

Abstract
We have investigated at the oligomeric level interactions between Aβ(25–35) and Tau(273–284), two important fragments of the amyloid-β and Tau proteins, implicated in Alzheimer’s disease. We are able to directly observe the coaggregation of these two peptides by probing the conformations of early heteroligomers and the macroscopic morphologies of the aggregates. Ion-mobility experiment and theoretical modeling indicate that the interactions of the two fragments affect the self-assembly processes of both peptides. Tau(273–284) shows a high affinity to form heteroligomers with existing Aβ(25–35) monomer and oligomers in solution. The configurations and characteristics of the heteroligomers are determined by whether the population of Aβ(25–35) or Tau(273–284) is dominant. As a result, two types of aggregates are observed in the mixture with distinct morphologies and dimensions from those of pure Aβ(25–35) fibrils. The incorporation of some Tau into β-rich Aβ(25–35) oligomers reduces the aggregation propensity of Aβ(25–35) but does not fully abolish fibril formation. On the other hand, by forming complexes with Aβ(25–35), Tau monomers and dimers can advance to larger oligomers and form granular aggregates. These heteroligomers may contribute to toxicity through loss of normal function of Tau or inherent toxicity of the aggregates themselves.
Introduction
Senile plaques of amyloid-β (Aβ) and neurofibrillary tangles (NFT) of Tau are pathophysiological markers of Alzheimer’s disease (AD). Tau proteins include six different intracellular, intrinsically disordered isoforms consisting of two functional domains. The projection domain mediates interactions of microtubules with neural plasma membrane and cytoskeletal elements and is involved in signal transduction, while the Tau microtubule binding pseudorepeat domain (MTBR) regulates microtubule assembly from tubulin (for a comprehensive review, see ref (1)). Aggregation of Tau into NFT is promoted in cases of hyperphosphorylation or deficiency in dephosphorylation.1−3 More than 37 mutations of Tau isoforms have been shown to induce neurodegenerative diseases (e.g., frontotemporal dementia);3,4 however, there are no Tau mutations directly linked to any known familial form AD. While a “loss of function” hypothesis is often invoked to explain the role of Tau aggregation in AD, the latter does not fully account for the etiology of the disease, and does not address the role of Aβ in AD, or its possible interaction with Tau.5
Unlike Tau which resides in the neurons, the Aβ peptide is produced in the extracellular space from the proteolytic cleavage of a large transmembrane amyloid β-protein precursor protein (APP). An amyloid cascade hypothesis has been proposed that stipulates that AD is caused by the aggregation of Aβ,5 with β-rich toxic oligomers6,7 or fibrillar aggregates8 signaling cell apoptosis or decreasing synaptic plasticity. However, this hypothesis has been challenged given the lack of strong correlation between the amount of Aβ produced, or senile plaques deposited, and neuronal loss or cognitive impairment.9,10 The observation that Aβ monomers or soluble oligomers permeate cell membranes11,12 and form highly functional multimeric aggregates6 capable of interfering with normal cellular activities is offering a new research direction in AD. Yet another hypothesis, and the one that we will focus on in this work, is that the interaction of Aβ and Tau may be significant in AD. Indeed, while the Aβ peptide is produced in the extracellular space, soluble Aβ oligomers have been shown to exist in the intracellular space as well13 and interact with a variety of proteins14 including Tau.15−21 Exposure of Tau to Aβ oligomers22 could lead to loss of microtubule integrity,15 formation of new, insoluble aggregates,23 or an enhancement of NFT formation.8 Tau has been shown to induce Aβ toxicity in hippocampal neurons,24 while Aβ-mediated Tau phosphorylation has been discovered in both hippocampal and cholinergic neurons.19,25−27
The full length Aβ is a 39–43-residue peptide with Aβ(1–40) and Aβ(1–42) being the two most abundant isoforms. The full length Tau, depending on its isoforms, can be as long as 400 residues, and studying complexes of Aβ and Tau poses a real challenge, both from an experimental and computational standpoint. We then turn in this work to the study of a model system that captures the essential features of Aβ and Tau assembly. We use a combination of ion-mobility mass spectrometry (IM-MS), atomic force microscopy (AFM), and computational modeling to investigate the interactions between an Aβ and a Tau fragment: Aβ(25–35) and Tau(273–284). These two peptide fragments contain important regions of the full Aβ and Tau proteins, and we and others have studied early oligomer conformations and aggregation propensities of both peptides.28−33 Aβ(25–35) is an amphipathic peptide with similar aggregation propensity and toxicity to the full length Aβ(1–42).34,35 Takashima et al.27 have shown that embryonic rat hippocampal neurons undergo progressive degeneration and that Tau phosphorylation is enhanced after exposure to Aβ(25–35), similar to the result obtained with Aβ(1–40) by Busciglio and co-workers.25 Tau(273–284) is in the second repeat (R2) of MTBR and encompasses the PHF6* hexapeptide (VQIINK), one of the two hexapeptides known to play an important role in Tau aggregation.36−39 It has also been shown experimentally that Tau binds between the middle and C-terminal regions of Aβ(1–42) (the region of APP inserted into the cell membrane).23 A recent theoretical work by Nussinov and co-workers also suggests that Aβ oligomers interact more strongly with R2 of MTBR than R3 or R4.40 Although R2 appears in some but not all Tau isoforms (encoded only by exon 10 of the MAPT protein), changing the ratio of four-repeat Tau to three-repeat Tau is sufficient to cause serious degeneration in microtubule assembly.41
Of note is that both peptides contain a hydrophobic stretch of six-residue sequence, i.e., PHF6* in Tau(273–284) and GAIIGL from Aβ(25–35). These two hexapeptides have been shown to adopt steric zipper motifs that facilitate amyloid fibril formation.36,42,43 The sequences of the fragments are shown in Figure 1.
Figure 1.
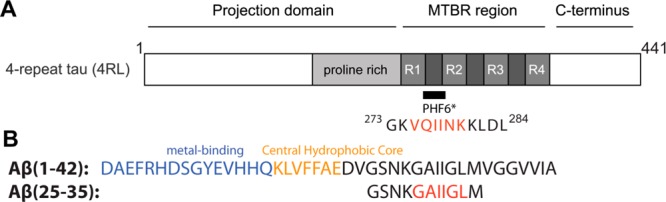
(A) Full length four-repeat Tau and location of PHF6* and the Tau(273–284) construct used in this study (see Larini et al.30 Adapted in part from ref (30). Copyright 2013 Royal Society of Chemistry.). The longest Tau isoforms contain either three or four imperfect repeats in the microtubule-binding pseudorepeat domain (MTBR). (B) Full length Aβ(1–42) with different regions and the peptide sequence of Aβ(25–35). All peptides studied here have acetylated N-termini and amidated C-termini. The hydrophobic hexapeptide fragments are color coded in red.
Materials and Methods
All peptides have blocked termini to minimize the effect of terminal charges. Lyophilized samples of Ac-Tau(273–284)-NH2 were purchased from Genscript (Piscataway, NJ, USA). Ac-Aβ(25–35)-NH2 was synthesized by FMOC (N-(9-fluorenyl)methoxycarbonyl) chemistry. The peptide was purified by reverse-phase high-performance liquid chromatography (HPLC) and characterized by mass spectrometry and amino acid analysis to confirm the purity and integrity of the peptide (>95% purity). Working stock solutions (2 mM for the Ac-Tau(273–284)-NH2 and 500 μM for Ac-Aβ(25–35)-NH2) were prepared by dissolving the lyophilized peptide in filtered deionized water. This stock was divided into several tubes, flash-frozen in liquid nitrogen, and stored at −80 °C until use. The concentration of peptide samples in IM-MS and AFM experiments is 200 μM. Polydisperse heparin of 11 kDa was from Sigma-Aldrich (St. Louis, MO, USA) and stocked at 1 mM concentration in water at room temperature.
Ion-Mobility Mass Spectrometry
In IM-MS experiments, stock solutions were diluted in water to a desired concentration, loaded into gold coated nano-electrospray ionization (ESI) capillaries, and electrosprayed on a home-built instrument.44 Ions were pulled through a 200 cm long, helium filled drift cell under the influence of a weak electrical field and the drag force created from collisions with buffer gas ions. Particular species with specific m/z can be mass-selected, and their arrival time distributions can be measured at different pressure to drift voltage ratios (P/V), allowing the determination of reduced mobility K0 and experimental collision cross-sections σ.45
Atomic Force Microscopy
AFM images were collected using an Asylum MFP-3D-SA system (Asylum Research, Santa Barbara, CA, USA). Details about AFM protocol have been described previously.46 All images were collected at 1 Hz using 512 × 512 scan points. Images were processed using Igor Pro software and were modified by masking fibrils and then applying a first-order flatten to height images (“Magic Mask” in MFP3D software). All height and length measurements of single fibrils and granular aggregates were conducted using Igor Pro, and each reported value is the average of 20 measurements.
Molecular Dynamics Simulations
Temperature-based replica exchange molecular dynamics (T-REMD) simulations were performed using the GROMACS 4.6.3 package47,48 with a combination of the OPLS-AA force field49−52 and TIP3P water.53 In these simulations, the initial conformation of Ac-Aβ(25–35)-NH2 is a β-hairpin obtained from T-REMD simulations of the same peptide using the same force field (data not shown), and also consistent with a previous simulation of free terminal Aβ(25–35) by Larini and Shea.31 An initial Ac-Tau(273–284)-NH2 was chosen from the T-REMD simulations described in ref (30). Structures obtained from these simulations were minimized in the gas phase to mimic the dehydration process that occurred when solution-phase structures enter a solvent-free environment. Their collision cross-sections were computed using the trajectory method available from the Mobcal package.54,55
Results and Discussion
Tau(273–284) Binds to Aβ(25–35) with High Affinity To Form Heteroligomers
All IM-MS experiments are performed on a home-built IM-MS instrument described previously.44 The mass spectra of Ac-Aβ(25–35)-NH2, Ac-Tau(273–284)-NH2, and a mixture of both at 1:1 ratio are shown in Figure 2. Water is used in place of buffer to slow down aggregation kinetics, as well as minimize the charge screening effect of buffer ions that may affect aggregation.
Figure 2.

ESI–mass spectra of (A) Ac-Aβ(25–35)-NH2, (B) Ac-Tau(273–284)-NH2, and (C, D) a mixture of the two peptides at 1:1 ratio. The concentration of each peptide is 200 μM in water. Each peak is annotated with [n + k]/z where n is the oligomer number of Ac-Aβ(25–35)-NH2, k is the oligomer number of Ac-Tau(273–284)-NH2, and z is the net charge of the complex. A representative ATD of the peak at 705 m/z (k/z 1/2) is shown in panel B to illustrate that dimer is the largest Tau oligomer formed under these conditions. Note that the dimer is relatively minor compared to the monomer.
The mass spectrum of Ac-Aβ(25–35)-NH2 (Figure 2A) contains two major peaks at 550 m/z (n/z 1/2) and 1101 m/z (n/z 1/1), where n is the Aβ(25–35) oligomer size and z is the charge. Less intense peaks are observed at 734 m/z (n/z 2/3), 826 m/z (n/z 3/4), and 1407 m/z (n/z 9/7). The arrival time distributions (ATDs) at m/z other than 1101 contain either a single oligomer species or a single family of conformations (see Supporting Information Figure S1A–C,E). ATDs of the n/z 1/1 peak contain multiple features from dimer to hexamer (Supporting Information Figure S1D). The progression of oligomer cross-sections σ as a function of oligomer size n (see Figure 3A) shows a positive deviation from the isotropic oligomer model starting at the trimer stage,56 indicating the formation of extended oligomers. Based on our earlier studies on free terminal Aβ(25–35),31,32 we can ascribe these extended oligomers to β-rich oligomers. Formation of β-rich oligomers is consistent with the fibril formation propensity of the peptide probed by AFM (see the next section).
Figure 3.
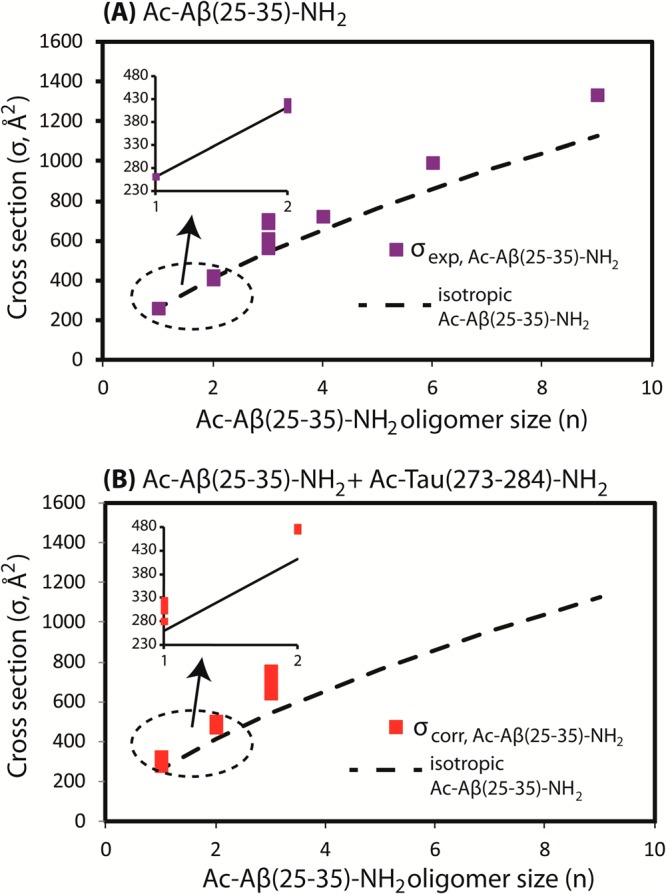
(A) Experimental cross-section of Ac-Aβ(25–35)-NH2 (σexp, Å2) as a function of n. (B) Correlated cross-section of Ac-Aβ(25–35)-NH2 + Ac-Tau(273-284)-NH2 (σcorr, Å2) obtained from the experimental cross-section of heteroligomers. The isotropic cross-sections of Ac-Aβ(25–35)-NH2 are shown by the dashed line.
The mass spectrum of Ac-Tau(273–284)-NH2 (Figure 2B) consists of two peaks at 470 m/z (k/z 1/3) and 705 m/z (k/z 1/2) where k is the oligomer number of Ac-Tau(273–284)-NH2. Peaks at higher m/z are not observed, consistent with the fact that peptide oligomerization occurs at a much slower rate compared to the same peptide in buffer solution.30 The dimer is the largest oligomer observed (the ATD of the peak at 705 m/z is shown in the inset of Figure 2B). In the absence of aggregation-promoting factors (e.g., heparin), this peptide is unable to form any structural aggregates.30,39 Furthermore, previous simulations and experimental cross-sections (σexp = 310–313 Å2 for z = +2 and +3) suggest that the Tau monomer is in a relatively compact conformation (i.e., β-turn or β-hairpin). Simulations suggest that aggregation is controlled by a conformational transition of the monomers which later associate to form extended dimers.30
The mass spectrum of the mixture (Figure 2C) shows strong peaks for Ac-Tau(273–284)-NH2 monomer, but the peaks associated with Ac-Aβ(25–35)-NH2 are strongly suppressed, replaced by several new peaks composed of peptides from both fragments. These mass spectral peaks are annotated with [n + k]/z where n is the oligomer number of Ac-Aβ(25–35)-NH2, k is the oligomer number of Ac-Tau(273–284)-NH2, and z is the net charge of the complex. Cross-sections for the mixed aggregate peaks were determined by their ATDs. The experimental cross-sections are accurate to 1%.44 Figure 3B qualitatively represents the correlated cross-section of Ac-Aβ(25–35)-NH2 monomer and oligomers extracted from the experimental cross-section of the heteroligomers (see Supporting Information section S2.1 and Table S1),32 assuming that the Tau conformation is relatively compact (as indicated from simulation30). We note that σcorr (n) can be considered as representative values for the ratios σexp(n,k)/σiso(n,k) between the experimental cross-sections of heteroligomers and the isotropic cross-section model of heteroligomers. Hence if σexp(n,k) ≫ σiso(n,k), we expect a large deviation of σcorr(n) from the isotropic cross-section of Aβ(25–35) oligomer, σiso(n), indicating that the heteroligomers are nonisotropic.
Under this assumption, we observe that the cross-section deviation occurs as early as heterodimers (n = 1, k = 1) and the trend continues for larger oligomers (n = 3, k = 2 and n = 2, k = 3). The observation indicates that these heteroligomers adopt extended conformations.56 This conclusion is generally true regardless of our assumption that the Tau conformation is compact. However, the largest heteroligomers observed in this IM-MS experiment are hexamers, whereas, in the pure Ac-Aβ(25–35)-NH2 sample, hexamers and nonamers can be unambiguously detected. Thus, it appears that Ac-Tau(273–284)-NH2 can trap Ac-Aβ(25–35)-NH2 in smaller oligomeric forms.
The significant intensities of the heterodimer peaks (i.e., [1 + 1]/4 at 627 m/z and [1 + 1]/3 at 837 m/z) indicate that the Tau fragment has a high affinity to bind Ac-Aβ(25–35)-NH2 monomer relative to forming homo-Tau dimers (Kd in micromolar range; see Supporting Information section S2.2).
T-REMD simulation57 of the heterodimer reveals the existence of at least two major families of structures: compact structures (∼40%, in which both Tau and Aβ peptide chains are relatively compact) and extended structures (∼10%, where both chains are extended in antiparallel configuration) and other structures in between (see Supporting Information Figure S3).
In terms of cross-sections, two populated clusters of extended antiparallel heterodimers have average cross-sections of σtheory = 553 and 525 Å2 (clusters F and H shown in Supporting Information Figure S3, respectively), which are in good agreement with the [1 + 1]/4 species with the cross-section of σexp = 559 Å2. Similarly, the most populated compact heterodimer clusters have average cross-sections of σtheory = 496 and 493 Å2 (clusters A and B, respectively), which agree well with the [1 + 1]/3 species, σexp = 492 Å2. We note that the theoretical cross-section obtained from the trajectory method available from the Mobcal package,54,55 from a specific structure, is good to less than 5%.58,59
Figure 4A shows a schematic representation of the alignment of two Tau(273–284) peptides, a mixed Tau(273–284)/Aβ(25–35), and two Aβ(25–35) peptides within a single-layer ideal β-sheet. In the context of a fragment such as Tau(273–284) or full length Tau, the hydrophobic PHF6* region is blocked between K274 at one end and K280/281 at the other end, making associations between the hydrophobic PHF6* cores more restricted. In the Ac-Tau(273–284)-NH2 extended dimer conformation probed in the simulation of Larini et al.,30 the two PHF6* segments are shifted away from each other. The extended dimer is stabilized by a weak hydrogen bonding network between backbone atoms of hydrophobic residues such as isoleucine and hydrophilic charged residues such as lysine, consistent with the scheme shown in Figure 4A.
Figure 4.
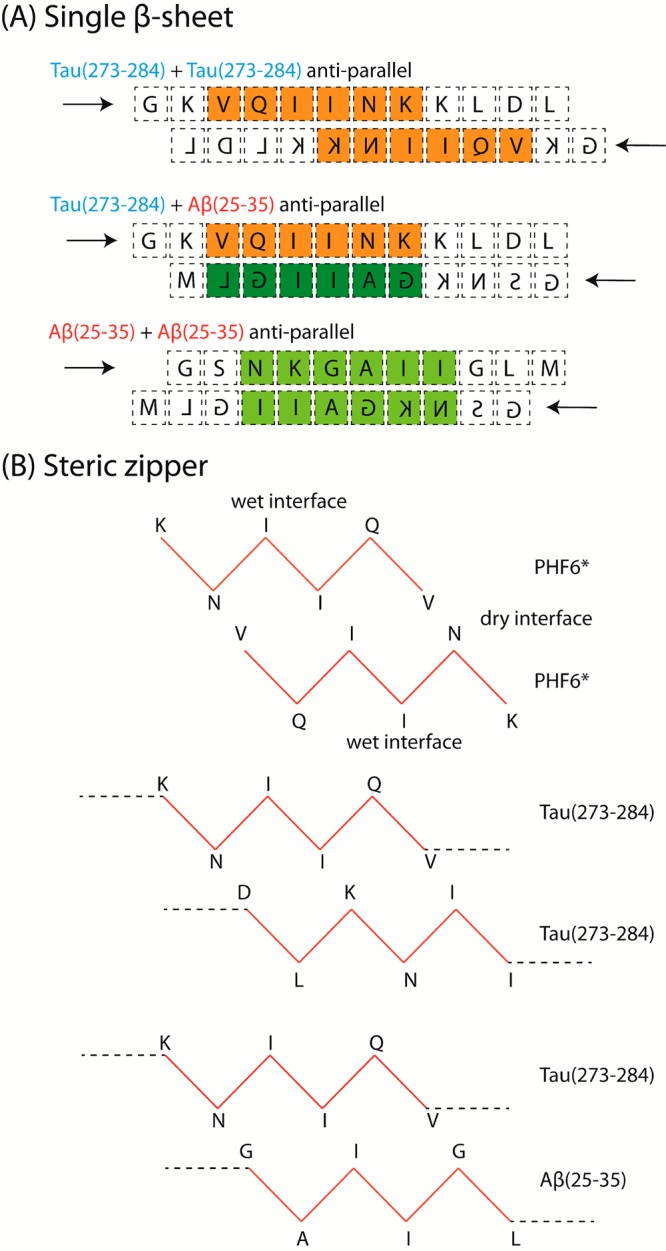
Proposed sequence alignments of the peptides in (A) single extended β-sheet and (B) steric zipper conformation.
Since the two PHF6* segments are not within an optimal distance to create a dry interface, the extended dimer structure does not support steric zipper formation optimal for fibrillization.46 On the other hand, the GAIIGL segment of Ac-Aβ(25–35)-NH2 can behave similarly to PHF6*, and the adjacent residues around this segment are less charged than those of Tau. In addition, since Ac-Aβ(25–35)-NH2 is one residue shorter than Ac-Tau(273–284)-NH2 and the GAIIGL segment is near the C-terminus, Figure 4A also proposes that a better alignment between GAIIGL and PHF6* can be achieved in antiparallel fashion.
Simulations suggest that both the homodimer of Tau(273–284) and of Aβ(25–35) and the heterodimer of Tau(273–284) and Aβ(25–35) can adopt antiparallel, extended structures with the hydrogen bonding networks shown in Figure 4A. The distributions of distances between hydrophobic cores (between one PHF6* and another PHF6* in homo-Tau(273–284) dimer, and between PHF6* and GAIIGL in the heterodimer) suggest that the hydrophobic cores in the heterodimer can interact more strongly (i.e., within a 5.0 Å distance) than PHF6* in the homo-Tau(273–284) dimer (see Supporting Information Figure S2 and Figure 5C).
Figure 5.
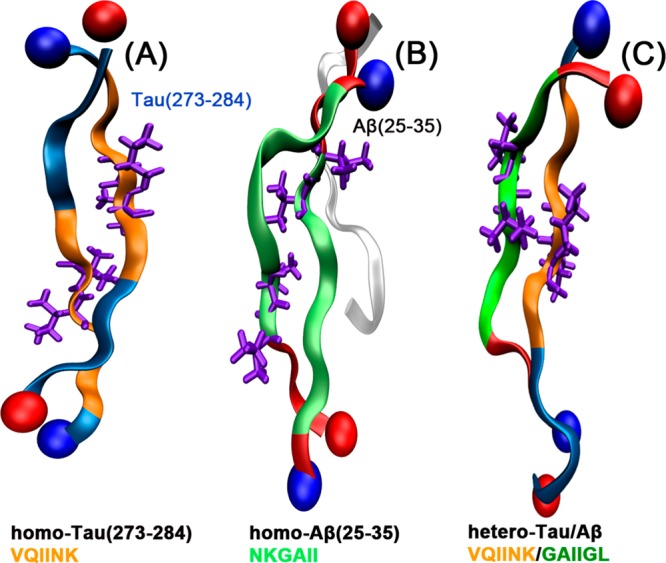
Representative structures of the most populated extended clusters obtained from the simulations of (A) blocked terminal homo-Tau(273–284) dimers (see ref (30)), (B) free terminal homo- Aβ(25–35) trimers (see ref (31)) and, (C) blocked terminal hetero-Tau(273–284)/Aβ(25–35) dimers. The interacting hydrophobic cores are highlighted using the same color codes shown in Figure 4. The isoleucine pairs within the hydrophobic stretches are shown in licorice.
According to Figure 4B, the interactions between two β-sheets within a heterodimer steric zipper maintains interactions at the dry interface similar to when PHF6* is considered alone, with no charged residues in the dry interface. The homo-Tau(273–284) steric zipper, on the other hand, is much less stable, since it has to accommodate charged residues such as lysine and aspartic acid within the dry interface and hydrophobic residues such as isoleucine and leucine at the wet interface. Therefore, it is reasonable to believe that interactions between Ac-Aβ(25–35)-NH2 and Ac-Tau(273–284)-NH2 can promote β-rich oligomers better than when Ac-Tau(273–284)-NH2 is incubated separately.
As previously examined from both experiment and theory,31,32 the aggregation process of Aβ(25–35) involves a structural transition from dimer to trimer stages, in which a compact dimer composed of mainly β-hairpin monomers converts into extended β-rich structures, as shown in Figure 5B. In these populated β-sheet oligomers (trimer or larger) the hydrophobic cores are NKGAII whose locations are more toward the middle of the chains, providing access to steric zipper formation, and eventually fibril formation. NKGAII β-sheets can interact in both face-to-face and back-to-back fashions (Protein Data Bank (PDB) ID 3Q2X).42 Due to the versatility of these interaction motifs, it is likely that these NKGAII segments can provide a strong steric zipper interface for Aβ(25–35) to quickly aggregate into fibrils.
The GAIIGL steric zipper (PDB ID 3PZZ) is composed of antiparallel β-sheets interacting face-to-back. This class 6 zipper43 is often found in weakly aggregating systems such as the islet amyloid polypeptide fragment NFLVHSS (PDB ID 3FTH),60 and the Enkephalin mutants YVVFV (4OLR) and YVVFL (4ONK).61 Thus, we can predict that the heterosystem of Aβ(25–35) and Tau(273–284) stabilized by PHF6*/GAIIGL interactions would self-assemble with slower kinetics into a different aggregate morphology as compared to pure Aβ(25–35).
Heteroligomers Composed of Aβ(25-35) Monomer and Oligomers but Not of Tau Oligomers Larger than Dimer
In Figure 2C, two types of heteroligomers larger than dimers are observed: (1) those with more Aβ than Tau peptide chains, which will be referred to as hetero-Aβ oligomers and (2) those with more Tau than Aβ, referred to as hetero-Tau oligomers.
The hetero-Aβ oligomer peaks (i.e., [2 + 1]/4 at 903 m/z, [3 + 2]/7 at 875 m/z, and [3 + 2]/6 at 1016 m/z) are abundant in intensity indicating that Ac-Tau(273–284)-NH2 can effectively replace Ac-Aβ(25–35)-NH2 peptide chains to form dimer, trimer, and pentamer, and such processes are favorable. Similar hetero-Tau oligomers are also observed (i.e., [1 + 2]/4 at 979 m/z and [2 + 3]/5 at 1280 m/z). At first glance, the compositions of these hetero-Tau oligomers appear to be similar to those of hetero-Aβ oligomers, which may suggest that the Aβ and Tau peptides can randomly associate to form large oligomers. However, as seen in Figure 6A,C, the ATDs of hetero-Aβ oligomers (i.e., trimer and pentamer) present multiple features and a variety of compact and extended conformations with similar intensities. On the other hand, the ATDs of hetero-Tau oligomers contain a dominant feature with few minor features (Figure 6B,D). A possible explanation is that since homoligomers of Ac-Tau(273–284)-NH2 larger than dimers are not stable and populated in water, the number of possible arrangements that hetero-Tau oligomers can adopt should always be less than that of hetero-Aβ oligomers.
Figure 6.
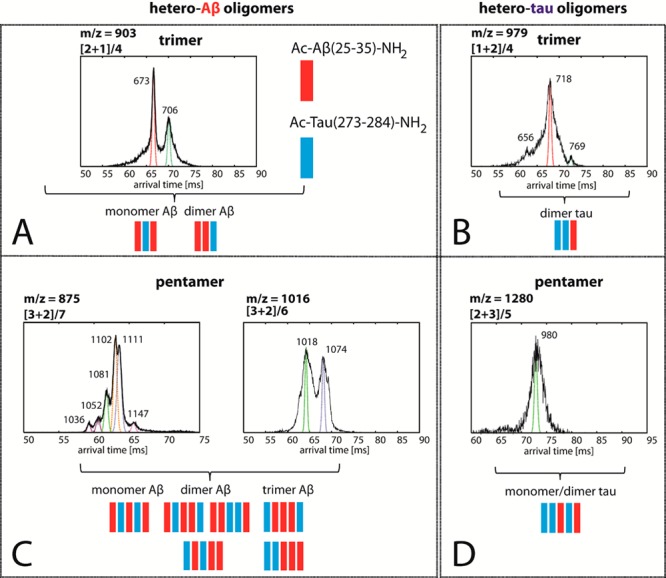
Representative ATDs of hetero-Aβ and hetero-Tau trimer (A,B) and pentamer (C,D) peaks. Each feature is annotated with an experimental cross-section (σ, Å2). Possible configurations of the heteroligomers are shown with Ac-Aβ(25–35)-NH2 in red and Ac-Tau(273–284)-NH2 in blue.
The hetero-Aβ trimer ATD has two peaks with cross-sections of σexp = 673 and 706 Å2 which mechanistically can be assigned to a trimer composed of two Aβ monomers and a Tau monomer intercalating in between (i.e., ABA) and a trimer of an Aβ dimer and a Tau monomer (i.e., AAB configuration where A is an Ac-Aβ(25–35)-NH2 and B is an Ac-Tau(273–284)-NH2 monomer). It is worth noting that if both ABA and AAB adopt similar conformations (i.e., extended structures) then the cross-section σ(AAB) should be larger than that of σ(ABA), since the former structure would have more Tau side chains exposed to the outside (Tau is larger in size than Aβ), which increases the collision cross-section. Both features are populated since the Aβ monomer and dimer in the sample are abundant (see Supporting Information Figure S1) . On the other hand, the hetero-Tau trimer has three features, but the shortest and longest arrival time features are very minor. Statistically the BBA/ABB structures are more likely than the BAB structures (i.e., the BBA/ABB can be formed starting with either AB heterodimer or BB homodimer, whereas the BAB can only be formed from the heterodimer) suggesting this is the structure of the major peak, but this cannot be determined with certainty.
Global stabilization of each quaternary structure often involves not only strong pairwise interactions between adjacent chains but also a network of noncovalent contacts among different subunits. Consequently, the preceding mechanistic description of heteroligomers appears to be too simple. However, the T-REMD simulations of hetero-Aβ and hetero-Tau trimers qualitatively support such a description. As seen in Figure 7A, the radial distribution function (RDF g(r)) computed for the distances between the Aβ monomers within the hetero-Aβ trimers populates several peaks near 0.5 and 1.0 nm. The distance of 0.5 nm indicates that the two Ac-Aβ(25–35)-NH2 peptides are adjacent to each other (AAB conformations), whereas that of 1.0 nm suggests the Tau peptide is intercalating in between (ABA conformations). Clustering analysis shows that the conformation of each chain within a structure type can be either compact or extended, which gives rise to a diverse set of conformations. The representative structures and the theoretical cross-sections of the major clusters obtained from the simulation of hetero-Aβ trimer are shown in Supporting Information Figure S4. While the Tau monomer is relatively extended, the two Ac-Aβ(25–35)-NH2 chains can adopt either β-hairpin-like or extended structures. Although the largest experimental cross-section of the hetero-Aβ trimer is ∼10% larger than the reported theoretical cross-sections, if we consider the broad distributions of theoretical cross-sections, the theory and experiment are in good agreement. Some oligomers may be kinetically but not thermodynamically stable. Kinetically stable oligomers have been observed and discussed in our recent study of free terminal Aβ(25–35) aggregation.32
Figure 7.
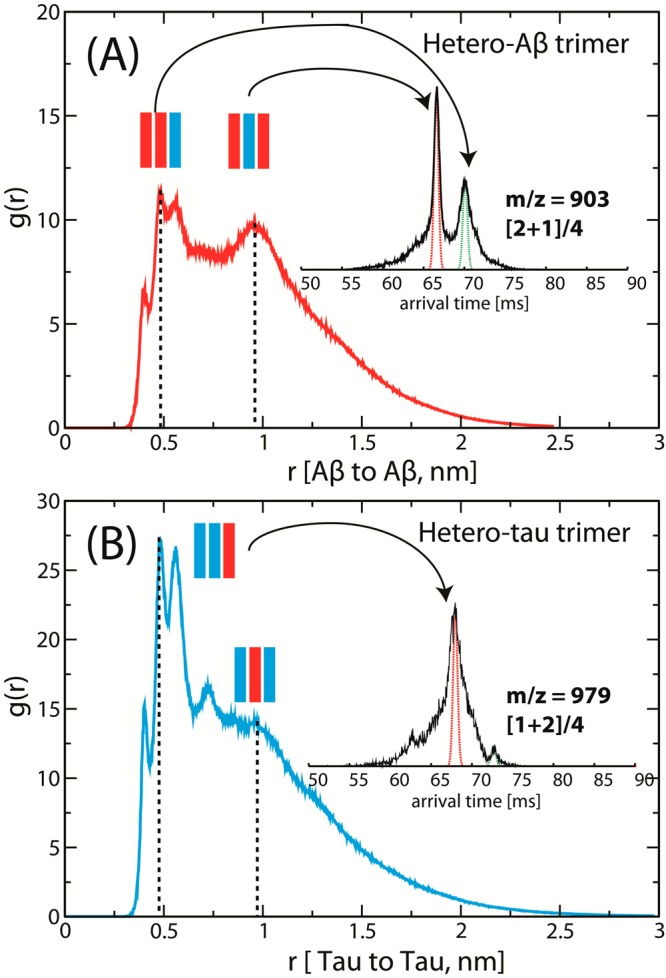
Radial distribution functions RDFs g(r) computed for the distances r (nm) (A) between the Ac-Aβ(25–35)-NH2 monomers within the hetero-Aβ trimer and (B) between the Ac-Tau(273–284)-NH2 monomers within the hetero-Tau trimer.
For the hetero-Tau trimer, all of the dominant peaks of g(r) are located near 0.5 nm, suggesting that the BBA conformations are dominant, in agreement with the assignment made in Figure 6B. Clustering analysis of the hetero-Tau trimers obtained from T-REMD reveals that a majority of extended structures is of BBA type. Some compact, disordered structures are also observed (see Supporting Information Figure S5).
As the size of the heteroligomers grows, a clear distinction between hetero-Aβ and hetero-Tau oligomers is observed in the IM-MS data. For the heteropentamer, those of Aβ contain multiple features in the ATDs corresponding to different species (i.e., there are six possible configurations of hetero-Aβ pentamer). Since the size of the systems is expensive for high-level MD simulations, we constructed a series of fully extended hetero-Aβ pentamers corresponding to the configurations shown schematically in Figure 6 (see Supporting Information Figure S6 for the structures obtained from simulation and their theoretical cross-sections). These antiparallel β-sheet models have cross-sections in good agreement with the experiment cross-sections for both charge states (z = +6 and +7), suggesting the hetero-Aβ pentamers adopt extended, β-sheet-like structures.
In contrast, the formation of the single hetero-Tau pentamer is conformationally restricted, as manifested in the ATD containing only one narrow feature. This feature has a smaller cross-section and lower charge state than the hetero-Aβ pentamers, suggesting that the conformation is relatively more compact.
Ac-Tau(273–284)-NH2 Acting as an Inhibitor That Drives Ac-Aβ(25–35)-NH2 Self-Assembly into Both Granular and Heterofibrillar Aggregates
Both the IM-MS data and the T-REMD simulations support extended conformations of both Ac-Aβ(25–35)-NH2 and Ac-Tau(273–284)-NH2 as early as the heterodimer stage. Figure 8 shows the differences in quantities and morphologies of aggregates in the pure Aβ(25–35), Tau(273–284), and mixture samples. From these data, we conclude that the formation of heteroligomers does not lead to enhanced fibril formation. The behavior of Ac-Tau(273–284)-NH2 is similar to an inhibitor affecting the aggregation process of Ac-Aβ(25–35)-NH2, in which Tau monomer (and some dimer) competitively binds to Ac-Aβ(25–35)-NH2 monomer and oligomers. For the sample of pure Ac-Aβ(25–35)-NH2, abundant fibrillar aggregates are observed (Figure 8A and Supporting Information Figure S7C–F), whereas less fibrils but more granular aggregates are seen in the mixture sample (Figure 8B) and finally no aggregates are found in the pure Tau(273–284) sample (Figure 8C). This raises two questions:
Figure 8.
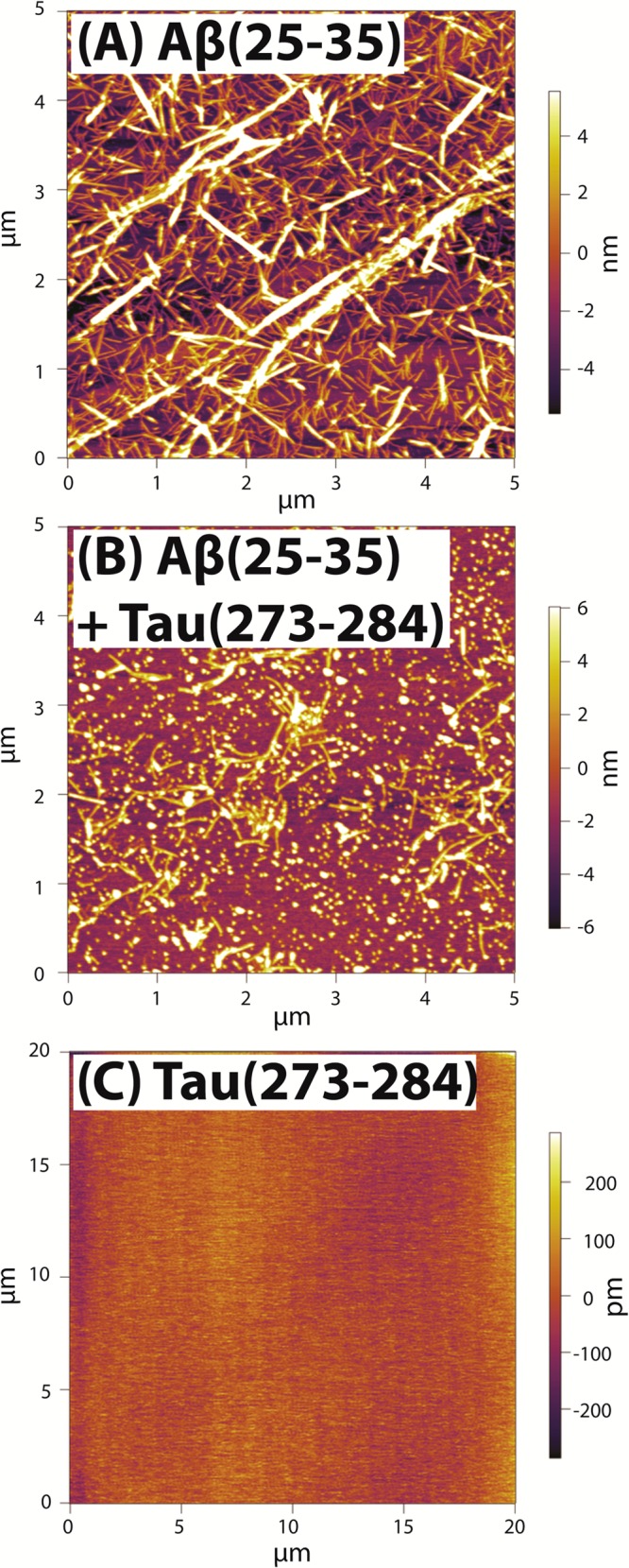
Representative AFM images of (A, B) 1 week incubated 200 μM Ac-Aβ(25–35)-NH2 in water without Ac-Tau(273–284)-NH2 and with Ac-Tau(273–284)-NH2 at the ratio of 1:1 and (C) 2 week incubated 200 μM Ac-Tau(273–284)-NH2 in water.
(1) What is the nature of the granular aggregates and fibril-like structures observed in the AFM image of the mixture?
(2) Through which mechanisms does Ac-Tau(273–284)-NH2 limit the strong aggregation of Ac-Aβ(25–35)-NH2 and why do the extended conformations of heteroligomers, observed by experiment and by theoretical modeling, not promote abundant formation of fibrils?
To answer the first question, we consider the following possibilities. The granular aggregates can be either (a) homo-Tau aggregates, (b) early globular aggregates of homo-Aβ(25–35), or (c) a type of new aggregate produced by the heteroligomers. Granular aggregates have been observed as intermediates in some Tau fragment aggregation62 but not specifically for Ac-Tau(273–284)-NH2 in the absence of aggregation-promoting factor (see Figure 8C). In the presence of 11 kDa polydisperse heparin at 4:1 peptide:heparin ratio, aggregation of Ac-Tau(273–284)-NH2 in water occurs within 1 h to form abundant well-defined aggregates (see Supporting Information Figure S7A,B) and no granular aggregates are detected. Thus, the granular aggregates observed in the mixture sample should not be from the Tau itself; this eliminates option a of homo-Tau oligomers.
We now consider option b: homo-Aβ oligomers. We note that, in our experiment, fresh Ac-Aβ(25–35)-NH2 forms large, β-rich oligomers within a short time after sample preparation and forms fibrillar aggregates within an hour (Supporting Information Figure S7C,D), suggesting that there is not a noticeable morphology transition occurring during the aggregation of Ac-Aβ(25–35)-NH2. A few small aggregates are sparsely distributed at the edges of the AFM slide (Supporting Information Figure S7G,H). However, these aggregates vary widely in length and have on average a lower height than the granular aggregates observed in the mixture sample which suggests a different composition (see Table 1). We consider the possibility that Ac-Tau(273–284)-NH2 acts as an inhibitor to efficiently disassemble Ac-Aβ(25–35)-NH2 fibrils into globular aggregates. In other words, the interactions between Ac-Tau(273–284)-NH2 and Ac-Aβ(25–35)-NH2 must be significantly stronger than Aβ(25–35) with itself in order to successfully compete with existing interactions between Aβ chains inside the fibrils. To examine whether Ac-Tau(273–284)-NH2 can disaggregate Ac-Aβ(25–35)-NH2 fibrils, fresh Tau was added to a week-old sample containing extensive fibrillar aggregates of Ac-Aβ(25–35)-NH2. No decrease in fibrillar aggregate abundance was observed. From the data, it is clear that Ac-Tau(273–284)-NH2 does not disassemble Ac-Aβ(25–35)-NH2 aggregates, which eliminates b as an option (see Supporting Information Figure S8).
Table 1. Dimension (Height (nm) and Length (nm)) of the Aggregates Observed by AFM for the Samples of Pure Ac-Aβ(25-35)-NH2 and 1:1 Mixture of Ac-Aβ(25-35)-NH2 and Ac-Tau(273-284)-NH2 at 200 μM.
Finally, we turn to option c, the possibility of forming mixed aggregates. As seen in IM-MS and in simulations, Ac-Tau(273–284)-NH2 and Ac-Aβ(25–35)-NH2 can interact. It is hence reasonable to assume that Ac-Tau(273–284)-NH2 can decorate existing Ac-Aβ(25–35)-NH2 oligomers and protofibrils by occasionally adding on the growing aggregate in place of new Aβ chains. The more Aβ that are replaced by Tau, the less prone the oligomers are to grow further since there are structural differences between hetero-Aβ and hetero-Tau oligomers. Therefore, option c is the most probable. Compact hetero-Tau and some hetero-Aβ oligomers can trap Ac-Aβ(25–35)-NH2 oligomers in β-hairpin or β-turn conformations, leading to the formation of granular aggregates. Since Ac-Tau(273–284)-NH2 monomer is larger than Ac-Aβ(25–35)-NH2 monomer in size, the fibrils grown from heteroligomers would be more likely to have larger dimensions than the pure Ac-Aβ(25–35)-NH2 fibrils. Additional AFM measurements show that both heights and lengths of the fibrils and granular features formed in the mixture sample are greater than those of the pure Ac-Aβ(25–35)-NH2 (see Table 1). In these measurements, only the dimension of individual fibrils is taken into account, and the larger features which are probably clusters of several fibrils are not considered.
For the second question, we first consider the mechanisms through which Aβ(25–35) can aggregate. The simulations of Ac-Aβ(25–35)-NH2 by Wei et al.28 show the existence of a V-shaped parallel dimer, an out of register antiparallel dimer, and an intertwined β-hairpin conformation at small populations as potential seeds for amyloid formation. The major driving force for the first two families of structures is hydrophobic interactions between GAIIGL segments. Larini and Shea,31 in simulations of Aβ(25–35), suggest that the transition from β-hairpin Aβ(25–35) monomer to extended structures of oligomers is regulated by a competition between electrostatics and hydrophobic effects. Electrostatics dominate for dimer formation to produce mainly compact structures composed of two tilted β-hairpin monomers, whereas hydrophobic interactions facilitate the β-sheet trimers and tetramers. They also propose the role of β-hairpin structure in a kinetic growth model as a means to stabilize flat β-sheets and prevent newly formed oligomers from hydrophobic collapse. When Ac-Tau(273–284)-NH2 and Ac-Aβ(25–35)-NH2 form extended dimers, or Ac-Tau(273–284)-NH2 intercalates between Ac-Aβ(25–35)-NH2 hairpins at the trimer or pentamer, Tau limits the influences of β-hairpin structure in the growth of Aβ(25–35) fibrils and subsequently decreases the aggregation propensity. By replacing the hydrophobic core of NKGAII/NKGAII with PHF6*/GAIIGL, and limiting the role of Aβ(25–35) hairpins, Ac-Tau(273–284)-NH2 decreases the aggregation propensity of Ac-Aβ(25–35)-NH2. The hetero-Aβ oligomers are relatively extended, but not as aggregation-prone as the homo-Aβ oligomers, due to the mismatches in side-chain interdigitations and their heterogeneity. The presence of a few Tau chains within the heterocomplexes may decrease the ability to self-associate, but the system is still able to grow further into fibrils, which is consistent with what has been observed in the AFM image of the mixture (Figure 8B).
It has been long predicted that heterotypic β-sheet and steric zipper structures can exist. Eisenberg and co-workers have shown that the macroscopic morphologies and abundances of fibrillar aggregates formed in binary mixtures of different Aβ segments are distinct from the component segments.42 However, no X-ray crystal structures of heterotypic β-sheet and steric zipper have been solved. One of the possible explanations is that even in a binary system of two short peptides, the tendency of homotypic structure formation is dominant, and crystallization is highly dependent upon the kinetics and growth conditions which are very selective. The two factors often promote the crystallization of one type or the other, but not heterotypic crystals. Thus, IM-MS appears to be a versatile technique to approach this problem. Previous work has shown that the experimental cross-sections of aggregating peptide oligomers are in good agreement with X-ray crystal structures.56 Furthermore, even a small amount of heteroligomers can be detected with this technique.
Summary and Conclusions
(1) Using a combination of experimental and theoretical techniques, we are able to identify the interactions promoting and stabilizing Ac-Aβ(25–35)-NH2/Ac-Tau(273–284)-NH2 heteroligomers. The formation of heteroligomers triggers conformation transitions from compact to extended forms for both peptides as early as the dimer stage. Ac-Tau(273–284)-NH2 monomer prefers to associate with an Ac-Aβ(25–35)-NH2 monomer over another Tau monomer as shown by the populations of heterodimer and homo-Tau dimer in the mixture. The intercalation of Ac-Tau(273–284)-NH2 monomers between Ac-Aβ(25–35)-NH2 monomers (and oligomers) is a probable mechanism allowing large heteroligomers to grow with a wide range of configurations and conformations.
(2) The heterogeneity of the heteroligomers and mismatches in side-chain interdigitations limit (but do not fully eliminate) aggregation. The hetero-Tau oligomers can be trapped at relatively small sizes, producing granular aggregates. On the other hand, the architectures of hetero-Aβ oligomers contain β-rich content, which later can grow into heterofibrils, as illustrated in Figure 9.
Figure 9.
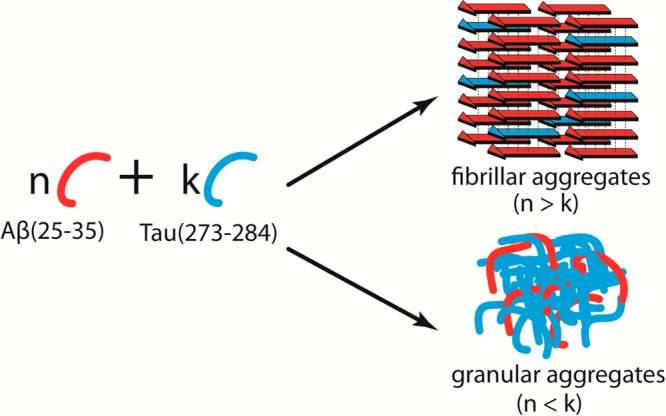
Illustration of how fibrillar and granular aggregates can be formed in the mixture of Ac-Aβ(25–35)-NH2 and Ac-Tau(273–284)-NH2. The morphologies of the aggregates are determined by the population of the individual peptides.
(3) A possible source of toxicity of early Aβ oligomers may lie in their ability to bind Tau monomers, thereby decreasing the population of Tau needed to regulate microtubule dynamics. Other possible pathways to toxicity lie in the structures of the heteroligomers formed by the interactions of different Tau to Aβ fragments, which awaits a further systematic investigation.
(4) IM-MS can probe conformations and abundances of heteroligomers, demonstrating its capacity as a promising screening method to identify important segments from different aggregating proteins (e.g., Aβ, Tau, prion, islet amyloid polypeptide) that can interact with each other to form heterotypic structures. These studies will provide insights into synergy effects of protein markers responsible for aggregation-related diseases, which are currently challenging to unravel.
Acknowledgments
We thank Ms. Margaret Condron and Prof. David B. Teplow for supplying the Ac-Aβ(25-35)-NH2 peptide. T.D.D. thanks Dr. Nichole E. LaPointe and Ms. Kaitlin McDermott for useful discussion. We gratefully acknowledge support from the National Science Foundation Grant CHE-1301032 (M.T.B.) for personnel support, and the National Institutes of Health Grant 1RO1AG047116-01 (M.T.B.) for material support and partial personal support. N.J.E. acknowledges the National Science Foundation Graduate Student Research Fellowship. S.K.B. acknowledges funding from the MURI and DURIP programs of the U.S. Army Research Laboratory and U.S. Army Research Office under Grant Nos. DAAD 19-03-1-0121 and W911NF-09-1-0280 for purchase of the AFM instrument. We acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing HPC resources through the XSEDE Grant No. TG-MCA05S027 (J.-E.S.) and also support from the Center for Scientific Computing at the CNSI and MRL: NSF MRSEC (Grant DMR- 1121053) and NSF Grant CNS-0960316.
Supporting Information Available
Text giving full descriptions of experimental and theoretical protocols and accompanying references, additional figures from the analysis of T-REMD simulation data, additional AFM images, and ATDs of pure Ac-Aβ(25-35)-NH2 and pure Ac-Tau(273-284)-NH2, and a table listing collision cross-sections for oligomers of Aβ(25-35). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Buee L.; Bussiere T.; Buee-Scherrer V.; Delacourte A.; Hof P. R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Rev. 2000, 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Binder L. I.; Guillozet-Bongaarts A. L.; Garcia-Sierra F.; Berry R. W. Tau, Tangles, and Alzheimer’s Disease. Biochim. Biophys. Acta 2005, 1739, 216–223. [DOI] [PubMed] [Google Scholar]
- Wolfe M. S. Tau Mutations in Neurodegenerative Diseases. J. Biol. Chem. 2009, 284, 6021–6025. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G. Tau Mutations in Frontotemporal Dementia Ftdp-17 and Their Relevance for Alzheimer’s Disease. Biochim. Biophys. Acta 2000, 1502, 110–121. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J.-E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem. 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Liu C.; Sawaya M. R.; Whitelegge J. P.; Park J.; Zhao M. L.; Pensalfini A.; Soriaga A. B.; Landau M.; Teng P. K.; Cascio D.; Glabe C.; Eisenberg D. Atomic View of a Toxic Amyloid Small Oligomer. Science 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J.; Chen F.; van Dorpe J.; Nitsch R. M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Aβ 42 Fibrils. Science 2001, 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Lee H. G.; Zhu X. W.; Castellani R. J.; Nunomura A.; Perry G.; Smith M. A. Amyloid-β in Alzheimer Disease: The Null Versus the Alternate Hypotheses. J. Pharmacol. Exp. Ther. 2007, 321, 823–829. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P.; Herrmann F. R.; Bussiere T.; Bouras C.; Kovari E.; Perl D. P.; Morrison J. H.; Gold G.; Hof P. R. Tangle and Neuron Numbers, but Not Amyloid Load, Predict Cognitive Status in Alzheimer’s Disease. Neurology 2003, 60, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Williams T. L.; Serpell L. C. Membrane and Surface Interactions of Alzheimer’s Aβ Peptide—Insights into the Mechanism of Cytotoxicity. FEBS J. 2011, 278, 3905–3917. [DOI] [PubMed] [Google Scholar]
- Bokvist M.; Lindstrom F.; Watts A.; Grobner G. Two Types of Alzheimer’s β-Amyloid (1–40) Peptide Membrane Interactions: Aggregation Preventing Transmembrane Anchoring Versus Accelerated Surface Fibril Formation. J. Mol. Biol. 2004, 335, 1039–1049. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M.; Green K. N.; Oddo S. Intracellular Amyloid-β in Alzheimer’s Disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [DOI] [PubMed] [Google Scholar]
- Olzscha H.; Schermann S. M.; Woerner A. C.; Pinkert S.; Hecht M. H.; Tartaglia G. G.; Vendruscolo M.; Hayer-Hartl M.; Hartl F. U.; Vabulas R. M. Amyloid-Like Aggregates Sequester Numerous Metastable Proteins with Essential Cellular Functions. Cell 2011, 144, 67–78. [DOI] [PubMed] [Google Scholar]
- King M. E.; Kan H. M.; Baas P. W.; Erisir A.; Glabe C. G.; Bloom G. S. Tau-Dependent Microtubule Disassembly Initiated by Prefibrillar β-Amyloid. J. Cell Biol. 2006, 175, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J.; Dickson D. W.; Lin W. L.; Chisholm L.; Corral A.; Jones G.; Yen S. H.; Sahara N.; Skipper L.; Yager D.; Eckman C.; Hardy J.; Hutton M.; McGowan E. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. Soluble Amyloid β-Protein Dimers Isolated from Alzheimer Cortex Directly Induce Tau Hyperphosphorylation and Neuritic Degeneration. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H.; Luedtke J.; Kumar Y.; Biernat J.; Dawson H.; Mandelkow E.; Mandelkow E. M. Amyloid-β Oligomers Induce Synaptic Damage Via Tau-Dependent Microtubule Severing by TTLL6 and Spastin. EMBO J. 2013, 32, 2920–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W. H.; Bastianetto S.; Mennicken F.; Ma W.; Kar S. Amyloid-β Peptide Induces Tau Phosphorylation and Loss of Cholinergic Neurons in Rat Primary Septal Cultures. Neuroscience 2002, 115, 201–211. [DOI] [PubMed] [Google Scholar]
- Ma Q. L.; Yang F.; Rosario E. R.; Ubeda O. J.; Beech W.; Gant D. J.; Chen P. P.; Hudspeth B.; Chen C.; Zhao Y.; Vinters H. V.; Frautschy S. A.; Cole G. M. β-Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate Via C-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. J. Neurosci. 2009, 29, 9078–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokutake T.; Kasuga K.; Yajima R.; Sekine Y.; Tezuka T.; Nishizawa M.; Ikeuchi T. Hyperphosphorylation of Tau Induced by Naturally Secreted Amyloid-B at Nanomolar Concentrations Is Modulated by Insulin-Dependent AKT-GSK3β Signaling Pathway. J. Biol. Chem. 2012, 287, 35222–35233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner L. M.; Gotz J. Amyloid-Beta and Tau—A Toxic Pas De Deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Guo J.-P.; Arai T.; Miklossy J.; McGeer P. L. Aβ and Tau Form Soluble Complexes That May Promote Self Aggregation of Both into the Insoluble Forms Observed in Alzheimer’s Disease. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M.; Dawson H. N.; Binder L. I.; Vitek M. P.; Ferreira A. Tau Is Essential to β-Amyloid-Induced Neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J.; Lorenzo A.; Yeh J.; Yankner B. A. β-Amyloid Fibrils Induce Tau-Phosphorylation and Loss of Microtubule-Binding. Neuron 1995, 14, 879–888. [DOI] [PubMed] [Google Scholar]
- Ferreira A.; Lu Q.; Orecchio L.; Kosik K. S. Selective Phosphorylation of Adult Tau Isoforms in Mature Hippocampal Neurons Exposed to Fibrillar Aβ. Mol. Cell. Neurosci. 1997, 9, 220–234. [DOI] [PubMed] [Google Scholar]
- Takashima A.; Honda T.; Yasutake K.; Michel G.; Murayama O.; Murayama M.; Ishiguro K.; Yamaguchi H. Activation of Tau Protein Kinase I/Glycogen Synthase Kinase-3beta by Amyloid-β Peptide (25–35) Enhances Phosphorylation of Tau in Hippocampal Neurons. Neurosci. Res. 1998, 31, 317–323. [DOI] [PubMed] [Google Scholar]
- Wei G.; Jewett A. I.; Shea J. E. Structural Diversity of Dimers of the Alzheimer Amyloid-β(25–35) Peptide and Polymorphism of the Resulting Fibrils. Phys. Chem. Chem. Phys. 2010, 12, 3622–3629. [DOI] [PubMed] [Google Scholar]
- Wei G. H.; Shea J. E. Effects of Solvent on the Structure of the Alzheimer Amyloid-β (25–35) Peptide. Biophys. J. 2006, 91, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larini L.; Gessel M. M.; Lapointe N. E.; Do T. D.; Bowers M. T.; Feinstein S. C.; Shea J. E. Initiation of Assembly of Tau(273–284) and Its ΔK280 Mutant: An Experimental and Computational Study. Phys. Chem. Chem. Phys. 2013, 15, 8916–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larini L.; Shea J. E. Role of B-Hairpin Formation in Aggregation: The Self-Assembly of the Amyloid-β (25–35) Peptide. Biophys. J. 2012, 103, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleiholder C.; Do T. D.; Wu C.; Economou N. J.; Bernstein S. S.; Buratto S. K.; Shea J.-E.; Bowers M. T. Ion Mobility Spectrometry Reveals the Mechanism of Amyloid Formation of Aβ(25–35) and Its Modulation by Inhibitors at the Molecular Level: Epigallocatechin Gallate and Scyllo-Inositol. J. Am. Chem. Soc. 2013, 135, 16926–16937. [DOI] [PubMed] [Google Scholar]
- Kittner M.; Knecht V. Disordered Versus Fibril-Like Amyloid-β (25–35) Dimers in Water: Structure and Thermodynamics. J. Phys. Chem. B 2010, 114, 15288–15295. [DOI] [PubMed] [Google Scholar]
- Clementi M. E.; Marini S.; Coletta M.; Orsini F.; Giardina B.; Misiti F. Aβ(31–35) and Aβ(25–35) Fragments of Amyloid Beta-Protein Induce Cellular Death through Apoptotic Signals: Role of the Redox State of Methionine-35. FEBS Lett. 2005, 579, 2913–2918. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Walencewiczwasserman A. J.; Kosmoski J.; Cribbs D. H.; Glabe C. G.; Cotman C. W. Structure-Activity Analyses of β-Amyloid Peptides - Contributions of the Beta-25–35 Region to Aggregation and Neurotoxicity. J. Neurochem. 1995, 64, 253–265. [DOI] [PubMed] [Google Scholar]
- Moore C. L.; Huang M. H.; Robbennolt S. A.; Voss K. R.; Combs B.; Gamblin T. C.; Goux W. J. Secondary Nucleating Sequences Affect Kinetics and Thermodynamics of Tau Aggregation. Biochemistry 2011, 50, 10876–10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. K.; Lee V. M. Y. Characterization of Two VQIXXK Motifs for Tau Fibrillization in Vitro. Biochemistry 2006, 45, 15692–15701. [DOI] [PubMed] [Google Scholar]
- Goux W. J.; Kopplin L.; Nguyen A. D.; Leak K.; Rutkofsky M.; Shanmuganandam V. D.; Sharma D.; Inouye H.; Kirschner D. A. The Formation of Straight and Twisted Filaments from Short Tau Peptides. J. Biol. Chem. 2004, 279, 26868–26875. [DOI] [PubMed] [Google Scholar]
- von Bergen M.; Barghorn S.; Li L.; Marx A.; Biernat J.; Mandelkow E. M.; Mandelkow E. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. J. Biol. Chem. 2001, 276, 48165–48174. [DOI] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. Synergistic Interactions between Repeats in Tau Protein and Aβ Amyloids May Be Responsible for Accelerated Aggregation Via Polymorphic States. Biochemistry 2011, 50, 5172–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S. F.; Leboeuf A. C.; Massie M. R.; Jordan M. A.; Wilson L.; Feinstein S. C. Three- and Four-Repeat Tau Regulate the Dynamic Instability of Two Distinct Microtubule Subpopulations in Qualitatively Different Manners. Implications for Neurodegeneration. J. Biol. Chem. 2005, 280, 13520–13528. [DOI] [PubMed] [Google Scholar]
- Colletiera J.-P.; Laganowskya A.; Landau M.; Zhao M.; Soriaga A. B.; Goldschmidt L.; Flot D.; Cascio D.; Sawaya M. R.; Eisenberg D. Molecular Basis for Amyloid-β Polymorphism. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16938–16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; MacFarlane H. T.; Madsen A. Ø.; Riekel C.; Eisenberg D. Atomic Structures of Amyloid Cross-β Spines Reveal Varied Steric Zippers. Nature 2007, 447, 453–457. [DOI] [PubMed] [Google Scholar]
- Kemper P. R.; Dupuis N. F.; Bowers M. T. A New, Higher Resolution, Ion Mobility Mass Spectrometer. Int. J. Mass. Spectrom. 2009, 287, 46–57. [Google Scholar]
- Mason E. A.Transport Properties of Ions in Gases, 99th ed.; John Wiley & Sons: New York, 1988. [Google Scholar]
- Do T. D.; Economou N. J.; LaPointe N. E.; Kincannon W. M.; Bleiholder C.; Feinstein S. C.; Teplow D. B.; Buratto S. K.; Bowers M. T. Factors That Drive Peptide Assembly and Fibril Formation: Experimental and Theoretical Analysis of Sup35 NNQQNY Mutants. J. Phys. Chem. B 2013, 117, 8436–8446. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. Gromacs 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–437. [DOI] [PubMed] [Google Scholar]
- Spoel D. V. D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Damm W.; Halgren T. A.; Murphy R. B.; Smondyrev A. M.; Friesner R. A.; Jorgensen W. L. OPLS_2002: A New Version of the OPLS-AA Force Field. Abstr. Pap. Am. Chem. Soc. 2002, 224, U471–U471. [Google Scholar]
- Halgren T. A.; Murphy R. B.; Jorgensen W. L.; Friesner R. A. Extending the OPLS-AA Force Field for Ligand Functionality. Abstr. Pap. Am. Chem. Soc. 2000, 220, U277–U277. [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. The Opls Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A.; Tirado-Rives J.; Jorgensen W. L. OPLS-AA/L Force Field for Proteins: Using Accurate Quantum Mechanical Data. Abstr. Pap. Am. Chem. Soc. 2000, 220, U279–U279. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. Structural Information from Ion Mobility Measurements: Effects of the Long Range Potential. J. Phys. Chem. A 1996, 100, 16082–16086. [Google Scholar]
- Shvartsburg A. A.; Jarrold M. F. An Exact Hard-Spheres Scattering Model for the Mobilities of Polyatomic Ions. Chem. Phys. Lett. 1996, 261, 86–91. [Google Scholar]
- Bleiholder C.; Dupuis N. F.; Wyttenbach T.; Bowers M. T. Ion Mobility-Mass Spectrometry Reveals a Conformational Conversion from Random Assembly to β-Sheet in Amyloid Fibril Formation. Nat. Chem. 2011, 3, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar]
- Anderson S. E.; Bleiholder C.; Brocker E. R.; Stang P. J.; Bowers M. T.; Novel A. Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (III): Application to Supramolecular Coordination-Driven Assemblies with Complex Shapes. Int. J. Mass. Spectrom. 2012, 330, 78–84. [Google Scholar]
- Bleiholder C.; Contreras S.; Bowers M. T.; Novel A. Projection Approximation Algorithm for the Fast and Accurate Computation of Molecular Collision Cross Sections (IV). Application to Polypeptides. Int. J. Mass. Spectrom. 2013, 354, 275–280. [Google Scholar]
- Wiltzius J. J. W.; Landau M.; Nelson R.; Sawaya M. R.; Apostol M. I.; Goldschmidt L.; Soriaga A. B.; Cascio D.; Rajashankar K.; Eisenberg D. Molecular Mechanisms for Protein-Encoded Inheritance. Nat. Struct. Mol. Biol. 2009, 16, U973–U998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. D.; LaPointe N. E.; Sangwan S.; Teplow D. B.; Feinstein S. C.; Sawaya M. R.; Eisenberg D. S.; Bowers M. T. Factors That Drive Peptide Assembly from Native to Amyloid Structures: Experimental and Theoretical Analysis of [Leu-5]-Enkephalin Mutants. J. Phys. Chem. B 2014, 118, 7247–7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Sahara N.; Saito Y.; Murayama M.; Yoshiike Y.; Kim H.; Miyasaka T.; Murayama S.; Ikai A.; Takashima A. Granular Tau Oligomers as Intermediates of Tau Filaments. Biochemistry 2007, 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.