

Background: Lou Gehrig's disease is accompanied by misfolding and aggregation of proteins.
Results: The intramolecular reactivity of cysteines of superoxide dismutase 1 results in new disulfide bonds in unusual positions.
Conclusion: Thiol/disulfide exchange reactions are altered in mutated proteins.
Significance: Intramolecular reorganization of disulfides may be one of the mechanisms of protein misfolding related to neurodegenerative diseases.
Keywords: Amyotrophic Lateral Sclerosis (ALS) (Lou Gehrig disease), Atomic Force Microscopy (AFM), Cysteine-mediated Cross-linking, Disulfide, Neurodegenerative Disease, Protein Misfolding, Superoxide Dismutase (SOD), Atomic Force Spectroscopy
Abstract
Neurodegenerative diseases share a common characteristic, the presence of intracellular or extracellular deposits of protein aggregates in nervous tissues. Amyotrophic Lateral Sclerosis (ALS) is a severe and fatal neurodegenerative disorder, which affects preferentially motoneurons. Changes in the redox state of superoxide dismutase 1 (SOD1) are associated with the onset and development of familial forms of ALS. In human SOD1 (hSOD1), a conserved disulfide bond and two free cysteine residues can engage in anomalous thiol/disulfide exchange resulting in non-native disulfides, a hallmark of ALS that is related to protein misfolding and aggregation. Because of the many competing reaction pathways, traditional bulk techniques fall short at quantifying individual thiol/disulfide exchange reactions. Here, we adapt recently developed single-bond chemistry techniques to study individual disulfide isomerization reactions in hSOD1. Mechanical unfolding of hSOD1 leads to the formation of a polypeptide loop held by the disulfide. This loop behaves as a molecular jump rope that brings reactive Cys-111 close to the disulfide. Using force-clamp spectroscopy, we monitor nucleophilic attack of Cys-111 at either sulfur of the disulfide and determine the selectivity of the reaction. Disease-causing mutations G93A and A4V show greatly altered reactivity patterns, which may contribute to the progression of familial ALS.
Introduction
Amyotrophic Lateral Sclerosis (ALS)5 is a fatal neurodegenerative disease that causes progressive paralysis and death within 3–5 years of diagnosis (1). Many familial cases of ALS are linked to any of the more than 150 described mutations in the superoxide dismutase 1 (SOD1) gene (2). However, the mechanisms by which apparently unrelated mutations in SOD1 lead to the same lethal phenotype remain unknown. The effects of the mutations in the stability and catalytic activity of SOD1 are heterogeneous (3–5). Indeed, the lethality of mutated enzymes has been proposed to arise from the gain of a toxic function, rather than loss of function (6, 7). This idea is strengthened by the fact that SOD1 knock-out mice do not develop symptoms of the disease (8).
SOD1 is a redox-active enzyme that catalyzes the disproportionation of two molecules of superoxide anion (O2⨪) into O2 and H2O2. Human SOD1 (hSOD1) is a homodimer in which each subunit is composed of eight antiparallel beta strands zipped by hydrogen bonds and building up a Greek key motif (9). hSOD1 contains four cysteines at positions 6, 57, 111, and 146 (Fig. 1A). A conserved disulfide bond between Cys-57 and Cys-146 determines a structural loop that contains more than one-half of the molecule (Fig. 1, A and B). The presence of Cys-111 in this loop (Fig. 1, A and B) is a unique hallmark of the enzyme in primates, humans, and chicken (10).
FIGURE 1.

Construction of AFM-ready polyproteins to study hSOD1. A, representation of a single monomer of hSOD1 (Protein Data Bank, accession number 2C9V). The N terminus (Nt) and the C terminus (Ct) are indicated. The four cysteine residues appear as yellow sticks and are labeled according to their position in the sequence of hSOD1. Cys-57 and -146 form an intramolecular disulfide bond, which determines a loop containing Cys-111. The segment of the loop between Cys-57 to Cys-111 is colored in blue, and the rest of the loop, up to Cys-146, is colored in green. For clarity in the representation, metal ions are not shown. B, schematic representation of the loop defined by the disulfide in hSOD1. C, scheme of the polyprotein construct I273-SOD-I273. A single molecule of hSOD1 (blue) is placed between six modules of I27 protein (yellow). In our experiments, single polyprotein molecules are attached between the AFM cantilever and a gold-covered surface. Mechanical force is generated by retracting the gold-covered surface using a piezoelectric actuator. Mechanical response of the I27 protein domains fingerprints successful events where hSOD1 was subject to mechanical force. D, time-dependent superoxide generation by xanthine oxidase (black circles). We measure activity of SOD from the inhibition of superoxide formation. Our reference is hSOD1 extracted from human erythrocytes (black squares, 0.2 IU and black triangles 10 IU). Catalytic activity is conserved in polyproteins containing one molecule of hSOD1 WT (open circles), and also for the mutants G93A (open squares) and A4V (open inverted triangles). Solid lines fit a single exponential function y0 + Ae(−1/τ)x where y0 values range from 0.18 to 2.08; A from 0.16 to 2.17 and 1/τ from 0.05 to 0.34.
Formation of insoluble aggregates containing SOD1 is a common pathophysiological characteristic of ALS (6, 11). Non-native disulfide formation contributes to form and/or stabilize SOD1 aggregates that accompany progression of the disease (12–15). Indeed, it has been suggested that scrambling of the disulfide and the free cysteines in SOD1 results in formation of insoluble aggregates (16). Interestingly, the free cysteines in SOD1, in particular Cys-111, contribute to the toxicity of mutant forms of SOD1 (14, 17). However, little is known about how non-native disulfides involving Cys-6 and Cys-111 are formed in vivo.
Mitochondria are one of the main sources of misfolded hSOD1 and a key cellular organelle to induce motoneuron death (18). Albeit hSOD1 is preferentially cytosolic, it also reaches the intermembrane space of mitochondria as an apoenzyme (19, 20). In this journey, hSOD1 is mechanically unfolded as it is threaded through the TOM complex (21, 22). We have recently shown that mechanical unfolding of proteins can result in the exposure of reactive cysteines, which then can engage in redox reactions (23–25). Similarly, cysteines in hSOD1 are exposed to the solution as the protein is translocated into the intermembrane space of mitochondria, resulting in a much enhanced reactivity that could lead to formation of non-native disulfides. Remarkably, the mitochondrial fraction of the spinal cord of mutant SOD transgenic mice has been shown to contain insoluble SOD1 multimers that are crosslinked via disulfide bonds (26).
Although key to the interplay between thiol biochemistry and hSOD1 aggregation, the reactivity of the cysteines in hSOD1 remains poorly understood. Classic biochemistry techniques are of limited value to study reversible chemical reactions that involve several competing pathways, such as those originating from the four cysteines of hSOD1. In this report, we use recently developed single-molecule atomic force microscopy (AFM) techniques (23) to monitor directly the reactivity of Cys-111 toward the conserved disulfide in hSOD1, one of the reactions that triggers disulfide rearrangement associated with ALS. In our experiments, hSOD1 is unfolded mechanically to probe the reactivity of its cysteine residues. In vivo, equivalent reactions can be enabled during mitochondrial import or due to thermal fluctuations that transiently unfold hSOD1 (27, 28). We find that hSOD1 mutants that cause familial ALS show altered reactivity. Hence, aberrant thiol/disulfide chemistry in hSOD1 may influence the onset and progression of ALS.
EXPERIMENTAL PROCEDURES
Protein Purification
The expression and purification of polyproteins I273-SOD-I273, I273-G93A-I273, and I273-A4V-I273 were accomplished following standard protocols that have been described elsewhere (23). The gene coding for hSOD1 was a generous gift from Professor J.E. Esquerda, University of Lleida (Spain). To obtain the mutations G93A and A4V, we applied the QuikChange Multi Site-Directed Mutagenesis kit from Stratagene (La Jolla, CA). We constructed polyproteins by successive digestion and ligation of DNA fragments (29), (30). BamHI and KpnI sites were used to insert cDNAs into the expression vector pQE80L (Qiagen, Valencia, CA). Protein production in Escherichia coli Origami B cells was induced at A600 1.0 with 1 mm isopropyl-β-d-thiogalactoside overnight at 23 °C. SOD polyproteins were purified from the soluble fraction after sonication and two passes through a French press. We employed His-tag affinity chromatography using Talon resin (Mountain View, CA), followed by chromatography using a Superdex 200 column (Amersham Biosciences, Pittsburgh, PA) in HE buffer (10 mm HEPES, 150 mm NaCl, 1 mm EDTA, pH 7.2) or H buffer (10 mm HEPES, 150 mm NaCl, pH 7.2), which lacks EDTA. We did not observe any difference between proteins purified with or without EDTA in our experimental results, due to the metal ions being tightly chelated by the enzyme. Indeed, very harsh conditions are required to remove the metal ions from the enzyme (see below) (31).
Single Molecule Force Clamp Spectroscopy
Force spectroscopy experiments were done as previously reported (23). In summary, aliquots (1–10 μl) of a solution of polyproteins I273-SOD-I273, I273-G93A-I273, or I273-A4V-I273 (0.2 mg/ml, in HE or H buffer) were deposited onto a gold coverslip for 1 to 3 min before adding degassed H buffer with varying concentration of TCEP. We used the equipartition theorem to calibrate silicon nitride cantilevers (MLCT, Bruker, Camarillo, CA) (32). Measured spring constants were typically in the 14–19 pN nm−1 range. To pick up single polyproteins, the cantilever was pressed against the gold surface containing the polyproteins at contact forces of 2 nN for 1–2 s. Then, the piezo actuator was retracted. If a tether was established, we ramped the force between 0 and 250 pN during 5 s and then force was kept at 250 pN for 20 s or until the tether broke. We used a custom built AFM set-up that uses electronic feedback to control the force with a 5-ms response time (32, 33). Experiments were done at room temperature (22–23 °C) in a fluid cell filled with additional H buffer with TCEP. To assure that SOD1 molecules had been subject to mechanical force, we only analyzed traces containing between four and six 25 nm steps, corresponding to the unfolding of I27 modules. We measured the size of all the steps that were detected after the first step due to I27 unfolding or after the partially unfolded hSOD1 (see Fig. 3, A–C). For simplicity, in the final histograms we did not include the steps due to the unfolding of I27 modules. The data points and the error bars (S.E.) in Fig. 6B were estimated by bootstrapping (34), (35). To this end, several thousand artificial data sets were generated randomly from the experimentally observed reaction pathways. The frequency of each pathway was calculated for every data set, which allowed us to obtain a distribution of frequencies. The mean and the standard deviation of such distributions are reported in Fig. 6B.
FIGURE 3.

Mechanical unfolding exposes disulfide 57–146 enabling reduction by TCEP. Mechanical unfolding of hSOD1 and cleavage of its disulfide bond were monitored using force-clamp spectroscopy. Single I273-SOD-I273 polyproteins were initially subject to a linear increase in force from 0 to 250 pN at 50 pN/s, and then force was held constant at 250 pN. The recordings presented in all panels were obtained in a solution containing 2.5 mm TCEP. Mechanical force triggers mechanical unfolding of the protein modules in the polyprotein. I27 acts as a fingerprint protein that allows us to select traces where hSOD1 has been subjected to force. Mechanical unfolding of hSOD1 renders the disulfide accessible to TCEP, and enables thiol/disulfide exchange reactions involving the disulfide in hSOD1. In panels A, B, and C, the lower trace shows the force pulse, while the upper traces correspond to the length of the polyprotein. Each I27 unfolding event produces a 25-nm step (black dots). Occasionally, we detect unfolding of hSOD1 at low forces (marked by gray dots). An example of each one of the three patterns of steps that we found is shown in A through C. A, after extending the six I27 modules, an additional extension of 31 nm (red) is observed; B, after the extension of six I27 modules, two additional steps of 19 nm (blue) and 12 nm (green) are detected; C, the 12-nm step appears before the 19-nm step. D, distribution of step sizes induced by the mechanical extension of I273-SOD-I273 in the presence of TCEP. To build the histogram, we pooled results obtained at different concentrations of TCEP. We did not include in the histogram steps of 25 nm coming from the extension of I27 modules. The distribution reveals four peaks centered at: 12 nm (green), 19 nm (blue), 22 nm (gray), and 31 nm (red). The rest of the steps recorded. which could not be assigned and probably represent nonspecific interactions, are shown in white. n = 871 events.
FIGURE 6.
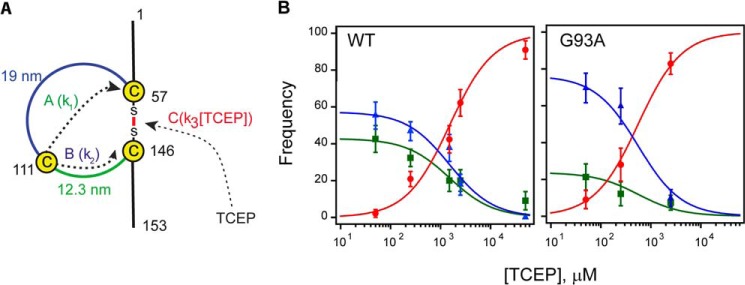
The intramolecular isomerization of the disulfide bond of hSOD1 competes with intermolecular reduction by TCEP. A, scheme of available reaction pathways. Pathway A: the attack of Cys-111 on Cys-57 results in a new disulfide bond between Cys-57 and Cys-111, releasing 35 amino acids (12-nm step). Pathway B: the attack of Cys-111 on Cys-146 forms a new disulfide bond between Cys-146 and Cys-111, releasing 54 amino acids (19-nm step). Pathway C: the disulfide bond is cleaved by TCEP releasing 89 amino acids (31 nm step). Notice that in pathways A and B the disulfide bond between Cys-111 and Cys-57, or Cys-111 and Cys-146, is eventually attacked by TCEP producing an additional step of 19 or 12 nm. k1, k2, and k3 represent the rate constants associated with each pathway. B, frequencies of the different reaction pathways at different TCEP concentrations; the first step after the fingerprint reveals the reaction pathway that accounts for the cleavage of the disulfide bond. Green squares correspond to step size of 12 nm (pathway A), blue triangles to 19 nm steps (pathway B), and red circles to 31 nm steps (pathway C). Left panel: frequencies in wild type SOD1 construct, n > 45. Right panel: frequencies in the G93A mutant, n > 24. Solid lines were obtained using the kinetic model described in “Experimental Procedures.”
Kinetic Analysis
In the presence of TCEP, disulfide 57–146 can be cleaved following three competing reaction pathways (Fig. 6A). The frequencies of appearance of the three pathways are given by Equations 1–3,
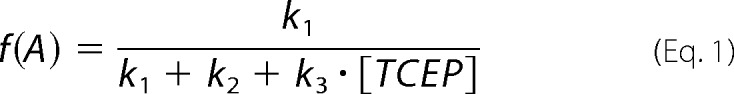 |
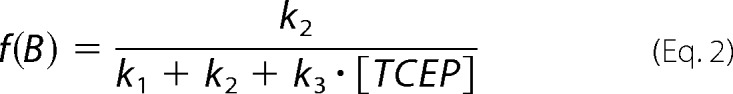 |
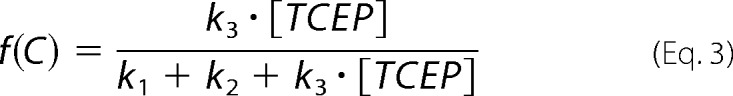 |
where k1, k2, and k3 are the corresponding rate constants. Since f(A) + f(B) + f(C) = 1, we can rewrite f(C) as (1 − a)/(a + [TCEP]) where a = (k1 + k2)/k3. We fitted the experimental data to this expression to obtain the value a for hSOD1 WT (1519 ± 264 μm) and G93A (559 ± 147 μm) (Fig. 6B, red lines). Considering the regioselectivity of the intramolecular reaction, b = k1/(k1+k2), we can rewrite f(A) = b·a/(a + [TCEP]) and f(B) = (1 − b)·a/(a + [TCEP]). Regioselectivity was calculated from the number of events observed for each reaction pathway, b = n(A)/(n(A) + n(B)), and errors were estimated by bootstrapping (see above). We used the experimentally determined regioselectivities to plot f(A) and f(B) in Fig. 6B for WT and G93A proteins (green and blue lines).
Blocking of Cys-111 by Incubation with Maleimide
Following adsorption to the gold-coated coverslips, the polyprotein was incubated for 90 min with 250 mm maleimide freshly dissolved in Na-borate buffer, 50 mm, pH 8.5 (36). The preparation was washed with H buffer, initially with no TCEP added and finally recorded in the same solution in the presence of 2.5 mm TCEP.
Preparation of the Apo Form of hSOD1
We followed a procedure adapted from McCord and Fridovich (31). After the polyprotein was adsorbed to a gold coverslip, 5 μl of a solution of 100 mm Na-acetate buffer, 1 mm EDTA, pH 3.8 were added. After incubation for at least 5 min, the sample was rinsed with recording solution containing 2.5 mm TCEP.
Remetallation of the Apoenzyme
The apo form of hSOD1 prepared as above was rinsed with a solution containing 2.7 mm CuSO4, 0.9 mm ZnCl2, 150 mm NaCl, Tris 50 mm, pH 6.8, and then incubated in the same buffer for 30 min (31). Then, the sample was rinsed with 10 mm Tris, pH 6.8, and finally washed again with 150 mm NaCl, 10 mm HEPES, pH 7.2. Finally, 2.5 mm TCEP was added before starting the AFM experiments.
Determination of Superoxide Dismutase Activity
SOD1 activity was measured using the assay of Enzo Life Sciences (Exeter, UK). This system measures the coupled reaction between Xanthine Oxidase, which generates superoxide radical, and SOD, which converts superoxide into water and peroxide. The superoxide anion reacts with the dye WST-1, which is converted to a colored WST-1 formazan product that absorbs light at 450 nm. SOD scavenges superoxide anion and thereby reduces the rate of formation of WST-1 formazan product.
Western Blot
Standard SDS-PAGE was performed in 4–20% acrylamide in Tris-glycine gels (Bio-Rad). Samples were boiled for 5 min in sample buffer containing 5% β-mercaptoethanol prior to electrophoresis. The fractionated proteins were electrically transferred onto nitrocellulose membranes using a semidry apparatus (Bio-Rad). The membranes were stained with 0.2% Ponceau S and washed three times with distilled water. Nonspecific binding was blocked by incubating with 0.1% Tween 20 and 5% skimmed milk in PBS for 1 h. Then, the membranes were incubated with sheep polyclonal antibody against human SOD1 (Calbiochem, Darmstadt, Germany) at dilution of 1:5000 in blocking solution overnight at 4 °C. Rabbit anti-sheep-HRP secondary antibody, at dilution 1:2000 in blocking solution 1 h at 37 °C, was used. The membranes were washed three times in Tris-buffered saline containing 0.05% Tween 20. Positive bands were detected using an enhanced chemiluminescence system.
RESULTS
Disulfide 57–146 Is Buried in the Folded State of hSOD1
Thiol/disulfide exchange reactions involving disulfide 57–146 and the free cysteines of hSOD1 have been proposed to contribute to aggregation and misfolding in ALS (15, 16, 26, 37–39). However, description of these reactions at the molecular level is still missing.
Since solvent accessibility determines the reactivity of disulfide bonds (40), we first examined the solvent accessibility of disulfide 57–146 in hSOD1 using force-clamp spectroscopy. When a protein is pulled using force clamp, its mechanical unfolding generates step changes in the length of the polypeptide. The size of an unfolding step depends exclusively on the number of amino acids that are released by force following protein unfolding (23). Disulfide bonds cannot be broken by forces in the picoNewton (pN) range. Hence, if present in a protein, a disulfide can be detected from the limited extension of the protein upon mechanical unfolding (40). We exploited this property to determine whether disulfide 57–146 in hSOD1 is solvent accessible and can be cleaved by incubation with reducing agents.
To be able to study hSOD1 using force clamp, we engineered and purified to homogeneity the polyprotein I273-SOD-I273 (Fig. 2). This chimeric protein consists of one hSOD1 domain flanked by six I27 modules (Fig. 1C). Mechanical unfolding of the I27 domains provides a mechanical fingerprint that identifies successful single-molecule recordings (29, 41). SOD1-containing polyproteins retain superoxide dismutase activity (Fig. 1D) implying that the functional fold of hSOD1 is preserved in the polyprotein. Polyproteins containing ALS-linked mutations G93A and A4V in hSOD1 also retain enzymatic activity (Fig. 1D).
FIGURE 2.
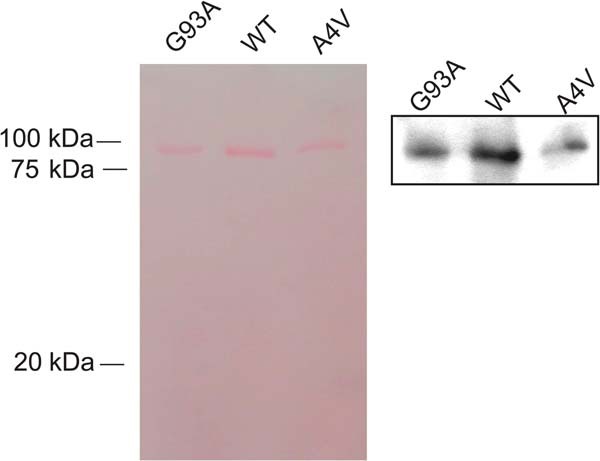
Western blot of polyproteins containing SOD1. Western blot of polyprotein I273-SOD-I273 containing a monomer of wild type hSOD1 (WT), or mutants G93A or A4V. In the left panel the membrane was stained with Ponceau S. Bands correspond to the expected mobility of six subunits of I27 and one molecule of hSOD1. In the right panel, membrane was stained using a polyclonal antibody against hSOD1.
In our AFM experiments, we include 50 μm-50 mm of the reducing agent TCEP in the bathing solution. We apply a linear increase in force from 0 to 250 pN to single I273-SOD-I273 polyproteins and then keep the force constant at 250 pN for up to 20 s. This protocol triggers the unfolding and extension of the I27 domains. I27 unfolding events are detected as step changes in length of 25 nm (Fig. 3, A–C, black dots) (25). Given the design of the I273-SOD-I273 polyprotein, we are certain that hSOD1 has been subject to force in traces where at least four I27 unfolding events are detected (Fig. 1C). We therefore selected traces that showed four 25-nm steps and analyzed the length of the remaining steps. We find four different populations of steps centered at 12, 19, 22, and 31 nm (Fig. 3D), which arise from the mechanical unfolding of hSOD1 and the reactivity of its conserved disulfide bond, as explained below. These populations of events do not depend on the nature of the reducing agent, since experiments in the presence of dithiothreitol induce a very similar pattern of step lengths (Fig. 4).
FIGURE 4.
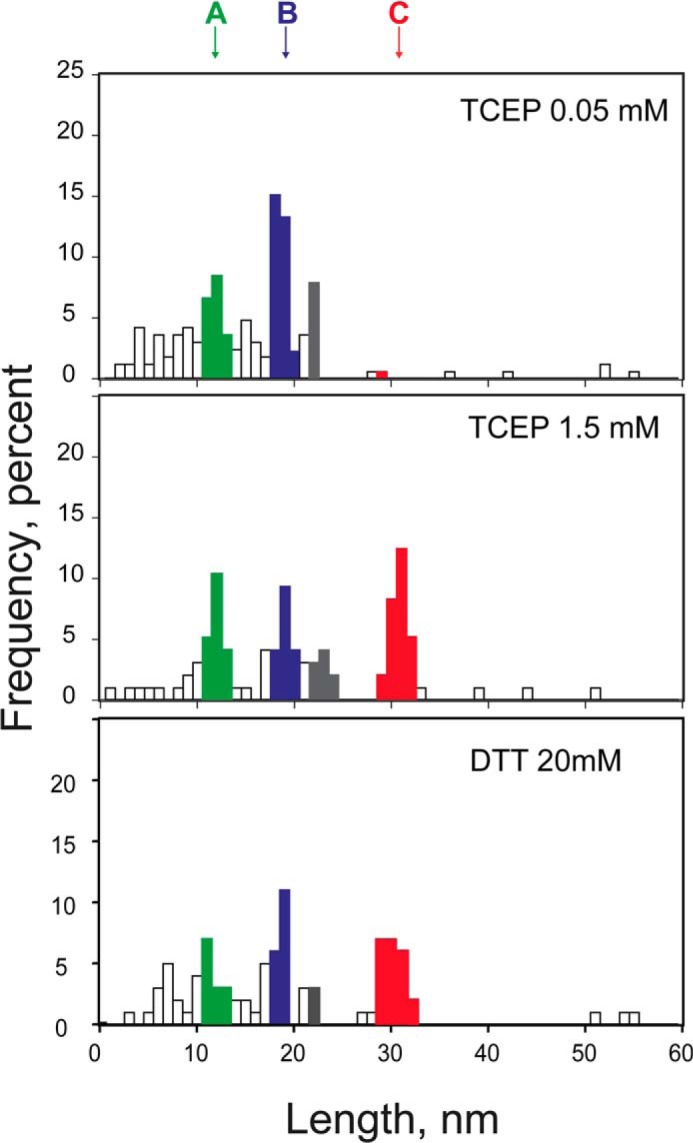
Effects of concentration and different reducing agents on thiol/disulfide exchange reactions in hSOD1. Distribution of step sizes found at 0.05 mm TCEP (n = 66), 1.5 mm TCEP (n = 95), and 20 mm DTT (n = 89).
Since the size of mechanical unfolding steps is determined by the number of amino acids released to force, we could predict the step size associated with the mechanical unfolding of hSOD1, both in the reduced and oxidized states. If disulfide 57–146 was exposed, it would be reduced by TCEP before the molecule is picked up by the cantilever (AFM experiments are conducted over time scales of hours). In this scenario, all 153 amino acids should be extended after mechanical unfolding. Considering that the distance between N and C termini of folded hSOD1 is 0.9 nm, we predict that the mechanical unfolding of reduced hSOD1 would result in a step size of 48.7–53.1 nm between 100 pN and 250 pN (23). In the alternative scenario, disulfide 57–146 is buried, and therefore would remain unreactive to TCEP. In this case, the disulfide bond would prevent the unfolding of 89 amino acids and the step size associated with hSOD1 unfolding would be 20.5–22.4 nm between 100 pN and 250 pN. These values match the population of steps at 22 nm in Fig. 3D (colored in gray). We do not detect a significant population of steps at ∼50 nm that would be compatible with the mechanical unfolding of reduced hSOD1, indicating that disulfide 57–146 remains buried in the fold of hSOD1 and therefore unreactive. The 22 nm step appears in 9.3% of the traces, and mostly at low forces (gray dots in Fig. 3, A and B). In the remaining traces, unfolding of hSOD1 probably occurs at a force below our detection limit. As explained in the next section, the other populations of steps in Fig. 3D are the result of chemical reactions involving disulfide 57–146. These reactions are triggered by mechanical unfolding of hSOD1, which gives further support to our conclusion that disulfide 57–146 is buried and unreactive in the folded structure of hSOD1.
Detection of Thiol/Disulfide Exchanges from the Length Signature of the Reactions
Mechanical unfolding of hSOD1 induces the formation of a polypeptide loop containing Cys-111, which is held by disulfide 57–146 (Fig. 1B). We have shown before that an equivalent loop behaves as a molecular “jump rope” where the free cysteine remains close to the disulfide, enabling intramolecular reactions between the cysteine and the disulfide (23). We took advantage of this configuration to study the reactivity of Cys-111 against the conserved disulfide in hSOD1 (Figs. 3, 5, and 6).
FIGURE 5.
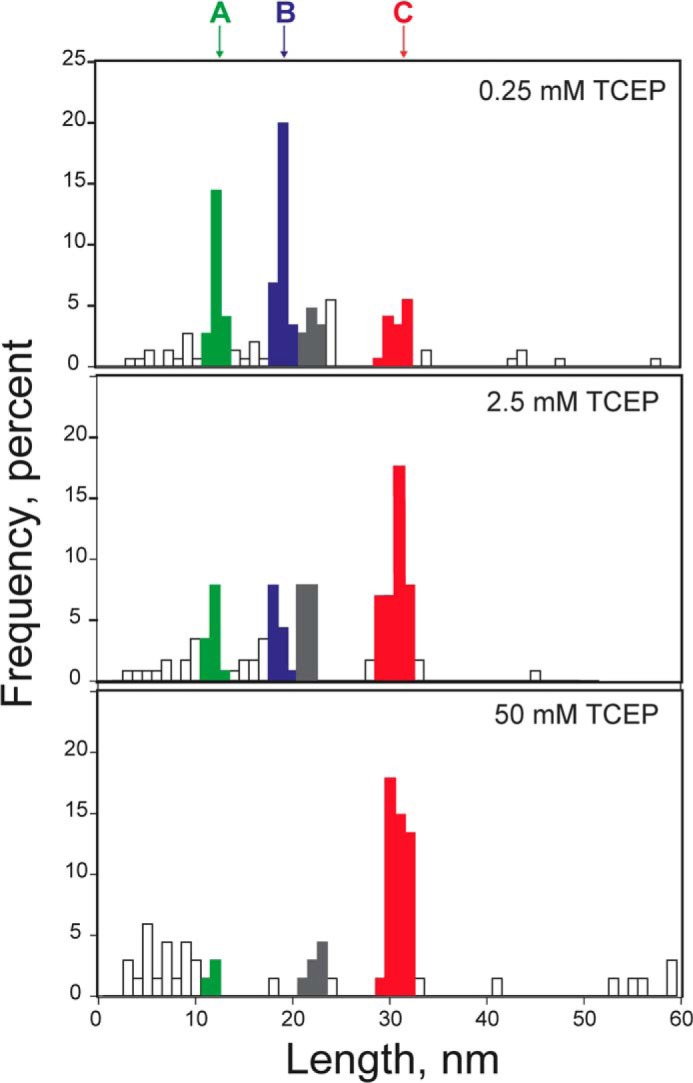
Distribution of step sizes at different concentrations of TCEP. The amplitude of peaks (A, 12 nm; B, 19 nm; C, 31 nm) varies according to the concentration of TCEP. At low concentrations of TCEP (0.25 mm), peaks centered at 12 and 19 nm predominate over the peak at 31 nm, as the intramolecular reaction of Cys-111 with the disulfide dominates over the intermolecular cleavage of the disulfide by TCEP. At high TCEP concentrations, the peak at 31 nm is higher than peaks at 12 and 19 nm (data obtained from n = 165 upper panel, n = 115, middle panel, n = 67 lower panel). The population of events coming from the unfolding of hSOD1 is colored in gray.
In our experiments, the intramolecular reactions compete with the intermolecular cleavage of the disulfide by TCEP, for a total of three competing reactions (Fig. 6A). Since the segments of the loop delimited by Cys-111 have different lengths, it is possible to distinguish unambiguously the three reaction pathways. The cleavage of disulfide 57–146 by TCEP releases 89 amino acids, which corresponds to an increase in length of 30.7 nm at 250 pN (23). This value corresponds to one of the populations in the length histogram (Fig. 3D, red, see Fig. 3A for an example trace). The intramolecular attack of Cys-111 on the disulfide at position 57 frees 35 amino for a theoretical extension of 12.3 nm; whereas the alternative intramolecular reaction involving position 146 is predicted to generate a step of 19 nm (Fig. 6A). Both sizes match the remaining two populations of steps in Fig. 3D (blue and green).
Intramolecular reaction between Cys-111 and disulfide 57–146 triggers the formation of new disulfides between Cys-111 and Cys-57 or Cys-146. Such newly formed disulfides can be cleaved by TCEP reaching a fully extended polypeptide, which is detected as a final step in our recordings. In Fig. 3B, a trace is presented where the initial reaction of Cys-111 on Cys-146 generates a step of 19 nm, which is followed by cleavage of disulfide 111–146 producing a step of 12 nm. In Fig. 3C, a trace is shown where Cys-111 attacks the disulfide at position 57 (12 nm). Subsequent reduction of disulfide 57–111 is accompanied by a final 19-nm step. Hence, using force-clamp we can unambiguously identify individual thiol/disulfide exchanges involving Cys-111 and the conserved disulfide in hSOD1.
Regioselectivity and Competition between Inter- and Intramolecular Reactions
Inter- and intramolecular reaction pathways are mutually exclusive and therefore in competition (Fig. 6A). We never observed steps of 31 nm (reporting on intermolecular reduction of the disulfide) and 12/19 nm (reporting on intramolecular reactions) in the same single-molecule trace, which is in agreement with our assignment of step sizes. To further test the validity of our model, we compared the distribution of step sizes obtained at increasing concentrations of TCEP. Since the rate of intermolecular attack of TCEP depends on the concentration of TCEP, while the rate of intramolecular reactions is independent of the concentration of TCEP, we predicted that the intermolecular pathway would be favored at high concentrations of the reducing agent. Results showed that at low TCEP concentrations, the steps reporting on intramolecular pathways are dominant (12 and 19 nm steps, Fig. 5). In contrast, the intermolecular pathway is favored at higher concentrations of TCEP (31 nm steps, Fig. 5). Hence, our results are in agreement with the kinetic model in Fig. 6A.
Using both the size of the steps and their order of appearance, we could assign unambiguously experimental traces to one of the three possible reaction pathways to cleave disulfide 57–146. Fig. 6B represents the frequency of each pathway of disulfide reduction at different concentrations of TCEP, with solid lines representing fits to the experimental data using the kinetic model in Fig. 6A (“Experimental Procedures”). Results in Fig. 6B confirm that the intermolecular attack by TCEP is favored at higher concentrations of reducing agent. Also, we detect that the frequency of pathway A (green) is slightly lower than that of pathway B (blue). Pooling together all experimental traces, we conclude that nucleophilic attack of Cys-111 at position 146 is 1.3 times more frequent than attack on position 57 (n = 168) in wild-type hSOD1.
Disease-linked Variants of hSOD1 Show Altered Reactivity Patterns
Since aberrant thiol/disulfide exchange reactions involving hSOD1 have been proposed to occur in ALS (14, 16, 42–50), we applied our single-molecule methods to study the reactivity of Cys-111 and the conserved disulfide 57–146 in diseased-linked forms of hSOD1. We concentrated our efforts in hSOD1 G93A, which was used to produce a widely employed transgenic mice model of ALS (51) and hSOD1 A4V, which is the most prevalent mutation associated with ALS in North America (52).
The regioselectivity of disulfide isomerization in hSOD1 G93A is greatly increased (Fig. 6B). In this mutant, the attack at position 146 is 3.2 times more frequent than at position 57 (n = 55), a 2.5-fold increase with respect to the wild-type protein. To investigate whether the mutation G93A is accompanied by a decrease in the overall reactivity of Cys-111, we calculated the ratio k2/k3 = a·(1 − b) (see “Experimental Procedures”) as a measure of the propensity of Cys-111 to attack Cys-146. For WT, we obtained k2/k3 = 869 + 162 μm, while the value for the G93A mutant is k2/k3 = 427 + 117 μm. Hence, our results show that mutation G93A both decreases the overall reactivity of Cys-111, and biases the intramolecular reactivity toward the attack on position 146. These effects could be explained by two non-exclusive mechanisms. First, the mutation could induce structural perturbations that result in alterations of the chemical environment of Cys-111. In this regard, it has been shown that mutation G93A leads to noticeable changes in the structure and dynamics of hSOD1 (53, 54). In addition, the mutation may render Cys-111 more sensitive to oxidation (47, 48), which would make Cys-111 unreactive. Indeed, equivalent loss of reactivity of Cys-111 can be reproduced in hSOD1 WT by preincubation with maleimide, an alkylating agent that targets free, accessible thiols (Fig. 7, A and B).
FIGURE 7.

Effects of mutations G93A and A4V on thiol/disulfide exchange reactions in hSOD1. Distribution of step sizes induced by the mechanical extension of I273-SOD-I273 in the presence of 2.5 mm TCEP for different protein preparations. Colors and nomenclature are the same as in Fig. 3. A, for mutant G93A, the intramolecular pathways A and B are less populated than for the wild-type protein (n = 96). B, the lack of reactivity of Cys-111 can be reproduced in wild-type hSOD1 by pre-incubation with the thiol alkylating agent maleimide (n = 69). C, for mutant A4V, the steps reporting on the reduction/isomerization of the disulfide bond are rare, suggesting that the disulfide bond in hSOD1 A4V is accessible to TCEP in solution (n = 111). D, similar results were obtained with apo hSOD1 (n = 51).
Mutation A4V abolishes the pattern of step sizes observed for the wild-type protein, with no clear populations of steps (Fig. 7C). We propose that in hSOD1 A4V the disulfide bond is exposed to the solvent, making it sensitive to reduction by TCEP before the molecule is picked up by the AFM cantilever. Since there would be no disulfide by the time the mutant protein is pulled, no steps involving reactivity of the disulfide bond can be detected. It has been proposed that structural disorder in metal-free (apo) hSOD1 proteins leads to an increased reactivity of the disulfide bond, probably arising from higher exposure to the solvent (55). We found that apo hSOD1, as the A4V mutant, does not show steps that can be assigned to reactions involving the disulfide (Fig. 7D). This result supports the hypothesis that structural destabilization of hSOD1 A4V leads to the exposure of the disulfide, which can be easily reduced by the TCEP in the solution. The absence of a population of events reporting on the mechanical unfolding of apo hSOD1 or the A4V mutant, which would be expected at ∼50 nm if no disulfide bond is present (see above), supports the idea that the apo enzyme and the mutant A4V are significantly destabilized and they unfold at forces below our detection limit. In 15 out of 21 traces in which the apoenzyme was remetalized with Cu2+ and Zn2+, the steps due to the disulfide bond reduction were recovered (Fig. 8), indicating that exposure of the disulfide bond in apo hSOD1 is reversible upon incorporation of Cu2+ and Zn2+.
FIGURE 8.

Recovery of the intramolecular disulfide bond after remetalizing apo hSOD1. The polyprotein I273SODI273 was treated to remove the metals and then incubated with solutions containing Cu2+ and Zn2+ to reintroduce these metals in hSOD1. Cleavage of the disulfide bond could be detected in pulling experiments in the presence of TCEP. Black dots indicate the unfolding of the six I27 modules, whereas the gray dot marks the unfolding of hSOD1 up to the disulfide bond. Red arrowhead indicates the rupture of disulfide bond 57–146, rendering a step of 31 nm.
DISCUSSION
The presence of disulfide bonds in cytosolic proteins, such as SOD1, is rather uncommon (9). Our single-molecule data show that hSOD1 is able to maintain the disulfide bond in a highly reducing environment. This property is probably important for the stability of the protein in the cytosol, since the reduction of the disulfide induces misfolding of parts of the molecule (42). Interestingly, hSOD1 contains free cysteine residues that can cleave the disulfide triggering disulfide isomerization reactions. Disulfide isomerization may lead to disulfide cross-linked hSOD1 aggregates, which have been detected in spinal cords of model ALS mice (12). Indeed, these abnormal configurations have been implicated in the gain of toxic function of hSOD1 (13, 14, 47). In order to examine thiol/disulfide exchange reactions that are involved in the aggregation of hSOD1, there is a need to develop reliable assays that can probe directly the reactivity of the cysteines in hSOD1.
We applied single-molecule methods using force clamp AFM spectroscopy (34, 56) to study the effect of disease-linked mutations in the reactivity of Cys-111 and the conserved disulfide of hSOD1. In our assay, hSOD1-containing polyproteins are stretched using an AFM cantilever. The resulting pulling force triggers the unfolding of the hSOD1 molecule, exposing the disulfide to the bathing solution. In the presence of reducing agents, we monitored the intermolecular reduction of the disulfide in competition with intramolecular disulfide isomerization reactions mediated by Cys-111.
Cys-111 is only present in SOD from primates and its role in ALS pathophysiology has been extensively investigated. Cys-111 binds Cu2+ during the biosynthesis of SOD1 (10) and contributes to keep this metal in the correct position (57). Cys-111 is placed on the surface of the molecule where it can be oxidized changing the conformation of the wild type molecule in a way similar to several point mutations (58). In addition, Cys-111 reacts with sulfide (59), and can bridge the two molecules of the homodimer through a polysulfane moiety (60). Hence, Cys-111 appears to function as an antenna sensitive to the redox environment, and has been related to aggregation of mutant forms of SOD1. For instance, glutathionylation of Cys-111 increases the proportion of highly fibrylogenic hSOD1 monomers (61, 62). Indeed, removal of Cys-111 reduces the formation of aggregates (17, 57, 63). Our novel assay probes directly the reactivity of the key Cys-111. We find that Cys-111 is highly reactive against the disulfide in the unfolded state of hSOD1. We speculate that isomerization reactions in hSOD1 could be triggered in vivo following rare unfolding events induced by thermal fluctuations, or as a consequence of mechanical unfolding during translocation into the intermembrane space of mitochondria.
We show that in disease-linked hSOD1 mutants, the reactivity pattern of Cys-111 and the conserved disulfide is altered. Thiol/disulfide exchange reactions originating at Cys-111 are deeply perturbed in hSOD1 G93A, enhancing the regioselective attack on Cys-146 and decreasing the overall reactivity of Cys-111. Interestingly, oxidation of Cys-111 is favored in murine models of ALS that express hSOD1 G93A (64). We hypothesize that in hSOD1 G93A, Cys-111 becomes more sensitive to redox modifications, probably because of increased solvent accessibility. This increased exposure to the solvent may lead to intermolecular thiol/disulfide exchange reactions and aggregation (15, 26, 38). It is also possible that structural perturbations caused by the mutation result in decreased reactivity of Cys-111 (54) (53). Remarkably, our results show that the structural constraints introduced by the addition of a single methyl group to a distant residue can be enough to significantly bias intramolecular reactions. Such alteration of reactivity could result in the accumulation of one kind of non-native disulfides, which may contribute to the gain of toxic function that leads to ALS.
We did not observe any signature originating from the disulfide in hSOD1 A4V, strongly suggesting that the bond is exposed to the solvent and is readily cleaved by the reducing agent before the molecule can be probed by the AFM. Our results suggest that the A4V mutation may perturb the structure of hSOD1, rendering the disulfide more sensitive to reduction and accelerating misfolding and aggregation of the protein (37, 39). In agreement with this view, high-resolution structural studies have concluded that there is significant structural destabilization associated with the A4V mutation in hSOD1 (65, 66).
It remains unknown why many apparently unrelated mutations in hSOD1 converge in the ALS phenotype (9, 11). Our novel force-clamp assay can be used to systematically examine the thiol/disulfide chemistry of diseased-linked forms of hSOD1, as done here for A4V and G93A, to gain a deeper understanding of the molecular pathogenesis of ALS. Here, we have found that A4V and G93A showed different reactivity patterns from wild-type protein. It will be interesting to examine whether other hSOD1 variants share the same reactivity profiles.
Our results suggest that conformational changes associated with mutations A4V and G93A cause changes in the thiol chemistry of hSOD, which could favor misfolding and aggregation through formation of non-native inter- and intramolecular disulfide bonds. We propose that wild-type hSOD1 may occasionally sample those toxic conformations, maybe accounting for some forms of sporadic ALS. We show, at the single-molecule level, evidence that the disulfide bond of monomeric hSOD1 is susceptible to isomerization. The isomerization reactions are strongly affected by the mutations A4V and G93A. Since the isomerization reaction competes with the intermolecular reduction of the disulfide by redox metabolites, the extent of isomerization reactions is predicted to be sensitive to the redox state of motoneurons. During translocation to the intermembrane space of mitochondria, the reduced cysteine residues in hSOD1 experiences a more oxidizing environment than in the cytosol. We therefore speculate that mitochondria may be hot spots in which hSOD1, and especially diseased-linked mutants, become misfolded and prone to oligomerize.
This work was supported in whole or in part by National Institutes of Health Grants HL66030 and HL61228 (to J. M. F.) and Grants SAF2011-28485 from the Spanish Ministry of Economy and Competitiveness and European Union FEDER PR2011-0339 from the Spanish Ministry of Education, and Government of Catalonia SGR2009/152 (to C. S.).
- ALS
- amyotrophic lateral sclerosis
- SOD
- superoxide dismutase
- AFM
- atomic force microscopy.
REFERENCES
- 1. Robberecht W., Philips T. (2013) The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264 [DOI] [PubMed] [Google Scholar]
- 2. Andersen P. M., Al-Chalabi A. (2011) Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615 [DOI] [PubMed] [Google Scholar]
- 3. Rodriguez J. A., Shaw B. F., Durazo A., Sohn S. H., Doucette P. A., Nersissian A. M., Faull K. F., Eggers D. K., Tiwari A., Hayward L. J., Valentine J. S. (2005) Destabilization of apoprotein is insufficient to explain Cu, Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borchelt D. R., Lee M. K., Slunt H. S., Guarnieri M., Xu Z. S., Wong P. C., Brown R. H., Jr., Price D. L., Sisodia S. S., Cleveland D. W. (1994) Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8292–8296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayward L. J., Rodriguez J. A., Kim J. W., Tiwari A., Goto J. J., Cabelli D. E., Valentine J. S., Brown R. H., Jr. (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277, 15923–15931 [DOI] [PubMed] [Google Scholar]
- 6. Bruijn L. I., Houseweart M. K., Kato S., Anderson K. L., Anderson S. D., Ohama E., Reaume A. G., Scott R. W., Cleveland D. W. (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854 [DOI] [PubMed] [Google Scholar]
- 7. Boillée S., Vande Velde C., Cleveland D. W. (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59 [DOI] [PubMed] [Google Scholar]
- 8. Reaume A. G., Elliott J. L., Hoffman E. K., Kowall N. W., Ferrante R. J., Siwek D. F., Wilcox H. M., Flood D. G., Beal M. F., Brown R. H., Jr., Scott R. W., Snider W. D. (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47 [DOI] [PubMed] [Google Scholar]
- 9. Valentine J. S., Doucette P. A., Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 [DOI] [PubMed] [Google Scholar]
- 10. Liu H., Zhu H., Eggers D. K., Nersissian A. M., Faull K. F., Goto J. J., Ai J., Sanders-Loehr J., Gralla E. B., Valentine J. S. (2000) Copper(2+) binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry 39, 8125–8132 [DOI] [PubMed] [Google Scholar]
- 11. Chattopadhyay M., Valentine J. S. (2009) Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox. Signal 11, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Furukawa Y., Fu R., Deng H. X., Siddique T., O'Halloran T. V. (2006) Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karch C. M., Prudencio M., Winkler D. D., Hart P. J., Borchelt D. R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7774–7779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Niwa J., Yamada S., Ishigaki S., Sone J., Takahashi M., Katsuno M., Tanaka F., Doyu M., Sobue G. (2007) Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. J. Biol. Chem. 282, 28087–28095 [DOI] [PubMed] [Google Scholar]
- 15. Roberts B. L., Patel K., Brown H. H., Borchelt D. R. (2012) Role of disulfide cross-linking of mutant SOD1 in the formation of inclusion-body-like structures. PLoS One 7, e47838. 10.1371/journal.pone.0047838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toichi K., Yamanaka K., Furukawa Y. (2013) Disulfide scrambling describes the oligomer formation of superoxide dismutase (SOD1) proteins in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 288, 4970–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cozzolino M., Amori I., Pesaresi M. G., Ferri A., Nencini M., Carrì M. T. (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pickles S., Vande Velde C. (2012) Misfolded SOD1 and ALS: zeroing in on mitochondria. Amyotroph. Lateral Scler. 13, 333–340 [DOI] [PubMed] [Google Scholar]
- 19. Field L. S., Furukawa Y., O'Halloran T. V., Culotta V. C. (2003) Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J. Biol. Chem. 278, 28052–28059 [DOI] [PubMed] [Google Scholar]
- 20. Kawamata H., Manfredi G. (2008) Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 17, 3303–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neupert W., Brunner M. (2002) The protein import motor of mitochondria. Nat. Rev. Mol. Cell Biol. 3, 555–565 [DOI] [PubMed] [Google Scholar]
- 22. Shariff K., Ghosal S., Matouschek A. (2004) The force exerted by the membrane potential during protein import into the mitochondrial matrix. Biophys. J. 86, 3647–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alegre-Cebollada J., Kosuri P., Rivas-Pardo J. A., Fernández J. M. (2011) Direct observation of disulfide isomerization in a single protein. Nat. Chem. 3, 882–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alegre-Cebollada J., Kosuri P., Giganti D., Eckels E., Rivas-Pardo J. A., Hamdani N., Warren C. M., Solaro R. J., Linke W. A., Fernández J. M. (2014) S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 156, 1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kosuri P., Alegre-Cebollada J., Feng J., Kaplan A., Inglés-Prieto A., Badilla C. L., Stockwell B. R., Sanchez-Ruiz J. M., Holmgren A., Fernández J. M. (2012) Protein folding drives disulfide formation. Cell 151, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nordlund A., Oliveberg M. (2006) Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: parallels to precursors in amyloid disease. Proc. Natl. Acad. Sci. U.S.A. 103, 10218–10223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das A., Plotkin S. S. (2013) Mechanical Probes of SOD1 Predict Systematic Trends in Metal and Dimer Affinity of ALS-Associated Mutants. J. Mol. Biol. 425, 850–874 [DOI] [PubMed] [Google Scholar]
- 29. Alegre-Cebollada J., Badilla C. L., Fernández J. M. (2010) Isopeptide bonds block the mechanical extension of pili in pathogenic Streptococcus pyogenes. J. Biol. Chem. 285, 11235–11242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carrion-Vazquez M., Marszalek P. E., Oberhauser A. F., Fernandez J. M. (1999) Atomic force microscopy captures length phenotypes in single proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11288–11292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCord J. M., Fridovich I. (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055 [PubMed] [Google Scholar]
- 32. Popa I., Kosuri P., Alegre-Cebollada J., Garcia-Manyes S., Fernandez J. M. (2013) Force dependency of biochemical reactions measured by single-molecule force-clamp spectroscopy. Nat. Protoc. 8, 1261–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schlierf M., Li H., Fernandez J. M. (2004) The unfolding kinetics of ubiquitin captured with single-molecule force-clamp techniques. Proc. Natl. Acad. Sci. U.S.A. 101, 7299–7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wiita A. P., Perez-Jimenez R., Walther K. A., Gräter F., Berne B. J., Holmgren A., Sanchez-Ruiz J. M., Fernandez J. M. (2007) Probing the chemistry of thioredoxin catalysis with force. Nature 450, 124–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Efron B. (1987) The Jackknife, the Bootstrap, and Other Resampling Plans, Society for Industrial and Applied Mathematics [Google Scholar]
- 36. Zimmermann J. L., Nicolaus T., Neuert G., Blank K. (2010) Thiol-based, site-specific and covalent immobilization of biomolecules for single-molecule experiments. Nat. Protoc. 5, 975–985 [DOI] [PubMed] [Google Scholar]
- 37. Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Lindberg M., Oliveberg M., Marklund S. L. (2006) Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 129, 451–464 [DOI] [PubMed] [Google Scholar]
- 38. Wang J., Xu G., Borchelt D. R. (2006) Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J. Neurochem. 96, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Furukawa Y., Kaneko K., Yamanaka K., O'Halloran T. V., Nukina N. (2008) Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 283, 24167–24176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wiita A. P., Ainavarapu S. R., Huang H. H., Fernandez J. M. (2006) Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci. U.S.A. 103, 7222–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carrion-Vazquez M., Oberhauser A. F., Fowler S. B., Marszalek P. E., Broedel S. E., Clarke J., Fernandez J. M. (1999) Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. U.S.A. 96, 3694–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tiwari A., Hayward L. J. (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278, 5984–5992 [DOI] [PubMed] [Google Scholar]
- 43. Khare S. D., Ding F., Dokholyan N. V. (2003) Folding of Cu, Zn superoxide dismutase and familial amyotrophic lateral sclerosis. J. Mol. Biol. 334, 515–525 [DOI] [PubMed] [Google Scholar]
- 44. Ray S. S., Nowak R. J., Strokovich K., Brown R. H., Jr., Walz T., Lansbury P. T., Jr. (2004) An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry 43, 4899–4905 [DOI] [PubMed] [Google Scholar]
- 45. Tiwari A., Xu Z., Hayward L. J. (2005) Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J. Biol. Chem. 280, 29771–29779 [DOI] [PubMed] [Google Scholar]
- 46. Ding F., Dokholyan N. V. (2008) Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 105, 19696–19701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oztug Durer Z. A., Cohlberg J. A., Dinh P., Padua S., Ehrenclou K., Downes S., Tan J. K., Nakano Y., Bowman C. J., Hoskins J. L., Kwon C., Mason A. Z., Rodriguez J. A., Doucette P. A., Shaw B. F., Selverstone Valentine J. (2009) Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS One 4, e5004. 10.1371/journal.pone.0005004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tiwari A., Liba A., Sohn S. H., Seetharaman S. V., Bilsel O., Matthews C. R., Hart P. J., Valentine J. S., Hayward L. J. (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 284, 27746–27758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kayatekin C., Zitzewitz J. A., Matthews C. R. (2010) Disulfide-reduced ALS variants of Cu, Zn superoxide dismutase exhibit increased populations of unfolded species. J. Mol. Biol. 398, 320–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bouldin S. D., Darch M. A., Hart P. J., Outten C. E. (2012) Redox properties of the disulfide bond of human Cu,Zn superoxide dismutase and the effects of human glutaredoxin 1. Biochem. J. 446, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X. (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 52. Juneja T., Pericak-Vance M. A., Laing N. G., Dave S., Siddique T. (1997) Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology 48, 55–57 [DOI] [PubMed] [Google Scholar]
- 53. Shipp E. L., Cantini F., Bertini I., Valentine J. S., Banci L. (2003) Dynamic properties of the G93A mutant of copper-zinc superoxide dismutase as detected by NMR spectroscopy: implications for the pathology of familial amyotrophic lateral sclerosis. Biochemistry 42, 1890–1899 [DOI] [PubMed] [Google Scholar]
- 54. Galaleldeen A., Strange R. W., Whitson L. J., Antonyuk S. V., Narayana N., Taylor A. B., Schuermann J. P., Holloway S. P., Hasnain S. S., Hart P. J. (2009) Structural and biophysical properties of metal-free pathogenic SOD1 mutants A4V and G93A. Arch. Biochem. Biophys. 492, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Banci L., Bertini I., Durazo A., Girotto S., Gralla E. B., Martinelli M., Valentine J. S., Vieru M., Whitelegge J. P. (2007) Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. U.S.A. 104, 11263–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang J., Fernández J. M. (2009) Mechanochemistry: one bond at a time. ACS Nano. 3, 1628–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Prudencio M., Lelie H., Brown H. H., Whitelegge J. P., Valentine J. S., Borchelt D. R. (2012) A novel variant of human superoxide dismutase 1 harboring amyotrophic lateral sclerosis-associated and experimental mutations in metal-binding residues and free cysteines lacks toxicity in vivo. J. Neurochem. 121, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J. P., Brady S. T., Brown R. H., Jr. (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Beus M. D., Chung J., Colón W. (2004) Modification of cysteine 111 in Cu/Zn superoxide dismutase results in altered spectroscopic and biophysical properties. Protein Sci. 13, 1347–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. You Z., Cao X., Taylor A. B., Hart P. J., Levine R. L. (2010) Characterization of a covalent polysulfane bridge in copper-zinc superoxide dismutase. Biochemistry 49, 1191–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., Chen X., Caplow M., Dokholyan N. V. (2009) Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Redler R. L., Wilcox K. C., Proctor E. A., Fee L., Caplow M., Dokholyan N. V. (2011) Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry 50, 7057–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karch C. M., Borchelt D. R. (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J. Biol. Chem. 283, 13528–13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 65. Cardoso R. M., Thayer M. M., DiDonato M., Lo T. P., Bruns C. K., Getzoff E. D., Tainer J. A. (2002) Insights into Lou Gehrig's disease from the structure and instability of the A4V mutant of human Cu,Zn superoxide dismutase. J. Mol. Biol. 324, 247–256 [DOI] [PubMed] [Google Scholar]
- 66. Hough M. A., Grossmann J. G., Antonyuk S. V., Strange R. W., Doucette P. A., Rodriguez J. A., Whitson L. J., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc. Natl. Acad. Sci. U.S.A. 101, 5976–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]