

Background: The regulation of RAF kinases is a highly complex process.
Results: Loss of B-RAF in alveolar epithelial type II cells does not inhibit oncogenic C-RAF BxB-driven lung tumor initiation but diminishes MAPK signaling and tumor growth.
Conclusion: B-RAF cooperates with oncogenic C-RAF BxB in lung tumorigenesis.
Significance: Inhibition of RAFs dimerization might be a new therapeutic option for oncogenic C-RAF-driven tumors.
Keywords: Cell Biology, Lung Cancer, Mitogen-activated Protein Kinase (MAPK), Mouse Genetics, Raf Kinase, Signal Transduction
Abstract
Tumors are often greatly dependent on signaling cascades promoting cell growth or survival and may become hypersensitive to inactivation of key components within these signaling pathways. Ras and RAF mutations found in human cancer confer constitutive activity to these signaling molecules thereby converting them into an oncogenic state. RAF dimerization is required for normal Ras-dependent RAF activation and is required for the oncogenic potential of mutant RAFs. Here we describe a new mouse model for lung tumor development to investigate the role of B-RAF in oncogenic C-RAF-mediated adenoma initiation and growth. Conditional elimination of B-RAF in C-RAF BxB-expressing embryonic alveolar epithelial type II cells did not block adenoma formation. However, loss of B-RAF led to significantly reduced tumor growth. The diminished tumor growth upon B-RAF inactivation was due to reduced cell proliferation in absence of senescence and increased apoptosis. Furthermore, B-RAF elimination inhibited C-RAF BxB-mediated activation of the mitogenic cascade. In line with these data, mutation of Ser-621 in C-RAF BxB abrogated in vitro the dimerization with B-RAF and blocked the ability to activate the MAPK cascade. Taken together these data indicate that B-RAF is an important factor in oncogenic C-RAF-mediated tumorigenesis.
Introduction
The Ras-mitogen-activated protein kinase (MAPK)7 pathway participates in the control of many important cellular processes including survival, proliferation, apoptosis, and differentiation. The pathway starts at the cell membrane with activation of the EGF receptor (EGFR) or other receptor tyrosine kinases (RTKs) that recruit and activate the Grb2-Sos complex by direct or indirect mechanisms. Grb2 is a small adapter protein that forms a complex with the Ras guanine nucleotide exchange factor (GEF) Sos, thereby linking RTK stimulation at the cell membrane to activation of the small GTPase Ras inside the cell (1). Active Ras recruit RAFs serine/threonine kinases at the plasma membrane where they become activated. Active RAFs then phosphorylate MEK that in turn phosphorylate ERK, which can ultimately affect several transcription factors and other substrates (2). The MAPK pathway has been found to be hyperactivated in around 30% of all cancer; two members of this pathway, Ras and RAF, are known oncogenes (2). The RAF family consists of three members, A-, B-, and C-RAF. Among these, B-RAF is the most frequently mutated RAF oncogene in human cancer (3). Missense point mutations are the most frequent type of RAF mutations. Recently, chromosomal translocations involving either B- or C-RAF in a small percentage of several type of cancers were found (4, 5). Notably, these gene fusions encode for RAF proteins retaining the kinase domain but losing the N-terminal RAS-binding domain, suggesting that the mutant proteins may be constitutively active (4).
Previously, our laboratory has generated a mouse lung tumor model by targeting an active C-RAF (C-RAF BxB) to type II cells using the SP-C promoter. C-RAF BxB lacks the regulatory NH2-terminal sequences including the Ras interaction domain (6). SP-C-driven C-RAF BxB expression yields several adenomas that were well differentiated, poorly vascularized, and did not progress to metastasis (6). In vitro experiments have shown that B-RAF constitutively heterodimerizes with C-RAF BxB by targeting its kinase domain and by activating it in the cytoplasm (7, 8). These data may suggest that the C-RAF BxB/B-RAF heterodimerization is an important event for the oncogenic properties of C-RAF BxB.
In this project, we aimed to evaluate in vivo and in vitro the role of B-RAF in C-RAF BxB-mediated tumorigenesis. Toward this aim, we have generated compound mice in which we could specifically knock-out B-RAF in C-RAF BxB-expressing alveolar epithelial type II cells. Conditional inactivation of B-RAF did not block oncogenic C-RAF-driven adenoma formation however, it reduced the tumor growth. The reduced tumor growth was caused by diminished cell proliferation in absence of augmented cell death or senescence mechanisms. Moreover, B-RAF elimination led to reduced C-RAF BxB-mediated activation of the MAPK signaling. Furthermore, the mutation of a residue of C-RAF BxB (Ser-621) that is necessary for 14-3-3-mediated dimerization abolished an efficient binding of C-RAF BxB to B-RAF and abrogated its kinase activity. Altogether, our data indicate that both in vivo and in vitro B-RAF cooperates with C-RAF BxB in activating the MAPK cascade, and therefore it is an important factor in oncogenic C-RAF-mediated tumor growth.
EXPERIMENTAL PROCEDURES
Transgenic Animals
All animal experiments were performed according to the regulations of the Bavarian State authorities. Mice were housed in a pathogen-free environment with a 12-hour dark/light circle and with the access to food and water ab libitum. The generation of B-RAFflox/flox transgenic mice have been previously described (9). In B-RAFflox/flox mice, loxP sites flank exon 3 of the b-raf gene. SpC-rtTA, Tet-O-cre, and SpC-C-RAF BxB23 (hereafter SpC-C-RAF BxB) transgenic mice have been previously described (6) (10). B-RAFflox/flox, SpC-rtTA, and SpC-C-RAF BxB mice were routinely bred in the C57BL/6 background whereas Tet-o-cre animals were kept in the FVB/N background. B-RAFflox/flox and B-RAFflox-wt mice were mated with SpC-rtTA and Tet-o-cre mice to obtain triple transgenic SpC-rtTA/Tet-o-cre/B-RAFflox/flox and SpC-rtTA/Tet-o-cre/B-RAFflox/wt. Triple transgenic mice were mated with SpC-C-RAF BxB mice to obtain quadruple transgenic animals SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox, and SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/wt. Exon 3 deletion of the b-raf gene in normal and in tumor-bearing lungs was carried out by induction of compound mice with doxycyclin (DOX) (Sigma) containing food (500 mg/kg body weight; Ssniff, Soest, Germany) throughout gestation and until euthanization. DOX-exposed SpC-rtTA/Tet-o-cre/B-RAFflox/flox mice (hereafter triple B-RAF Δ/Δ) were compared with uninduced SpC-rtTA/Tet-o-cre/B-RAFflox/flox mice (hereafter triple B-RAF flox/flox). DOX-exposed SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox mice (hereafter B-RAFΔ/Δ) were compared with DOX-exposed SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/wt littermates (hereafter B-RAFΔ/wt) and uninduced SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox mice (hereafter B-RAF flox/flox). Genotypes were identified by PCR from genomic tail DNA as described previously (9, 10).
Reagents and Antibodies
For immunohistochemistry/immunofluorescence, primary antibodies against the following proteins were used: BRAF (H-145, Santa Cruz Biotechnology, 1:100); Active Caspase-3 (Asp175) (#9664, Cell Signaling, 1:200); Ki67 (VPK452, MM1 Vector laboratories, 1:50); pan-Cytokeratin (Z0622, Dako, 1:500); phospho-ERK (#4376, Cell Signaling, 1:100); phospho-H3 (S10) (#9701, Cell Signaling, 1:100); Pro SP-C (gift of Jeffrey A. Whitsett); Raf1 (E10, Santa Cruz Biotechnology, 1:100). TUNEL staining was carried out using the Dead End Colorimetric TUNEL Kit (Promega) following the manufactureŕs instructions. For immunoblot analysis antibodies against the following proteins were used: BRAF (H-145, Santa Cruz Biotechnology, 1:2000); CCSP (T18, Santa Cruz Biotechnology, 1:1000), C-RAF (E10, Santa Cruz Biotechnology, 1:500); ERK 1/2 (#4695, Cell Signaling, 1:1000); phospho-ERK (#4376, Cell Signaling, 1:1000); HA (#3724, Cell Signaling, 1:5000) β-actin (I10, Santa Cruz Biotechnology, 1:3000). HA-tagged C-RAF proteins, including wild type and S621D mutant, were expressed from pcDNA3 (Invitrogen) vector constructs.
Histology and Immunostaining
Animals were sacrificed by overdose of anesthetic and perfused with phosphate buffer saline (PBS). The lungs were dissected, fixed overnight with 4% paraformaldehyde in PBS and paraffin embedded. Sections of 6-μm thickness were deparaffinized, rehydrated in graded series of alcohol and hematoxylin-eosin (H&E) stained. For immunohistochemical staining sections were deparaffinized and rehydrated. Antigen retrieval was carried out by heating the tissue sections for 6–20 min in a microwavable vessel containing 10 mm citrate buffer (pH 6) using a microwave oven. Endogenous peroxidase activity was quenched by incubating the tissue slides with 0.3–3% H2O2 in PBS or methanol for 30 min at room temperature. Unspecific binding of the antibodies was blocked by incubating the tissue sections with a blocking solution containing 3–5% normal serum and 0.1% Triton X-100 in PBS for 1 h at room temperature. After primary antibody incubation overnight at 4 °C, tissue sections were incubated for 90 min at room temperature with biotinylated secondary antibody (DakoCytomation) 1:200 diluted in blocking solution. ABC reagent ((Vectastain Elite ABC Kit, Vector Labs) was prepared and applied to tissue sections according to the manufacturer's instructions, and then developed in diaminobenzidine (DAB) (0.8% H2O2 in1 ml DAB). Tissue slides were then counterstained with hematoxilin, dehydrated, and mounted. The phospho-ERK staining was essentially carried out as previously described except that TBS with 1% Tween 20 was used instead of PBS. B-RAF/C-RAF double immunofluorescence staining was basically done as previously described except that goat anti-mouse-Cy5 and donkey anti rabbit-Cy3, diluted (1:200) in blocking solution, were used as secondary antibody. Moreover counterstaining was done by using 4′,6-diamidino-2-phenylindole (DAPI) and by mounting with Mowiol (Roth).
Immunohistopathological Analysis
Lung and tumor areas were determined using AxioVision software (Carl Zeiss Microscopy GmbH). Number and size (in μm2) of lung tumors were determined by analyzing 5–7 randomly selected pictures (taken at 10× magnification) in H&E-stained lung sections. Tumor burden was expressed as the sum of the tumor area divided by the total lung area. For each lung, two nonconsecutive slides (at least 100 ∼μm apart) were used for quantification. At least five mice for each genotype and age were analyzed: 140 tumors from B-RAF flox/flox, 188 tumors from B-RAF flox/wt and 168 tumors from B-RAFΔ/Δ at the age of two months were analyzed. 430 tumors from B-RAF flox/flox, 329 tumors from B-RAF flox/wt and 314 tumors from B-RAFΔ/Δ at the age of seven months were analyzed. For quantitation of active caspase 3, TUNEL, Ki67, and phosphoH3 immunostaining 15 randomly selected tumors were counted for each lung from five mice for each group. At least 6300 (± 300) cells at the age of two months and at least 13000 (± 3000) cells at the age of seven months were counted for each group. The intensity of the MAPK signal in tumor bearing lungs was assessed by estimating the percentage of tumor cells with phospho-ERK nuclear staining. A total of 40 tumors from four mice were quantified for each group. At least 4500 tumors at the age of two months and at least 6500 tumors at the age of seven months were counted for each group. The degree of B-RAF protein lost was evaluated by counting the fraction of C-RAF/B-RAF double positive tumor cells in double immunofluorescence stained lung sections of B-RAFΔ/Δ and B-RAF flox/flox control mice. At least 3000 C-RAF-positive cells from a total of 40 tumors from four mice were quantified for each group.
Molecular Analysis of Recombination Efficiency
DNA was isolated from total lung of B-RAF flox/flox and B-RAFΔ/Δ compound mice. Analysis of the conditional b-raf allele was done using standard PCR as previously reported (9). Briefly, the primer pair for detection of the floxed allele (flox) was brafgs12 (5′-TGT AGC CTC GGC TGT GGA ACT C) and brafgas8 (5′- GAG ACC AAA CCA AGG ACC TCT G) yielding a fragment of 326 bp. The primer pair for detection of the deletion allele (Δ) was 5xhoI (5′-CCT GAA AGC TGC TAG TAG AAG AC) and brafgas8 (5′-GAG ACC AAA CCA AGG ACC TCT G) yielding a fragment of 421 bp.
Alveolar Type II Cells Isolation
Alveolar epithelial cells were isolated as described previously with some modifications (11). Mice were anesthetized and lungs were perfused with PBS then filled with ∼1.5 ml sterile Dispase (BD Biosciences) and 0.5 ml low-melting agarose (Sigma-Aldrich) via the trachea. After gelling, lungs were removed and incubated further in Dispase for 40 min at room temperature with rotation. The trachea and surrounding connective tissue were removed, and lung tissue was minced in DMEM/2.5% HEPES with 0.01% DNase (50 μg/ml; Sigma) and incubated with shaking for further 10 min at 37 °C. Digested tissue was washed with PBS and single cells separated by successive filtration through 100- and 40-μm nylon filters and collected by centrifugation (12, 13). To remove the immune cells, single cell suspension was incubated with CD32 (3 μl/lung) and CD45 (7 μl/lung) antibodies for 30 min at 37 °C. Cell-antibodies mixture was centrifuged, resuspended in 7 ml of medium, and incubated with PBS-pre-equilibrated streptavidin magnetic beads for 30 min on rolling table. After purification of all bound cells, the supernatant containing all epithelial cells was plated on fibronectin-coated plates. Once the cells adhered to the plates, they were washed several times with PBS to get rid of erythrocytes. The cells were kept for maximum 3 days in culture.
Whole Lung Protein Extraction and Immunoblotting
Mice were anesthetized and the lungs were perfused with PBS injected through the right ventricle and snap-frozen in liquid nitrogen. Lungs were lysed in ice-cold RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% deoxycholate and 1% Nonidet P-40) with protease and phosphatase inhibitor cocktails (Roche) using an Ultra-Turrax (IKA) homogenizer in a cold environment. After centrifuging for 10 min at full speed at 4 °C, supernatant-containing proteins was analyzed for protein concentration using the BCA protein assay kit (Thermo Scientific). The remaining protein extracts were mixed with 3× Laemmli buffer, heated at 95 °C and vortexed for 10 s. 30 μg protein lysates were separated by SDS-PAGE on polyacrylamide gels and transferred onto 0.2-μm nitrocellulose membranes (Bio-Rad). After blocking with 5% nonfat-dry-milk in NET-gelatin (50 mm Tris/HCl pH 7.4, 5 mm EDTA, 0.05% Triton X-100, 150 mm NaCl, 0,25% gelatin) blots were incubated overnight at 4 °C with primary antibodies, washed three times with NET-gelatin and incubated for 1 h at room temperature with peroxidase-coupled secondary antibodies (GE Healthcare). After three washes with NET-gelatin, blots were subsequently developed using an ECL kit (Thermo Scientific).
Cell Transfection, Protein Extraction, Immunoprecipitation, and Kinase Assay
Human embryonic kidney, HEK293 cells (American Type Culture Collection) were seeded in 100-mm dishes. 24 h after seeding cells were transfected with 7.5 μg of plasmid coding for different C-RAF constructs (such as pcDNA3, pcDNA3-C-RAF BxB, and pcDNA3-C-RAF BxB S621D) using the HP DNA Transfection Reagent (Roche). 48 h after transfection cells lysed in ice-cold Nonidet P-40 (Nonidet P-40) buffer supplied with proteases and phosphatases inhibitor cocktails (Roche) for 30 min at 4 °C. Supernatants were cleared by centrifugation and protein concentration was measured using the BCA protein assay kit (Thermo Scientific). Immunoprecipitation of HA-C-RAF BxB proteins was carried out by incubating 500 mg protein lysates with HA-specific antibody for 1 h at 4 °C. After adding 30 μl Protein-G-agarose the incubation was extended for additional 2 h at 4 °C. The agarose beads were washed twice with Nonidet P-40 buffer and one time with kinase assay buffer. Kinase assays were carried out directly with immunoprecipitated C-RAF BxB proteins using recombinant MEK and ERK-2 as substrates in 25 mm HEPES, pH 7.6, 150 mm NaCl, 25 mm β- glycerophosphate, 10 mm MgCl2, 1 mm dithiothreitol, and 1 mm sodium vanadate buffer (in a 50 μl total volume). After incubating the mixture for 30 min at 30 °C, the reaction was stopped by adding 50 μl of Laemmli buffer prior to boil it at 95 °C for 5 min. Proteins were separated by SDS-PAGE on polyacrylamide gels, and the kinase activity was measured by immunoblotting with phospho-ERK antibody.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism4 software (Graphpad Software, Inc, San Diego, CA). Student's t test (two-tailed) was used to compare two groups (p < 0.05 was considered significant).
RESULTS
Conditional Ablation of b-raf in C-RAF BxB-expressing Lung Type II Cells in Transgenic Mice
To examine whether B-RAF is necessary for oncogenic-C-RAF-mediated lung tumorigenesis SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox and SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/wt quadruple transgenic mice were induced with doxycyclin (DOX) throughout gestation and until the day of scarification (Fig. 1A). Lung-specific recombination of the floxed B-RAF allele was detected in SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox compound mice that were exposed to DOX (Fig. 1B). Consistent with lung specific allele recombination, immunoblot analysis of protein lysates from isolated alveolar epithelial type II cells of DOX-induced and control transgenic mice display reduction of B-RAF protein expression in B-RAFΔ/Δ compound animals (Fig. 1C). Remaining B-RAF protein in lysates from type II cells of B-RAFΔ/Δ mice are probably due to contamination of other lung epithelial cells such as Clara cells that express the Clara Cell Secretory Protein (CCSP) (Fig. 1C). To assess the effect of b-raf loss on lung tumorigenesis, compound mice were DOX-treated throughout gestation for 2, 7, and 12 months respectively; and subsequently examined by histology. Interestingly, all compound animals displayed lung tumors (Fig. 2, A and B). It is known that both rtTA and Cre in the presence of DOX show potential off-target toxicity, therefore as control we have also included DOX-induced SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/wt compound mice which have an heterozygote floxed B-RAF allele (Fig. 2, A and B). We found no difference in the tumor incidence of B-RAFΔ/Δ compound mice and age-matched controls at 2 and 7 months of age (Fig. 2B). Quantification of single lung tumors in 12 months old B-RAFΔ/Δ and control animals was not possible because in the lung of B-RAF Δ/wt and B-RAF flox/flox animals the adenomas were confluent (Fig. 2A). However, we observed a significant reduction in the lung tumor growth only in B-RAF Δ/Δ compound animals (Fig. 2, C and D). Although reduced in size, the tumors of B-RAFΔ/Δ animals were well differentiated as judged by their cell morphology and immunostaining of two tumor markers (pan-Cytokeratine and Pro-SPC) (Fig. 3). The reduced tumor growth in lung tumors could be a consequence of toxic effects caused by the embryonic elimination of b-raf. Therefore, to ascertain whether B-RAF expression is required for normal lung tissue, SpC-rtTA/Tet-o-cre/B-RAFflox/flox triple transgenic mice were fed with DOX- containing food throughout gestation for a period of 2 months and subsequently examined by histology. Notably, the genomic deletion of B-RAF had no effect on lung development and structure of the adult lung (Fig. 4A). Consistent with alveolar type II cell specific-inactivation of the b-raf gene, immunostaining of paraffin embedded lung sections from DOX-induced and control triple transgenic mice showed loss of B-RAF expression only in alveolar cells of triple B-RAFΔ/Δ compound animals (Fig. 4B). Notably, the loss of B-RAF expression did not change the proliferation rate of alveolar cells (Fig. 4B). B-RAF has been shown to play a major role in regulating ERK phosphorylation, we therefore immunoblotted lung protein lysates from 2-month-old triple B-RAF flox/flox and triple B-RAFΔ/Δ compound mice for phospho-ERK. Interestingly we did not observe significant variation in the levels of ERK phosphorylation (Fig. 5). These data indicate that in mice B-RAF is a dispensable factor for lung development and for C-RAF BxB-mediated lung tumorigenesis. Nevertheless, B-RAF seems to play an essential role for lung tumor expansion.
FIGURE 1.

Respiratory-epithelium specific deletion of b-raf in the lung of transgenic mice. A, schematic representation of b-raf deletion strategy in normal alveolar epithelial type II cells of triple transgenic animals (SpC- rtTA/Tet-O-cre/B-RAFflox/flox) and in oncogenic-C-RAF-expressing type II cells of quadruple transgenic animals (SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox). In utero treatment with doxycycline induced Cre-mediated deletion of loxP-flanked exon 3 of the b-raf gene, termed B-RAFΔ/Δ. DOX-untreated mice are termed B-RAF flox/flox. B, PCR genotyping of compound transgenic mice (as above). DNA samples from lung, spleen and tail of 2-month-old untreated and DOX-treated compound animals (2 mice for each group) were analyzed for floxed (flox/flox) or inactivated (Δ/Δ) allele of b-raf. C, lung type II cells protein lysates from 2-month DOX-treated and control compound mice were gel separated and immunoblotted with the indicated antibodies, expression levels of B-RAF were quantified by densitometric analysis and normalized by comparison to the β actin loading control, each column represents one sample; genotype and induction status are as indicated.
FIGURE 2.

Oncogenic C-RAF BxB cooperates with B-RAF in lung tumor growth but not in tumor incidence. A, representative pictures of H&E stained lung sections from DOX-induced (B-RAFΔ/Δ) compound mice and aged-matched controls (B-RAFΔ/wt and B-RAFflox-flox), M, months, red arrows point to lung adenomas. B–D, analysis of tumor incidence, tumor area (μm2) and tumor burden in compound mice (as above, for details on quantification methods see “Experimental Procedures”). Mean values are + S.E.; t test, ns, not significant; *, p < 0.05; ***, p < 0.0005.
FIGURE 3.
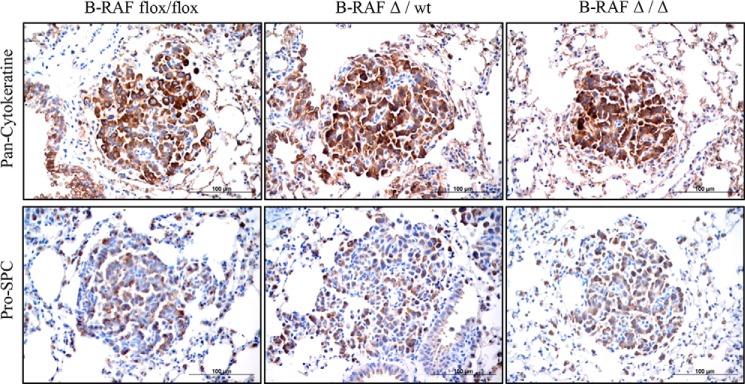
Pan-Cytokeratine and Pro-SPC expression in lung tumors of DOX-induced and controls compound mice. Representative pictures of paraffin embedded lung sections from 7 months old DOX-treated (B-RAFΔ/Δ) and controls animals that were stained with the indicated markers (brown). Hematoxylin was used as a counterstain.
FIGURE 4.

Embryonic ablation of B-RAF in alveolar epithelial cells has no effect on lung development and homeostasis. A, representative pictures of H&E stained lung sections from 2-month-old DOX-induced (triple B-RAFΔ/Δ) and controls compound mice (triple B-RAFflox-flox). B, representative pictures of paraffin-embedded lung sections from 2-month-old DOX-treated (triple B-RAFΔ/Δ), and controls animals (triple B-RAFflox-flox) that were stained with the indicated markers (brown), red arrowheads point to B-RAF-positive alveolar cells, green arrowheads point to Ki67-positive alveolar cells. B indicates bronchioles. Hematoxylin was used as a counterstain.
FIGURE 5.
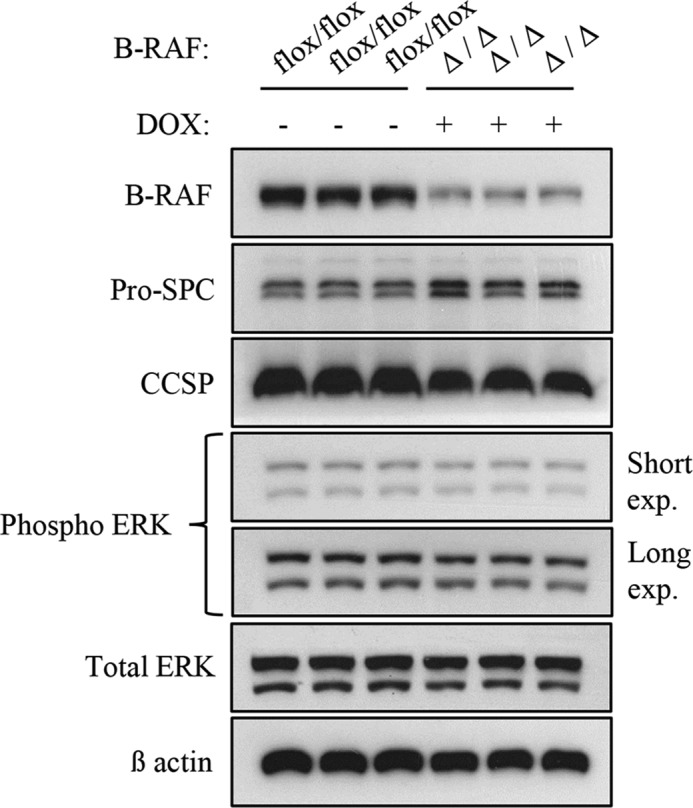
Loss of B-RAF expression in alveolar epithelial cells has no effect on MAPK signal intensity. Protein lysate from 2-month-old DOX-treated (triple B-RAFΔ/Δ) and controls compound mice (triple B-RAFflox-flox) were gel separated and immunoblotted with the indicated antibodies. Genotype and induction status are as indicated.
Reduced Cell Proliferation in Lung Tumors Lacking B-RAF
The reduced tumor growth in oncogenic-C-RAF driven lung tumors, which lack B-RAF, could be either a consequence of reduced cell proliferation or an increase in apoptosis or even a combination of both. We therefore screened tumor bearing lung sections from B-RAFΔ/Δ and B-RAFflox/flox mice with markers of both proliferation and apoptosis. Ki67 and phospho-H3 staining clearly showed a strong reduction of cycling cells upon B-RAF ablation in C-RAF BxB induced lung tumors (Fig. 6, A and B). TUNEL and active caspase 3 staining did not exhibit any difference between B-RAFΔ/Δ and control animals (Fig. 7, A and B). No decrease in the number of lung tumors between 2 and 7 months and the absence of apoptosis argue against cell death mechanisms being responsible for the slow tumor growth after B-RAF elimination (Figs. 2, A and B and 7, A and B). Alternatively, the reduction of lung tumor growth in DOX-induced compound mice might be due to transgene shut down upon b-raf knock out in type II cells. Hence, we co-stained paraffin embedded lung sections from B-RAFΔ/Δ and B-RAFflox/flox mice for B-RAF and C-RAF BxB. Quantification of B-RAF/C-RAF BxB double positive tumor cells showed that in B-RAFΔ/Δ mice 90% of C-RAF BxB-expressing cells lack B-RAF expression (Fig. 8, A and B). Another cell autonomous mechanism that restricts tumor growth is oncogene-induced senescence, an irreversible form of growth arrest (14). To determine the status of senescence in C-RAF BxB-driven lung tumors upon B-RAF elimination we stained lung sections from DOX-induced and control mice for different senescence markers. Staining with antibodies against p16INK4a, HP1γ, and p21WAF1 showed no difference in the expression between tumors lacking B-RAF and controls (Fig. 9A). Consistently, immunofluorescence staining for p19ARF didn't show any difference between DOX-induced and control animals (Fig. 9B). Taken together these data indicate that loss of B-RAF in C-RAF BxB-transformed alveolar epithelial lung type II cells reduces tumor cell proliferation in absence of augmented cell death and senescence.
FIGURE 6.

Reduced proliferation in C-RAF BxB lung tumors of DOX-induced compound mice. A and B, SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox compound mice were DOX-induced for 2 and 7 months and compared with aged-matched controls. Representative pictures and quantification of Ki67 (A), phospho-H3 (B) staining (brown) of paraffin-embedded lung sections. Genotype and induction status are as indicated. Red arrows point to positively stained cells. Hematoxylin was used as a counterstain. A total of 75 tumors from five mice for each group were analyzed. Mean values are + S.E.; t test, ns, not significant, ***, p < 0.0005.
FIGURE 7.

No evidence for increased cell death in C-RAF BxB lung tumors of DOX-induced compound mice. A and B, SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox compound mice were DOX-induced for 2 and 7 months and compared with aged-matched controls. Representative pictures and quantification of TUNEL (A), cleaved caspase-3 (B) staining (brown) of paraffin-embedded lung sections. Genotype and induction status are as indicated. Red arrows point to positively stained cells. Hematoxylin was used as a counterstain. A total of 75 tumors from five mice for each group were analyzed. (Mean values are + S.E.; t test; ns, not significant).
FIGURE 8.

Absence of B-RAF protein expression in C-RAF BxB-expressing lung tumors of DOX-induced compound mice. A, representative pictures of paraffin-embedded lung sections from 2-month-old DOX-treated (B-RAFΔ/Δ) and control animals (B-RAFflox/flox) that were double immunofluorescence stained with the indicated markers. Sections were counterstained with DAPI (blue). B, quantitation of B-RAF/C-RAF BxB double positive tumor cells in the stained sections (see A). A total of 40 tumors from four mice for each group were analyzed. Mean values are + S.E.; t test, ***, p < 0.0005.
FIGURE 9.

Lack of evidence for senescence in C-RAF BxB lung tumors upon B-RAF ablation. A, representative pictures of paraffin embedded lung sections from 7-month-old DOX-treated (B-RAFΔ/Δ) and controls animals (B-RAFflox/flox) that were stained with the indicated markers (brown). B, representative pictures of paraffin-embedded lung sections from 7-month-old DOX-treated (B-RAFΔ/Δ) and control animals (B-RAFflox/flox) that were immunofluorescence stained for p19ARF (red), sections were counterstained with DAPI (blue), insets represent segment magnifications of nucleus indicated by arrow. Positive control is a lung section from a 3-week-old SpC-C-RAF BXB11/Bmi1−/− transgenic mouse, which displays increased expression of p19ARF (24).
B-raf Cooperates with Oncogenic C-RAF in the Activation of the MAPK Cascade in Vivo and in Vitro
Previous studies have demonstrated that B-RAF and C-RAF BxB constitutively heterodimerize and this may be important for the activation of mitogenic signaling and for cellular transformation and expansion (7, 8). To test whether in our in vivo system the elimination of B-RAF reduces C-RAF BxB-mediated mitogenic signal we immunoblotted protein lysates from total lung of B-RAF flox/flox and B-RAFΔ/Δ at the age of 2, 7, and 12 months, respectively. Notably, protein lysates from DOX-induced mice showed reduced phospho-ERK levels in comparison to control animals at all ages analyzed (Fig. 10A). Since the lung is a heterogenous cell population, these results could simply reflect relative changes in cell populations after B-RAF elimination. We therefore immunostained lung sections from B-RAFΔ/Δ and B-RAFflox/flox mice for phospho-ERK. Quantification of nuclear phospho-ERK in lung tumors clearly showed a strong reduction of mitogenic signal intensity in DOX-induced compound mice (Fig. 10B). It has been shown that C-RAF/B-RAF heterodimerization is, among other, controlled by mitogenic signal and enhanced by 14-3-3 proteins (2). C-RAF display two major 14-3-3 binding sites, Ser-259 and Ser-621. In C-RAF BxB, which has a deletion in the N-terminal part of the protein, only the S621 is present. To investigate the role of the Ser-621 in C-RAF BxB/B-RAF heterodimerization and activation of the MAPK signaling, HEK 293 cells were transiently transfected with HA-tagged C-RAF BxB- and C-RAF BxB S621D-expressing plasmids. 48 h after transfection HA-C-RAF BxB and HA-C-RAF BxB S621D proteins were immunoprecipitated, and their kinase activity was measured. As previously shown for the full-length C-RAF protein (7) the mutation of Ser-621 in C-RAF BxB compromised the heterodimerization with B-RAF, suggesting that 14-3-3 binding to this site may be required for efficient heterodimerization (Fig. 11). Consistent with the reduced ability to heterodimerize the C-RAF BxB S621D mutant is also devoid of kinase activity (Fig. 11). All in all these data indicate that C-RAF BxB needs to dimerize with B-RAF both in vivo and in vitro to activate the mitogenic cascade and therefore to promote tumor cell expansion.
FIGURE 10.

B-RAF cooperates with C-RAF BxB in the activation of the MAPK cascade. A, total lung protein lysates from 2, 7, and 12 months old DOX-treated and control compound mice were gel separated and immunoblotted with the indicated antibodies; M, months, Genotype and induction status are as indicated. B, SpC-C-RAF BXB/SpC- rtTA/Tet-O-cre/B-RAFflox/flox compound mice were DOX-induced for 2 and 7 months and compared with aged-matched controls. Representative pictures and quantification of nuclear phospho-ERK staining (brown) of paraffin-embedded lung sections. Genotype and induction status are as indicated. Red arrows point to cells with nuclear staining. Hematoxylin was used as a counterstain. A total of 40 tumors from four mice for each group were analyzed. Mean values are + S.E.; t test, ***, p < 0.0005.
FIGURE 11.

Ser-621 of C-RAF BxB is necessary for its dimerization with B-RAF and for its kinase activity. HA-tagged C-RAF proteins were immunoprecipitated (IP HA) from protein lysates of HEK 293 cells that were transfected with the indicated expression plasmids. The kinase activity of immunoprecipitated C-RAF BxB proteins was measured as the ability to phosphorylate purified ERK-2 in vitro as reported under “Experimental Procedures.” IPs, kinase assays, and total lysates were gel separated and immunoblotted with the indicated antibodies.
DISCUSSION
Recent studies have shown that RAFs dimerization is necessary for normal Ras-dependent RAF kinase activation and contributes to the pathogenic function of disease-associated mutant RAF (15). In the present study, we employed triple and quadruple transgenic compound mice with a lung-specific DOX-inducible b-raf gene ablation to investigate the dependence of normal and oncogenic-C-RAF expressing lung type II cells, on B-RAF. Embryonic deletion of b-raf in SpC- rtTA/Tet-O-cre/B-RAF flox/flox triple compound mice did not cause detectable changes in lung by histology. Moreover, loss of B-RAF in normal alveolar type II cells didńt change phospho-ERK levels and proliferation rate of lung cells. Interestingly, the abrogation of b-raf in C-RAF BxB-expressing lung type II cells did not yield a significant reduction in tumor initiation events as defined by the number of tumors. However, tumors lacking B-RAF displayed pronounced decrease in tumor growth rate in absence of augmented apoptosis and senescence. We also show that C-RAF BxB-expressing lung tumor cells exhibited reduced ERK phosphorylation levels in absence of B-RAF. These in vivo data suggest that C-RAF BxB/B-RAF dimerization is necessary for full ERK phosphorylation and tumor cell proliferation. In vitro experiments confirmed these data as mutation in a 14-3-3 binding site of C-RAF BxB (S621D) compromised C-RAF BxB dimerization with B-RAF and completely abrogated its kinase activity.
In the mouse model we employed, both the cre-mediated inactivation of the b-raf gene and the C-RAF BxB expression are driven by the SpC promoter which is expressed in the lung already at embryonic stage (16). However, elimination of B-RAF did not impair normal lung development and did not reduce the number of lung tumors. Hence, these data suggest that oncogenic C-RAF mediated tumor initiation is B-RAF independent and that embryonic elimination of B-RAF does not reduce the number of potential tumor initiating cells. It is conceivable that the reduced tumor growth in oncogenic-C-RAF driven lung cancer that lack B-RAF is caused by a reduction in signaling through the mitogenic cascade. These results are in line with previous biochemical data showing that B-RAF heterodimerizes with wild type and C-RAF BxB by targeting its kinase domain and by activating it in the cytosol in a RAS-independent manner (7) (8). Alternatively, the reduction in ERK phosphorylation that we observed could be simply due to an overall reduction of RAF activity in the absence of B-RAF. However, elimination of B-RAF in normal lung cells did not change ERK phosphorylation levels and their proliferation rate. These results are in line with a previous report showing that the systemic ablation of both b-raf and c-raf in normal adult mice doesńt lead to significant variation in the levels of MEK and ERK phosphorylation (17). Moreover, Blasco et al. demonstrated in mouse models that C-RAF, but not B-RAF, is essential for the development of oncogenic-K-Ras-driven NSCLC (17). Interestingly, in this work, the elimination of B-RAF expression had no effect on the levels of MEK and ERK phosphorylation suggesting that other RAF proteins can maintain mitogenic signaling (17). Instead, our data underline the importance of B-RAF in C-RAF BxB-mediated neoplastic transformation. B-RAF seems to be necessary for C-RAF BxB in the activation of the mitogenic cascade.
Others and we have previously shown that the BAY 43–9006 compound is a very potent inhibitor of the catalytic domain of C- RAF and wild type B-RAF (18). However, it was not efficient in the inhibition of lung tumor growth in SpC-C-RAF BxB transgenic mice (19). This can be explained by our current study, since the dimerization process with B-RAF seems to be necessary for proper lung tumor growth. On the other hand, the PLX4032 compound (a new B-RAF V600E inhibitor) inhibits tumors with mutant RAFs but it does not suppress the growth of tumors with mutant Ras or wild type B-RAF. Indeed, this inhibitor can activate the MAPK cascade through homo- or heterodimerization mechanisms of B- and C-RAF in a Ras-dependent manner (20) (21) (22). It is conceivable that the BAY 43-9006 inhibitor treatment did not block B-RAF/C-RAF BxB heterodimerization and therefore had no effects on the mitogenic signal intensity (19). In the present work, the complete ablation of the B-RAF protein renders RAFs heterodimerization impossible. This might explain why C-RAF BxB lung tumors lacking B-RAF showed a reduced intensity in mitogenic signal.
RAFs dimerization is a very complex process. Among the mechanisms that are involved in this process, 14-3-3 proteins play an important role in regulating RAFs dimerization (2). Consistent with this view, a mutation in the S621, a 14-3-3 binding site in C-RAF, reduced C-RAF BxB/B-RAF heterodimerization. Moreover, this mutation completely abolished the kinase activity of the protein. These observations are in line with our hypothesis supporting the role of C-RAF BxB/B-RAF dimer formation as a crucial event in MAPK signaling activation. However, the S621 of C-RAF is also an autophosphorylation site that, if mutated, renders the kinase inactive (23). It will be difficult to clarify whether the protein is inactive because it is unable to dimerize or because it is not able to autophosphorylate the Ser-621.
Recently, chromosomal translocations involving either B- or C-RAF in a small percentage of several types of cancers were found (4) (5). Notably, these gene fusions encode for RAF proteins that lack the N-terminal regulatory domain and retain the kinase domain, suggesting that the mutant proteins may be constitutively active (4). Our data suggest that in addition to the use of RAF inhibitors, blocking RAF dimerization might be a valid therapeutic approach to inhibit RAF signaling in tumors harboring this kind of mutations.
Acknowledgments
We thank Jeffrey A. Whitsett for SpC-rtTA mice and Pro-SPC antibody, Hermann Bujard for Tet-O-Cre mice. We are grateful to Fatih Ceteci, Semra Ceteci, and Gabriel Kirchgraber for technical help and helpful discussion.
This work was supported by an institutional grant from the Max Planck Society (to U. R. R.) and by the UGMLC and Loewe Program as well as DZL (The German Center for Lung Research).
- MAPK
- mitogen-activated protein kinase
- RTK
- receptor tyrosine kinase
- DAB
- diaminobenzidine
- DOX
- doxycycline
- rtTA
- reverse tetracycline transactivator
- CRE
- Cre-recombinase
- H&E
- hematoxylin and eosin.
REFERENCES
- 1. Davis M. J., Schlessinger J. (2012) The genesis of Zelboraf: targeting mutant B-Raf in melanoma. J. Cell Biol. 199, 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rushworth L. K., Hindley A. D., O'Neill E., Kolch W. (2006) Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 26, 2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A. (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 4. Palanisamy N., Ateeq B., Kalyana-Sundaram S., Pflueger D., Ramnarayanan K., Shankar S., Han B., Cao Q., Cao X., Suleman K., Kumar-Sinha C., Dhanasekaran S. M., Chen Y. B., Esgueva R., Banerjee S., LaFargue C. J., Siddiqui J., Demichelis F., Moeller P., Bismar T. A., Kuefer R., Fullen D. R., Johnson T. M., Greenson J. K., Giordano T. J., Tan P., Tomlins S. A., Varambally S., Rubin M. A., Maher C. A., Chinnaiyan A. M. (2010) Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med. 16, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McMahon M. (2010) RAF translocations expand cancer targets. Nat. Med. 16, 749–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerkhoff E., Fedorov L. M., Siefken R., Walter A. O., Papadopoulos T., Rapp U. R. (2000) Lung-targeted expression of the c-Raf-1 kinase in transgenic mice exposes a novel oncogenic character of the wild-type protein. Cell Growth Differ. 11, 185–190 [PubMed] [Google Scholar]
- 7. Weber C. K., Slupsky J. R., Kalmes H. A., Rapp U. R. (2001) Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 61, 3595–3598 [PubMed] [Google Scholar]
- 8. Garnett M. J., Rana S., Paterson H., Barford D., Marais R. (2005) Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 20, 963–969 [DOI] [PubMed] [Google Scholar]
- 9. Pfeiffer V., Götz R., Xiang C., Camarero G., Braun A., Zhang Y., Blum R., Heinsen H., Nieswandt B., Rapp U. R. (2013) Ablation of BRaf impairs neuronal differentiation in the postnatal hippocampus and cerebellum. PLoS One 8, e58259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ceteci F., Ceteci S., Karreman C., Kramer B. W., Asan E., Götz R., Rapp U. R. (2007) Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer Cell 12, 145–159 [DOI] [PubMed] [Google Scholar]
- 11. Corti M., Brody A. R., Harrison J. H. (1996) Isolation and primary culture of murine alveolar type II cells. Am. J. Respir. Cell Mol. Biol. 14, 309–315 [DOI] [PubMed] [Google Scholar]
- 12. Ahlbrecht K., Schmitz J., Seay U., Schwarz C., Mittnacht-Kraus R., Gaumann A., Haberberger R. V., Herold S., Breier G., Grimminger F., Seeger W., Voswinckel R. (2008) Spatiotemporal expression of flk-1 in pulmonary epithelial cells during lung development. Am. J. Respir. Cell Mol. Biol. 39, 163–170 [DOI] [PubMed] [Google Scholar]
- 13. Unkel B., Hoegner K., Clausen B. E., Lewe-Schlosser P., Bodner J., Gattenloehner S., Janssen H., Seeger W., Lohmeyer J., Herold S. (2012) Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. J. Clin. Investig. 122, 3652–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collado M., Gil J., Efeyan A., Guerra C., Schuhmacher A. J., Barradas M., Benguría A., Zaballos A., Flores J. M., Barbacid M., Beach D., Serrano M. (2005) Tumour biology: senescence in premalignant tumours. Nature 436, 642. [DOI] [PubMed] [Google Scholar]
- 15. Freeman A. K., Ritt D. A., Morrison D. K. (2013) Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 49, 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perl A. K., Tichelaar J. W., Whitsett J. A. (2002) Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 11, 21–29 [DOI] [PubMed] [Google Scholar]
- 17. Blasco R. B., Francoz S., Santamaría D., Cañamero M., Dubus P., Charron J., Baccarini M., Barbacid M. (2011) c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 19, 652–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilhelm S. M., Carter C., Tang L., Wilkie D., McNabola A., Rong H., Chen C., Zhang X., Vincent P., McHugh M., Cao Y., Shujath J., Gawlak S., Eveleigh D., Rowley B., Liu L., Adnane L., Lynch M., Auclair D., Taylor I., Gedrich R., Voznesensky A., Riedl B., Post L. E., Bollag G., Trail P. A. (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109 [DOI] [PubMed] [Google Scholar]
- 19. Kramer B. W., Götz R., Rapp U. R. (2004) Use of mitogenic cascade blockers for treatment of C-Raf induced lung adenoma in vivo: CI-1040 strongly reduces growth and improves lung structure. BMC Cancer 4, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poulikakos P. I., Zhang C., Bollag G., Shokat K. M., Rosen N. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heidorn S. J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J. S., Springer C. J., Pritchard C., Marais R. (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hatzivassiliou G., Song K., Yen I., Brandhuber B. J., Anderson D. J., Alvarado R., Ludlam M. J., Stokoe D., Gloor S. L., Vigers G., Morales T., Aliagas I., Liu B., Sideris S., Hoeflich K. P., Jaiswal B. S., Seshagiri S., Koeppen H., Belvin M., Friedman L. S., Malek S. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 [DOI] [PubMed] [Google Scholar]
- 23. Mischak H., Seitz T., Janosch P., Eulitz M., Steen H., Schellerer M., Philipp A., Kolch W. (1996) Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol. 16, 5409–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker M., Korn C., Sienerth A. R., Voswinckel R., Luetkenhaus K., Ceteci F., Rapp U. R. (2009) Polycomb group protein Bmi1 is required for growth of RAF driven non-small-cell lung cancer. PLoS ONE 4, e4230. [DOI] [PMC free article] [PubMed] [Google Scholar]