

Abstract
Background
Gray leaf spot (GLS) caused by Cercospora zeae-maydis (Czm) or Cercospora zeina (Cz) is a devastating maize disease and results in substantial yield reductions worldwide. GLS resistance is a quantitatively inherited trait. The development and cultivation of GLS-resistant maize hybrids are the most cost-effective and efficient ways to control this disease.
Results
We previously detected a major GLS resistance QTL, qRgls2, in bin 5.03–04, which spans the whole centromere of chromosome 5 encompassing a physical distance of ~110-Mb. With advanced backcross populations derived from the cross between the resistant Y32 and susceptible Q11 inbred lines, a sequential recombinant-derived progeny testing strategy was adapted to fine map qRgls2. We narrowed the region of qRgls2 from an initial ~110-Mb to an interval of ~1-Mb, flanked by the markers G346 and DD11. qRgls2 showed predominantly additive genetic effects and significantly increased the resistance percentage by 20.6 to 24.6% across multiple generations. A total of 15 genes were predicted in the mapped region according to the 5b.60 annotation of the maize B73 genome v2. Two pieces of the mapped qRgls2 region shared collinearity with two distant segments on maize chromosome 4.
Conclusions
qRgls2, a major QTL involved in GLS resistance, was mapped to a ~1-Mb region close to the centromere of chromosome 5. There are 15 predicted genes in the mapped region. It is assumed that qRgls2 could be widely used to improve maize resistance to GLS.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-014-0230-6) contains supplementary material, which is available to authorized users.
Keywords: Maize, GLS, QTL, Fine-mapping, Candidate genes
Background
Gray leaf spot (GLS) is a destructive fungal disease and poses a serious threat to maize production worldwide. The yield loss caused by GLS varies with different environmental conditions and cultivars. For instance, epidemic GLS could result in >50% yield loss in the United States [1], 20–50% in Brazilian Central Region [2], and 20 to 60% in South Africa [3].
Both Cercospora zeae-maydis (Czm) and Cercospora zeina (Cz) are considered to be the casual pathogens to GLS [4]. Cercospora spores overwinter on corn debris left in the field, until conidia begin to develop in warm temperature and high humidity in the next growing season [5,6]. After initial pathogen infection, GLS lesions appear first on the bottom leaves. In the early stages, it is hard to distinguish GLS symptoms from those of other foliar diseases, such as Northern corn leaf blight and Southern corn leaf blight. Some mature GLS lesions, however, have unique features and are characterized by their distinct rectangular shapes parallel to the veins [7].
Compared with regular methods for disease control (fungicide spraying, conventional tillage, and intercropping), cultivation of GLS-resistant hybrids is a cost-effective and environmentally friendly way to reduce yield loss due to gray leaf spot [8]. GLS resistance is a canonical quantitatively-inherited trait [9]. Moreover, general combining ability is predominant in GLS resistance, implying that additive genetic effects are more important than non-additive effects for resistance development [10-12]. Thus, QTL identification would accelerate the breeding of resistant hybrids. For example, the simple sequence repeat (SSR) markers linked to QTLs in bins 4.03 and 4.04 were used to select GLS-resistant maize [13]. So far, a quite number of QTLs associated with GLS resistance have been reported using various parental lines and mapping groups [7,14-19]. Out of 57 resistance QTLs, seven consensus QTLs were found on chromosome bins 1.06, 2.06, 3.04, 4.06, 4.08, 5.03, and 8.06 [20]. QTLs for GLS resistance were reported to show stable genetic contributions to GLS resistance in different environments [2]. Some resistance QTLs were localized in the region associated with multiple-disease resistance, suggesting an intriguing broad-spectrum resistance [21]. In our previous study, we used the GLS-resistant line Y32 and the GLS-susceptible line Q11 to develop mapping populations for QTL analysis. One of the major QTLs in bin 8.01–03 is restricted to an ~1.4-Mb region. Another major QTL, qRgls2 in bin 5.03–04, is located within an ~110-Mb region spanning the whole centromere of chromosome 5 [22].
Using a recombinant-derived progeny testing strategy, we ultimately mapped qRgls2 to a ~1-Mb interval on chromosome 5, which was close to the heterochromatin portion around the centromere. The mapped qRgls2 region shows syteny with two distant segments on chr.4 in maize as well as one segment on chr.2 in rice. Our results provide useful information for qRgls2 cloning, and the markers developed around qRgls2 can be readily used for breeding GLS-resistant maize.
Methods
Plant materials
A highly GLS-resistant inbred line, Y32, was developed from the tropical population Suwan1 and used as a donor parent. The highly GLS-susceptible line Q11 was used as a recurrent receptor parent (Figure 1). The two parental lines were crossed to produce the F1 hybrid, which was continuously selfed to generate F2, F2:3, and F3:4 populations. The F1 and F2 populations were planted in Kunming (Yunnan province, China). The 161 F2:3 families derived from 161 F2 individuals were evaluated for GLS resistance in Baoshan and Dehong (Yunnan province, China). In the initial QTL mapping, the mean disease scale of each F2:3 family was used to describe the disease state of the parental F2 individual [22]. In the winter nursery of 2010/2011, the F3:4 families was planted in Jinghong (Yunnan province, China) to screen for recombinants using flanking markers bnlg1046 and umc1171. Then, recombinants were identified and backcrossed to Q11 to produce BC1F4 progeny, which were planted in Baoshan to evaluate GLS disease severity. We planted the BC1F4 progeny of each F3:4 recombinant into one plot and all plots were randomly distributed. Every plot has eight rows, 3.5 m in length and 0.5 m in width each row. Each row has 15 holes. Totally, 120 seeds were sowed per plot. Because of the severity of GLS in the recurrent parent Q11, it was impossible to backcross BC1F4 individuals to Q11. Therefore, we selfed each BC1F4 individual as well as the BC2F5 and BC3F6 populations in Baoshan. In 2011/2012, we planted BC1F4:5 families in the winter nursery in Jinghong to screen for more recombinants. The resultant recombinants were then backcrossed to Q11 to develop BC2F5 progeny. The BC2F5 population was grown in Boshan to investigate its GLS resistance. The BC2F5 progeny of each BC1F4:5 recombinants were planted in one plot with ten rows. There are 16 holes each row. Totally, 160 seeds were sowed per plot. In 2012/2013, the BC2F5:6 families were grown in Sanya (Hainan province, China) to obtain recombinants. In 2013, BC3F6 plants derived from BC2F5:6 recombinants were grown in Boshan for fine-mapping of qRgls2. We arranged 12 rows per plot for each BC3F6 progeny. Totally, 192 seeds were sowed per plot. The development of the mapping populations and the screening of recombinants are depicted in Additional file 1: Figure S1.
Figure 1.

GLS symptoms of resistant line Y32 (left) and susceptible line Q11 (right). The ovals show enlarged images of the leaves. Y32 had a disease scale of 3; Q11 had a disease scale of 7.
Disease scoring in the field
The fine-mapping populations were planted in Baoshan and infected naturally with causal fungus Cercospora zeina (Cz) [23]. We scored GLS symptoms three times at intervals of 1 week, beginning 2 weeks after pollination. The number and size of disease spots on the leaves of the entire plant were used to evaluate GLS severity. Disease severity was rated using the following scale: 1 (highly resistant), 3 (resistant), 5 (intermediate resistant/susceptible), 7 (susceptible), and 9 (highly susceptible) [22].
Development of high-density markers in the qRgls2 region
According to the B73 reference genome v2.0 assembly (B73 RefGen_v2) (http://www.maizesequence.org/index.html), the physical distance of the confidence interval for qRgls2 is ~110-Mb and covers the whole centromere of chromosome 5. SSR and insertions or deletions (InDels) polymorphism (IDP) markers located in the qRgls2 region were retrieved from the Maize Genetics and Genomics Database (http://www.maizegdb.org). Because of the low density of these existing markers, we then developed new markers within the QTL region. We downloaded the sequences of the qRgls2 region from B73 RefGen_v2 (http://www.maizesequence.org/index.html) and mined possible SSR sequences using SSRHunter1.3 software [24]. Single-copy SSR sequences were obtained after BLASTn comparison with the maize high-throughput genome sequence (HTGS) database, and SSR markers were developed using PRIMER5.0 software [25]. For primer design, we searched for single-copy sequences in the qRgls2 region based on the B73 genome, and we then amplified those sequences from the genomes of the two parents, Y32 and Q11. PCR products were separately cloned into the pGEM-T vector for sequencing. To confirm that the correct sequences were obtained, we projected the sequenced amplicons on the B73 genome using ContigExpress Project software (http://www.contigexpress.com/index.html). Sequence alignment of the two parents revealed InDels that were then developed into IDP markers. Single-nucleotide polymorphisms (SNPs) were used to develop SNP markers. Finally, to ensure that markers were located in the qRgls2 region, we ran a BLAST analysis on the Gramene website (http://www.maizesequence.org/index.html). For SSR and IDP markers, PCR products were analyzed electrophoretically on a 2% agarose gel or 6% polyacrylamide gel. For SNP markers, PCR products were cloned into a pGEM-T vector and sequenced to determine sequence variation.
Estimation of the genetic effect of qRgls2
Sequential fine-mapping of qRgls2 was carried out using recombinants and their progeny. The progeny derived from a given recombinant were divided into two genotypes based on their sequences at the qRgls2 region: the heterozygous Q11/Y32 genotype and the homozygous Q11/Q11 genotype. The disease scales 1 and 3 were classified as resistant, scales 5, 7, and 9 as susceptible. The resistance percentage for each genotype was an estimate of the proportion of resistant plants within the population. The relative difference in the resistance percentage between two genotypes represents the genetic contribution of the introgression region to GLS disease resistance.
A statistical model for declaration of qRgls2
A linear regression model yi = α + βxi + εi was used to test whether a significant association exists between the disease scales and the genotypes in the recombinant-derived progeny. In the backcross population, variable xi represents the genotype of the donor segment, xi = 1, when the marker genotype is Aa (heterozygous Q11/Y32), or xi = 0, when the marker genotype is aa (homozygous Q11/Q11). Yi represents the phenotypic value for the ith individual, α is an intercept, β is the regression coefficient for yi on xi, and εi is random error [26].
The significance of the regression coefficient was evaluated using the t-test. A P-value of ≤0.05 indicated the presence of a significant association, and the donor segment covered the qRgls2 locus. In contrast, a P-value of >0.05 revealed no significant correlation, and the donor did not contain the qRgls2 locus. The statistical analysis was run with R 2.15.3 software (http://cran.r-project.org/).
Results
Development of high-density markers in the qRgls2 region
In the initial QTL mapping, qRgls2 was mapped to a large chromosomal region because of the small mapping population and low-density markers. High-resolution mapping of qRgls2 is essential for the isolation of genes involved in GLS resistance. We thus mined SSRs in the qRgls2 region based on the B73 reference genome and identified 826 single/low-copy SSRs. Based on their flanking regions, we designed 826 primer pairs to amplify both parental lines Y32 and Q11. Polymorphic PCR products were observed in 67 out of 826 primer pairs. Of these 67 SSR markers, only 18 were selected to saturate the qRgls2 region (Table 1), as the other 49 SSR markers were tightly linked to these 18 markers. To develop IDP and SNP markers, we searched B73 reference genome and selected 144 single/low-copy segments in the qRgls2 region to design primers. These primers were used to amplify the two parental genome DNA and the amplicons were sequenced. Based on sequence variations between Y32 and Q11, we developed 15 IDP markers and one SNP marker. Finally, in the light of amplification efficiency and physical location, 4 of the 15 IDP markers and one SNP marker were used to saturate the candidate interval (Table 1).
Table 1.
Newly developed molecular markers in the qRgls2 region on chromosome 5
| Location * (Mb) | Marker | Forward primer (5′–3′) | Reverse primer (5′–3′) | Annealing temperature (°C) | Type |
|---|---|---|---|---|---|
| 67.05 | G414 | TGCTTCCAAACTCCTCCCTA | GCCTTGAGGGTCACCTTTC | 60 | SSR |
| 67.51 | G520 | CACACCACACCAATGCAAAT | CACAGCCATGTTCAGGTCAG | 60 | SSR |
| 69.40 | B96 | CCCTGGGCGCAAAGCAAAGG | TCAGACGGTAGTGCAAGGCACC | 58 | SSR |
| 70.80 | G386 | CAGCACCCTGCTGGTTATTT | GCGGGTTGAAACCGTAGTAG | 58 | SSR |
| 70.90 | IDP36 | TCCTCCTGGCAGTCTAGGAA | TCCGTTTTGTTCTGTTGTGC | 60 | IDP |
| 71.40 | G350 | ACCTCTTCGACGCAACACTC | CGTCGATGAACCTCCGTAGA | 60 | SSR |
| 72.70 | G366 | GCCTGGAAGCTCGTAGTTGT | GTCCAGTCCGTCCCATAAAA | 60 | SSR |
| 73.05 | G346 | CACAGAAGCGTTTCCTTCG | GCTCTGGCTCTGGTTCTAGC | 58 | SSR |
| 73.11 | DD3 | GTGTTTCGCCTCTGGATTTC | AAAAACTGCGTTGCCAGTCT | 58 | IDP |
| 74.05 | Q22 | GGTGCTCCATTGATTGACCT | CGCCCTGTTCTTATTTGCTC | 58 | IDP |
| 74.1 | DD11 | GGAAACAATGGCACACTTCA | GCTTGCATTAGGCTGTTCCT | 58 | SNP |
| 74.50 | G286 | TGTCGTCGTTCCATTACGAG | CGGTTTCCGAAAATGAAGAG | 60 | SSR |
| 74.70 | IDP41 | TGAAGGCTCCAGCTAATGGT | CCGAGGCACGATAAACATCT | 60 | IDP |
| 80.19 | G241 | GATATGGAGGCCCTCTCTCC | ATGATCTCGGTGGTTTCAGG | 58 | SSR |
| 80.41 | G51-1 | ATGCACTGATGGGGAGTGAT | TGTTCTCTGAGCACCAGACG | 60 | SSR |
| 80.83 | G206 | ATCGGCAGATAAAAGCAGGA | CGGGATAAGGGAGGATGATT | 58 | SSR |
| 83.08 | G192 | TTGATGGGCTTAACATTGTCC | TTCGGTTAGGGTGGATTGAG | 60 | SSR |
| 85.87 | G64 | GGAAAAGGGAATGGATGGAT | GGAAGGATCAAGGGAAAGGA | 58 | SSR |
| 95.87 | L19 | AACTCTGGACTCGCTAGGCA | CCGGATGAAGCTAACTGCA | 58 | SSR |
| 131.46 | xl57 | CCGCTCCGCGTATAAAGTAG | CTCTGAAGCCAGGACGGTAG | 60 | SSR |
| 138.12 | G5 | CAATGACTTCCGCACCAGTA | CTCGTTGCCGGTCTCTATGT | 58 | SSR |
| 146.08 | 35-1 | CTCCTTGTTTGGGCCTAATG | TAGGATAAGAGCCCGTGAGC | 60 | SSR |
| 162.00 | xl12 | TCCTCCCTCCCTTGATGAAT | CAGGGAGGCTCAGTAATGGA | 60 | SSR |
*Location: The physical location according to B73 RefGen_v2.
Fine-mapping of qRgls2
qRgls2 was mapped to an interval of ~110-Mb, flanked by the markers umc1784 and umc1171 (Additional file 2: Figure S2) in the initial QTL mapping [22]. It explained 18.9–23.9% of the total phenotypic variation in the mapping population [22]. We conducted a one-way analysis of variance (ANOVA) in the F2:3 families using the qRgls2-tagged marker G386 (Additional file 3: Table S1). The homozygous Y32/Y32 or heterozygous Q11/Y32 genotype showed a lower disease scale than the homozygous Q11/Q11 genotype, indicating the presence of qRgls2 in the mapped region (Additional file 3: Table S1). Therefore, we conducted sequential fine-mapping of qRgls2 from 2011, 2012, and 2013.
A total of 22 recombinants screened from the 1,258 progeny of 52 F3:4 families were backcrossed to Q11 to generate 22 corresponding BC1F4 progeny. The sizes of their donor regions were estimated by genotyping at nine markers, including four newly developed markers (B96, xl57, 35-1, and xl12) (Figure 2A). In the summer of 2011, 1,688 BC1F4 individuals were planted (Figure 2A). All BC1F4 plants were scored for GLS disease severity and genotyped at the introgression region. The presence of a significant correlation (P ≤ 0.05) between genotypes and disease scales indicated that the introgression segment covered the qRgls2 locus.
Figure 2.

Sequential fine-mapping of the major QTL qRgls2 in recombinant maize cultivated from 2011 to 2013. The 22 F4 (A), 32 BC1F5 (B), and 42 BC2F6 (C) recombinants were classified into 7, 18, and 17 types, respectively. The genomic architecture for each type is depicted as black, white, and gray rectangles, representing heterozygous Q11/Y32, homozygous Q11/Q11, and mixed regions (in mixed regions recombination occurs, but the exact breakpoint is uncertain), respectively. Table on the right: Increased resistance percentage is defined as the difference in resistance percentage between genotypes Q11/Y32 and Q11/Q11 that resulted from the donor region of Y32; The total number of plants refers to all progeny of a given recombinant type; List of markers used to detect the presence/absence of the donor region; A P-value of ≤0.05 indicates the regression coefficient between genotype and disease scale is significant within the progeny derived from a given recombinant type. This suggests the presence of qRgls2 on the donor region, and the parental recombinant was deduced to be GLS resistant (R). A P-value of >0.05 indicates that no significant correlation between genotype and disease scale is present, suggesting that qRgls2 is absent from the donor region of the parental recombinant, which is therefore marked as GLS susceptible (S). Analysis of both the deduced phenotype and the donor region for all recombinants enabled us to narrow qRgls2 from an ~110-Mb to an ~1-Mb region, flanked by the markers G346 and DD11.
For a given F3:4-derived BC1F4 progeny, there are two genotypes, Q11/Q11 and Q11/Y32, at the introgression segment. The recombinants were considered to have the same genotype if they shared the same introgression segment. Thus, the 22 F3:4 recombinants were classified into seven types (types I, II, III, IV, V, VI, and VII). Types I and II carried the introgression segments downstream of the umc1747 and bnlg1287 markers, respectively, whereas type VII carried the introgression segments upstream of B96. No significant correlation between genotype and disease scale was detected in the BC1F4 progeny for each of these three types (P > 0.05), indicating that qRgls2 was absent in their introgression segments. In contrast, types III and IV carried the introgression segments downstream of B96 and umc1784, respectively, and types V and VI had the introgression segments upstream of umc1171 and umc1747, respectively. The regression coefficients between genotype and disease scale were significant (P < 0.01) for types III, IV, V, and VI, implying the presence of the resistance QTL, qRgls2, in their introgression segments. Thus the qRgls2 region could be narrowed to an interval between the markers B96 and xl57 (Figure 2A) with a physical distance of ~62-Mb according to B73 RefGen_v2.
Similar analysis was conducted in the BC1F5-derived 2,850 BC2F5 progeny in 2012. Sixteen markers were used to genotype 32 BC1F5 recombinants and to group them into 18 types. Of these, 12 (G386, G286, IDP41, G241, G51-1, G206, G192, G64, umc1591, L19, umc1563, and bnlg1287) were located between B96 and xl57 and two (G5 and umc1747) were located downstream of xl57. Within the mapped qRgls2 region, types I–IX had downstream introgression segments that did not carry the resistance QTL, qRgls2 (P > 0.05). Type IX had the longest introgression segments of these, indicating that qRgls2 was not present downstream of G241. Types X to XVIII carried the introgression segments that harbored the resistance QTL, qRgls2 (P < 0.05). Type XVIII carried the introgression segment upstream of IDP41, suggesting the presence of qRgls2 in the introgression segment upstream of IDP41. The findings from types IX and XVIII thus restrict the right border of qRgls2 to IDP41. Type X had the closest crossing-over point to the left of the qRgls2 locus, thus restricting the left border of qRgls2 to G386. In summary, these three types had the closest recombination breakpoints to qRgls2 and restricted qRgls2 to an interval of ~3.9-Mb between markers G386 and IDP41 (Figure 2B).
In 2013, a total of 5,631 BC3F6 plants derived from 42 BC2F6 recombinants were planted. Moreover, a total of 14 markers, ten (G386, IDP36, G350, G366, G346, DD3, Q22, DD11, G286, and IDP41) within the qRgls2 region and four (G414, G520, B96, and AY104079) located upstream of G386, were used to genotype 42 BC2F6 recombinants and to group them into 17 types. Within the newly mapped qRgls2 region, types I to V and types XV to XVII did not have qRgls2 in their introgression segments (P > 0.05), suggesting qRgls2 was present between the left marker G346 and the right marker DD11. Types VI to XIV had the resistance QTL qRgls2 in their introgression segments (P < 0.05) and thus restricted qRgls2 to the G366 and G286 interval. These types, with and without qRgls2, unanimously mapped qRgls2 into the G346/DD11 interval with the physical distance of ~1-Mb according to the B73 RefGen_v2 (Figure 2C).
Genetic contribution of qRgls2 to GLS resistance
The genetic effect of qRgls2 was estimated in F2:3, BC1F4, BC2F5, and BC3F6 populations, respectively. As expected, plants carrying the qRgls2 regions showed higher GLS resistance than those without qRgls2. In F2:3 families, the percentages of resistant individuals were estimated to be 52.5%, 72.6%, and 80.5% for Q11/Q11, Q11/Y32, and Y32/Y32 genotypes, respectively (Figure 3). The resistance percentage in the BC1F4 population in 2011 was 74.9% in plants with qRgls2 versus 54.3% in plants without qRgls2 (Figure 3). Similarly, phenotyping of the BC2F5 population in 2012 suggested consistent resistance improvement associated with qRgls2 (70.6% in plants carrying the qRgls2 segments versus 46.9% in plants lacking the qRgls2 segments; Figure 3). Finally, in the BC3F6 population, individuals carrying the QTL regions showed a resistance percentage of 56.1%, whereas individuals lacking the QTL segments only showed 31.5% disease resistance (Figure 3). These results, which were derived from multiple populations over many years, indicated qRgls2 enhances GLS resistance by 20.6 to 24.6% and that this genetic effect is passed on to subsequent generations.
Figure 3.
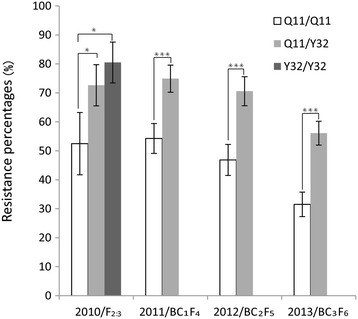
Estimation of the genetic effect of qRgls2 on GLS resistance. Resistance percentages of different genotypes at qRgls2 in the F2:3, BC1F4, BC2F5, and BC3F6 populations are shown. The P-value of the resistance percentage between the genotypes Q11/Q11 and Q11/Y32 from 2010 to 2013 was 0.04, 2.39E-08, 5.57E-08, 4.02E-13, respectively. The P-value between the genotypes Q11/Q11 and Y32/Y32 was 0.02 in 2010.
Genomic architecture and gene discovery in the qRgls2 region
The genomic sequence between G346 and DD11 was retrieved from B73 RefGen_v2 (http://www.maizesequence.org/index.html). This region is predicted to contain 15 genes according to the 5b.60 annotation of the maize B73 genome v2 (http://www.maizesequence.org/index.html) (Table 2).
Table 2.
Predicted genes in the mapped qRgls2 interval and syntenic genes in maize and rice
| Fine-mapped qRgls2 on maize Chr. 5 | Syntenic gene on Maize Chr. 4 | Rice | ||
|---|---|---|---|---|
| Gene ID | Predicted function | Syntenic gene | Predicted function | |
| GRMZM2G030013 | KH domain–containing protein participates in RNA binding in post-transcription | NA | Os02t0125500 | KH domain–containing protein |
| GRMZM2G477236 | lil3 protein, a light harvesting–like protein, plays an essential role in chlorophyll and tocopherol biosynthesis | GRMZM2G027640 | Os02t0125700 | lil3 protein |
| GRMZM2G175137 | RNA polymerase II transcription factor B subunit 4 | GRMZM2G027209 | NA | NA |
| GRMZM2G099827 | Microtubule-associated protein with anther-specific expression | GRMZM2G027187 | NA | NA |
| GRMZM5G868966 | Unknown | GRMZM5G820374 | NA | NA |
| GRMZM2G157068 | Protein kinase | GRMZM2G053868 | Os02t0126400 | Protein kinase |
| GRMZM2G456088 | Pentatricopeptide repeat (PPR) superfamily protein, participates in post-transcriptional processes in plastids | AC216235.2_FGT010 | Os02t0126500 | PPR domain–containing protein |
| GRMZM2G157046 | Leaf-expressed protein with unknown function | GRMZM2G053830 | Os02t0126600 | Unknown |
| GRMZM2G157026 | Golgi SNARE 12 protein | GRMZM5G838961 | Os02t0126800 | Golgi SNAP receptor complex member |
| GRMZM2G156983 | Phosphatidate cytidylyltransferase | GRMZM2G053711 | NA | NA |
| AC189771.3_FG001 | Anther-specific expression with unknown function | NA | NA | NA |
| GRMZM2G009065 | Inflorescence-expressed gene with unknown function | NA | NA | NA |
| GRMZM2G039254 | PPR superfamily protein | NA | Os02t0127600 | PPR domain–containing protein |
| GRMZM2G038791 | Ribose-phosphate pyrophosphokinase | NA | Os02t0127700 | Ribose-phosphate pyrophosphokinase |
| GRMZM2G153178 | Unknown | NA | Os02t0128100 | Unknown |
NA: Not available; Chr.:Chromosome.
qRgls2 is adjacent to the centromere of chromosome 5, and the whole region exhibits very low gene density and recombination frequencies as compared with the average across the whole genome [27]. In eukaryotes, genes often show uneven distribution along the chromosomes, and they organize in clusters with different gene densities and sizes [28]. This characteristic is also seen in the qRgls2 region. The 15 predicted genes are unequally scattered throughout the mapping region. Ten genes are clustered on the left end, three genes are located at the right end, and only two genes are located in the middle portion, which occupies ~800-kb and ranges from 73,235,500 to 74,036,000 bp based on the B73 RefGen_v2 (Figure 4). We analyzed the sequence of this 800-kb gene-desert region using the RepeatMasker (http://www.repeatmasker.org) and found that 92.79% of the sequence consists of transposable elements (90.06% retroelements, 2.59% transposons, and 0.14% unclassified; Additional file 4: Table S2). This percentage is higher than that across the whole maize genome (~85%) [27].
Figure 4.
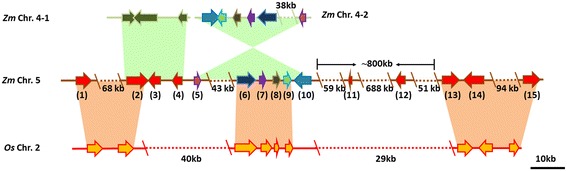
Genomic architecture of the qRgls2 region and its syntenic blocks in maize and rice. Maize chromosome 4 has two syntenic blocks: Zm Chr. 4-1 ranges from 238,744,350 to 238,761,826 bp (~17.5 kb), and Zm Chr. 4-2 ranges from 13,562,579 to 13,631,290 bp (~68.7 kb). In rice, the syntenic block Os Chr.2 is located on chromosome 2, from 1,334,563 to 1,456,346 bp (~121 kb). The syntenic blocks in maize are depicted as green zones; syntenic blocks between maize and rice are depicted as pink zones. The arrows indicate genes and their orientations; the dotted lines indicate regions without genes. Each pair of reversely syntenic genes with identical sequence is tinted with the same color. The 15 predicted genes in the qRgls2 region in chromosome 5 are GRMZM2G030013 (1), GRMZM2G477236 (2), GRMZM2G175137 (3), GRMZM2G099827 (4), GRMZM5G868966 (5), GRMZM2G157068 (6), GRMZM2G456088 (7), GRMZM2G157046 (8), GRMZM2G157026 (9), GRMZM2G156983 (10), AC189771.3_FG001 (11), GRMZM2G009065 (12), GRMZM2G039254 (13), GRMZM2G038791 (14), and GRMZM2G153178 (15). Zm: Zea mays; Os: Oryza sativa; Chr.: chromosome.
We then searched for duplicated genomic fragments using the Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication) and found some genes in the qRgls2 interval that share high synteny with rice genes located on chromosome 2 (Figure 4). Moreover, we detected two syntenic blocks in maize that are present separately on the short and long arms of chromosome 4 [29]. The one (Zm Chr. 4-1) present on the long arm is a part of the large duplicated region between chromosomes 4 and 5, which is assumed to originate from whole-genome duplication followed by genome rearrangement from tetraploid to diploid [30]. The gene content and order within this syntenic block are well conserved, and only a few sequence variations were observed between the coding regions of homologous genes (Figure 4). Intriguingly, the other syntenic block (Zm Chr. 4-2), which is located on the short arm, shows perfect reverse collinearity, with the coding sequences of six genes being identical in reverse order in two collinear blocks (Figure 4, Additional file 5: Data Set 1). This inverted synteny may derive from a recent genomic duplication and rearrangement, which took place long after the ancient whole-genome duplication.
Discussion
GLS resistance is a quantitatively inherited trait and thus hinders breeding of resistant maize varieties [14]. Host resistance is the most cost-effective and efficient way to control GLS disease [8]. Therefore, discovery of a resistance QTL and introgression into elite inbred lines via marker-assisted selection (MAS) would greatly increase GLS resistance [18]. The qRgls2 locus overlaps with previously reported QTLs [8,20], implying the qRgls2 region may exist in other mapping populations. Moreover, we showed that the qRgls2 locus is heritable and could stably enhance resistance percentages by >20% across multiple generations. These findings suggest that qRgls2 could be used to improve maize resistance to GLS, and high-density markers around the qRgls2 region will be useful for MAS.
Because of the low density of publicly available markers around qRgls2, we retrieved single/low-copy sequences to develop either SSR or IDP or SNP markers to saturate the qRgls2 region. This strategy, however, turned out to be inefficient. Only a small fraction of the designed primers could be converted into markers. In the future, using genome sequences from diverse maize inbred lines, we can compare in silico the mined single/low-copy sequences, for instance between B73 and Mo17, and select those with sequence variations.
Sequential recombinant-derived progeny testing is a powerful method for fine-mapping of resistance QTLs [31]. This strategy can be modified to accommodate different situations. In the current study, we evaluated GLS resistance during the growing season in Baoshan in advanced backcross populations. However, it was impossible for us to produce backcross populations in Baoshan because of severe infections of the recurrent parent line Q11. Therefore, all individuals were selfed, and the resulting self-progeny of newly identified recombinants were grown in winter nurseries (Jinghong or Sanya) where no GLS was prevalent. Heterozygous individuals were then selected and backcrossed to Q11. If the recombinant-derived progeny segregated at the qRgls2 region, we were able to estimate the relative difference of GLS resistance between two genotypes and to fine-map the qRgls2 locus. In addition, the advanced backcross progeny shared similar genetic backgrounds with very low background noise [31]. This is very helpful for accurately estimating the genetic contribution of the donor region of a given recombinant to GLS resistance. During screening of recombinants, we were aware that the recombination frequency seems to increase in the qRgls2 region as backcrossing advanced. The genetic distance per Mb was estimated to be 1.59E-4, 3.06E-4, and 3.78E-3 in F3:4, BC1F4:5 and BC2F5:6 populations, respectively. We hope to obtain some key recombinants in the qRgls2 locus when screening more advanced populations. The key recombinants in or around the qRgls2 locus is critical to narrow a QTL to a QTG (quantitative trait gene), or even to a QTN (quantitative trait nucleotide) for qRgls2.
There are 15 functional genes in qRgls2 region according to the 5b.60 annotation of the maize B73 genome v2. We also evaluated B73 for its resistance to GLS in Boshan and found B73 is highly susceptible to GLS. Thus, none of the 15 genes is likely to be the candidate for qRgls2. The resistance gene in the Y32 may be a resistance allele to one of the 15 predicted genes or totally a new gene that is absent in B73. Fortunately, we have constructed the Y32 bacterial artificial chromosome (BAC) library and obtained the positive BAC clones covering the qRgls2 region. Sequence analysis of the qRgls2 region of Y32 could reveal all candidate genes, including the resistance gene underlying qRgls2.
The ratio of genetic to physical distance in the mapped qRgls2 region is only ~0.24 cM/Mb, which is much lower than the average ratio of 2.1 cM/Mb across the entire maize genome [32]. Chromosomal recombination occurs more frequently at chromosomal ends as compared with centromere regions in maize [27]. Additionally, gene density is much higher at chromosomal ends than in the centromere regions [28]. The qRgls2 region is located in the centromere region of chromosome 5 and is characterized by both low gene density (~73 kb/gene) and a low recombination frequency. Furthermore, the 15 predicted genes are unequally distributed in the mapped region. These findings render it very difficult to further screen the key recombinants for fine-mapping, although we continue to expand the mapping population. The same situation was reported for the cloning of Ghd7, which is involved in the regulation of the heading date in rice [33]. The candidate gene was mapped to the final 0.31-cM interval in the centromere region of chromosome 7, corresponding to the physical distance of 2,284 kb. Gene annotation identified the candidate gene for Ghd7, which was finally cloned via functional complementation [33]. It seems that several approaches are required to clone the genes in the centromeric region. Apart from continued fine-mapping, we will also use transcriptome sequencing and association mapping to confirm the candidate gene for qRgls2.
Conclusions
The development and cultivation of resistant maize hybrids are the most environmentally friendly and cost-effective ways to improve maize resistance to GLS. We identified a major QTL, qRgls2, for GLS resistance and narrowed its location from an initial ~110-Mb to a ~1-Mb region. qRgls2 mainly acted in an additive manner and showed very stable genetic effects across multiple generations. The qRgls2 region is very close to the centromere of chromosome 5 and is characterized by low and unequal gene density. A total of 15 genes were predicted in the final mapped interval, according to the 5b.60 annotation of the maize B73 genome v2. Our findings provide a solid base for map-based cloning of the GLS resistance gene underlying qRgls2 in maize. The high-density markers developed around qRgls2 will be useful in MAS for GLS-resistant breeding.
Acknowledgements
This study was financially supported by the National High-tech and Development Program of China, Grant No: 2012AA10A306 and 2012AA101104. We thank Zhijian Xie, Xinyi Pang, Fuyan Jiang, Kun Wang, Shiqiang Li and Haiyan Li for field maize cultivation and pollination.
Abbreviations
- GLS
Gray leaf spot
- Czm
Cercospora zeae-maydis
- Cz
Cercospora zeina
- SSR
Simple sequence repeat
- B73 RefGen_v2
B73 reference genome v2.0 assembly
- Mb
Megabase pairs
- PCR
Polymerase chain reaction
- QTL
Quantitative trait loci
- IDP
InDel polymorphism
- SNP
Single-nucleotide polymorphisms
- MAS
Marker-assisted selection
- QTG
Quantitative trait gene
- QTN
Quantitative trait nucleotide
- BAC
Bacterial artificial chromosome
Additional files
Flow chart of QTL identification and fine-mapping. Individual plants from the F2:3 families, BC1F4 progeny, BC2F5 progeny, and BC3F6 progeny (as indicated by boxes) were used to evaluate the GLS disease scale. F2:3 families were used for QTL identification, and the other populations were used for fine-mapping of the major QTL qRgls2. The year, the number of plants/families and locations in each set of experiments was carried out is noted.
Detection of the QTL qRgls2 across four replicate plots. Logarithm of odds (LOD) profiles (A) and additive genetic effects (B) of the QTL qRgls2 for GLS resistance. The QTL was detected based on data collected from 161 F2:3 families that were grown in Baoshan (two replicates, BS1 and BS2) and Dehong (two replicates, DH1 and DH2) in 2010.
Multiple comparisons of disease scales at marker G386.
Sequence analysis of gene-desert region using RepeatMasker.
Alignment of the coding regions for six pairs of syntenic genes. These syntenic genes showed no sequence divergence.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LX carried out molecular genetic studies and statistical analysis, performed phenotypic evaluation, field maize cultivation and pollination. MLX supervised the research, designed the experiments and involved in data analysis. YZ participated in field maize cultivation and pollination. SQS carried out field maize cultivation and pollination. WC, JT and XMF provided us with parental materials and helped with field cultivation. MZ helped with genotype detection. TZ helped with phenotypic evaluation. LX wrote the draft manuscript and MLX edited and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ling Xu, Email: xu_ling2006@126.com.
Yan Zhang, Email: zhangyan4023@gmail.com.
Siquan Shao, Email: shaosiquan@126.com.
Wei Chen, Email: chenweirick@126.com.
Jing Tan, Email: tanjingli@sina.com.
Mang Zhu, Email: zhumang2013@126.com.
Tao Zhong, Email: zhongtaomvp@163.com.
Xingming Fan, Email: xingmingfan@163.com.
Mingliang Xu, Email: mxu@cau.edu.cn.
References
- 1.Lipps P: Gray leaf spot: a global threat to corn production.APSNet Feature 1998. doi:10.1094/APSnetFeature-1998-0598.
- 2.Juliatti FC, Pedrosa MG, Silva HD, Correa da Silva JV. Genetic mapping for resistance to gray leaf spot in maize. Euphytica. 2009;169(2):227–238. doi: 10.1007/s10681-009-9943-2. [DOI] [Google Scholar]
- 3.Ward J, Laing M, Nowell D. Chemical control of maize grey leaf spot. Crop Protect. 1997;16(3):265–271. doi: 10.1016/S0261-2194(96)00097-X. [DOI] [Google Scholar]
- 4.Korsman J, Meisel B, Kloppers F, Crampton BG, Berger DK. Quantitative phenotyping of grey leaf spot disease in maize using real-time PCR. Eur J Plant Pathol. 2012;133(2):461–471. doi: 10.1007/s10658-011-9920-1. [DOI] [Google Scholar]
- 5.Shim WB, Dunkle LD. Identification of genes expressed during cercosporin biosynthesis in Cercospora zeae-maydis. Physiol Mol Plant Pathol. 2002;61(4):237–248. doi: 10.1006/pmpp.2002.0437. [DOI] [Google Scholar]
- 6.Paul P, Munkvold G. Influence of temperature and relative humidity on sporulation of Cercospora zeae-maydis and expansion of gray leaf spot lesions on maize leaves. Plant Dis. 2005;89(6):624–630. doi: 10.1094/PD-89-0624. [DOI] [PubMed] [Google Scholar]
- 7.Ward JM, Stromberg EL, Nowell DC, Nutter FW., Jr Gray leaf spot: a disease of global importance in maize production. Plant Dis. 1999;83(10):884–895. doi: 10.1094/PDIS.1999.83.10.884. [DOI] [PubMed] [Google Scholar]
- 8.Lehmensiek A, Esterhuizen A, Van Staden D, Nelson S, Retief A. Genetic mapping of gray leaf spot (GLS) resistance genes in maize. Theor Appl Genet. 2001;103(5):797–803. doi: 10.1007/s001220100599. [DOI] [Google Scholar]
- 9.Bubeck D, Goodman M, Beavis W, Grant D. Quantitative trait loci controlling resistance to gray leaf spot in maize. Crop Sci. 1993;33(4):838–847. doi: 10.2135/cropsci1993.0011183X003300040041x. [DOI] [Google Scholar]
- 10.Derera J, Tongoona P, Pixley KV, Vivek B, Laing MD, van Rij NC. Gene action controlling gray leaf spot resistance in Southern African maize germplasm. Crop Sci. 2008;48(1):93–98. doi: 10.2135/cropsci2007.04.0185. [DOI] [Google Scholar]
- 11.Brito AH, Davide LMC, Pinho RGV, Carvalho RP, Reis MC. Genetic control of resistance to gray leaf spot of maize in tropical germplasm. Crop Breed Appl Biotechnol. 2012;12(2):145–150. doi: 10.1590/S1984-70332012000200008. [DOI] [Google Scholar]
- 12.Nzuve F, Githiri S, Mukunya D, Gethi J. Combining abilities of maize inbred lines for grey leaf spot (GLS), grain yield and selected agronomic traits in Kenya. J Plant Breed Crop Sci. 2013;5(3):41–47. doi: 10.5897/JPBCS12.056. [DOI] [Google Scholar]
- 13.Veiga AD, Pinho RGV, Resende LV, Pinho ÉVRV, Balestre M, Pereira LA. Quantitative trait loci associated with resistance to gray leaf spot and grain yield in corn. Ciência e Agrotecnologia. 2012;36:31–38. [Google Scholar]
- 14.Saghai Maroof MA, Yue YG, Xiang ZX, Stromberg EL, Rufener GK. Identification of quantitative trait loci controlling resistance to gray leaf spot disease in maize. Theor Appl Genet. 1996;93(4):539–546. doi: 10.1007/BF00417945. [DOI] [PubMed] [Google Scholar]
- 15.Clements MJ, Dudley J, White D. Quantitative trait loci associated with resistance to gray leaf spot of corn. Phytopathology. 2000;90(9):1018–1025. doi: 10.1094/PHYTO.2000.90.9.1018. [DOI] [PubMed] [Google Scholar]
- 16.Gordon SG, Bartsch M, Matthies I, Gevers HO, Lipps PE, Pratt RC. Linkage of molecular markers to Cercospora zeae-maydis resistance in maize. Crop Sci. 2004;44(2):628–636. [Google Scholar]
- 17.Danson J, Lagat M, Kimani M, Kuria A. Quantitative trait loci (QTLs) for resistance to gray leaf spot and common rust diseases of maize. Afr J Biotech. 2008;7(18):3247–3254. [Google Scholar]
- 18.Benson, Marie J. Plant and Animal Genome XXI Conference: 2013. 2013. Genetic mapping of gray leaf spot disease resistance loci and the elucidation of mechanisms underlying these loci. [Google Scholar]
- 19.Balint Kurti PJ, Wisser R, Zwonitzer JC. Use of an advanced intercross line population for precise mapping of quantitative trait loci for gray leaf spot resistance in maize. Crop Sci. 2008;48(5):1696–1704. doi: 10.2135/cropsci2007.12.0679. [DOI] [Google Scholar]
- 20.Shi LY, Li XH, Hao ZF, Xie CX, Ji HL, Lü XL, Zhang SH, Pan GT. Comparative QTL mapping of resistance to gray leaf spot in maize based on bioinformatics. Agric Sci China. 2007;6(12):1411–1419. doi: 10.1016/S1671-2927(08)60002-4. [DOI] [Google Scholar]
- 21.Chung CL, Poland J, Kump K, Benson J, Longfellow J, Walsh E, Balint-Kurti P, Nelson R. Targeted discovery of quantitative trait loci for resistance to northern leaf blight and other diseases of maize. Theor Appl Genet. 2011;123(2):307–326. doi: 10.1007/s00122-011-1585-9. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Xu L, Fan X, Tan J, Chen W, Xu M. QTL mapping of resistance to gray leaf spot in maize. Theor Appl Genet. 2012;125(8):1797–1808. doi: 10.1007/s00122-012-1954-z. [DOI] [PubMed] [Google Scholar]
- 23.Liu K, Xu X. First report of gray leaf spot of maize caused by Cercospora zeina in china. Plant Dis. 2013;97(12):1656–1656. doi: 10.1094/PDIS-03-13-0280-PDN. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Wan JM. SSRHunter: development of a local searching software for SSR sites. Hereditas. 2005;27(5):808. [PubMed] [Google Scholar]
- 25.Ren L, Zhu B, Zhang Y, Wang H, Li C, Su Y, Ba C. The research of applying primer premier 5.0 to design PCR primer. J Jinzhou Med Coll. 2004;25:43–46. [Google Scholar]
- 26.Doerge R, Zeng Z, Weir B: Statistical issues in the search for genes affecting quantitative traits in experimental populations.Statistical Sci 1997, 195–219.
- 27.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 28.Sidhu D, Gill KS. Distribution of genes and recombination in wheat and other eukaryotes. Plant Cell Tiss Org Cult. 2005;79(3):257–270. doi: 10.1007/s11240-005-2487-9. [DOI] [Google Scholar]
- 29.Soderlund C, Bomhoff M, Nelson WM. SyMAP v3. 4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011;39(10):e68–e68. doi: 10.1093/nar/gkr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet. 2007;3(7):e123. doi: 10.1371/journal.pgen.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Q, Zhang D, Xu M. A sequential quantitative trait locus fine-mapping strategy using recombinant-derived progeny. J Integr Plant Biol. 2012;54(4):228–237. doi: 10.1111/j.1744-7909.2012.01108.x. [DOI] [PubMed] [Google Scholar]
- 32.Yao H, Zhou Q, Li J, Smith H, Yandeau M, Nikolau BJ, Schnable PS. Molecular characterization of meiotic recombination across the 140-kb multigenic a1-sh2 interval of maize. Proc Natl Acad Sci. 2002;99(9):6157–6162. doi: 10.1073/pnas.082562199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet. 2008;40(6):761–767. doi: 10.1038/ng.143. [DOI] [PubMed] [Google Scholar]