

Abstract
Background
Complex regional pain syndrome (CRPS) is a painful condition with ~50,000 annual new cases in the United States. It is a major cause of work-related disability, chronic pain after limb fractures and persistent pain after extremity surgery. Additionally, CRPS patients often experience cognitive changes, anxiety and depression. The supraspinal mechanisms linked to these CRPS-related comorbidities remain poorly understood.
Methods
We used a previously-characterized mouse model of tibia fracture/cast immobilization showing the principal stigmata of CRPS (n=8–20/group) observed in humans. Our central hypothesis was that fracture/cast mice manifest changes in measures of thigmotaxis (indicative of anxiety) and working memory reflected in neuroplastic changes in amygdala, perirhinal cortex, and hippocampus.
Results
We demonstrate that nociceptive sensitization in these mice is accompanied by altered thigmotactic behaviors in the zero maze but not open field assay, and working memory dysfunction in novel object recognition and social memory but not in novel location recognition. Furthermore, we found evidence of structural changes and synaptic plasticity including changes in dendritic architecture and decreased levels of synaptophysin and brain derived neurotrophic factor in specific brain regions.
Conclusions
Our findings provide novel observations regarding behavioral changes and brain plasticity in a mouse model of CRPS. In addition to elucidating some of the supraspinal correlates of the syndrome, this work supports the potential use of therapeutic interventions that not only directly target sensory input and other peripheral mechanisms, but also attempt to ameliorate the broader pain experience by modifying its associated cognitive and emotional comorbidities.
INTRODUCTION
Complex regional pain syndrome (CRPS) is a painful, disabling and often chronic condition with an estimated 50,000 new cases in the United States each year.1 It is characterized by severe pain-related changes (allodynia and hyperalgesia), trophic changes (abnormalities in hair and nail growth), neurovascular abnormalities (abnormal sweating, edema and skin discoloration), and motor changes (tremor).2–4 Despite the fact that CRPS has been a documented clinical entity for at least 150 years, we have only a partial understanding of the supporting mechanisms, and no broadly effective treatments. Current therapies including physical, interventional, pharmacological, rehabilitative, and alternative are limited in their effectiveness, and none are curative,5,6 leaving more than 80% of chronic CRPS patients severely disabled.7
Similar to other chronic pain conditions, CRPS encompasses neurocognitive changes as well as alterations in mood and anxiety that affect quality of life, level of disability and need for additional healthcare.8,9 More broadly, a recent meta-analysis of 24 separate studies on cognition in pain patients concluded that there was a consistent moderate effect of pain on working memory.10 In a related study, it was observed that approximately 20% of the new patients to a multidisciplinary pain center showed clinically significant impairment on neurocognitive testing.11 While the preponderance of the CRPS literature focuses on peripheral changes that accompany the signs and symptoms of this syndrome, more recent studies have used neuroimaging techniques to study changes within the central nervous system (CNS); the existence of pain-associated cognitive and emotional co-morbidities suggests that alterations in brain structure and function may contribute to sustained pain, emotional changes and neurocognitive features in chronic CRPS patients.12,13
Various rodent models that mimic common signs and symptoms observed in human CRPS patients exist. For instance, the chronic post-ischemic pain model is characterized by chronic mechanical allodynia, microvascular injury, and chronic ischemia,14 while the needlestick distal nerve injury is accompanied by allodynia, dystonia, and epidermal neurite loss15 and the focal neuritis model displays changes in sympathetic and sensory neuronal discharge.16 To better understand the changes in cognition and emotion in CRPS and to examine the structural and biochemical brain correlates of these changes, we utilized a mouse tibial fracture model characterized by nociceptive sensitization, bone demineralization, edema and warmth similar to the changes found in CRPS I patients.17–19
Our primary aim of this study was to measure the behavioral phenotypes of anxiety and memory dysfunction that accompany ongoing mechanical allodynia in our model. These studies were then followed by a second line of investigation in order to examine the neuroplastic changes that accompany these behavioral phenotypes. To that end, histological analysis of dendritic architecture in addition to the assessment of synaptophysin and brain derived neurotrophic factor (BDNF) protein levels were performed on tissue from the amygdala, perirhinal cortex and hippocampus. These three centers have well established roles in emotional disorders and cognitive difficulties.20–24
The results of this study support the hypothesis that common features of chronic CRPS including pain, anxiety and memory deficits are accompanied by structural and biochemical changes in specific brain centers.
MATERIALS AND METHODS
Animals
A total of four cohorts of mice were used. Male C57/B6J mice aged 12–14 weeks were purchased from a commercial supplier (Jackson Labs; Sacramento, CA) and were allowed to habituate to the animal facility for a minimum of 10 days prior to the experiments. Mice were housed in groups of four on a 12 h light/dark cycle and an ambient temperature of 22 ± 3°C, with food and water available ad libitum. All animal procedures and experimental designs were approved by the Veterans Affairs Palo Alto Health Care System Institutional Animal Care and Use Committee (Palo Alto, CA) and followed the “animal subjects” guidelines of the International Association for the Study of Pain.
Limb fracture and cast immobilization
Mice were randomly allocated to the control or the fracture/cast group. Mice were anesthetized with 1.5% isoflurane and underwent a distal tibial fracture in the right leg. Briefly, a hemostat was used to make a closed fracture of the right tibia just distal to the middle of the tibia. Then the hindlimb was wrapped in casting tape (Scotchcast Plus, 3M; St Paul, MN) so the hip, knee and ankle were all fixed. The cast extended from the metatarsals of the hindpaw up to a spica formed around the abdomen. A window was left open over the dorsal paw and ankle to prevent constriction when post-fracture edema developed.18 After the procedure, the mice were given subcutaneous buprenorphine (0.05 mg/kg) and enrofloxacin (5mg/kg) for the next two days, as well as normal saline (1.5 ml once) for postoperative analgesia, prevention of infection and prevention of dehydration. Mice were inspected daily to ensure the cast was positioned properly through the three week period of cast immobilization. Mice were provided with chow pellets postoperatively ad libitum; dietary gels were also made available on the cage floor for mice having undergone surgery. At three weeks after surgery, the mice were briefly anesthetized using isoflurane and the casts were removed.
Naïve age- and sex-matched mice were used as control. We have chosen to use naïve mice instead of sham (cast immobilization only) animals because cast immobilization results in an intermediate phenotype that shows signs of transient mechanical allodynia.17 This is consistent with the clinical situation where limited CRPS-like changes are observed when casts are applied to healthy human limbs.25 In the tibia fracture/cast immobilization model of CRPS, both the fracture and the immobilization are needed for the sustained, CRPS-like phenotype in mice, and as such, the cast immobilization is a crucial part of the model rather than an inevitable procedure following fracture.
Behavioral testing
The experimenter was blind to the identity and experimental condition of the animals throughout the behavioral experiments and data analysis. Mice were habituated to handling by the experimenter for a few minutes each day for seven days before initiation of the behavioral tests. Each of the two experimental cohorts included 16 (control) and 20 (fracture/cast) animals and, except for mechanical sensitivity, all behavioral tests were carried out seven weeks after fracture (completed by nine weeks after fracture) in the following order: open field (OF), novel location recognition (NLR), novel object recognition (NOR), zero maze and social memory. Eight (control) and 11 (fracture/cast) animals were randomly chosen to be tested at the 14 and 18 week timepoints (see supplemental digital content 1). Sample size determination was guided by prior experience with these behavioral assays.
Animals were allowed to habituate in their home cages in the experimental room for 1 h prior to testing. All behavioral studies were carried out in a clean and quiet room. OF, zero maze, NOR, NLR, and social memory tests were recorded and analyzed by TopScan Lite (CleverSys; Reston, VA) in real time.
Mechanical hypersensitivity
Calibrated monofilaments (Stoelting Co.; Wood Dale, IL) were applied to the plantar surface of the hindpaw and the 50% threshold to withdraw (grams) was calculated as previously described.26 The stimulus intensity ranged from 0.004 to 1.7g, corresponding to filament numbers (1.65, 2.36, 2.44, 2.83, 3.22, 3.61, 3.84, 4.08, 4.17, and 4.31). For each animal, the actual filaments used within the aforementioned series were determined based on the lowest filament to evoke a positive response followed by five consecutive stimulations using the up-down method. The filament range and average interval were then incorporated along with the response pattern into each individual threshold calculation. Mechanical sensitivity was assessed on the plantar surface of the left hindpaw (response = flexion reflex).
Open field
The OF arena measured 45 × 45 × 45 cm and was made of opaque plastic material. Luminosity inside the OF was measured to be 5 lux. The mice were placed into the arena and allowed to explore for 10 min. Total locomotor activity and the time spent in the central portion (11% of total area) were determined for each mouse. The time spent in the center area was used as an index of thigmotaxis, a measure often used for evaluating general anxiety levels in rodents.27
Zero maze
The use of zero maze to measure thigmotaxis, indicative of anxiety, was carried out following previously published methods.28 The maze has an outer diameter of 24 inches, inner diameter of 20 inches, is 24 inches above the floor and the closed quadrants have 6 inch tall walls. Luminosity inside the open quadrant was measured to be 20 lux, while that inside the closed quadrant was measured to be 10 lux. Mice were placed facing one of the closed quadrants of the elevated zero maze to begin the 5 min test period. Total time spent in open and closed quadrants as well as total distance traveled were recorded.
Novel location recognition and novel object recognition
NLR and NOR tests were carried out in the same arena as the one used for the OF test to assess working memory.29 Exploration behavior was used to assess object location and recognition. Mice were placed into the arena from the middle of the south wall, with the north wall of the arena having a large visual cue (28 cm × 21.5 cm [W × H] sheet with alternating black and white columns [column width = 1.5 cm]). Each mouse was presented with two identical objects (OBJ1 and OBJ2) for 10 min (habituation). The two identical objects were placed side by side with equal distance from the east and west walls and were 30 cm away from the visual cue. The next day, mice were first given three 10 min trials with identical setting as the habituation day and the time spent exploring the objects was recorded. In the fourth 10 min trial, OBJ1 was moved toward the visual cue (novel location), and the time spent exploring OBJ1 in the new location was recorded. In the fifth 10 min trial, OBJ2 (the familiar object) was replaced by a new object (OBJ3, novel object), and the time spent exploring OBJ3 was recorded. During the inter-trial intervals of 5 min, mice were returned to their home cage. Objects were replaced with replicates after each trial, and 4% (vol/vol) acetic acid was used to clean and remove potential odors. Exploration behavior in the first 5 min was used to assess NLR and NOR. For NLR, the data were calculated as exploration ratio of time (seconds) spent exploring object in the novel location versus that for exploring object in its original location. The exploration ratio of time spent exploring the novel object versus that for exploring the familiar object is calculated in NOR. The exploration ratio for novel and familiar location/object is calculated as Tnovel/ (Tnovel + Tfamiliar) and Tfamiliar/(Tnovel + Tfamiliar), respectively, where T equals time. Mice that consistently failed to explore objects for a minimum of 5 s during the training sessions were excluded (a priori exclusion criterion) from the NOR and NLR tests (n=4).
Social Memory
Assessment of social recognition memory was carried out according to previously published methods.30–32 The social testing apparatus is a rectangular, three-chambered box. Each chamber measures 43 cm × 23 cm × 20 cm (W × H × L) in size (two interaction chambers and one neutral chamber). Dividing walls are made from clear Plexiglas (Taps Plastic; Mountain View, CA), with rectangular openings (10 cm × 5 cm) allowing access into each chamber. In each interaction chamber a stimulus cage (round wire cage, made in-house) was placed during the social testing. During testing, a “stranger” mouse was placed in one or both stimulus cages. The wire mesh separating the interaction chamber from the stimulus cage allows nose contact between test mouse and stranger mouse, while preventing aggressive interactions and also prevents the “stranger” mouse from initiating the social contact. All test mice were experimentally naive to the social interaction test and are habituated to the test room for at least 30 min prior to assessment. Stranger mice were male C57BL/6J housed in groups of four per cage and have previously been habituated to the round wire cages 24–48 h prior to testing for 15 min each side (30 min total time). Adaptation phase: Baseline adaptation was done by placing the test mouse in the central neutral chamber for a 5 min habituation period, followed by the partitions being raised and allowing the mouse to explore freely all three chambers for a 10 min period. During baseline assessments there was no “stranger” mouse in either of the stimulus cages. Time spent in each chamber was used as baseline data to assess general activity, willingness to circumnavigate the testing apparatus and to assure that no mouse exhibited a side preference. Locomotor activity and the total number of entries (transitions between chambers of the apparatus) during the 10 min baseline period were assessed. Social preference (sociability) test: Following baseline data collection, each mouse was returned to the neutral central chamber for 5 min with partitions closed. During this time, an unfamiliar mouse (stranger #1) was placed within one of the stimulus cages of the interaction chamber. The “doors” to the partitions were raised, allowing the test mouse to move freely throughout the three chambers for a 10 min test session. Location of the `stranger mouse' was alternated between the left and right stimulus cages on consecutive sessions. Measurements of explorations or sniffing directed towards a “stranger mouse” (nose within 1cm of the wire mesh and directed toward the stranger mouse) were collected in 5 min time bins. The data for the initial 5 min bin was then compared to exploration and sniffing behavior exhibited by the test mouse towards the empty stimulus cage wire mesh in the second interaction chamber. Memory testing phase: At the end of the first 10 min, each mouse was returned to its home cage and tested in a second 10 min session to quantify social memory. A second, unfamiliar mouse (stranger mouse #2) was placed into the previously empty stimulus cage. Stranger #1 and stranger #2 mice originate from different home cages and have never come into physical contact with each other or the test subject. Measurements were collected in an identical fashion to the social interaction test. Mice that consistently failed to explore each of the three chambers for a minimum of 90 s (15% of the total test time) during the initial 10 min exploratory period (when no stranger mice is present) were excluded (a priori exclusion criterion) from the social preference and social memory tests (n=4).
Brain histology and dendritic analysis
All analyses were done blind to the identity and experimental condition of the animal/tissue. Mice were randomly chosen from a cohort used for behavioral studies. They were anesthetized by 300 ul of ketamine/xylazine cocktail, followed by transcardiac perfusion with 0.9% saline solution at seven weeks after injury. Following the quick and careful removal from the skull, fresh brains were immersed in 10 ml of Golgi-cox stain for 10 days (room temperature and in the absence of light) and transferred to a solution of 30% sucrose for three days at 4°C. Coronal sections were cut at room temperature at 100 μm thickness on a vibratome (Leica VA 1200S; Leica Biosystems, Buffalo Grove, IL) and immersed in 0.3% gelatin prior to mounting on a slide. The stain was visualized by immersing the tissue in the developing solution for 5–10 min. Slides were then rinsed in distilled water, dehydrated through graded ethanol, mounted on slides with permount (Fisher Scientific SP15; Pittsburgh, PA) mounting medium and cover slipped.
Brain sections located within bregma −1.2 to −2.2 mm were imaged with a 20X objective at 3 μm steps using bright field imaging on a Zeiss LSM 700 microscope (Thornwood, NY). The Mouse Brain in Stereotaxic Coordinates33 was used as a guide to select appropriate brain sections and to identify areas covering perirhinal cortex and amygdala. The indentation (rhinal fissure) between the ectorhinal cortex and perirhinal cortex was used as a guide to draw a 100 μm × 50 μm rectangular box as the perirhinal cortex proper. On the other hand, the lateral ventricle was used as a guide to draw a circle of 500 μm in diameter to include lateral, basolateral, and basomedial amygdaloid nuclei. Images were analyzed as a 3 μm-step z-stack of 20 slices (20X objective magnification) using bright field imaging (Zeiss LSM 700).
Only Golgi-stained neurons with completely impregnated dendrites and spines were used. Two images per animal (within the designated boxed areas) were randomly chosen for analysis. All neurons in the selected images were numbered and identified using the “Mark and Count” tool in Image J (Bethesda, MD). Then, 10 IDs were randomly selected for further analysis in each z-stack image. Soma area was quantified using the Image J software. Dendritic length, number, and arborization were measured using manual tracing in Neuron J (Bethesda, MD). Sholl analysis was carried out using Fiji (Bethesda, MD) with the Sholl radius set at 5 μm (See supplemental digital content 2 for an illustration of this method). Due to the dense neuronal population in the hippocampus, Sholl analysis was not possible.
Furthermore, in all three brain regions studied, the total number of neurons and spine density were quantified using the “Mark and Count” tool in Image J. Dendritic spines in the amygdala, perirhinal cortex, and granule cells of the hippocampal dentate gyrus were identified based on specific morphological characteristics as reported previously.34 To determine spine density, images were acquired with 100X oil lens and secondary dendrites with a minimum of 10 μm in length were sampled. The segment of secondary dendrites within 10 μm of the branching point or 10 μm of dendritic terminals were excluded. Spine density is expressed as number of spines per 10 μm dendrite.
Protein quantification
All analysis was done blind to the identity and experimental condition of the animal/tissue. A separate cohort of mice was used for the biochemical measurements. Mice were anesthetized (isoflurane) and sacrificed by decapitation at seven weeks after injury. Following the quick and careful removal from the skull, the left amygdala, perirhinal cortex and hippocampus were extracted according to bregma coordinates and stored at −80°C until use.
Synaptosome preparation
Tissue was homogenized in 1XSyn-PER reagent (Thermo Scientific 87793; Pittsburgh, PA) containing “Halt” protease inhibitor cocktail (Thermo Scientific), centrifuged at 1000 ×g for 10 min to remove cell debris, and the supernatant was transferred to a new tube where it was centrifuged at 17,500 xg for 20 min. The pellets, containing synaptosomes, were gently re-suspended in 40 ul of the reagent (Syn-PER + “Halt”).
Two-color fluorescent western blot analysis
To assess the protein levels of the synaptophysin (also known as p38 synaptic protein) in our synaptosome preparation, Western blot analysis was performed according to standard procedures. Briefly, after SDS-PAGE and blotting, proteins on the membranes were detected by overnight incubation at 4°C with the primary antibody (mouse monoclonal anti-synaptophysin: 1:2000; Sigma S5768; St. Louis, MO) followed by incubation with an IRDye 800CW goat anti-mouse IgG (H+L) (1:20,000; LI-COR Biosciences 926-32210; Lincoln, NE). β-actin was used as an internal control and was detected with the mouse monoclonal anti-β-actin antibody (1:5000; Abcam ab6276; Cambridge, MA) followed by incubation with an IRDye 680CW goat anti-mouse IgG (H+L) (1:20,000; LI-COR Biosciences 926-32220). The signals were detected and quantified using Odyssey (LI-COR Biosciences).
Protein extraction and ELISA
Tissues were homogenized using T-PER Protein Extraction Reagent (Thermo Scientific 87793) in the presence of proteinase and phosphatase inhibitors (Roche Applied Science 04906837001; San Francisco, CA) and centrifuged at 12,000 ×g for 2 min at 4°C. Supernatant fractions were then frozen at −80°C until use. An aliquot was subjected to protein assay (Bio-Rad 500-0001; Hercules, CA) to normalize BDNF levels. The mouse BDNF ELISA kit was used per manufacturer's instructions (Boster Immunoleader EK0309; Pleasanton, CA).
Statistical analysis
All data are expressed as mean ± SEM. Analysis of repeated parametric measures was accomplished using a two-way analysis of variance (ANOVA) followed by Bonferroni testing. For simple comparisons of two groups, a two-tailed Student t-test was employed. Welch's correction was used when the assumption of equal variances was not met. For Sholl analysis, individual two-tailed Student t-tests were used with the Holm-Sidak method for multiple comparisons. Significance was set at p < 0.05. (Prism 5; GraphPad Software, La Jolla, CA). Sample size for each experimental endpoint is indicated in the figure legends.
RESULTS
Tibia fracture is accompanied by long-lasting nociceptive sensitization in the injured limb
Ipsilateral (Fig. 1A) and contralateral (Fig. 1B) measurements of hindpaw mechanical thresholds were performed at three, four, seven, 14, and 18 weeks post-fracture. Mechanical sensitivity, assessed using Von Frey filaments, demonstrated a significant and persistent reduction in mechanical thresholds – up to 14 weeks post-fracture - in the injured limb compared to measurements of the contralateral limb or control non-fractured/non-casted mice (2-way ANOVA [fracture factor F value=27.8] followed by Bonferroni testing). Testing for mechanical sensitivity of the contralateral hindpaw failed to demonstrate any significant nociceptive changes across the 18 week time course (Fig. 1B) (2-way ANOVA; [fracture factor F value=2.96] followed by Bonferroni testing). Although mechanical sensitivity was measured in all behavioral cohorts, only animals that were randomly chosen (n=8 for control and 11 for fracture) to be tested on the 14 and 18 week timepoints are included in this analysis. Earlier data from our group showed that by seven weeks the edema and warmth characteristic of acute CRPS and found at earlier time points had resolved.18
Figure 1. Long-lasting mechanical hypersensitivity in fracture/cast mice.
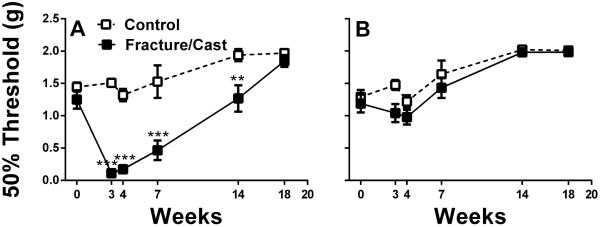
Fracture/cast mice display long-lasting mechanical hypersensitivity on the ipsilateral hindpaw 14 weeks after fracture (A). No significant differences in mechanical sensitivity were observed in the contralateral hindpaw for any of the groups (B). **p<0.01, *** p<0.001. n=8 (control) and n=11 (fracture/cast). Errors bars=S.E.M.
Fracture/cast mice exhibit signs of increased anxiety levels in the zero maze, but not open field assay in the absence of motor impairment
Open field testing
Fracture/cast mice did not differ from control in overall motor activity measured by the total distance traveled in the OF apparatus (fig. 2A and B; 2-tailed t-test, 31.94±2.05 m in control versus 28.01±1.70 m in fracture/cast mice, p=0.15). Similarly, they did not differ from control in the time spent in the center 11% of the OF (fig. 2C; 2-tailed t-test, 16.63±3.44 s in control versus 16.76±2.23 s in fracture/cast mice, p=0.98). Furthermore, time in center wasn't different between the two groups when the central area was designated to be 25% of the total area (2-tailed t-test, 39.32±5.74 s in controls and 38.03±3.74 s in fracture/cast mice, p=0.88; data not shown).
Figure 2. Fracture/cast mice show signs of anxiety.

Schematic representation of the open field apparatus highlighting the central square (A). Fracture/cast mice do not differ from control mice in the total distance traveled in the open field (B), nor do they differ in the time spent in the central square (C). Schematic representation of the zero maze apparatus (D). Fracture/cast mice do not differ from control mice in the total distance covered in the zero maze (E); however, they do differ in the time spent in the open quadrants (F). * p<0.05. n=16/group. Errors bars=S.E.M.
Zero Maze testing
Consistent with the results obtained from the OF assay, Fracture/cast mice did not differ from control in overall motor activity measured by the total distance traveled in the zero maze apparatus (fig. 2D and E; 2-tailed t-test, 12.01±0.85 m in control versus 11.08±0.76 m in fracture/cast mice, p=0.42). However, they spent less time in the open quadrants of the zero maze, a measure that is indicative of neophobic anxiety in mice (fig. 2F; 2-tailed t-test, 72.69±7.36 s in control versus 47.67±6.67 s in fracture/cast mice, p=0.02).
Example of track images of the OF and zero maze assays can be found in supplemental digital content 3.
Fracture/cast mice exhibit signs of memory impairment
Object location and recognition
Both control and fracture/cast groups performed equally well in the NLR test as they spent significantly greater time exploring the displaced object compared to the object that was not manipulated (fig. 3A and B; 2-way ANOVA [fracture factor F value~0; object factor F value=45.25] followed by Bonferroni testing). However, compared to control animals, the fracture/cast group performed poorly in the NOR test as they did not spend more time exploring the novel object compared to the familiar object (fig. 3C; 2-way ANOVA [fracture factor F value~0; object factor F value=4.65] followed by Bonferroni testing). The failure to differentiate novel from familiar objects is indicative of a working memory deficit.
Figure 3. Fracture/cast mice show signs of memory deficit.

Schematic representation of the open field apparatus highlighting the habituation (HAB), novel location recognition (NLR) and novel object recognition (NOR) testing configurations (A). Fracture/cast mice do not differ from control mice in the time spent exploring the displaced object (B), but they do differ in the time spent exploring a novel object (C). Schematic representation of the social memory testing apparatus (D). Fracture/cast mice do not differ from control mice in the total time spent socializing with a stranger mouse (E); however, fracture/cast mice do not spend more time socializing with a newly–introduced stranger (F). * p<0.05, *** p<0.001. n=16/group. Errors bars=S.E.M.
Social memory
Both control and fracture/cast groups performed equally well in the sociability test (fig. 3D) where they spent significantly more time exploring the chamber containing the stranger mouse relative to the chamber with the empty cage (fig. 3E; 2-way ANOVA [fracture factor F value=0.2; stranger mouse factor F value=54.13] followed by Bonferroni testing). However, the fracture/cast group did not perform as well as the control group in the social memory test. Fracture/cast animals did not spend more time exploring the new stranger mouse, a result that is indicative of diminished social memory (fig. 3F; 2-way ANOVA [fracture factor F value=0.005; stranger mouse factor F value=15.92] followed by Bonferroni testing).
Example of track images of the novel location recognition, novel object recognition, and social memory assays can be found in supplemental digital content 3.
Fracture/cast animals show altered dendritic architecture in the amygdala
In order to link the anxiety and memory deficits to structural changes in various brain regions, we quantified dendritic complexity in Golgi-Cox-stained sections. Sholl analysis revealed that, compared to control, fracture/cast mice show increased complexity in dendritic structure in the contralateral amygdala, as shown by the mean number of intersections of dendritic branches with consecutive 5 μm-spaced concentric Sholl rings (fig. 4A; individual 2-tailed t-tests with Holm-Sidak correction for multiple comparisons; area under the curve comparison: 2-tailed t-test, 238.5±13.92 in control versus 404.1±28.47 in fracture/cast mice, p=0.01). Furthermore, independent quantification of neuronal structure using direct counts revealed that the fracture/cast group has a significantly greater number of secondary nodes compared to control, indicative of increased dendritic branching (fig. 4B; 2-tailed t-test, 1.31±0.12 in control versus 2.75±0.35 in fracture/cast mice, p=0.01). No differences were seen in measures of dendritic length (fig. 4C; 2-tailed t-test, 243.7±15.08 μm in control versus 403.7±77.79 μm in fracture/cast mice, p=0.09, [Welch's correction applied]), average soma area (fig. 4D; 2-tailed t-test, 108.2±6.75 μm2 in control versus 109.5±7.43 μm2 in fracture/cast mice, p=0.9), total number of neurons (fig. 4E; 2-tailed t-test, 29.2±1.24 in control versus 30.71±2.81 in fracture/cast mice, p=0.67), or dendritic spine density (fig. 4F and 4G; 2-tailed t-test, 12.96±0.96 in control versus 11.86±1.11 in fracture/cast mice, p=0.49).
Figure 4. Dendritic architecture is changed in the amygdala of fracture/cast mice.

Fracture/cast mice display increased dendritic complexity in the amygdala as shown by the mean number of intersections of dendrite branches with consecutive 5μm-spaced concentric Sholl rings (A) and by calculating the area under the curve (AUC) for these two curves (A, inset). In addition, the fracture group has a significantly greater number of secondary nodes compared to control (B). No differences were seen in measures of total dendritic length (C), average soma area (D), total number of neurons (E), or dendritic spine density (F, G). Scale bar=1μm. * p<0.05, **p<0.01. n=5 (control) and n=7 (fracture/cast). Errors bars=S.E.M.
Fracture/cast animals show altered dendritic architecture in the perirhinal cortex
Similar to the results obtained in the amygdala, Sholl analysis revealed that, compared to control, fracture/cast mice show increased complexity in dendritic structure in the contralateral perirhinal cortex, as shown by the mean number of intersections of dendrite branches with concentrically-spaced Sholl rings (fig. 5A; individual 2-tailed t-tests with Holm-Sidak correction for multiple comparisons; area under the curve comparison: 2-tailed t-test, 304.9±32.33 in control versus 416.6±23.83 in fracture/cast mice, p=0.02). Independent quantification of neuronal structure revealed that the fracture/cast group has a significantly greater number of secondary nodes compared to control, indicative of increased dendritic branching (fig. 5B; 2-tailed t-test, 1.62±0.04 in control versus 2.57±0.29 in fracture/cast mice, p=0.03, [Welch's correction applied]). Furthermore, total dendritic length was shown to be greater in fracture/cast mice (fig. 5C; 2-tailed t-test, 280.4±12.69 μm in control versus 471.5±41.23 μm in fracture/cast mice, p=0.003, [Welch's correction applied]). No differences were seen in average soma area (fig. 5D; 2-tailed t-test, 205.3±15.23 μm2 in control versus 242.2±20.7 μm2 in fracture/cast mice, p=0.2), total number of neurons (fig. 5E; 2-tailed t-test, 17.2±0.80 in control versus 14.43±1.9 in fracture/cast mice, p=0.27), or dendritic spine density (fig. 5F and 5G; 2-tailed t-test, 18.04±1.4 in control versus 19.32±0.71 in fracture/cast mice, p=0.2).
Figure 5. Dendritic architecture is changed in the perirhinal cortex of fracture/cast mice.

Cast/fracture mice display increased dendritic complexity in the perirhinal cortex as shown by the mean number of intersections of dendrite branches with consecutive 5μm-spaced concentric Sholl rings (A) and by calculating the area under the curve (AUC) for these two curves (A, inset). In addition, the fracture group has a significantly greater number of secondary nodes compared to control (B) and a greater average of total dendritic length (C). No differences were observed in average soma area (D), total number of neurons (E), or dendritic spine density (F, G). Scale bar=1μm. * p<0.05, **p<0.01. n=5 (control) and n=7 (fracture/cast). Errors bars=S.E.M.
Fracture/cast animals show no obvious changes in dendritic morphology in the hippocampus
Unlike the amygdala and the perirhinal cortex, dendritic analysis was not undertaken in the hippocampus due high dendrite density. Instead, the number of neurons and the dendritic spine density were quantified. Our results show no significant change in the number of neurons (fig. 6A; 2-tailed t-test, 37.4±2.06 in control versus 39.5±3.96 in fracture/cast mice, p=0.67) or dendritic spines (fig. 6B and 6C; 2-tailed t-test, 13.88±1.87 in control versus 10.8±0.99 in fracture/cast mice, p=0.07).
Figure 6. No obvious dendritic changes in the hippocampus of fracture/cast mice.

Fracture/cast mice do not differ in the number of neurons (A) or dendritic spine density (B,C) in the hippocampus. Scale bar=1μm. n=5 (control) and n=7 (fracture/cast). Errors bars=S.E.M.
Fracture/cast animals show decreased levels of synaptophysin in the hippocampus
In order to complement the dendritic morphology analysis, we measured synaptophysin levels in synaptosomal preparations from the following brain regions: amygdala, perirhinal cortex, and hippocampus. Our results reveal that, despite our prior findings showing increased dendritic branching in the amygdala and perirhinal cortex, synaptophysin levels remained unchanged (compared to control) in both regions (fig. 7A; 2-tailed t-test, 1.53±0.15 in control versus 1.16±0.19 in fracture/cast mice, p=0.21, fig. 7B; 2-tailed t-test, 2.82±0.86 in control versus 2.13±0.22 in fracture/cast mice, p=0.26). In the hippocampus, however, the fracture mice exhibited decreased levels of synaptophysin, indicative of a reduction in the number of synapses35 (fig. 7C; 2-tailed t-test, 2.82±0.58 in control versus 1.01±0.07 in fracture/cast mice, p=0.001).
Figure 7. Decreased synaptophysin in the hippocampus of fracture/cast mice.
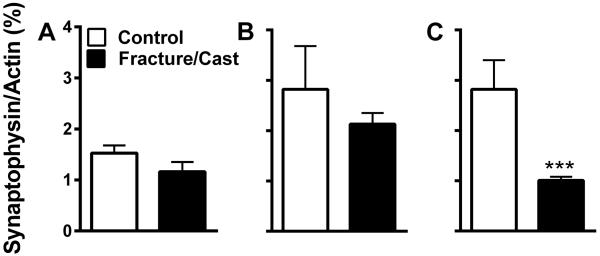
Compared to control mice, fracture/cast mice do not show differences in the levels of synaptophysin in the amygdala (A) or the perirhinal cortex (B). However, they do display decreased levels of synaptophysin in the hippocampus (C) measured by western blots of the synaptosomal preparation. *** p<0.001. n=5 (control) and n=7 (fracture/cast). Errors bars=S.E.M.
Fracture/cast animals show decreased BDNF levels in the perirhinal cortex and hippocampus
In order to test whether these morphological/synaptic changes are paralleled by changes in neurotrophic factors, we measured BDNF levels using ELISA. Our data shows that, in the amygdala, fracture/cast and control mice show similar levels of BDNF (fig. 8A; 2-tailed t-test, 1076±106 ng/g in control versus 1142±30.55 ng/g in fracture/cast mice, p=0.76). In contrast, compared to control mice, fracture/cast mice display decreased levels of BDNF in the perirhinal cortex (fig. 8B; 2-tailed t-test, 1398±66.53 ng/g in control versus 959.6±82.58 ng/g in fracture/cast mice, p=0.006) and the hippocampus (fig. 8C; 2-tailed t-test, 7312±346.3 ng/g in control versus 6335±214.6 ng/g in fracture/cast mice, p=0.03).
Figure 8. Decreased BDNF in the perirhinal cortex and hippocampus of fracture/cast mice.
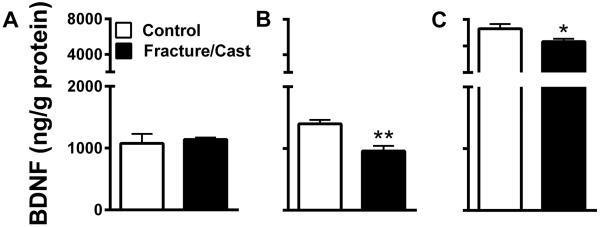
Fracture/cast mice do not differ from control in the levels of brain derived neurotrophic factor (BDNF) protein in the amygdala (A). However, they do display decreased levels of BDNF in the perirhinal cortex (B) and the hippocampus (C). * p<0.05, **p<0.01. n=5 (control) and n=7 (fracture/cast). Errors bars=S.E.M.
DISCUSSION
While pain itself is the most common complaint of the chronic pain patient, it is often associated with comorbid conditions such as cognitive impairment,36 memory deficits,37 depression,38 and anxiety.39,40 These affect up to 50% of chronic pain patients11,41,42 and impair both quality of life and response to treatment.43 In addition, CRPS patients reportedly demonstrate diminished capacity for tactile perceptual learning44 and altered performance in a gambling task, implying changes in emotional influences on decision making and risk taking.45 Neuropsychiatric testing on a group of 137 CRPS patients revealed deficits in 65% of patients, including subgroups having deficits in executive functioning, memory, and even global cognitive impairment.46 Furthermore, psychological factors such as depression and anxiety are strongly associated with CRPS severity.47,48 Taken together, these data emphasize that cognitive and affective impairments are significant clinical problems in chronic CRPS.
While most preclinical pain studies focus on mechanical sensitivity as the primary outcome, there has been increasing interest in the affective measures of pain and its associated comorbidities. For instance, there is evidence of anxiety49,50 and attention deficits51–55 in rodent models of pain, in addition to changes in mood50,56,57 and overall cognitive function58–60. Similar to what is seen in patients,61 some of these conditions subside once the pain is ameliorated.62 We have shown that selective signs of neophobic anxiety (in the form of altered thigmotaxis in the zero maze but not open field assay) and memory impairment (novel object recognition and social memory but not novel location recognition) are present in our fracture/cast mice.
What, then, is the neurobiological substrate of these behavioral changes? With the advent of noninvasive brain imaging techniques, we now have evidence of structural and functional brain changes directly associated with the pain experience in chronic neuropathy,63,64 fibromyalgia,48,65 migraine,66 chronic back pain67–69 and CRPS.70 While such studies have given us invaluable insight into the “pain” brain, there remains a need for a more detailed analysis of neuroplasticity in the affected areas. Based on our behavioral observations, we focused on three potential neurobiological substrates. The amygdala was chosen based on its role in processing threatening stimuli,20 mood,21 fear,71 emotional memory,22 anxiety,72,73 and the emotional-affective dimension of pain.74 The perirhinal cortex and hippocampus were chosen for their roles in memory. The participation of the hippocampus in memory has been described extensively (for example, see75,76), and can be studied using paradigms such as novel object location and social memory.77 There is accumulating evidence for the role of the perirhinal cortex in memory as well. Studies in humans, monkeys, and rodents suggest that this area is involved in signaling the familiarity of objects,78 in particular, novel object recognition23,79 and this may take place in a hippocampus-independent manner.24,78 It is noteworthy that, despite attributing certain behavioral outcomes to respective brain regions, there is considerable overlap between the roles that these regions play.80
One of the mechanisms of modulating synaptic plasticity is the differential localization of specific synaptic sites. As such, analysis of dendritic structure, morphology, length and arborization, in addition to quantification of spine density and levels of BDNF and the presynaptic protein synaptophysin are indicators of ongoing neural plasticity. Our behavioral observations of increased anxiety-related behaviors in the zero maze, impairment in novel object recognition, and impairment in social memory were paralleled by changes in the dendritic architecture of the amygdala and perirhinal cortex where data from our Golgi-Cox-stained sections showed increased dendritic branching and length in CRPS mice, without any changes in soma size. This is consistent with previous findings where dendritic architecture in the anterior cingulate cortex, somatosensory cortex and dorsal horn spinal cord neurons is altered in rodent pain models, 81,82 with the notable difference that dendritic spine density wasn't shown to change in our model. Remarkably, increased dendritic complexity is also associated with enriched environments,83 implying that these changes are not necessarily maladaptive in all cases. In the current study, dendritic analysis was not undertaken in the hippocampus due to the high dendrite density in that region making the Golgi-Cox-stained patterns difficult to analyze. Instead, quantification of spine density was carried out and revealed no statistically significant change in dendritic spine number. Furthermore, no change in the number of newborn neurons was observed between the fracture/cast and control group in this region (data not shown).
The dendritic architecture of a neuron is critical in determining the number and type of synaptic connections possessed by that neuron in addition to governing how synaptic inputs are integrated to produce a coded output. We therefore examined whether synaptic abundance parallels the increased dendritic branching. Our findings show that the histological changes indicative of increased dendritic complexity occur in the amygdala and perirhinal cortex in the absence of increased synaptophysin, an indirect measure of synaptic numbers.35 Furthermore, synaptophysin levels are decreased in hippocampi isolated from CRPS mice, and trended towards lower levels in homogenates of tissue from the perirhinal cortex and amygdala. These data agree with our histological measures of spine density. It is possible that although CRPS brains display patterns of increased dendritic branching, they don't necessarily form more synapses compared to control mice, a hypothesis that is consistent with the anxiety and memory deficits observed in this model. Alternatively, it is possible that dendritic branching is a consequence of, or compensation for, synaptic loss. These data are in agreement with previous findings showing that decreased levels of synaptophysin are accompanied by impairment in cognitive function and memory84 and vice versa.85 In contrast, some brain regions (such as the prefrontal cortex) display increased levels of synaptic proteins,86 including synaptophysin,87 following chronic painful conditions.
Finally, brain plasticity can be assessed by quantifying levels of neurotrophins such as BDNF. BDNF is a potent modulator of synaptic plasticity,88,89 due at least in part to co-localization with its cognate receptor TrkB at glutamatergic synapses.90 The role of BDNF in pain has been extensively studied in preclinical models at the spinal level, and increased spinal BDNF is thought to contribute to central sensitization.91 In addition, levels of BDNF increase in the periaqueductal gray and rostral ventromedial medulla in animal models of pain, possibly contributing to descending pain facilitation.92 In contrast, BDNF is decreased in the hippocampus following painful stimuli,93,94 but its levels haven't been measured in the more chronic stages of pain, where comorbidities often appear. We observed a significant decrease in BDNF levels in the perirhinal cortex and hippocampus in agreement with other rodent studies where depression and anxiety/stress are associated with signs of memory impairment95 and decreased levels of BDNF in the hippocampus.96,97 Finally, our findings failed to show changes in BDNF levels in the amygdala, despite previous studies linking increased BDNF to stress and trauma.98 It is possible that a more careful examination of the subregions of the amygdala is needed in order to address this disparity. Alternatively, it is possible that anxiety levels in our fracture/cast mice (detectable by zero maze and not open field) are not “high” enough to be associated with BDNF changes.
We believe that the approach of measuring neurocognitive and affective components of pain in conjunction with neuroplastic CNS changes in models such as the fracture/cast animals is crucial in understanding the multi-faceted nature of chronic pain. In this way we will build a “language of translation”, helping us to focus mechanistically-oriented animal studies on regions of the CNS implicated in human studies, and perhaps to focus human studies on approaches to treatment or prevention of the CNS changes that are optimized using animal models.
There are several limitations to the present set of studies. For example, we focused only on the hippocampus, amygdala and perirhinal cortex. While these regions are involved in anxiety and memory, areas such as the prefrontal cortex, the somatosensory cortex, and the thalamus are also important to examine since each of these has been associated with brain changes in preclinical models of pain99 and in chronic pain patients.48,67,100 In addition, we have not identified any of the specific mechanisms linking peripheral injury and immobilization with the CNS changes. Future studies examining the molecular underpinnings of the fracture/cast-related changes in the brain, perhaps with particular emphasis on epigenetic changes, are needed. Epigenetic mechanisms are well suited to modulate the long-lasting CNS changes that accompany chronic pain.86,99,101
Collectively, these data provide unique evidence for structural and biochemical changes in the brain that accompany selective measures of anxiety (changes in thigmotactic behaviors in the zero maze but not the open field assay) and working memory (novel object recognition but not novel location recognition) and social memory impairment in a mouse model of tibia fracture/cast immobilization. These findings have clear clinical implications for the CRPS patient, since, in addition to elucidating some of the supraspinal correlates of the syndrome, this work supports the potential use of therapeutic interventions that not only directly target sensory input and other peripheral mechanisms, but also attempt to ameliorate the pain experience by modifying its associated cognitive and emotional comorbidities.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Frances Davies, PhD (Palo Alto Institute of Research and Education, Palo Alto, CA) for technical assistance.
SUPPORT This study was supported by National Institute of Health grant NS072168, Bethesda, MD to Wade S Kingery, PhD, and J David Clark, MD/PhD (Palo Alto Institute of Research and Education, Palo Alto, CA), and the Department of Veterans Affairs merit review grant to Ting Ting Huang, PhD (Veterans Affairs Palo Alto Health Care System Palo Alto, CA).
ABBREVIATED TITLE
- CRPS
comorbidities and brain neuroplasticity
Footnotes
CONFLICT OF INTEREST The authors report no conflict of interest.
REFERENCES
- 1.de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: A population-based study. Pain. 2007;129:12–20. doi: 10.1016/j.pain.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Goebel A. Complex regional pain syndrome in adults. Rheumatology (Oxford) 2011;50:1739–50. doi: 10.1093/rheumatology/ker202. [DOI] [PubMed] [Google Scholar]
- 3.Birklein F, Riedl B, Sieweke N, Weber M, Neundorfer B. Neurological findings in complex regional pain syndromes: Analysis of 145 cases. Acta Neurol Scand. 2000;101:262–9. doi: 10.1034/j.1600-0404.2000.101004262x./. [DOI] [PubMed] [Google Scholar]
- 4.Sieweke N, Birklein F, Riedl B, Neundorfer B, Handwerker HO. Patterns of hyperalgesia in complex regional pain syndrome. Pain. 1999;80:171–7. doi: 10.1016/s0304-3959(98)00200-0. [DOI] [PubMed] [Google Scholar]
- 5.Cossins L, Okell RW, Cameron H, Simpson B, Poole HM, Goebel A. Treatment of complex regional pain syndrome in adults: A systematic review of randomized controlled trials published from June 2000 to February 2012. Eur J Pain. 2013;17:158–73. doi: 10.1002/j.1532-2149.2012.00217.x. [DOI] [PubMed] [Google Scholar]
- 6.Dirckx M, Stronks DL, Groeneweg G, Huygen FJ. Effect of immunomodulating medications in complex regional pain syndrome: A systematic review. Clin J Pain. 2012;28:355–63. doi: 10.1097/AJP.0b013e31822efe30. [DOI] [PubMed] [Google Scholar]
- 7.Subbarao J, Stillwell GK. Reflex sympathetic dystrophy syndrome of the upper extremity: Analysis of total outcome of management of 125 cases. Arch Phys Med Rehabil. 1981;62:549–54. [PubMed] [Google Scholar]
- 8.Harden RN, Bruehl S, Perez RS, Birklein F, Marinus J, Maihofner C, Lubenow T, Buvanendran A, Mackey S, Graciosa J, Mogilevski M, Ramsden C, Schlereth T, Chont M, Vatine JJ. Development of a severity score for CRPS. Pain. 2010;151:870–6. doi: 10.1016/j.pain.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 9.Lohnberg JA, Altmaier EM. A review of psychosocial factors in complex regional pain syndrome. J Clin Psychol Med Settings. 2013;20:247–54. doi: 10.1007/s10880-012-9322-3. [DOI] [PubMed] [Google Scholar]
- 10.Berryman C, Stanton TR, Jane Bowering K, Tabor A, McFarlane A, Lorimer Moseley G. Evidence for working memory deficits in chronic pain: A systematic review and meta-analysis. Pain. 2013;154:1181–96. doi: 10.1016/j.pain.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Landro NI, Fors EA, Vapenstad LL, Holthe O, Stiles TC, Borchgrevink PC. The extent of neurocognitive dysfunction in a multidisciplinary pain centre population. Is there a relation between reported and tested neuropsychological functioning? Pain. 2013;154:972–7. doi: 10.1016/j.pain.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Linnman C, Becerra L, Borsook D. Inflaming the brain: CRPS a model disease to understand neuroimmune interactions in chronic pain. J Neuroimmune Pharmacol. 2013;8:547–63. doi: 10.1007/s11481-012-9422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwenkreis P, Maier C, Tegenthoff M. Functional imaging of central nervous system involvement in complex regional pain syndrome. AJNR Am J Neuroradiol. 2009;30:1279–84. doi: 10.3174/ajnr.A1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coderre TJ, Xanthos DN, Francis L, Bennett GJ. Chronic post-ischemia pain (CPIP): A novel animal model of complex regional pain syndrome-type I (CRPS-I; reflex sympathetic dystrophy) produced by prolonged hindpaw ischemia and reperfusion in the rat. Pain. 2004;112:94–105. doi: 10.1016/j.pain.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Siegel SM, Lee JW, Oaklander AL. Needlestick distal nerve injury in rats models symptoms of complex regional pain syndrome. Anesth Analg. 2007;105:1820–9. doi: 10.1213/01.ane.0000295234.21892.bc. [DOI] [PubMed] [Google Scholar]
- 16.Bove GM. Focal nerve inflammation induces neuronal signs consistent with symptoms of early complex regional pain syndromes. Exp Neurol. 2009;219:223–7. doi: 10.1016/j.expneurol.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 17.Guo TZ, Offley SC, Boyd EA, Jacobs CR, Kingery WS. Substance P signaling contributes to the vascular and nociceptive abnormalities observed in a tibial fracture rat model of complex regional pain syndrome type I. Pain. 2004;108:95–107. doi: 10.1016/j.pain.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Guo TZ, Wei T, Shi X, Li WW, Hou S, Wang L, Tsujikawa K, Rice KC, Cheng K, Clark DJ, Kingery WS. Neuropeptide deficient mice have attenuated nociceptive, vascular, and inflammatory changes in a tibia fracture model of complex regional pain syndrome. Mol Pain. 2012;8:85. doi: 10.1186/1744-8069-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher JJ, Tajerian M, Guo T, Shi X, Li W, Zheng M, Peltz G, Kingery W, Clark JD. Acute and chronic phases of complex regional pain syndrome in mice are accompanied by distinct transcriptional changes in the spinal cord. Mol Pain. 2013;9:40. doi: 10.1186/1744-8069-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasan RO, Nguyen NK, Weger S, Sartori SB, Singewald N, Heilbronn R, Herzog H, Sperk G. The central and basolateral amygdala are critical sites of neuropeptide Y/Y2 receptor-mediated regulation of anxiety and depression. J Neurosci. 2010;30:6282–90. doi: 10.1523/JNEUROSCI.0430-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seymour B, Dolan R. Emotion, decision making, and the amygdala. Neuron. 2008;58:662–71. doi: 10.1016/j.neuron.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: From animal models to human behavior. Neuron. 2005;48:175–87. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Mumby DG, Glenn MJ, Nesbitt C, Kyriazis DA. Dissociation in retrograde memory for object discriminations and object recognition in rats with perirhinal cortex damage. Behav Brain Res. 2002;132:215–26. doi: 10.1016/s0166-4328(01)00444-2. [DOI] [PubMed] [Google Scholar]
- 24.Barker GR, Warburton EC. When is the hippocampus involved in recognition memory? J Neurosci. 2011;31:10721–31. doi: 10.1523/JNEUROSCI.6413-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terkelsen AJ, Bach FW, Jensen TS. Experimental forearm immobilization in humans induces cold and mechanical hyperalgesia. Anesthesiology. 2008;109:297–307. doi: 10.1097/ALN.0b013e31817f4c9d. [DOI] [PubMed] [Google Scholar]
- 26.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 27.Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur J Pharmacol. 2003;463:3–33. doi: 10.1016/s0014-2999(03)01272-x. [DOI] [PubMed] [Google Scholar]
- 28.Shepherd JK, Grewal SS, Fletcher A, Bill DJ, Dourish CT. Behavioural and pharmacological characterisation of the elevated “zero-maze“ as an animal model of anxiety. Psychopharmacology (Berl) 1994;116:56–64. doi: 10.1007/BF02244871. [DOI] [PubMed] [Google Scholar]
- 29.Mumby DG, Gaskin S, Glenn MJ, Schramek TE, Lehmann H. Hippocampal damage and exploratory preferences in rats: memory for objects, places, and contexts. Learn Mem. 2002;9:49–57. doi: 10.1101/lm.41302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res. 2008;187:207–20. doi: 10.1016/j.bbr.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inta D, Vogt MA, Perreau-Lenz S, Schneider M, Pfeiffer N, Wojcik SM, Spanagel R, Gass P. Sensorimotor gating, working and social memory deficits in mice with reduced expression of the vesicular glutamate transporter VGLUT1. Behav Brain Res. 2012;228:328–32. doi: 10.1016/j.bbr.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, Frahm J, Fischer J, Bourgeron T, Ehrenreich H, Brose N. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA. 2008;105:1710–5. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paxinos G, Franklin KBJ, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd edition Academic Press; San Diego: 2001. [Google Scholar]
- 34.Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol. 2006;198:401–15. doi: 10.1016/j.expneurol.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR. Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol. 1996;25:821–8. doi: 10.1007/BF02284844. [DOI] [PubMed] [Google Scholar]
- 36.McCracken LM, Iverson GL. Predicting complaints of impaired cognitive functioning in patients with chronic pain. J Pain Symptom Manage. 2001;21:392–6. doi: 10.1016/s0885-3924(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 37.Schnurr RF, MacDonald MR. Memory complaints in chronic pain. Clin J Pain. 1995;11:103–11. doi: 10.1097/00002508-199506000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: Antecedent or consequence of chronic pain? A review. Clin J Pain. 1997;13:116–37. doi: 10.1097/00002508-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Dersh J, Polatin PB, Gatchel RJ. Chronic pain and psychopathology: Research findings and theoretical considerations. Psychosom Med. 2002;64:773–86. doi: 10.1097/01.psy.0000024232.11538.54. [DOI] [PubMed] [Google Scholar]
- 40.McWilliams LA, Cox BJ, Enns MW. Mood and anxiety disorders associated with chronic pain: An examination in a nationally representative sample. Pain. 2003;106:127–33. doi: 10.1016/s0304-3959(03)00301-4. [DOI] [PubMed] [Google Scholar]
- 41.Geisser ME, Roth RS, Robinson ME. Assessing depression among persons with chronic pain using the Center for Epidemiological Studies-Depression Scale and the Beck Depression Inventory: A comparative analysis. Clin J Pain. 1997;13:163–70. doi: 10.1097/00002508-199706000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Gerrits MM, van Oppen P, van Marwijk HW, Penninx BW, van der Horst HE. Pain and the onset of depressive and anxiety disorders. Pain. 2014;155:53–9. doi: 10.1016/j.pain.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Bromley Milton M, Borsbo B, Rovner G, Lundgren-Nilsson A, Stibrant-Sunnerhagen K, Gerdle B. Is Pain Intensity Really That Important to Assess in Chronic Pain Patients? A Study Based on the Swedish Quality Registry for Pain Rehabilitation (SQRP) PLoS One. 2013;8:e65483. doi: 10.1371/journal.pone.0065483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maihofner C, DeCol R. Decreased perceptual learning ability in complex regional pain syndrome. Eur J Pain. 2007;11:903–9. doi: 10.1016/j.ejpain.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004;108:129–36. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 46.Libon DJ, Schwartzman RJ, Eppig J, Wambach D, Brahin E, Peterlin BL, Alexander G, Kalanuria A. Neuropsychological deficits associated with Complex Regional Pain Syndrome. J Int Neuropsychol Soc. 2010;16:566–73. doi: 10.1017/S1355617710000214. [DOI] [PubMed] [Google Scholar]
- 47.Kim SK, Eto K, Nabekura J. Synaptic structure and function in the mouse somatosensory cortex during chronic pain: In vivo two-photon imaging. Neural Plast. 2012;2012:640259. doi: 10.1155/2012/640259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen KB, Loitoile R, Kosek E, Petzke F, Carville S, Fransson P, Marcus H, Williams SC, Choy E, Mainguy Y, Vitton O, Gracely RH, Gollub R, Ingvar M, Kong J. Patients with fibromyalgia display less functional connectivity in the brain's pain inhibitory network. Mol Pain. 2012;8:32. doi: 10.1186/1744-8069-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Song Y, Yang J, Zhang Y, Zhao P, Zhu XJ, Su HC. The contribution of TNF-alpha in the amygdala to anxiety in mice with persistent inflammatory pain. Neurosci Lett. 2013;541:275–80. doi: 10.1016/j.neulet.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki T, Amata M, Sakaue G, Nishimura S, Inoue T, Shibata M, Mashimo T. Experimental neuropathy in mice is associated with delayed behavioral changes related to anxiety and depression. Anesth Analg. 2007;104:1570–7. doi: 10.1213/01.ane.0000261514.19946.66. [DOI] [PubMed] [Google Scholar]
- 51.Low LA, Millecamps M, Seminowicz DA, Naso L, Thompson SJ, Stone LS, Bushnell MC. Nerve injury causes long-term attentional deficits in rats. Neurosci Lett. 2012;529:103–7. doi: 10.1016/j.neulet.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 52.Millecamps M, Etienne M, Jourdan D, Eschalier A, Ardid D. Decrease in non-selective, non-sustained attention induced by a chronic visceral inflammatory state as a new pain evaluation in rats. Pain. 2004;109:214–24. doi: 10.1016/j.pain.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 53.Ford GK, Moriarty O, McGuire BE, Finn DP. Investigating the effects of distracting stimuli on nociceptive behaviour and associated alterations in brain monoamines in rats. Eur J Pain. 2008;12:970–9. doi: 10.1016/j.ejpain.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 54.Boyette-Davis JA, Thompson CD, Fuchs PN. Alterations in attentional mechanisms in response to acute inflammatory pain and morphine administration. Neuroscience. 2008;151:558–63. doi: 10.1016/j.neuroscience.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 55.Pais-Vieira M, Lima D, Galhardo V. Sustained attention deficits in rats with chronic inflammatory pain. Neurosci Lett. 2009;463:98–102. doi: 10.1016/j.neulet.2009.07.050. [DOI] [PubMed] [Google Scholar]
- 56.Yalcin I, Bohren Y, Waltisperger E, Sage-Ciocca D, Yin JC, Freund-Mercier MJ, Barrot M. A time-dependent history of mood disorders in a murine model of neuropathic pain. Biol Psychiatry. 2011;70:946–53. doi: 10.1016/j.biopsych.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 57.Leitl MD, Onvani S, Bowers MS, Cheng K, Rice KC, Carlezon WA, Jr, Banks ML, Negus SS. Pain-related depression of the mesolimbic dopamine system in rats: Expression, blockade by analgesics, and role of endogenous kappa-opioids. Neuropsychopharmacology. 2014;39:614–24. doi: 10.1038/npp.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kodama D, Ono H, Tanabe M. Increased hippocampal glycine uptake and cognitive dysfunction after peripheral nerve injury. Pain. 2011;152:809–17. doi: 10.1016/j.pain.2010.12.029. [DOI] [PubMed] [Google Scholar]
- 59.Gregoire S, Michaud V, Chapuy E, Eschalier A, Ardid D. Study of emotional and cognitive impairments in mononeuropathic rats: effect of duloxetine and gabapentin. Pain. 2012;153:1657–63. doi: 10.1016/j.pain.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 60.Ren WJ, Liu Y, Zhou LJ, Li W, Zhong Y, Pang RP, Xin WJ, Wei XH, Wang J, Zhu HQ, Wu CY, Qin ZH, Liu G, Liu XG. Peripheral nerve injury leads to working memory deficits and dysfunction of the hippocampus by upregulation of TNF-alpha in rodents. Neuropsychopharmacology. 2011;36:979–92. doi: 10.1038/npp.2010.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koffler SP, Hampstead BM, Irani F, Tinker J, Kiefer RT, Rohr P, Schwartzman RJ. The neurocognitive effects of 5 day anesthetic ketamine for the treatment of refractory complex regional pain syndrome. Arch Clin Neuropsychol. 2007;22:719–29. doi: 10.1016/j.acn.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 62.Vachon P, Millecamps M, Low L, Thompsosn SJ, Pailleux F, Beaudry F, Bushnell CM, Stone LS. Alleviation of chronic neuropathic pain by environmental enrichment in mice well after the establishment of chronic pain. Behav Brain Funct. 2013;9:22. doi: 10.1186/1744-9081-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Q, Inman RD, Davis KD. Neuropathic pain in ankylosing spondylitis: A psychophysics and brain imaging study. Arthritis Rheum. 2013;65:1494–503. doi: 10.1002/art.37920. [DOI] [PubMed] [Google Scholar]
- 64.Widerstrom-Noga E, Pattany PM, Cruz-Almeida Y, Felix ER, Perez S, Cardenas DD, Martinez-Arizala A. Metabolite concentrations in the anterior cingulate cortex predict high neuropathic pain impact after spinal cord injury. Pain. 2013;154:204–12. doi: 10.1016/j.pain.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson ME, Craggs JG, Price DD, Perlstein WM, Staud R. Gray matter volumes of pain-related brain areas are decreased in fibromyalgia syndrome. J Pain. 2011;12:436–43. doi: 10.1016/j.jpain.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Messina R, Rocca MA, Colombo B, Valsasina P, Horsfield MA, Copetti M, Falini A, Comi G, Filippi M. Cortical abnormalities in patients with migraine: A surface-based analysis. Radiology. 2013;268:170–80. doi: 10.1148/radiol.13122004. [DOI] [PubMed] [Google Scholar]
- 67.Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004;24:10410–5. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt-Wilcke T, Leinisch E, Ganssbauer S, Draganski B, Bogdahn U, Altmeppen J, May A. Affective components and intensity of pain correlate with structural differences in gray matter in chronic back pain patients. Pain. 2006;125:89–97. doi: 10.1016/j.pain.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 69.Seminowicz DA, Wideman TH, Naso L, Hatami-Khoroushahi Z, Fallatah S, Ware MA, Jarzem P, Bushnell MC, Shir Y, Ouellet JA, Stone LS. Effective treatment of chronic low back pain in humans reverses abnormal brain anatomy and function. J Neurosci. 2011;31:7540–50. doi: 10.1523/JNEUROSCI.5280-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mutso AA, Radzicki D, Baliki MN, Huang L, Banisadr G, Centeno MV, Radulovic J, Martina M, Miller RJ, Apkarian AV. Abnormalities in hippocampal functioning with persistent pain. J Neurosci. 2012;32:5747–56. doi: 10.1523/JNEUROSCI.0587-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–63. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Padival MA, Blume SR, Rosenkranz JA. Repeated restraint stress exerts different impact on structure of neurons in the lateral and basal nuclei of the amygdala. Neuroscience. 2013;246:230–42. doi: 10.1016/j.neuroscience.2013.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qin M, Xia Z, Huang T, Smith CB. Effects of chronic immobilization stress on anxiety-like behavior and basolateral amygdala morphology in Fmr1 knockout mice. Neuroscience. 2011;194:282–90. doi: 10.1016/j.neuroscience.2011.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–34. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- 75.Gerlai R. Behavioral tests of hippocampal function: Simple paradigms complex problems. Behav Brain Res. 2001;125:269–77. doi: 10.1016/s0166-4328(01)00296-0. [DOI] [PubMed] [Google Scholar]
- 76.Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci USA. 2004;101:14515–20. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56. doi: 10.1002/(SICI)1098-1063(2000)10:1<47::AID-HIPO5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 78.Ranganath C, Ritchey M. Two cortical systems for memory-guided behaviour. Nat Rev Neurosci. 2012;13:713–26. doi: 10.1038/nrn3338. [DOI] [PubMed] [Google Scholar]
- 79.Baxter MG, Murray EA. Opposite relationship of hippocampal and rhinal cortex damage to delayed nonmatching-to-sample deficits in monkeys. Hippocampus. 2001;11:61–71. doi: 10.1002/1098-1063(2001)11:1<61::AID-HIPO1021>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 80.Deacon RM, Bannerman DM, Rawlins JN. Anxiolytic effects of cytotoxic hippocampal lesions in rats. Behav Neurosci. 2002;116:494–7. doi: 10.1037//0735-7044.116.3.494. [DOI] [PubMed] [Google Scholar]
- 81.Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG. Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci. 2012;32:6795–807. doi: 10.1523/JNEUROSCI.1017-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao X, Yang Y, Zhang Y, Zhang XM, Zhao ZQ, Zhang YQ. Estrogen in the anterior cingulate cortex contributes to pain-related aversion. Cereb Cortex. 2013;23:2190–203. doi: 10.1093/cercor/bhs201. [DOI] [PubMed] [Google Scholar]
- 83.Greenough WT, Volkmar FR. Pattern of dendritic branching in occipital cortex of rats reared in complex environments. Exp Neurol. 1973;40:491–504. doi: 10.1016/0014-4886(73)90090-3. [DOI] [PubMed] [Google Scholar]
- 84.Lassmann H, Weiler R, Fischer P, Bancher C, Jellinger K, Floor E, Danielczyk W, Seitelberger F, Winkler H. Synaptic pathology in Alzheimer's disease: Immunological data for markers of synaptic and large dense-core vesicles. Neuroscience. 1992;46:1–8. doi: 10.1016/0306-4522(92)90003-k. [DOI] [PubMed] [Google Scholar]
- 85.Ya BL, Liu WY, Ge F, Zhang YX, Zhu BL, Bai B. Dietary cholesterol alters memory and synaptic structural plasticity in young rat brain. Neurol Sci. 2013;34:1355–65. doi: 10.1007/s10072-012-1241-4. [DOI] [PubMed] [Google Scholar]
- 86.Alvarado S, Tajerian M, Millecamps M, Suderman M, Stone LS, Szyf M. Peripheral nerve injury is accompanied by chronic transcriptome-wide changes in the mouse prefrontal cortex. Mol Pain. 2013;9:21. doi: 10.1186/1744-8069-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hung KL, Wang SJ, Wang YC, Chiang TR, Wang CC. Upregulation of presynaptic proteins and protein kinases associated with enhanced glutamate release from axonal terminals (synaptosomes) of the medial prefrontal cortex in rats with neuropathic pain. Pain. 2014;155:377–87. doi: 10.1016/j.pain.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 88.Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–45. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 89.Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 91.Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: Peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA. 1999;96:9385–90. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling: A novel mechanism for descending pain facilitation. J Neurosci. 2006;26:126–37. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duric V, McCarson KE. Neurokinin-1 (NK-1) receptor and brain-derived neurotrophic factor (BDNF) gene expression is differentially modulated in the rat spinal dorsal horn and hippocampus during inflammatory pain. Mol Pain. 2007;3:32. doi: 10.1186/1744-8069-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duric V, McCarson KE. Persistent pain produces stress-like alterations in hippocampal neurogenesis and gene expression. J Pain. 2006;7:544–55. doi: 10.1016/j.jpain.2006.01.458. [DOI] [PubMed] [Google Scholar]
- 95.Patki G, Solanki N, Atrooz F, Allam F, Salim S. Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res. 2013;1539:73–86. doi: 10.1016/j.brainres.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blugeot A, Rivat C, Bouvier E, Molet J, Mouchard A, Zeau B, Bernard C, Benoliel JJ, Becker C. Vulnerability to depression: From brain neuroplasticity to identification of biomarkers. J Neurosci. 2011;31:12889–99. doi: 10.1523/JNEUROSCI.1309-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duric V, McCarson KE. Hippocampal neurokinin-1 receptor and brain-derived neurotrophic factor gene expression is decreased in rat models of pain and stress. Neuroscience. 2005;133:999–1006. doi: 10.1016/j.neuroscience.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 98.Bennett MR, Lagopoulos J. Stress and trauma: BDNF control of dendritic-spine formation and regression. Prog Neurobiol. 2014;112:80–99. doi: 10.1016/j.pneurobio.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 99.Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One. 2013;8:e55259. doi: 10.1371/journal.pone.0055259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Linnman C, Becerra L, Lebel A, Berde C, Grant PE, Borsook D. Transient and persistent pain induced connectivity alterations in pediatric complex regional pain syndrome. PLoS One. 2013;8:e57205. doi: 10.1371/journal.pone.0057205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Uchida H, Matsushita Y, Ueda H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: Implications in the development of neuropathic pain. Neuroscience. 2013;240:147–54. doi: 10.1016/j.neuroscience.2013.02.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.