% (weight/volume) methyl 4-hydroxybenzoate solution in ethanol. This will act as a preservative. Add 3.75 ml of the solution to the above apple juice-agar mixture. Keep stirring for another 5 min.
Pour 125 ml apple juice-agar mixture with preservative into each 300 cm2 plastic or styrofoam plate. Let it cool to RT and solidify. Avoid the forming of air bubbles while pouring. Note: The apple juice-agar plates are ready to use immediately. Cover unused plates with clear wraps and store at 4 °C for up to a week.
2. Drosophila Embryo Collection and Pre-crosslinking Treatment
Load a population cage with approximately 40,000 to 50,000 flies from which embryos will be collected.
Make two plates each containing 125 ml of standard cornmeal-molasses medium12 and sprinkle approximately 4 g of dried baker's yeast on each plate. Place these plates into the population cage in a 25 °C incubator for 48 hr to fatten the flies.
Warm 2 apple juice-agar plates to 25 °C. Sprinkle approximately 1 g of dried baker's yeast on each plate. Place the 2 plates in the population cage to start collecting 0-1 hr-old embryos.
Take out the plates at the end of the 1 hr collection. Replace with 2 new plates if another round of collection is needed.
Add enough water to cover the plates and resuspend the embryos gently with a fine paint brush. Pour the resuspended embryos through a two layer sieve made out of stainless steel wire mesh. Note: The top layer is made of medium pore size (850 μm) mesh which collects large fly parts while letting through embryos. The bottom layer is made of fine mesh (75 μm) which collects embryos while letting through yeast and water.
Use the fine paint brush to transfer the collected embryos into a collection basket made with a 50 ml tube and fine nylon mesh. Rinse the embryos briefly with water then blot the mesh on paper towels to dry.
Immerse the embryos in 50% bleach with gentle swirling for 3 min. Rinse thoroughly with water to remove any remaining bleach. Blot the mesh on paper towels to dry.
Immerse the dechorionated embryos in isopropanol with gentle agitation for 15 sec or until the breakdown of multi-embryo clumps (this should not take longer than 30 sec). Blot the mesh on paper towels to dry thoroughly.
Immerse the embryos in heptane for 5 min while using a pipette to stream heptane over the embryos to keep them resuspended. Blot the mesh on paper towels to dry.
Rinse the embryos with streams of Phosphate Buffered Saline, pH 7.4 (PBS) containing 0.1% Triton X-100 (PBST) for 3 min.
Disassemble the collection basket and transfer the embryos along with the mesh to a 15 ml tube containing 10 ml PBST. Invert the tube gently to wash the embryos off the mesh.
Remove the mesh from the tube. Put the tube in a vertical position and let the embryos sink to the bottom of the tube by gravity. Note: Approximately 100 - 300 μl of embryos is expected for a 1 hr collection from one cage.
Remove the PBST and continue to perform crosslinking immediately.
3. In vivo Crosslinking of Collected Embryos
Prepare 0.2% formaldehyde in PBST immediately before use. Add 10 ml of the formaldehyde fixative to the embryos and incubate at 25 °C with gentle agitation on a rotary shaker for 10 min. Discard unused fixative by the end of the day.
- Quench the crosslinking reaction
- At the end of the crosslinking, let the embryos sink to the bottom of the tube.
- Remove the formaldehyde solution. Immediately add 10 ml of 0.25 M glycine in PBST to quench the crosslinking reaction. Incubate at 25 °C with gentle agitation on a rotary shaker for 5 min.
- Let the embryos sink to the bottom of the tube. Remove the quench solution. Wash the embryos with 10 ml of PBST three times.
- Completely remove the PBST wash solution using a pipette. Store embryos at -80 °C until use.
4. Protein Sample Preparation and Immunoprecipitation
Homogenize 500 μl crosslinked embryos using a Dounce homogenizer in 2 ml lysis buffer (0.5 M urea, 0.01% SDS, 2% Triton-X 100, 2 mM phenylmethanesulfonyl fluoride [PMSF] and protease inhibitor cocktail in PBS). Note: Perform this and the following steps at RT unless otherwise specified.
Transfer the lysate to 1.5 ml centrifuge tubes and gently rock on a rotating platform for 15 min to ensure thorough lysis. Centrifuge the lysate at 16,000 x g for 5 min to pellet cellular debris. Save the supernatant in a clean 15 ml tube.
Add 40 μl anti-HA agarose beads slurry to the supernatant and incubate on a rotating platform for 2.5 hr.
Pellet the beads by centrifugation at 2,500 x g for 5 min. Remove the supernatant but leave approximately 250 μl in the tube. Resuspend the beads in the remaining supernatant and transfer to a 1.5 ml spin column with 10-μm pore filter.
Centrifuge the spin column at 12,000 x g for 10 sec and discard the flow through.
Wash the beads by adding 200 μl wash buffer (0.1 M glycine, 1 M NaCl, 1% IGEPAL CA-630, 0.1% Tween-20, in PBS). Centrifuge the column at 12,000 x g for 10 sec and discard the wash. Repeat the wash two more times.
Add 40 μl elution buffer (2 mg/ml HA-peptide in PBS) to the beads and incubate at 37 °C for 15 min with gentle agitation every 5 min. Centrifuge the column at 12,000 x g for 10 sec and collect the eluate.
Confirm elution of the complex by western-blot analysis using anti-HA antibody13.
Representative Results
The effectiveness of the crosslinking and the successful purification of the crosslinked Tud protein complex were analyzed by SDS-PAGE on a 3% - 7% step gel (illustrated in Figure 1) followed by western blot (Figure 2).
The purpose of using the 3% - 7% step gel is based on the effective separation of crosslinked Tud protein complex from the remaining uncrosslinked Tud protein and concentration of the complex. Under our in vivo crosslinking conditions mentioned above, a large proportion of Tud protein could still remain uncrosslinked. This free Tud protein would copurify with the crosslinked complex during IP until separation by SDS-PAGE. Though large in size, uncrosslinked Tud protein (285 kDa) migrates across the 3% - 7% border without obvious retention. Whereas crosslinked Tud protein complex with larger molecular weight would have difficulty migrating across the gel border, resulting in the complex being "trapped" and concentrated at the border or "tailing" near the border and therefore, separating from the free Tud protein.
The inclusion of a proper control is critical to validate the specificity of this crosslinking protocol. Therefore, we used embryos expressing HA-tagged GFP protein14 to monitor the extent of the crosslinking. We have performed pilot experiments of different formaldehyde concentrations (0.1% - 2%) on embryos expressing HA-tagged GFP and part of the results (0.1% - 0.5%) are shown here (Figure 3). It is clear that the absolute majority of GFP protein in the treated embryos (up to 0.5% formaldehyde) remained uncrosslinked, which validated that our crosslinking protocol does not crosslink target protein to random surrounding molecules. In particular, the 0.2% formaldehyde treatment (Figure 3, lane 5 and 6) did not show any crosslinked products when compared to uncrosslinked control (Figure 3, lane 1 and 2) despite the extended exposure time of the film, which further justified our choice of 0.2% formaldehyde in this protocol. However, the GFP control embryos should always be crosslinked in parallel with the Tud complex crosslinking under the same conditions and control GFP protein samples should be run on the 3% - 7% step gel together with Tud complex samples following IP. In addition, both Tud complex and the corresponding GFP control 3% - 7% gel areas should be excised and submitted for mass spectrometry analysis to determine false positive candidates that will be present in both Tud complex band and the GFP control.
The effectiveness of the in vivo crosslinking was determined by comparing free Tud protein and Tud complex from uncrosslinked control and crosslinked embryos samples respectively (Figure 2A). Free Tud protein from uncrosslinked control embryos almost exclusively migrated across the 3% - 7% border with minimal amount retained near the border area (Figure 2A, lane 1). Contrary to that, the crosslinked embryos showed the presence of large molecular weight Tud crosslinked complex at the 3% - 7% gel border (Figure 2A, lane 2). This clearly showed that our crosslinking procedure successfully crosslinked approximately 50% of total Tud protein with its in vivo interacting partners (compare Figure 2A lane 1 and 2).
The competitive elution with HA-peptide confirmed the successful purification and strengthened the specificity of the purified Tud protein complex (Figure 2B). The elution fraction by HA-peptide contained the complex concentrated around the 3% - 7% border area and also free Tud protein (Figure 2B, lane 1). The following SDS sample buffer eluted mostly free Tud remaining on the beads and minimal amount of Tud complex (Figure 2B, lane 2).
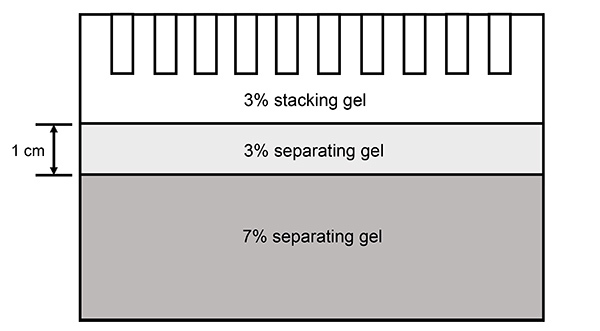 Figure 1. Diagram of the 3% - 7% step gel used for SDS-PAGE. The 3% - 7% step gel is manually cast in such a way that it contains three layers. From top to the bottom, those layers are 3% stacking gel, 3% separating gel and 7% separating gel. To work with the Mini-PROTEAN system (Bio-Rad) used in this manuscript, the height of the 3% separating gel was made to be approximately 1 cm as depicted in the diagram. Note: The 3% stacking gel and the 3% separating gel layers are extremely fragile so use extra caution when handling the gel to prevent distortion of these layers.
Figure 1. Diagram of the 3% - 7% step gel used for SDS-PAGE. The 3% - 7% step gel is manually cast in such a way that it contains three layers. From top to the bottom, those layers are 3% stacking gel, 3% separating gel and 7% separating gel. To work with the Mini-PROTEAN system (Bio-Rad) used in this manuscript, the height of the 3% separating gel was made to be approximately 1 cm as depicted in the diagram. Note: The 3% stacking gel and the 3% separating gel layers are extremely fragile so use extra caution when handling the gel to prevent distortion of these layers.
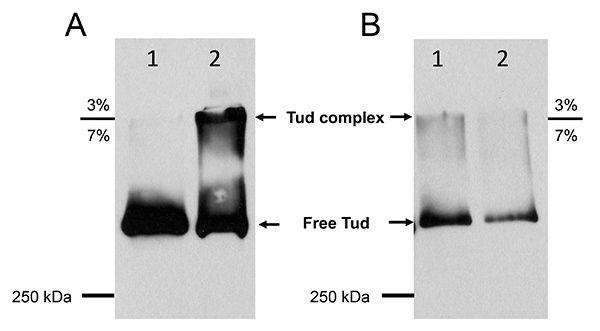 Figure 2. Western blot results of crosslinked embryos and control embryos lysates and purification of Tudor protein complex by IP. Panel A, lane 1, crude protein lysate of uncrosslinked control embryos; lane 2, crude protein lysate from formaldehyde crosslinked embryos. Panel B, lane 1, elution by HA-peptide after IP from crosslinked embryo lysate; lane 2, elution by 2 x SDS sample buffer after the initial HA-peptide elution. SDS-PAGE was run at 200 V for 3.5 hr. Anti-HA antibody (1:1,000) was used in western blot.
Figure 2. Western blot results of crosslinked embryos and control embryos lysates and purification of Tudor protein complex by IP. Panel A, lane 1, crude protein lysate of uncrosslinked control embryos; lane 2, crude protein lysate from formaldehyde crosslinked embryos. Panel B, lane 1, elution by HA-peptide after IP from crosslinked embryo lysate; lane 2, elution by 2 x SDS sample buffer after the initial HA-peptide elution. SDS-PAGE was run at 200 V for 3.5 hr. Anti-HA antibody (1:1,000) was used in western blot.
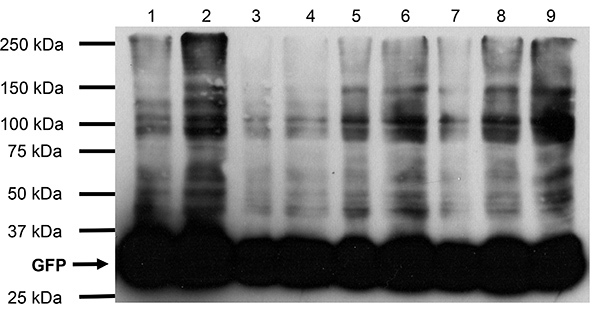 Figure 3. Western blot results of crosslinked embryos and control embryos expressing HA- tagged GFP. Lane 1, crude protein lysate of uncrosslinked control embryos; lane 2, same as lane 1 but twice the amount; lane 3, crude protein lysate of crosslinked embryos using 0.1% formaldehyde; lane 4, same as lane 3 but twice the amount; lane 5, crude protein lysate of crosslinked embryos using 0.2% formaldehyde; lane 6, same as lane 5 but twice the amount; lane 7, crude protein lysate of crosslinked embryos using 0.3% formaldehyde; lane 8, same as lane 7 but twice the amount. Lane 9, crude lysate of crosslinked embryos using 0.5% formaldehyde. SDS-PAGE was run at 200 V for 45 min in a 4% - 15% gradient gel. Anti-HA antibody (1:1,000) was used in western blot.
Figure 3. Western blot results of crosslinked embryos and control embryos expressing HA- tagged GFP. Lane 1, crude protein lysate of uncrosslinked control embryos; lane 2, same as lane 1 but twice the amount; lane 3, crude protein lysate of crosslinked embryos using 0.1% formaldehyde; lane 4, same as lane 3 but twice the amount; lane 5, crude protein lysate of crosslinked embryos using 0.2% formaldehyde; lane 6, same as lane 5 but twice the amount; lane 7, crude protein lysate of crosslinked embryos using 0.3% formaldehyde; lane 8, same as lane 7 but twice the amount. Lane 9, crude lysate of crosslinked embryos using 0.5% formaldehyde. SDS-PAGE was run at 200 V for 45 min in a 4% - 15% gradient gel. Anti-HA antibody (1:1,000) was used in western blot.
Discussion
Formaldehyde has been commonly used as a crosslinking reagent for identifying protein-protein and protein-nucleic acid interactions. Its good solubility and cell membrane permeability, together with the compatibility with downstream mass spectrometry procedures, make formaldehyde an ideal candidate agent for intracellular crosslinking applications3,15-17. In particular, it was successfully used to identify mRNAs associated with Vasa, a critical germ cell RNA helicase in Drosophila11. In addition, formaldehyde has one of the shortest spacer arms (2.3 – 2.7 Å) among commercially available crosslinkers, which implies that only molecules that exist in close proximity would be crosslinked under proper conditions, strengthening the specificity of the interacting partners of the target protein. For the reasons stated above, formaldehyde was chosen as the crosslinking agent in our protocol. However, this should not exclude the usefulness of other crosslinking agents in in vivo application for Drosophila embryos. However, we have tried to use dithiobis(succinimidyl propionate) (DSP), an N-hydroxysuccinimide (NHS) ester family crosslinker frequently used in combination with formaldehyde in ChIP assays17,18, to replace formaldehyde in our study, but no apparent Tud complex was obtained with this crosslinker On the other hand, DSP might be effective for other protein complexes which will need to be determined on case-by-case basis. In addition to in vivo crosslinking approach, in vitro crosslinkers from bismaleimidohexane (BMH) family have been shown to be effective in capturing protein–protein interactions in Drosophila embryos14,19, however, in these experiments the crosslinking occurred after embryo extracts were made.
The crosslinking time, temperature and the formaldehyde concentration are the three major factors that influence crosslinking efficiency. Our protocol has been tested using pilot experiments for each individual factor, therefore, it has been optimized for the crosslinking of Tud protein and its interacting partners in Drosophila embryos in vivo, while maintaining the balance between the specificity of the crosslinked complex and the risk of crosslinking nonspecific molecules. Nonspecific crosslinking leads to the presence of false positives in the complex and must be carefully controlled with an unrelated protein control, for example GFP, which was used in our experiments. In addition, the optimal crosslinking conditions may vary with different target molecules. Therefore, using our protocol as a starting point, the optimal crosslinking time, temperature and the formaldehyde concentration will have to be determined for a given protein complex of interest.
Once the optimal crosslinking conditions are established it is critical to follow them precisely each time the embryos are crosslinked to minimize nonspecific crosslinking. Additional crucial step is washing the beads after IP with high concentration of NaCl (1 M) and two detergents (IGEPAL CA-630 and Tween-20) to reduce nonspecific binding of unrelated proteins from embryonic extracts to the beads.
Our protocol depends on the high affinity between the target protein and the antibody used to pull down the protein complex during IP. Therefore, the antibody's epitope in the target protein should not be destroyed by crosslinking and must be accessible for the antibody during IP. In addition, the antibody-target protein interaction should withstand the stringent washes aimed at the reduction of nonspecific background proteins. These requirements for successful IP might prevent one from employing a particular antibody-epitope combination, however, it should be possible to find a suitable antibody-epitope pair for a protein of interest. In particular, HA-tag epitope and the corresponding antibody have been successfully used in this protocol.
The described protocol can be used for the isolation of native protein complexes during different developmental stages and in different tissues. Although we demonstrate this method for Drosophila, our protocol can be adapted for other organisms.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Jordan Davis, Yanyan Lin, Eric Schadler and Jimiao Zheng for their technical help with this study. This work was supported by NSF CAREER grant MCB-1054962 to A.L.A.
References
- Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murigneux V, Sauliere J, Roest Crollius H, Le Hir H. Transcriptome-wide identification of RNA binding sites by CLIP-seq. Methods. 2013. [DOI] [PubMed]
- Sutherland BW, Toews J, Kast J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. Journal of mass spectrometry : JMS. 2008;43:699–715. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- Creed TM, Loganathan SN, Varonin D, Jackson CA, Arkov AL. Novel role of specific Tudor domains in Tudor-Aubergine protein complex assembly and distribution during Drosophila oogenesis. Biochemical and biophysical research communications. 2010;402:384–389. doi: 10.1016/j.bbrc.2010.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkov AL, Wang JY, Ramos A, Lehmann R. The role of Tudor domains in germline development and polar granule architecture. Development. 2006;133:4053–4062. doi: 10.1242/dev.02572. [DOI] [PubMed] [Google Scholar]
- Boswell RE, Ptudor Mahowald A. a gene required for assembly of the germ plasm in Drosophila melanogaster. Cell. 1985;43:97–104. doi: 10.1016/0092-8674(85)90015-7. [DOI] [PubMed] [Google Scholar]
- Thomson T, Lasko P. Drosophila tudor is essential for polar granule assembly and pole cell specification, but not for posterior patterning. Genesis. 2004;40:164–170. doi: 10.1002/gene.20079. [DOI] [PubMed] [Google Scholar]
- Arkov AL, Ramos A. Building RNA-protein granules: insight from the germline. Trends in cell biology. 2010;20:482–490. doi: 10.1016/j.tcb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Arkov AL. Next generation organelles: Structure and role of germ granules in the germline. Molecular reproduction and development. 2012. [DOI] [PMC free article] [PubMed]
- Liu H, et al. Structural basis for methylarginine-dependent recognition of Aubergine by Tudor. Genes & development. 2010;24:1876–1881. doi: 10.1101/gad.1956010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Han H, Lasko P. Vasa promotes Drosophila germline stem cell differentiation by activating mei-P26 translation by directly interacting with a (U)-rich motif in its 3. UTR. Genes & development. 2009;23:2742–2752. doi: 10.1101/gad.1820709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KA. Drosophila melanogaster: Practical Uses in Cell and Molecular Biology. In: Goldstein LSB, Fyrberg EA, editors. Methods in Cell Biology. Vol. 44. Academic Press; 1995. pp. 13–32. [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Thomson T, Liu N, Arkov A, Lehmann R, Lasko P. Isolation of new polar granule components in Drosophila reveals P body and ER associated proteins. Mechanisms of development. 2008;125:865–873. doi: 10.1016/j.mod.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilescu J, Guo X, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- Klockenbusch C, Kast J. Optimization of formaldehyde cross-linking for protein interaction analysis of non-tagged integrin beta1. J Biomed Biotechnol. 2010;2010:9275–9285. doi: 10.1155/2010/927585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak DE, Tian B, Brasier AR. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques. 2005;39:715–725. doi: 10.2144/000112014. [DOI] [PubMed] [Google Scholar]
- Zeng PY, Vakoc CR, Chen ZC, Blobel GA, Berger SL. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques. 2006;41:694. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]
- Liu N, Dansereau DA, Lasko P. Fat facets interacts with vasa in the Drosophila pole plasm and protects it from degradation. Current biology : CB. 2003;13:1905–1909. doi: 10.1016/j.cub.2003.10.026. [DOI] [PubMed] [Google Scholar]