

Abstract
Bullous eruptions in patients with lupus erythematosus can be difficult to diagnose as bullous lesions can develop in lupus-specific lesions, and primary blistering disorders can also occur. Additionally, these patients tend to have multiple co-morbidities making them more likely to be on many medications that can lead to bullous drug reactions. A thorough history, the clinical presentation, and histopathological findings along with direct immunofluorescence can be helpful in diagnosing most cases. The authors report the case of a woman with a long history of systemic lupus erythematosus who initially presented in their clinic for diagnosis and management of erythema dyschromicum perstans and one year later developed bullae in atypical targetoid lesions on the extremities and trunk. They discuss several blistering disorders that have been reported in patients with lupus erythematosus with a focus on features that help distinguish erythema multiforme, fixed drug eruption, and lupus erythematosus from Stevens-Johnson syndrome/toxic epidermal necrolysis. In the patient described herein, the authors favor a diagnosis of Stevens-Johnson syndrome, but the classification between erythema multiforme major and Stevens-Johnson syndrome/toxic epidermal necrolysis cannot be made in some cases.
The diagnosis of bullous eruptions in patients with lupus erythematosus (LE) can be difficult to make as several different primary blistering disorders have been reported to occur in association with LE, including bullous pemphigoid, pemphigus vulgaris, dermatitis herpetiformis, epidermolysis bullosa acquisita, linear immunoglobulin A (IgA), porphyria cutanea tarda, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN).1-3 Bullous lesions can also occur in erythema multiforme (EM). These conditions must be differentiated from the bullous lesions that can occur in cutaneous lesions of LE, which can be due to extensive vacuolar degeneration of the basement membrane (BM) or from antibodies to type VII collagen in bullous systemic lupus erythematosus (BSLE).4 Patients with LE also tend to have multiple comorbidities, making them more likely to be on multiple medications that can lead to bullous drug eruptions. To add to the difficulty, many of these conditions may mimic SJS/TEN, which can be associated with significant morbidity and mortality. A thorough history, the clinical presentation, and histopathological findings along with direct immunofluorescence (DIF) can be used to diagnose most cases, but there are some cases where a clear diagnosis cannot be made. The authors report a case of a patient with systemic lupus erythematosus (SLE) who presented with a bullous eruption and focus on a discussion of features that help differentiate fixed drug eruption (FDE), LE, and erythema multiforme (EM) from SJS/TEN.
CASE REPORT
A 36-year-old African-American woman with a long history of SLE (diagnosed at age 12 with positive antinuclear antibody, anti-dsDNA antibody, seizures, nephritis, and arthritis), end stage renal disease (due to lupus nephritis) on peritoneal dialysis, antiphospholipid antibody, with history of cerebrovascular accident, on Coumadin presented to our clinic initially with a six-month history of oval-shaped brown lesions on her face, arms, and legs. Two biopsies were done one month apart, and both showed changes consistent with the clinical impression of erythema dyschromicum perstans (EDP), although postinflammatory hyperpigmentation from cutaneous LE or FDE could not be completely ruled out. The lesions did not respond to treatment with topical corticosteroids, and she was not interested in trying any systemic treatment as she was not bothered by the lesions. She was reassured of the benign nature of this condition and advised to return to the clinic as needed. One year later, she presented in clinic with a one-week history of raised lesions on her abdomen, back, arms, and legs. She felt this was different from the eruption with which she initially presented. Some of these lesions were developing into blisters. She also complained of some soreness in her mouth, but denied pain with swallowing or urination. She stated that she had developed soreness in her mouth with every hospital visit the last several months, which she believed could be due to irritation from endotracheal intubation for general anesthesia. She also reported a history of a few blisters on her extremities in two separate episodes two and four months prior to presentation, but none of these episodes were as extensive as the current eruption. She could not remember if the blisters in the previous outbreaks were in the same locations as the current lesions or if they were related to the previous periods of mouth soreness. A few days prior to presentation, she reported that she had felt warm and had generalized muscle aches thought to be possibly due to sinusitis. The sinusitis was being treated with cefdinir, and on the day of presentation she was on the last day of a 10-day course. One day prior to presentation, she felt joint aches and swelling in her fingers.
In addition to being treated for sinusitis, in the past six months, the patient had been hospitalized several times for abdominal pain thought to be due to chronic peritonitis, had been started on total parenteral nutrition, and was most recently hospitalized for abdominal pain due to a perforation from her peritoneal dialysis catheter. The latter was found during an exploratory laparotomy done two days before presenting in the clinic. A review of medications with the patient and review of medical records revealed that in addition to being on cefdinir for sinusitis at the time of presentation, she had also taken other cephalosporins (cefazolin and cefdinir) around two and four months prior to presentation, possibly being linked with the development of her previous blisters. She had taken a course of penicillin two weeks and also two months prior to presentation. Also included in her list of new medications were hydroxychloroquine and low-dose prednisone, which were started three months prior to presentation for SLE and sertraline five months prior for depression.
On physical exam, she had dark brown oval-shaped patches over her face, abdomen, arms, and legs (her stable lesions of the clinically favored diagnosis of EDP) with dark brown patches, some round, with overlying flaccid bullae on her arms, especially on her dorsal hands bilaterally, covering less than 10 percent total body surface area (Figure 1). Also present were scattered brown mildly hyperkeratotic plaques on her extremities and back (Figure 2). There was a crusted plaque on her left lower mucosal lip, with diffuse erythema and erosions of her labial mucosa. The following day, she was seen in the clinic, and she had developed dark brown round macules and patches with tense bullae in center on bilateral palms. All of her targetoid-appearing lesions were macular and had only two zones; some also had indistinct borders. No typical target lesions were found.
Figure 1.

Dark brown patches with overlying flaccid bullae on the dorsal hands
Figure 2.
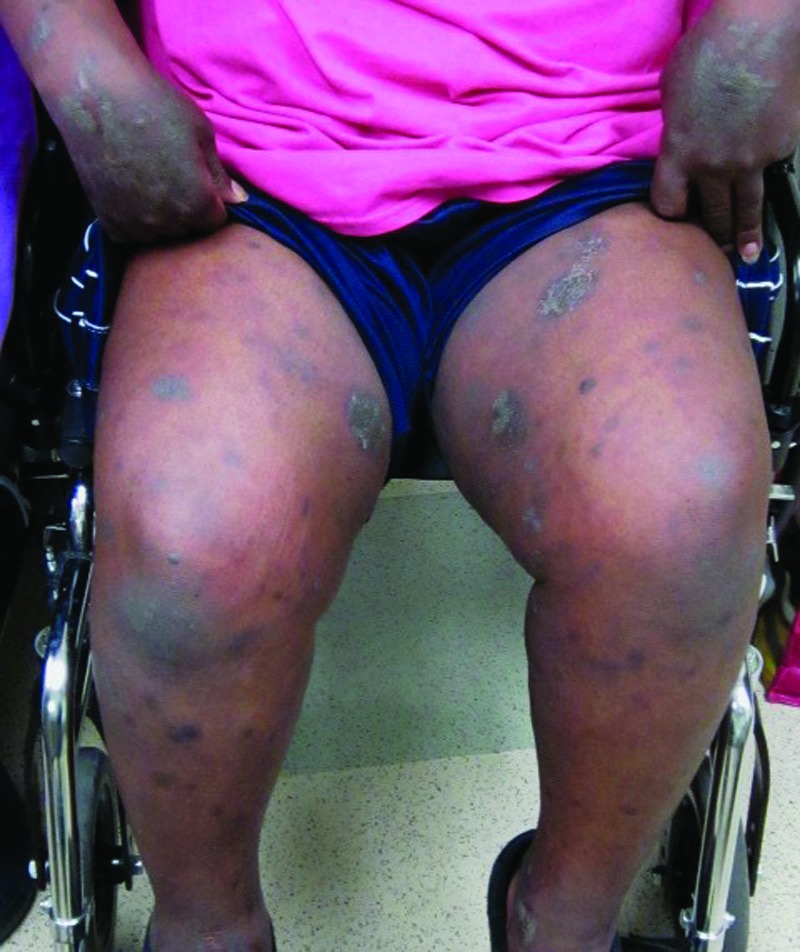
Dark brown patches and brown mildly hyperkeratotic plaques on the legs
Three 4mm punch biopsies were taken, which revealed full-thickness epidermal necrosis with separation of the epidermis from the papillary dermis, a moderate superficial perivascular inflammatory infiltrate of lymphocytes with numerous eosinophils (Figures 3 and 4). DIF for IgG, IgA, IgM, and C-3 was negative.
Figure 3.

H&E x200, showing full-thickness epidermal necrosis
Figure 4.

H&E x40, showing a superficial and deep perivascular infiltrate of lymphocytes with numerous eosinophils
The patient was seen by general surgery who did not feel the patient needed to be admitted. Wound care with an emollient was recommended. Two weeks later, she was seen for follow- up and noted to have resolution of the bullae with desquamating plaques on her trunk and extremities, especially her palms and ankles with resolution of the lesions in her oral mucosa and on her eyelids. The lesions consistent with the clinical impression of EDP persisted. She was advised to continue wound care with emollients and to avoid cephalosporins for likely diagnosis of EMM versus SJS.
DISCUSSION
There are many different causes of bullous eruptions in patients with SLE, and many of them can mimic SJS/TEN, making it difficult to make a specific diagnosis. Targetoid lesions are characteristic of EM, but they can be seen in SJS/TEN.5 Additionally SJS/TEN-like presentations of FDE, LE, and linear IgA have been reported.6-8 Linear IgA was ruled out with the patient’s negative DIF. This discussion focuses on the differences between bullous lesions in LE, EM, FDE, and SJS/TEN.
Bullous lesions in LE may occur due to extensive vacuolar degeneration of the BM in LE or due to antibodies to collagen VII in BSLE.4 In BSLE, both sun-exposed and nonexposed areas are involved, and there may be mucosal involvement. There is a predilection for the face, upper trunk, and proximal extremities. Tense blisters are seen on an erythematous or urticated base, resembling those seen in bullous pemphigoid, epidermolysis bullosa acquisita, and linear IgA. Histologically, a subepidermal blister is seen with a neutrophilic infiltrate in the upper dermis. There may be papillary microabscesses or a band-like neutrophilic infiltrate.9,10 On DIF, there is IgG, IgM, IgA, and C3 at the basement membrane zone (BMZ).10 In addition to bullae from BSLE, bullae can occur in lesions of cutaneous limited and SLE due to extensive vacuolar degeneration. This can lead to widespread bullae with clinical and histopathological features that can stimulate TEN.2,7 Compared to SJS/TEN, while there can be extensive skin involvement in SLE or acute cutaneous LE presenting with TEN-like lesions, there is no or limited mucosal involvement, no clear drug association, and the prognosis is better.11 More commonly, the skin lesions develop more slowly over weeks to months in TEN-like LE, rather than in hours to days like SJS/TEN.7,12 Additional features that may be helpful for a diagnosis of TEN-like LE include a history of recent LE exacerbation, photodistribution, and annular lesions. Features that help histopathologically are junctional vacuolar alteration, the presence of solitary necrotic keratinocytes at lower epidermal levels, moderate-to-dense periadnexal and perivascular lymphocytic infiltrates, a thickened BMZ, and the presence of plasma cells, melanophages, or mucin.7 Additionally, a positive DIF is usually seen in lesions of LE in the typical pattern of the underlying lupus (IgG and/or IgM, and less commonly IgA, granular deposits at dermal-epidermal junction and around hair follicles), but a negative DIF does not rule this out.2,11,13
Lesions in FDE are classically, sharply marginated, round-or oval-shaped lesions that can have bullae.8 There have been cases where widespread development of bullae has caused a TEN-like presentation.14 There are a few features that can help to differentiate between the two. Recurrent episodes of FDE typically occur in minutes to hours after re-exposure to the medication, whereas in SJS/TEN, recurrent episodes can be seen in as early as two days.5,15 Recurrent episodes of FDE occur in the same location as previous episodes, whereas lesions in SJS/TEN do not show a predilection for previously affected sites. When the diagnosis is unclear from the history or presentation, histological evaluation can help. In FDE, there tends to be a superficial and deep perivascular mixed infiltrate with lymphocytes, histiocytes, neutrophils, and eosinophils. However, eosinophils may predominate. In TEN, there is usually only a superficial perivascular infiltrate with lymphocytes and histiocytes. 16-18 It is typically characterized by little or no inflammation. However, some cases have shown a substantial number of eosinophils, which may be related to drug-related cases.19
In addition to difficulties with separating these lupus-specific lesions and FDE from SJS/TEN, it is particularly difficult at times to distinguish between EMM and SJS/TEN. Until recently, EM, SJS, and TEN were thought to be part of a spectrum. It is now believed that EM and SJS/TEN are likely two distinct clinical disorders with different causes, distribution of lesions, and prognosis.20-22 Histopathologically, they can look similar, so it is used more to rule out other causes, although EM may have a stronger infiltrate compared to SJS/TEN.22,23 It is most difficult to differentiate SJS from EMM, in which mucosal involvement is also found. The characteristic lesion of EM is the typical target lesion, which has a well-defined border and three distinct zones. Atypical target lesions with only two zones and/or a poorly defined border can also be present and are palpable in EM. Lesions are typically on the extremities and face. Atypical target lesions can also be present in SJS/TEN, but they are macular. It typically starts on the trunk, can spread to the neck/face and proximal upper extremities, and generally spares the distal extremities. Drugs have been found to be likely causes in 80 percent of cases of TEN, and in contrast to EM, infections are rare causes of SJS/TEN.12 More than 90 percent of cases of EM are caused by infections, most commonly by herpes simplex virus (HSV).7,24
In some cases, the diagnosis between EM or SJS/TEN cannot be made.20 Auquier-Dumant et al21 reviewed 552 patients with possible EMM or SJS/TEN and classified the cases into EMM, SJS, SJS-TEN overlap, TEN, or unclassified EM or SJS. Because of mixed features, they were unable to classify 92 cases (17%). A comparison of these groups showed that compared to patients with SJS, SJS-TEN overlap, or TEN, patients with EMM were younger, were more often male, had a higher rate of recurrence, less often had temperatures at or above 38.5°C, and less frequently had involvement of two or more mucous membranes. EMM cases also were never or rarely associated with collagen vascular diseases, HIV infection, or cancer compared to SJS, SJS-TEN overlap, and TEN combined. They looked at likely etiologies and found that the cases of SJS, SJS-TEN, and TEN when compared to EMM were more commonly caused by a drug that had been shown to be associated with SJS/TEN. Herpes was the main cause in EMM with 29 percent having a history of a recent outbreak of herpes compared to six percent in SJS. Table 1 shows the differences between EMM and SJS/TEN found in this study.21
TABLE 1.
Differentiating features between EMM vs. SJS/SJS-TEN/TEN found in the prospective study by Auquier-Dunant et al21
| EMM | SJS/SJS-TEN/TEN | |
|---|---|---|
| Median age | 24 | 45 |
| % male | 64 | 43 |
| Rate of recurrence | 30 | 3 |
| % with temperature ≥38.5°C | 32 | 54 |
| % with 2 or more mucous membranes involved | 71 | 85 |
| % caused by a drug shown to be significantly associated with SJS/TEN* | 5 | 43-48 |
| % caused by a drug shown to be associated with SJS/TEN** | 18 | 64-66 |
| % caused by recent HSV outbreak (within 4 weeks of onset of disease) | 29 | 6 in SJS, 0 in SJS-TEN and TEN |
| % associated with collagen vascular disease | 0 | 5 |
| % associated with HIV | 0 | 8 |
| % associated with cancer | 2 | 11 |
It can often be difficult to differentiate between FDE, EM, lupus-specific lesions, and SJS/TEN. Table 2 shows a summary of the clinical, histological, and DIF features discussed above that may help differentiate between the above entities. BSLE was ruled out in the authors’ patient with a negative DIF. Her lesions developed quickly, did not have lesions that were in a photodistribution, lacked histological features that favor LE, and there was a negative DIF. This helped to rule out a SJS/TEN-like LE eruption. The patient reported that her lesions started about three days after starting the cephalosporin, and the histopathology from our patient’s biopsies revealed a superficial perivascular infiltrate with lymphocytes and eosinophils. The fact that her lesions started days after re-exposure to the medication and the lack of a deep perivascular infiltrate and neutrophils helped in making FDE a less likely diagnosis. This narrowed our differential diagnosis to erythema multiforme major (EMM) and SJS/TEN.
Table 2.
Summary of characteristics of BSLE, SJS, and TEN-like lupus, FDE, EM, and SJS/TEN
| BSLE | SJS AND TEN-LIKE LUPUS | FDE | EM | SJS/TEN | |
|---|---|---|---|---|---|
| Cause | Antibodies to collagen VII | No clear drug association, may have history of recent LE exacerbation | Drug-induced | >90% caused by infections, most commonly HSV | Drugs cause in 80%, infections are rare |
| Distribution | Any cutaneous site with predilection for the face, upper trunk, and proximal extremities, can have mucosal involvement | Photodistribution, No or limited mucosal involvement | May occur on any site on skin/mucosa, most commonly palms, soles, lips, and glans penis16 | Typically lesions on extremities and face | Typically start on the trunk and can spread to the neck/face and proximal upper extremities, generally spare distal extremities |
| Lesion morphology | Tense bullae on an erythematous or urticated base | Preceded by characteristic cutaneous LE lesions. Develop over weeks to months | Sharply marginated round- or oval-shaped lesions that can have bullae. Usually few and localized, but can be multiple and disseminated | Typical target (well-defined border and three distinct zones). Atypical target lesions are palpable and have only two zones and/or a poorly defined border | Atypical target lesions if present have only two zones or a poorly defined border, but are macular. Develop over hours to days |
| Recurrent cases | Minutes-hours after re-exposure to medication. Occur in the same location as previous episodes | More frequently recurrent compared to SJS/TEN, recurrences due to HSV common | As early as 2 days. No predilection for previously affected sites | ||
| H&E | Subepidermal blister with neutrophilic infiltrate in upper dermis (papillary microabscesses or band-like infiltrate) | Junctional vacuolar alteration, presence of solitary necrotic keratinocytes at lower epidermal levels, moderate-to-dense periadnexal and perivascular lymphocytic infiltrate, thickened BMZ, presence of plasma cells, melanophages, mucin | Superficial and deep perivascular mixed infiltrate with lymphocytes, histiocytes, neutrophils, and eosinophils (eosinophils may predominate) | Superficial perivascular infiltrate with lymphocytes and histiocytes | Superficial perivascular infiltrate with lymphocytes and histiocytes. Typically characterized by little or no inflammation, but drug-related cases may have a substantial number of eosinophils |
| DIF | IgG, IgM, IgA, and C3 at the BMZ | Most characteristic— IgG and/or IgM (IgA less commonly) granular deposits at dermal-epidermal junction and around hair follicles, (neg DIF does not rule out LE) | Not well described (reports of linear IgG and C3 deposition along the BMZ, perivascular and stippled band of C3 at the BMZ, intercellular IgG and C3 reported.)25 | Not specific (C3 and IgM in blood vessels in the upper dermis, C3 may be found along BMZ and focally in Civatte bodies)26 | Not specific (C3 around blood vessels, IgM, and C3 at the dermoepidermal junction)26 |
The diagnosis was very difficult to determine in this patient, as she had features consistent with both EMM and SJS. She reported subjective fever and joint aches, and she had mucosal lesions. Systemic symptoms and mucosal involvement can be seen in both EMM and SJS. Factors that favor the diagnosis of SJS include the presence of atypical target lesions that were macular. Additionally, she reported crusting of her eyes, and she had oral lesions when she was seen in the clinic. SJS/TEN is more likely to have involvement of two or more mucosal sites. She was on a cephalosporin at presentation and reported that the skin lesions started around three days after starting her course, and the review of her medications revealed that she also had cephalosporins around the time she had her previous blistering eruptions. This makes cephalosporins the likely culprit in her case, and SJS is more commonly caused by a drug. Factors that favor the diagnosis of EMM include the fact that her lesions were mostly acral. It is unclear if her previously reported episodes of blistering were related, but if she in fact did have recurrent episodes, EM is more likely to be recurrent.21 There was a moderate amount of perivascular infiltrate, and there may be a heavier infiltrate in EM compared to SJS/TEN. Finally, there is a better prognosis for EMM, and the patient was doing well at follow-up with resolution of her bullae.
In the patient described herein, although the authors favor the diagnosis of SJS, they could not make a classification between EMM and SJS. Regardless of the exact diagnosis, cephalosporins were the likely cause, and the patient was 13. advised to avoid them in the future. This case highlights the challenge of diagnosing bullous eruptions in patients with LE as many blistering eruptions can occur, several of which can 14. mimic SJS/TEN, and patients with LE tend to have multiple comorbidities and medication exposure that can complicate the picture.
Footnotes
DISCLOSURE:The authors report no relevant conflicts of interest.
REFERENCES
- 1.Home NS, Narayan AR, Young RM, Frieri M. Toxic epidermal necrolysis in systemic lupus erythematosus. Autoimmun Rev. 2006;5(2):160–164. doi: 10.1016/j.autrev.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Mandelcorn R, Shear NH. Lupus-associated toxic epidermal necrolysis: a novel manifestation of lupus? J Am Acad Dermatol. 2003;48(4):525–529. doi: 10.1067/mjd.2003.107. [DOI] [PubMed] [Google Scholar]
- 3.Ting W, Stone MS, Racila D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13(12):941–950. doi: 10.1191/0961203304lu2037sa. [DOI] [PubMed] [Google Scholar]
- 4.Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29(4):649–653. doi: 10.1016/j.det.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 5.French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology, 2nd ed. St. Luois, MO: Mosby Elsevier; 2008. pp. 287–300. [Google Scholar]
- 6.Kakar R, Paugh H, Jaworsky C. Linear IgA bullous disease presenting as toxic epidermal necrolysis: a case report and review of the literature. Dermatology. 2013;227(3):209–213. doi: 10.1159/000353584. [DOI] [PubMed] [Google Scholar]
- 7.Ziemer M, Kardaun SH, Liss Y, Mockenhaupt M. Stevens-Johnson syndrome and toxic epidermal necrolysis in patients with lupus erythematosus: a descriptive study of 17 cases from a national registry and review of the literature. Br J Dermatol. 2012;166(3):575–600. doi: 10.1111/j.1365-2133.2011.10705.x. [DOI] [PubMed] [Google Scholar]
- 8.Lee AY. Fixed drug eruptions. Incidence, recognition, and avoidance. Am J Clin Dermatol. 2000;1(5):277–285. doi: 10.2165/00128071-200001050-00003. [DOI] [PubMed] [Google Scholar]
- 9.Yung A, Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol. 2000;41(4):234–237. doi: 10.1046/j.1440-0960.2000.00426.x. [DOI] [PubMed] [Google Scholar]
- 10.Grover C, Khurana A, Sharma S, Singal A. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58(6):492. doi: 10.4103/0019-5154.119973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67(3):417–421. doi: 10.1016/j.jaad.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Lee HY, Tey HL, Pang SM, Thirumoorthy T. Systemic lupus erythematosus presenting as Stevens-Johnson syndrome and toxic epidermal necrolysis: a report of three cases. Lupus. 2011;20(6):647–652. doi: 10.1177/0961203310385162. [DOI] [PubMed] [Google Scholar]
- 13.Lee LA. Lupus erythematosus. In: Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008. pp. 601–613. [Google Scholar]
- 14.Lin TK, Hsu MM, Lee JY. Clinical resemblance of widespread bullous fixed drug eruption to Stevens-Johnson syndrome or toxic epidermal necrolysis: report of two cases. J Formos Med Assoc. 2002;101(8):572–576. [PubMed] [Google Scholar]
- 15.Revuz J, Valeyrie-AUanore L. Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2nd ed. St. Louis, MO: Mosby Elsevier; 2008. pp. 301–320. [Google Scholar]
- 16.Baird BJ, De Villez RL. Widespread bullous fixed drug eruption mimicking toxic epidermal necrolysis. Int J Dermatol. 1988;27(3):170–174. doi: 10.1111/j.1365-4362.1988.tb04923.x. [DOI] [PubMed] [Google Scholar]
- 17.Ramos-Ceballos FI, Horn TD. Interface dermatitis. In: Barnhill RL, Crowson AN, Magro CM, Piepkorn MW, editors. Dermatopathology, 3rd ed. New York: McGraw-Hill Companies; 2010. pp. 36–63. [Google Scholar]
- 18.Rai R, Jain R, Kaur I, Kumar B. Multifocal bullous fixed drug eruption mimicking Stevens-Johnson syndrome. Indian J Dermatol Venereol Leprol. 2002;68(3):175–176. [PubMed] [Google Scholar]
- 19.Wetter DA, Camilleri MJ. Clinical, etiologic, and histopathologic features of Stevens-Johnson syndrome during an 8-year period at Mayo Clinic. Mayo ClinProc. 2010;85(2):131–138. doi: 10.4065/mcp.2009.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assier H, Bastugi-Garin S, Revuz J, Roujeau JC. Erythema multiforme with mucous membrane involvement and Stevens-Johnson syndrome are clinically different disorders with distinct causes. Arch Dermatol. 1995;131(5):539–543. [PubMed] [Google Scholar]
- 21.Auquier-Dunant A, Mockenhaupt M, Naldi L, et al. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol. 2002;138(8):1019–1024. doi: 10.1001/archderm.138.8.1019. [DOI] [PubMed] [Google Scholar]
- 22.Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johns on syndrome, and erythema multiforme. Arch Dermatol. 1993;129(l):92–96. [PubMed] [Google Scholar]
- 23.Watanabe R, Watanabe H, Sotozono C, et al. Critical factors differentiating erythema multiforme majus from Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) Eur J Dermatol. 2011;21(6):889–894. doi: 10.1684/ejd.2011.1510. [DOI] [PubMed] [Google Scholar]
- 24.Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called “RoweU’s syndrome”? Lupus. 2012;21(6):577–585. doi: 10.1177/0961203311430513. [DOI] [PubMed] [Google Scholar]
- 25.Hern S, Harman K, Clement M, Black MM. Bullous fixed drug eruption due to paracetamol with an unusual immunofluorescence pattern. Br J Dermatol. 1998;139(6):2. doi: 10.1046/j.1365-2133.1998.2576o.x. [DOI] [PubMed] [Google Scholar]
- 26.Brice SL, Huff JC, Weston WL. Erythema multiforme. CurrProbl Dermatol. 1990;2(l):5–25. [Google Scholar]