

Abstract
My career pathway has taken a circuitous route, beginning with a Ph.D. degree in electrical engineering from The Johns Hopkins University, followed by five postdoctoral years in biology at Hopkins and culminating in a faculty position in biological sciences at the University of Southern California. My startup package in 1973 consisted of $2,500, not to be spent all at once, plus an ancient Packard scintillation counter that had a series of rapidly flashing light bulbs to indicate a radioactive readout in counts/minute. My research pathway has been similarly circuitous. The discovery of Escherichia coli DNA polymerase V (pol V) began with an attempt to identify the mutagenic DNA polymerase responsible for copying damaged DNA as part of the well known SOS regulon. Although we succeeded in identifying a DNA polymerase, one that was induced as part of the SOS response, we actually rediscovered DNA polymerase II, albeit in a new role. A decade later, we discovered a new polymerase, pol V, whose activity turned out to be regulated by bound molecules of RecA protein and ATP. This Reflections article describes our research trajectory, includes a review of key features of DNA damage-induced SOS mutagenesis leading us to pol V, and reflects on some of the principal researchers who have made indispensable contributions to our efforts.
Keywords: DNA Polymerase, DNA Damage, DNA Synthesis, Mutagenesis, ATP, Translesion DNA Synthesis, SOS Mutagenesis, RecA Nucleoprotein Filament
Introduction
Never having taken a college-level course in biology, I was nonetheless appointed as an assistant professor in biological sciences at the University of Southern California in 1973. My undergraduate training at Queens College and Columbia University was primarily in physics and electrical engineering. My Ph.D. thesis in electrical engineering from The Johns Hopkins University was entitled Selective Hydrolysis of Adenosine Triphosphate Resulting from the Absorption of Laser Light in a Stretching Mode of the Terminal Phosphate Group, which meant that, at the very least, I needed to learn how to measure the hydrolysis of ATP → ADP + Pi. Maurice (Moishe) J. Bessman (Fig. 1), a professor of biology at Hopkins, provided a lab bench and personalized instruction for measuring inorganic phosphate release using the Lowry-Lopez colorimetric assay. The intellectual interest in Moishe's lab focused on understanding the biochemical basis of mutagenesis. Their exciting studies on DNA synthesis fidelity using bacteriophage T4 mutator and antimutator DNA polymerases proved infectious, so much so that when I received a Ph.D. degree in electrical engineering in 1968 from Hopkins, in lieu of a $30,000 salary offer from Bell Labs (Murray Hill, New Jersey), I chose instead to pursue the opportunity to learn and, more importantly, appreciate biochemical enzymology as a postdoctoral fellow with Moishe at a National Institutes of Health fellowship stipend of $6,500. From 1968 onward, the study of DNA polymerase fidelity became an enduring interest.
FIGURE 1.

Maurice J. Bessman (1973) shown brewing a cup of joe bearing his initials, “MJB” (see coffee label at rear).
Mutations for Worse or Better
Mutations are almost always bad, typically with negative consequences, in all forms of life. However, mutations can also be good, in fact essential, by serving as the engine that drives evolution. A specialized DNA polymerase family, the Y-family, has been studied for over a decade. With examples spanning bacteria to humans, the central role of Y-family polymerases appears to be copying past damaged DNA template bases that block replication. There is no free lunch, however, because the costs of fully replicating the genome are often mutations frequently targeted at lesion sites, but occasionally occurring within undamaged template regions. These Y-family enzymes are referred to as translesion synthesis (TLS)1 polymerases. This Reflections article describes the path leading to our discovery of the Y-family Escherichia coli DNA polymerase V (pol V) and to the further discovery of pol V properties that set it apart even from other Y-family polymerases.
The path to the discovery of pol V is closely linked to more than sixty years of experiments and concepts in the area referred to as “SOS error-prone DNA repair.” The logical place to begin is by reflecting on the insights that revealed the transcriptionally regulated error-prone DNA repair pathway in E. coli, the SOS regulon, which led us to pol V.
The Origin of SOS Error-prone DNA Repair
The year of the discovery of the structure of DNA, 1953, is coincidentally the year that Jean Weigle made a seminal observation showing that bacteriophage λ can be revitalized after killing it with UV light (1). Although the word “seminal” tends to be overused, especially when viewed alongside Watson and Crick, Weigle's experiment was nevertheless remarkable. It showed that UV light-irradiated bacteriophage λ, which were unable to generate progeny phage (i.e. form plaques when infecting E. coli), did in fact form plaques when the infected E. coli had also been exposed to UV light. Weigle further found that reactivation of bacteriophage λ was accompanied by a large increase in phage mutagenesis (1).
During the late 1960s to early 1970s, genetic studies by Evelyn Witkin (Fig. 2) were instrumental in showing that phage reactivation could be attributed to the presence of a DNA damage-inducible bacterial pathway that regulated the transcription of genes designed to repair the E. coli chromosome (2, 3). While eliminating DNA damage in the bacterial chromosome, newly induced bacterial repair and recombination proteins also repaired the phage DNA, enabling bacteriophage λ to generate progeny and subsequently lyse the cell. Miroslav Radman (Fig. 2) proposed in 1974 that this was a repair pathway of last resort, an “SOS” mutagenic pathway that could repair DNA in an error-free manner but could also allow unrepaired DNA to be copied by an error-prone process (4).
FIGURE 2.

Miroslav Radman (left) with Evelyn Witkin (right), Paris (2012).
DNA template lesions that would normally block replication fork progression could be copied via TLS, but at the cost of causing mutations targeted at DNA template lesions and elsewhere as well. A genetic locus was identified in the late 1970s that fit the error-prone role envisioned by Radman, the UV mutagenesis (umu) locus (5, 6). The locus was so named because when the umu genes were mutated, UV radiation did not produce chromosomal mutations in excess of spontaneous background levels. This locus was subsequently shown to encode two SOS-regulated genes, umuC and umuD (7, 8), which are used to form pol V (9).
Two proteins regulate the SOS response, LexA and RecA. LexA is a repressor that binds to more than 40 operators in the DNA damage-inducible regulon. Each operator has a consensus LexA-binding sequence, but with different neighboring sequences that determine repressor-operator binding affinities. Early protein induction, e.g. RecA (<1 min post-UV), accompanies weak LexA binding, whereas late protein induction, e.g. pol V (∼45 min), results from strong LexA binding to the umuDC operon. The late appearance of pol V provides the cell an early temporal window to feature error-free ways to excise DNA damage involving base excision, nucleotide excision, and homologous recombination. At later times, chromosomal damage that remains unrepaired is then subjected to pol V-catalyzed TLS in a last-ditch effort to restart replication.
RecA protein is induced rapidly following the exposure of E. coli to UV radiation or to chemicals that damage DNA. RecA assembles cooperatively on regions of single-stranded DNA (ssDNA) in the presence of ATP to form a nucleoprotein filament, often referred to as RecA*. RecA* plays so many different roles and is so important to the cell that I sometimes present a tongue-in-cheek seminar slide listing the three most salient aspects of life: death, taxes, and RecA nucleoprotein filaments. Roles for RecA* include initiating DNA strand invasion as a first step in homologous recombination (10) and serving as a coprotease facilitating the autocatalytic cleavage of the LexA repressor to induce the SOS response and, similarly, facilitating cleavage of the UmuD2 protein to its shorter mutagenically active form, UmuD′2 (11–13). UmuD′2 interacts with UmuC to form pol V (UmuD′2C) (9).
pol V copies undamaged DNA and performs TLS in the absence of any other E. coli DNA polymerase (9) but only when RecA* is present in the reaction. pol V (9) or UmuC (14) has minimal activity in the absence of RecA*. In 2009, we determined that the role of RecA* was to transfer a RecA monomer from the 3′-end of the nucleoprotein filament, which, along with a molecule of ATP, “activates” pol V to a mutasome complex, designated as pol V Mut (UmuD′2C-RecA-ATP) (15).
A Separate Role for RecA in SOS Mutagenesis
In 1989, Raymond Devoret (Fig. 3) and colleagues identified a recA mutant (originally named recA1730, which we now know to be recA(S117F)) that abolished SOS mutagenesis entirely (16). UV light-induced mutations, which typically increased by ∼100-fold in wild-type recA cells, remained at spontaneous background levels in recA1730 cells. The other RecA functions, including homologous recombination, conversion of UmuD to UmuD′, cleavage of the LexA protein, and induction of SOS, all occurred in cells that overexpressed RecA(S117F) (16), but SOS mutagenesis did not. Therefore, the absence of UV light-induced mutagenesis in this recA mutant background could not be attributed to the seemingly obvious reason that SOS could not be turned on. Instead, RecA must possess a separate mutagenic function. The identification of this new RecA function, especially in its connection to pol V, became the overriding research goal for my laboratory.
FIGURE 3.

Raymond Devoret a pris dîner à Sage American Grill and Oyster Bar, New Haven, Connecticut (2013).
Damaged Goods
We entered the SOS mutagenesis field serendipitously as a result of an “out of the blue” phone call in 1986 from Bruce Kaplan and Ramon Eritja at City of Hope in nearby Duarte, California. They asked if I could think of a biological use for a new reduced deoxyribose compound that they had recently synthesized in the form of a phosphoramidite incorporated in a DNA strand using their homemade DNA synthesizer. This was the first type of abasic site to be synthesized. Reduction at the C-1 position of the sugar rendered the potentially labile sugar stable as a rock, so the abasic lesion could be incorporated on a DNA template strand site-specifically and with high yield. The lesion-containing template annealed to an oligonucleotide 5′-32P-labeled primer strand could then be copied by any DNA polymerase. We could determine the fraction of extended primers that were blocked one base before the lesion, stopped opposite the lesion, and, most importantly, extended beyond the lesion, indicating successful TLS (17).
Parenthetically, we analyzed the polymerase-catalyzed 32P-labeled primer extension data using a brand-new technological marvel. Our lab had recently purchased a first edition PhosphorImager, which was developed by a startup company called Molecular Dynamics. This new technology allowed us to simultaneously visualize about twenty lanes of a polyacrylamide gel on which primers were separated electrophoretically according to length, so primers differing by just single-nucleotide additions were easily observed. The imaging provided high resolution and unheard-of sensitivity. A barely visible gel band, corresponding to polymerase extension directly opposite a lesion, and faint TLS extension bands continuing beyond the lesion could be resolved from the much darker gel band, which corresponded to a polymerase blocked at the lesion. The faint-to-dark band detection sensitivity was about ∼1000-fold, far exceeding the 30-fold range previously available using x-ray film scanning densitometry. A gel fidelity assay developed concurrently in our lab by my graduate students Michael Boosalis and Sandra Randall and my next-door faculty colleague John Petruska required the ratio of adjacent integrated gel band intensities as input to quantify nucleotide incorporation velocities. We showed that by determining the ratio of right and wrong incorporations, we could obtain the fidelity for any polymerase, including those with 3′ → 5′-exonuclease proofreading (18).
A Digression into DNA Polymerase II
RecA had yet to appear on our radar screen when we began looking for a biochemical basis for SOS mutagenesis in 1986. Devoret's paper demonstrating a separate role for RecA in SOS mutagenesis would be published three years later (16). Our initial foray into SOS was to take a bottom-up approach by incubating crude lysates prepared from untreated and nalidixic acid-treated E. coli with primer-template DNA containing an abasic template lesion. We observed a 7-fold increase in an unknown polymerase activity in DNA-damaged cell lysates exposed to nalidixic acid compared with undamaged cell lysates (19). DNA polymerase II (pol II) emerged as the enzyme activity responsible for the DNA damage-induced TLS activity based on its similar molecular mass and enzymatic properties to those of pol II reported by Jerry Hurwitz in 1972 (20).
However, the 7-fold activity induction number turned out to be especially important. By inserting a lacZ reporter gene at various locations on the E. coli chromosome, Graham Walker (Fig. 4) had previously identified a series of individual loci distributed along the chromosome that were induced in response to DNA damage, which he called damage-inducible (din) genes (21). This important study provided a salient piece of information: one of the induced genes, dinA, showed a 7-fold increase in transcription. The close correspondence of transcriptional induction to the polymerase activity induction suggested to us that dinA could be the structural gene for pol II, which was confirmed by showing that the nucleotide sequences for dinA and our structural gene for pol II were the same (22). A concurrent paper identifying dinA as the gene for pol II was published by Shinagawa and colleagues (23).
FIGURE 4.

Graham Walker, Cambridge, Massachusetts (2013).
Trials and Tribulations of UmuC and the Identification of a UmuD′2C Complex
The power of E. coli genetics to investigate complex biochemical processes was clearly evident in the reconstitution of a minimal system that was able to catalyze TLS in vitro. The complement of genetic requirements included the UV mutagenesis proteins UmuC and UmuD′ and the RecA protein. A primer-template DNA, including a damaged base on the template strand, could be used as a substrate. Based on genetic evidence available in the mid-1980s, the first TLS model was formulated by Bryn Bridges and his graduate student Roger Woodgate (Fig. 5). The Bridges-Woodgate two-step model (Fig. 6A) proposed that the role of the UmuDC proteins was to shepherd DNA polymerase III (pol III) past replication-blocking lesions by reducing polymerase fidelity, e.g. possibly by inhibiting exonucleolytic proofreading (24). It was suggested that in the first step, pol III could insert a nucleotide opposite a template lesion but could go no further. It required the Umu proteins and RecA* to copy past the lesion, after which there would be clear sailing on undamaged template DNA until further damage was encountered. Whatever the mechanism for TLS might turn out to be, it seemed clear that the Umu proteins working along with RecA* were required for pol III-catalyzed TLS.
FIGURE 5.

Roger Woodgate, Bryn Bridges, Myron F. Goodman, and Mengjia Tang (left to right): four SOS error-prone generations, Hilton Head, South Carolina (1999).
FIGURE 6.
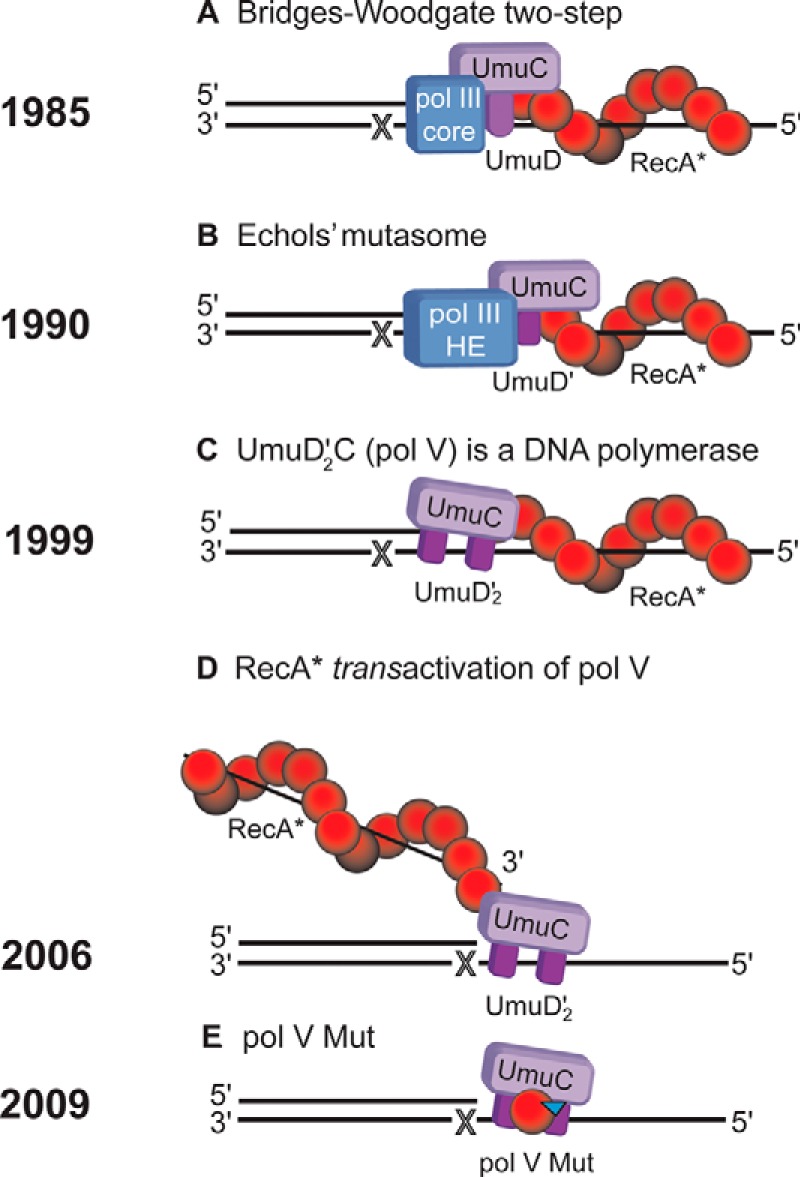
Evolving models for SOS error-prone TLS involving UmuD′2C (pol V) and RecA*. A, pol III core (composed of α-polymerase, ϵ-proofreading exonuclease, and θ-subunits) copies past a damaged template site X, requiring the presence of UmuDC and RecA* situated in cis on the damaged template strand being copied. B, pol III HE (composed of pol III core + β-sliding clamp + γ-clamp-loading complex) catalyzes TLS, requiring the presence of UmuD′C and cis-RecA*. C, pol V (composed of UmuD′2C) catalyzes TLS, requiring the presence of cis-RecA*. D, pol V activated by a RecA* located in trans to the primer-template DNA catalyzes TLS. E, pol V Mut (composed of pol V + RecA (red circle) + ATP (inverted blue triangle)) catalyzes TLS in the absence of RecA*.
Following the discovery in 1988 that SOS mutagenesis required cleavage of UmuD to UmuD′ (11–13), an updated TLS model was proposed by Harrison (Hatch) Echols (Fig. 7) and me (Fig. 5). This next generation model included UmuD′ in place of UmuD and the pol III holoenzyme complex (pol III HE) in place of pol III (Fig. 6B) (25). The stage was almost, but not quite, set to reconstitute TLS in vitro. pol III HE, RecA*, and UmuD′ were available as purified proteins. However, UmuC was lacking. In 1989, Hatch and his postdoctoral student Roger Woodgate, who was to become a longtime and current collaborator of ours, reported the purification of UmuC and its interaction with UmuD and UmuD′ (26). UmuD and UmuD′ were soluble in aqueous solution. In contrast, UmuC was insoluble and confined to inclusion bodies, prompting Roger to denature the protein in urea and renature it by slowly dialyzing out the urea. The result was a refolded form of UmuC that was soluble in aqueous solution. A similar denaturation-renaturation strategy had proved successful for purifying the insoluble ϵ-proofreading subunit of E. coli pol III, resulting in a water-soluble active 3′-exonuclease (27). However, extension of a primer past a template lesion (i.e. TLS) was marginal when renatured UmuC was used along with pol III HE, UmuD′, and RecA* (28).
FIGURE 7.

Hatch Echols, Berkeley, California (circa 1980).
Sadly, my close friend Hatch passed away on April 11, 1993. Several days earlier, I was invited by his wife, Carol Gross, to visit Hatch in their Berkeley home. During this very touching and brief visit, Hatch asked that I continue trying to reconstitute a biochemical system for TLS.
One could study the renatured form of ϵ with confidence because it was known that the native ϵ had a clearly defined 3′-exonuclease activity when present as a component of the soluble pol III core. However, in the case of UmuC, an alternative approach might be better suited to investigate a protein with no known biochemical function. Irina Bruck (a graduate student at the University of Southern California), Roger and I rebooted the study on UmuC, taking an alternative approach that opened the way to identify UmuC, not by itself but rather as part of soluble heterotrimeric complex, UmuD′2C (29).
By coexpressing the UmuD′C proteins, overproduced from a high-expression promoter, and purifying the UmuC protein from a strain carrying a deletion of the entire umuDC operon, we obtained a sizable amount of a soluble 46-kDa protein that we identified as UmuC by Western blotting and microsequencing. At the end of a multistep purification process, UmuC remained soluble but, as we soon discovered, only because it was accompanied throughout each step in the purification by UmuD′. During the purification, UmuC remained bound to UmuD′ as a stable 70-kDa heterotrimeric complex, UmuD′2C (29). Having UmuD′2C in hand, we then embarked on reconstituting an SOS TLS system in vitro that included the minimal requirements determined genetically, the UmuC and UmuD′ proteins, RecA*, primer-template DNA, and, of course, a DNA polymerase that was almost surely pol III HE (25). However, that turned out not to be the case as described next.
TLS Requires the Umu Proteins and RecA, But Not pol III
The approach to reconstitute TLS with purified proteins dictated by the genetics was straightforward. Simply take a primer-template DNA having a 5′-32P-labeled primer and a template containing an abasic lesion and incubate it with purified RecA, UmuD′2C, and pol III HE. The expectation was that pol III HE would extend the 32P-labeled primer up to the template lesion, but not beyond, in the absence of either RecA or UmuD′2C. The hope was that the addition of both RecA and UmuD′2C would allow pol III to insert a deoxynucleotide opposite the noncoding lesion and then continue primer extension to reach the end of the template strand. The hoped-for result was attained except for one unforeseen observation. Extension of the primer, copying undamaged template DNA to reach the lesion and then copying past the lesion (TLS), occurred in the absence of pol III core (30)! The wholly surprising result was that something other than pol III was able to avidly synthesize DNA. TLS was not the only unusual activity observed. Experiments leaving out one of the dNTP substrates made it pretty obvious that deoxynucleotide misincorporations were also occurring to an unprecedented extent opposite normal template bases.
It had occurred to us that UmuD′2C might be a new type of low-fidelity DNA polymerase. I had mentioned at a 1998 Taos conference that “DNA processing takes place in accordance with the ‘four Rs’: Replication, Recombination, Repair, REDUNDANCY,” and added that “if UmuD′2C walks like a duck and quacks like a duck, then perhaps it is a duck, disguised as a new type of DNA polymerase.” We titled the 1998 PNAS paper describing TLS “Reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein” (30). Bob Lehman, who communicated the paper to PNAS, suggested that we were being overly conservative by not stating flat out in the title that UmuD′2C was a new SOS-induced error-prone DNA polymerase. In hindsight, I obviously should have taken Bob's advice, but as Dick Vermeil, the superb head football coach for our “hated” rival school UCLA, had aptly said in a quote paraphrased from the poet Henry David Thoreau, “Never look back unless you are planning to go that way.”
There was, however, a mitigating factor in my chickening out. The specific activity of the low-fidelity TLS activity was miniscule, nowhere near that of pol I, II, or III, and perhaps a small contaminant from one or more of the pols could be present. Thanks to an exquisitely sensitive neutralizing antibody against pol I provided to us by Larry Loeb at the University of Washington, we could confidently rule out pol I as a contaminant. The deliberate addition of pol III inhibited TLS, suggesting that it was unlikely to be present in the preparation, but we could not entirely eliminate an adventitious presence of pol II. We had no antibody against pol II and no pol II deletion strain to exploit. Notably, as recounted above under “A Digression into DNA Polymerase II,” pol II was itself pretty adept at TLS. pol II was also induced by the SOS regulon in vivo in response to DNA damage (19, 22). Here, I reflect on that, back in the day, circa 1998–2004, we were fortunate to recover ∼2–4 mg of purified UmuD′2C from 30 liters of cells, even when overproduced. To obtain more workable amounts of UmuD′2C, Roger Woodgate fired up the 200-liter fermenter at the National Institutes of Health, shipping many grams of spun-down cell pellets to us on dry ice. This ensured a quarterly profit for FedEx lasting for several years. It also gave rise to heavy-duty KP (kold room patrol) by graduate students Irina Bruck, Mengjia Tang (Fig. 5), and Xuan Shen. Under such trying circumstances, a minor polymerase contaminant would not be too surprising.
A concurrent paper by Zvi Livneh and colleagues showed that when UmuC had a maltose-binding protein attached to its N terminus, it was soluble in aqueous solution; maltose-binding protein-UmuC along with UmuD′, RecA*, and pol III HE performed TLS. However, in contrast with our data, pol III HE was a necessary component in their reconstituted system (31). Absent pol III, there was no detectable TLS in Livneh's system. A PNAS Commentary discussing the two papers was written by Graham Walker and entitled “Skiing the black diamond slope: progress on the biochemistry of translesion DNA synthesis” (32). Graham's title referred specifically to a statement made by Hatch to a packed audience during a replication and repair “skiing” meeting held in Taos, New Mexico, in 1992, where he referred to UmuC as the “black diamond slope of DNA biochemistry.”
Black double diamond slope is what I remember Hatch having said. Either way, one or two diamonds, by mid-1998, the difficulties with UmuC and, more generally, with the biochemical reconstitution of TLS were at long last being bypassed (pun intended). In 1999, the heterotrimeric UmuD′2C complex was unequivocally shown to be a bona fide DNA polymerase (9), with the polymerase activity residing in the UmuC subunit (9, 14). The year 2001 witnessed the identification of a new polymerase family, the Y-family polymerases (33), that grew rapidly. A recent count identified 13 family members spanning from microorganisms to humans (34, 35).
A Fly in the cis-RecA* Filament Ointment
The model for TLS that Hatch and I proposed in 1990 (Fig. 6B) had not strayed far from the 1985 Bridges-Woodgate model (Fig. 6A), although the proteins involved were modified according to new biochemical data, including dispensing with pol III HE and replacing it with pol V (UmuD′2C) (Fig. 6C) (9). However, the core feature of the model remained: TLS was assumed to require the presence of a RecA* nucleoprotein filament assembled on the region of ssDNA located in cis at the 5′-side of the damaged template base (Fig. 6, A–C). Positioning of RecA* on the template strand proximal to the lesion surely made sense. When asked why he robbed banks, Willie Sutton famously replied, “Because that's where the money is.” The same localization principle logically accounted for positioning of RecA*. Although assembly of RecA* in cis on the template strand being copied was a reasonable supposition, there was no direct evidence to support it.
We were soon led to rethinking where and how RecA* functioned during TLS because of a talk that I gave in November 2000 at the National Academy of Sciences Colloquium on “Links Between Recombination and Replication: Vital Roles of Recombination” at University of California, Irvine. Projected on the screen were PAGE data depicting 32P-labeled primer elongation bands visualized at single-nucleotide resolution with remarkable clarity using the aforementioned PhosphorImager. Although the data showed that TLS was greatest with a RecA/ssDNA ratio of ∼1 RecA monomer/3–5 nucleotides, which is consistent with the optimal ratio for RecA* filament formation, TLS was also clearly observed at ∼1 RecA monomer/50 nucleotides. Following my talk, Mike Cox (Fig. 8) mentioned to me privately that RecA* would be unlikely to assemble on the region of ssDNA downstream of the template lesion at 1 RecA monomer/50 nucleotides.
FIGURE 8.

Mike Cox, Madison, Wisconsin (circa 2009). This is how he looks today.
I was grateful that Mike spared me a public “gotcha,” all the more so because his point proved to be spot-on. When one makes primer-template DNA by annealing a relatively short primer to a longer template strand, thermodynamics dictates that a portion of the DNA remains single-stranded. Furthermore, because we typically used an excess of primers over template to optimize the yield of the primer-template DNA duplex, there was always an excess of primer strands in solution to bind with RecA. Although the ratio of ∼1 RecA monomer/50 nucleotides was not sufficiently high for RecA nucleoprotein filament assembly on the primer-template DNA duplex, the ability to observe pol V-catalyzed TLS (36) suggested that perhaps RecA* forming on the shorter free primer strands was somehow able to facilitate lesion bypass by pol V by acting in trans (Fig. 6D) (37). We viewed these data as a potential “fly in the cis-RecA* filament ointment.” Perhaps RecA* assembled in trans on a ssDNA strand that was not being copied by pol V.
Transactivation of pol V by RecA*
Compelling evidence for transactivation of pol V-catalyzed DNA synthesis, including TLS, was obtained by Katharina (Kathi) Schlacher, a graduate student who came to our lab initially with the limited objective of performing a Master's thesis project for a year and defending in Austria, which she did. To our good fortune, Kathi then decided to return to the lab to initiate the transactivation studies, which I am pleased to say won her the “Best Ph.D. Dissertation of 2006” award from the University of Southern California. The key to proving that pol V can be activated via the assembly of RecA* on ssDNA that is not being copied was to design a primer-template DNA that could not support template filament formation. The primer-template was formed as a stable DNA hairpin with just a 3-nucleotide overhang region at its 5′-end to serve the template strand (see e.g. Fig. 9B, left) (37). A RecA monomer has a 3-nucleotide footprint on ssDNA, leaving no space on the template strand to form a cis-RecA* filament downstream of UmuD′2C. Instead, RecA* filaments were assembled on separate ssDNA molecules, which acted in trans on the primer-template DNA duplex hairpins. The 3-nucleotide template overhangs were copied but only with trans-RecA* present (37). The reaction of pol V with the primer-template DNA hairpins was second-order, i.e. linearly proportional to the concentration of trans-RecA*, leaving little (if any) room for doubt that RecA* assembled on a DNA different from the one being copied was responsible for pol V activity (Fig. 6D).
FIGURE 9.

pol V Mut (UmuD′2C-RecA-ATP) is a stand-alone DNA polymerase able to synthesize DNA in the absence of RecA*. A, pol V Mut is formed by incubating pol V with RecA* bound to resin and removing the resin-bound RecA*, leaving pol V Mut in solution. B, pol V Mut formed with wild-type RecA copies DNA, whereas pol V Mut formed with Devoret's SOS nonmutagenic RecA1730(S117F) cannot copy DNA. Red circle, RecA; inverted blue triangle, ATP or ATPγS. nt, nucleotides.
The historical positioning of RecA* downstream of pol V on the template strand being copied has always been based on an Occam's razor-like rationale that because RecA* was required for TLS, it had to act at a blocked replication fork (38). What had not been contemplated is that the role of RecA* might actually be indirect: that it is not interacting with a RecA* filament, be it located either in cis or in trans, that activates pol V, but instead, RecA* is needed to transfer a RecA monomer from its 3′-proximal tip to pol V and thereby activate it for DNA synthesis (15).
Captured Alive, the Active Form of pol V Is UmuD′2C-RecA-ATP
Deciphering the role of RecA* remained the key biochemical challenge, a mystery that had persisted since the mid-1980s with the Bridges-Woodgate model (Fig. 6A) (24) and crystallized by Devoret's discovery of an essential role for RecA* in damage-induced mutagenesis, distinct from its roles in recombination and the coproteolytic cleavage of the LexA repressor and UmuD (16). Mixing pol V with trans-RecA* resulted in an active enzyme capable of copying undamaged and damaged templates (37), but mixing pol V with RecA did not (39). An experiment was needed to nail down unambiguously how and why RecA* acts during pol V-catalyzed TLS.
We addressed the “how and why” by incubating pol V with RecA* in the absence of primer-template DNA and then isolating a possibly modified form of the polymerase with RecA* no longer present (Fig. 9A). The strategy entailed assembling RecA* filaments on ssDNA oligonucleotides that were stably attached to resins or beads (15). Following pol V incubation with immobilized RecA* and removal of the RecA* by centrifugation, we found that pol V had undergone a major modification. A single RecA molecule had been transferred from the 3′-proximal tip of RecA* to UmuD′2C, which, along with the binding of a molecule of ATP (or ATPγS), converted the virtually inactive pol V to the stand-alone activated pol V Mut (UmuD′2C-RecA-ATP) (Fig. 9A). This was the first time that pol V was observed to synthesize DNA without RecA* (Fig. 9B) (15). A new model for TLS involved pol V Mut with RecA* no longer directly in the picture (Fig. 6E).
pol V Mut was active with a bound wild-type RecA monomer, whereas pol V Mut was dead when formed with Devoret's UV light-nonmutable RecA(S117F) mutant (Fig. 9B) (15). pol V Mut(S117F) had no measurable DNA synthesis activity in vitro even though it was also transferred to UmuD′2C from the 3′-RecA* filament tip to form a stable, yet inactive, pol V Mut complex (15). It was gratifying to give a biochemical face to Devoret's critically important genetic identification of an independent mutagenic function for RecA*, which had provided the impetus for our work.
After submitting a manuscript to Nature, a multiangle light scattering (MALS) instrument arrived at the University of Southern California in the two weeks intervening between receipt of two referees' reports and the third report, which requested (i.e. demanded) more convincing evidence for the existence of a stable complex composed of a RecA monomer bound to UmuD′2C. MALS measures the absolute molecular mass of a pure protein in aqueous solution, subject only to a few nonrestrictive assumptions regarding protein globular shape (40). A mass standard for either calibration or comparison is not even required. Using MALS, we identified a light scattering peak centered at 113 kDa corresponding to a complex of UmuD′2C-RecA. Using SDS-PAGE, we showed that the protein composition in the 113-kDa peak contained UmuC, UmuD′, and RecA.
The Unexpected Identification of pol V Mut as DNA-dependent ATPase
Thanks to MALS and the third reviewer, pol V Mut was captured alive as UmuD′2C-RecA (15). However, a question remained: what about ATP? Was ATP required for pol V Mut activity? The short answer is that when present as UmuD′2C-RecA, pol V Mut lacking ATP cannot synthesize DNA. To be active, it must have a molecule of ATP (or ATPγS) bound to form UmuD′2C-RecA-ATP (15). The unresolved question was why. The answer that we arrived at while I was writing these Reflections is that pol V Mut function is regulated by an intrinsic DNA-dependent ATPase activity (41). The binding of a single ATP molecule to pol V Mut is required to bind the polymerase to primer-template DNA. DNA synthesis, including TLS, then takes place. ATP hydrolysis triggers the dissociation of pol V Mut from the DNA. pol V Mut appears to function as an on-off toggle switch. Once activated, it performs a single round of DNA synthesis. Following ATP hydrolysis, the enzyme dissociates from primer-template DNA in a deactivated form (Fig. 10). No such ATPase activity or autoregulatory mechanism has previously been found for a DNA polymerase.
FIGURE 10.
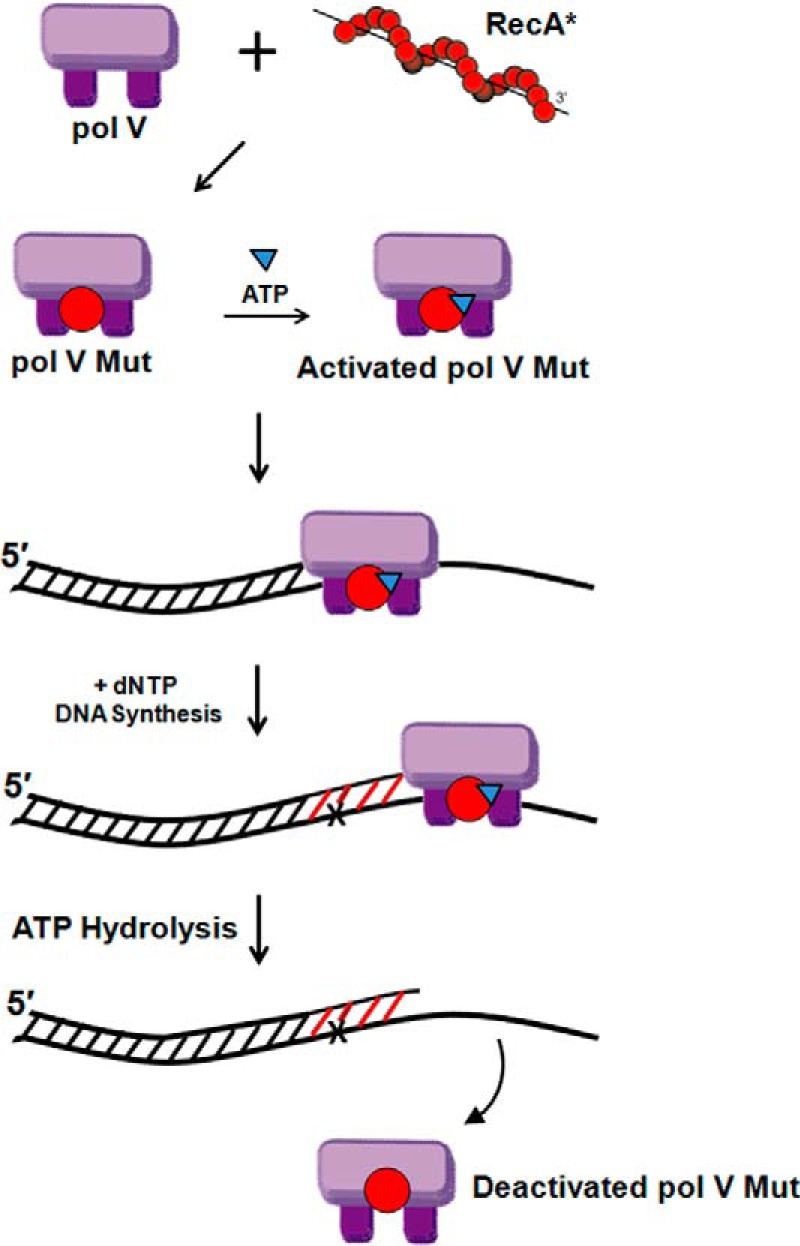
Model for pol V Mut regulation by RecA and ATP. Transfer of a RecA molecule (red circle) from RecA* and the subsequent binding of an ATP molecule (inverted blue triangle) to UmuD′2C form activated pol V Mut (UmuD′2C-RecA-ATP). A bound ATP molecule is required for polymerase binding to primer-template DNA. pol V copies an undamaged region of DNA, inserts a deoxynucleotide opposite a lesion (X), and continues DNA synthesis beyond the lesion (TLS). pol V Mut is deactivated after dissociating from the primer-template DNA. Dissociation is dependent on pol V Mut-dependent ATP hydrolysis.
What is the biological basis for the complex regulation of pol V by ATP? Mutagenic DNA synthesis during the SOS response is seemingly an act of cellular desperation that would best be limited as to when and where it is used. The regulation of pol V Mut is needed to limit mutations, especially in rapidly dividing cells (42). Mutational lethality can be avoided by restricting low-fidelity DNA synthesis to short DNA segments confined to replication forks blocked by DNA damage and to replication restart at stalled replication forks on undamaged DNA. Keeping pol V Mut processivity in check appears to be role of the internal ATPase that triggers polymerase dissociation from primer-template DNA (41). In essence, the enzyme has evolved to do the absolute minimum to get cellular DNA synthesis restarted.
Acknowledgments
I thank Matty Scharff and Phuong Pham for providing valuable and constructive criticisms on the manuscript and Jeff Bertram and Phuong for aid with the illustrations. I thank Evelyn Witkin, Michael Devoret, Mike Cox, Maurice Bessman, Graham Walker, and Stuart Linn for generously sending photographs. I am indebted to past and present students and postdoctoral fellows who provided invaluable intellectual support for the SOS-pol V studies while having interests that differed from pol V. These other projects investigated the fidelity of DNA polymerases and, more recently, the mutator mechanisms of APOBEC dC deaminases. The NIGMS, NIEHS, and NCI have provided generous sustenance. I especially thank Dan Shaughnessy (NIEHS), Dick Pelroy (NCI), and the late Paul B. Wolfe (NIGMS) for their gracious support and advice throughout my forty years and counting in the business.
Footnotes
- TLS
- translesion synthesis
- pol V
- DNA polymerase V
- ssDNA
- single-stranded DNA
- pol III HE
- pol III holoenzyme complex
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- MALS
- multiangle light scattering.
REFERENCES
- 1. Weigle J. J. (1953) Induction of mutation in a bacterial virus. Proc. Natl. Acad. Sci. U.S.A. 39, 628–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Witkin E. M. (1967) The radiation sensitivity of Escherichia coli B: a hypothesis relating filament formation and prophage induction. Proc. Natl. Acad. Sci. U.S.A. 57, 1275–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witkin E. M. (1969) Ultraviolet-induced mutation and DNA repair. Annu. Rev. Microbiol. 23, 487–514 [DOI] [PubMed] [Google Scholar]
- 4. Radman M. (1974) Phenomenology of an inducible mutagenic DNA repair pathway in Escherichia coli: SOS repair hypothesis. in Molecular and environmental aspects of mutagenesis (Prakash L., Sherman F., Miller M., Lawrence C., Tabor H. W., eds) pp. 128–142, Charles C. Thomas, Springfield, IL [Google Scholar]
- 5. Kato T., Shinoura Y. (1977) Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol. Gen. Genet. 156, 121–131 [DOI] [PubMed] [Google Scholar]
- 6. Steinborn G. (1978) Uvm mutants of Escherichia coli K12 deficient in UV mutagenesis. I. Isolation of uvm mutants and their phenotypical characterization in DNA repair and mutagenesis. Mol. Gen. Genet. 165, 87–93 [DOI] [PubMed] [Google Scholar]
- 7. Kitagawa Y., Akaboshi E., Shinagawa H., Horii T., Ogawa H., Kato T. (1985) Structural analysis of the umu operon required for inducible mutagenesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 82, 4336–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perry K. L., Elledge S. J., Mitchell B. B., Marsh L., Walker G. C. (1985) umuDC and mucAB operons whose products are required for UV light- and chemical-induced mutagenesis: UmuD, MucA, and LexA proteins share homology. Proc. Natl. Acad. Sci. U.S.A. 82, 4331–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang M., Shen X., Frank E. G., O'Donnell M., Woodgate R., Goodman M. F. (1999) UmuD′2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. U.S.A. 96, 8919–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox M. M. (2003) The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 57, 551–577 [DOI] [PubMed] [Google Scholar]
- 11. Nohmi T., Battista J. R., Dodson L. A., Walker G. C. (1988) RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl. Acad. Sci. U.S.A. 85, 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burckhardt S. E., Woodgate R., Scheuermann R. H., Echols H. (1988) UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc. Natl. Acad. Sci. U.S.A. 85, 1811–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shinagawa H., Iwasaki H., Kato T., Nakata A. (1988) RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 85, 1806–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reuven N. B., Arad G., Maor-Shoshani A., Livneh Z. (1999) The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 274, 31763–31766 [DOI] [PubMed] [Google Scholar]
- 15. Jiang Q., Karata K., Woodgate R., Cox M. M., Goodman M. F. (2009) The active form of DNA polymerase V is UmuD′2C-RecA-ATP. Nature 460, 359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dutreix M., Moreau P. L., Bailone A., Galibert F., Battista J. R., Walker G. C., Devoret R. (1989) New recA mutations that dissociate the various RecA protein activities in Escherichia coli provide evidence for an additional role for RecA protein in UV mutagenesis. J. Bacteriol. 171, 2415–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Randall S. K., Eritja R., Kaplan B. E., Petruska J., Goodman M. F. (1987) Nucleotide insertion kinetics opposite abasic lesions in DNA. J. Biol. Chem. 262, 6864–6870 [PubMed] [Google Scholar]
- 18. Creighton S., Bloom L. B., Goodman M. F. (1995) Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 262, 232–256 [DOI] [PubMed] [Google Scholar]
- 19. Bonner C. A., Randall S. K., Rayssiguier C., Radman M., Eritja R., Kaplan B. E., McEntee K., Goodman M. F. (1988) Purification and characterization of an inducible Escherichia coli DNA polymerase capable of insertion and bypass at abasic lesions in DNA. J. Biol. Chem. 263, 18946–18952 [PubMed] [Google Scholar]
- 20. Wickner R. B., Ginsberg B., Berkower I., Hurwitz J. (1972) Deoxyribonucleic acid polymerase II of Escherichia coli. I. The purification and characterization of the enzyme. J. Biol. Chem. 247, 489–497 [PubMed] [Google Scholar]
- 21. Kenyon C. J., Walker G. C. (1980) DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 77, 2819–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonner C. A., Hays S., McEntee K., Goodman M. F. (1990) DNA polymerase II is encoded by the DNA damage-inducible dinA gene of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 87, 7663–7667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwasaki H., Nakata A., Walker G. C., Shinagawa H. (1990) The Escherichia coli polB gene, which encodes DNA polymerase II, is regulated by the SOS system. J. Bacteriol. 172, 6268–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bridges B. A., Woodgate R. (1985) The two-step model of bacterial UV mutagenesis. Mut. Res. 150, 133–139 [DOI] [PubMed] [Google Scholar]
- 25. Echols H., Goodman M. F. (1990) Mutation induced by DNA damage: a many protein affair. Mutat. Res. 236, 301–311 [DOI] [PubMed] [Google Scholar]
- 26. Woodgate R., Rajagopalan M., Lu C., Echols H. (1989) UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc. Natl. Acad. Sci. U.S.A. 86, 7301–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scheuermann R. H., Echols H. (1984) A separate editing exonuclease for DNA replication: the ϵ subunit of Escherichia coli DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 81, 7747–7751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajagopalan M., Lu C., Woodgate R., O'Donnell M., Goodman M. F., Echols H. (1992) Activity of the purified mutagenesis proteins UmuC, UmuD′, and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III. Proc. Natl. Acad. Sci. U.S.A. 89, 10777–10781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bruck I., Woodgate R., McEntee K., Goodman M. F. (1996) Purification of a soluble UmuD′C complex from Escherichia coli. Cooperative binding of UmuD′C to single-stranded DNA. J. Biol. Chem. 271, 10767–10774 [DOI] [PubMed] [Google Scholar]
- 30. Tang M., Bruck I., Eritja R., Turner J., Frank E. G., Woodgate R., O'Donnell M., Goodman M. F. (1998) Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc. Natl. Acad. Sci. U.S.A. 95, 9755–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reuven N. B., Tomer G., Livneh Z. (1998) The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol. Cell 2, 191–199 [DOI] [PubMed] [Google Scholar]
- 32. Walker G. C. (1998) Skiing the black diamond slope: progress on the biochemistry of translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 95, 10348–10350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohmori H., Friedberg E. C., Fuchs R. P., Goodman M. F., Hanaoka F., Hinkle D., Kunkel T. A., Lawrence C. W., Livneh Z., Nohmi T., Prakash L., Prakash S., Todo T., Walker G. C., Wang Z., Woodgate R. (2001) The Y-family of DNA polymerases. Mol. Cell 8, 7–8 [DOI] [PubMed] [Google Scholar]
- 34. Sale J. E., Lehmann A. R., Woodgate R. (2012) Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goodman M. F., Woodgate R. (2013) Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 5, a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pham P., Seitz E. M., Saveliev S., Shen X., Woodgate R., Cox M. M., Goodman M. F. (2002) Two distinct modes of RecA action are required for DNA polymerase V-catalyzed translesion synthesis. Proc. Natl. Acad. Sci. U.S.A. 99, 11061–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlacher K., Cox M. M., Woodgate R., Goodman M. F. (2006) RecA acts in trans to allow replication of damaged DNA by DNA polymerase V. Nature 442, 883–887 [DOI] [PubMed] [Google Scholar]
- 38. Patel M., Jiang Q., Woodgate R., Cox M. M., Goodman M. F. (2010) A new model for SOS-induced mutagenesis: how RecA protein activates DNA polymerase V. Crit. Rev. Biochem. Mol. Biol. 45, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schlacher K., Leslie K., Wyman C., Woodgate R., Cox M. M., Goodman M. F. (2005) DNA polymerase V and RecA protein, a minimal mutasome. Mol. Cell 17, 561–572 [DOI] [PubMed] [Google Scholar]
- 40. Wyatt P. J. (1993) Light scattering and the absolute characterization of macromolecules. Anal. Chim. Acta 272, 1–40 [Google Scholar]
- 41. Erdem A. L., Jaszczur M., Bertram J. G., Woodgate R., Cox M. M., Goodman M. F. (2014) DNA polymerase V activity is autoregulated by a novel intrinsic DNA-dependent ATPase. eLIFE 3, e02384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Corzett C. H., Goodman M. F., Finkel S. E. (2013) Competitive fitness during feast and famine: how SOS DNA polymerases influence physiology and evolution in Escherichia coli. Genetics 194, 409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]