

Background: Glycans are synthesized in the Golgi by glycosyltransferase complexes, but their functional organization is unclear.
Results: Organizational interplay and trafficking of glycosyltransferase complexes involves dynamic transitions between enzyme homo- and heteromers.
Conclusion: Organellar microenvironmental factors play a crucial role in the functional organization of the Golgi glycosylation pathways.
Significance: Organizational interplay between glycosyltransferase complexes provides new insights into glycosylation and associated diseases.
Keywords: Fluorescence Resonance Energy Transfer (FRET), Glycobiology, Glycosylation, Glycosyltransferase, Golgi, Protein Complex
Abstract
Glycosylation of proteins and lipids takes place in the Golgi apparatus by the consecutive actions of functionally distinct glycosidases and glycosyltransferases. Current evidence indicates that they function as enzyme homomers and/or heteromers in the living cell. Here we investigate their organizational interplay and show that glycosyltransferase homomers are assembled in the endoplasmic reticulum. Upon transport to the Golgi, the majority of homomers are disassembled to allow the formation of enzyme heteromers between sequentially acting medial-Golgi enzymes GnT-I and GnT-II or trans-Golgi enzymes GalT-I and ST6Gal-I. This transition is driven by the acidic Golgi environment, as it was markedly inhibited by raising Golgi luminal pH with chloroquine. Our FRAP (fluorescence recovery after photobleaching) measurements showed that the complexes remain mobile Golgi membrane constituents that can relocate to the endoplasmic reticulum or to the scattered Golgi mini-stacks upon brefeldin A or nocodazole treatment, respectively. During this relocation, heteromers undergo a reverse transition back to enzyme homomers. These data unveil an unprecedented organizational interplay between Golgi N-glycosyltransferases that involves dynamic and organelle microenvironment-driven transitions between enzyme homomers and heteromers during their trafficking within the early secretory compartments.
Introduction
Glycosylation is the most abundant and diverse modification of cellular proteins and lipids, affecting more than half of all proteins expressed in a eukaryotic cell (1, 2). It is crucial not only for many basic cellular functions but also for various biological recognition events needed, e.g. for binding between cells, extracellular matrix components, and certain pathogens (3). Thus, it contributes not only to the development and maintenance of multicellular life but also to the onset and progression of various human diseases, such as inflammation, diabetes, and cancers (4–7).
The Golgi apparatus is the main site of glycan synthesis in eukaryotes. It houses dozens of functionally distinct glycosylation enzymes that sequentially remove (glycosidases) or link (glycosyltransferases) monosaccharides together via distinct glycosidic bonds by utilizing specific sugar nucleotides as donors (8, 9). Their concerted functioning is estimated to give rise to a huge repertoire of distinct glycan structures (∼100–500 × 103), of which some thousands seem to carry structural information important for their functions (10). How these enzymes, many of which compete with each other for the same acceptor and also show overlapping distributions in the Golgi (11), can synthesize such a vast array of different glycan structures without any template has only recently started to be understood. A number of studies in yeast, plant, and human cells have shown that Golgi glycosyltransferases typically form functionally relevant enzyme complexes between consecutively acting enzymes within each glycosylation pathway (12–17). Complex formation is thought to increase both the fidelity and efficiency of glycan synthesis first by minimizing intervention by competing enzymes and second, by reducing the number of acceptor recognition events via “substrate channeling” to the next enzyme for further processing.
However, the same enzymes that form functionally relevant enzyme heteromers have also been shown to form homomeric complexes in the cells (18–23). This is rather puzzling, as it raises concerns about their functional relevance for glycan synthesis. Homomer formation may modulate the binding affinity and/or selectivity against different acceptor and donor substrates or regulate enzyme stability and self-glycosylation (24, 25). Enzyme homomers and heteromers may also represent temporally or spatially distinct enzyme species that utilize the same interaction surfaces for binding. In support of this view, we previously showed that GalT-I2 self-binding can be inhibited by a heteromeric enzyme, ST6Gal-I (15). Glycosyltransferase homomers may also represent “dead end” enzyme species that persist because they cannot form heteromers in the absence of a relevant enzyme partner. This is the case when a single glycosyltransferase is overexpressed in the cell.
To clarify how or if Golgi N-glycosyltransferase homomers and heteromers are interrelated, here we investigated their assembly and organizational interplay mainly by using various live cell imaging approaches. We also addressed their trafficking and suggested recycling between the ER and Golgi to ascertain whether the observed complexes are compatible with the preferred “cisternal progression” or “rapid partitioning” Golgi trafficking models, both of which implicate that Golgi enzymes are mobile membrane constituents that continuously recycle between the Golgi and the ER (26–30). In contrast, the “stable compartments” model assumes that oligomerization results in stable enzyme assemblies that are physically too large to fit into post-Golgi transport vesicles (31–34). Our results show that the Golgi glycosyltransferase homomers and heteromers are interdependent and mobile membrane constituents that are assembled in the ER and the Golgi, respectively, and that undergo dynamic organellar microenvironment-dependent transitions between these two physical states during their recycling between the Golgi and the ER.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
All glycosyltransferase expression plasmids were prepared from commercially available full-length cDNA clones (Imagenes GmbH, Berlin, Germany). Golgi-localized pcDNA3-based bimolecular fluorescence complementation (BiFC) constructs possessing C-terminal monomeric yellow fluorescent protein (Venus, mVen) fragments were constructed as described earlier (15, 35). ER-retained glycosyltransferase constructs were prepared by PCR amplification and insertion of the amplified cDNAs after the N-terminal HA and FLAG tags in the original pCMV-based BiFC plasmid, kindly donated to us by Dr. Hu (Purdue University, IN). ER-retained pcDNA3-based HA-, FLAG-, or myc-tagged constructs were made with complementary annealing primers (i.e. for myc with HindIII; sense, 5′-AAGCTTATGGAACAAAAACTCATCTCAGAAGAGGATCTGA-3′; antisense, 5′-AGCTTATGGAACAAAAACTCATCTCAGAAGAGGATCTGA-3′). The annealed tag was inserted in-frame to the N terminus of each construct. Golgi-localized pcDNA3-based monomeric Venus (mVen) and Cerulean (mCer) fluorescence resonance energy transfer (FRET) plasmids as well as C-terminal-tagged HA and myc epitope-tagged plasmids were prepared as described earlier (15). The mCherry (mChe) FRET vector was constructed by first amplifying mChe cDNA from a pmCherry-Golgi plasmid kindly donated by Dr. Vesa Olkkonen (Biomedicum, Helsinki, Finland) with the following primers: (forward, 5′-AAAAATCTAGAAGCATCGCCACCATGGTGAGCAAGGGCG3′; reversed, AAAAGGGCCCTTACTTGTACAGCTCGTCCATG-3′). The amplified cDNA was then inserted to pcDNA3 using the XbaI and ApaI restriction sites, after which the glycosyltransferase cDNAs were subcloned in-frame with the XbaI site. All constructs were sequence-verified with the ABI3500xL Genetic Analyzer before use.
Cell Cultivation and Treatments
COS-7 cells were grown in DMEM supplemented with 10% FBS (HyClone, Thermo Scientific, Waltham, MA) as described earlier (35). Briefly, 1 day after plating the cells were transfected using 0.5 μg of each plasmid cDNA and FuGENE 6TM transfection reagent according to the supplier's protocol (Promega, Fitchburg, WI). After 24 h of transfection, cells were processed either for fluorescence microscopy, BiFC, fluorescence recovery after photobleaching (FRAP), or FRET measurements (see below). When appropriate, chloroquine (CQ, 40 μm, Sigma) was added to the culture medium for 4 h before the measurements. Nocodazole (1 μg/ml, Sigma) was added to cells 8 h after transfection and 16 h (overnight) before BiFC measurements. In recovery assays nocodazole was added to the cells 16 h (overnight) after transfection and treated for 4 h before the removal of the drug. Cells were then allowed to recover for an additional 4 h. Samples were collected at each time point. Brefeldin A (BFA, 10 μg/ml, Sigma) was added to cells 16 h after post-transfection (overnight). Cycloheximide was added to the cells 1 h before the analyses at a final concentration of 100 μm.
Fluorescence Microscopy
Cells were prepared for immunofluorescence microscopy as described previously (35). Shortly, after fixation, cells were permeabilized with 0.1% saponin in PBS and stained with the anti-GM130 (610822, BD Biosciences), mono- and polyclonal anti-HA (Sigma), anti-FLAG (Sigma), anti-myc (Abcam, Cambridge, UK), anti-protein disulfide isomerase (5B5, M877, Dakopatts a/s, Denmark), rabbit anti-N-terminal GFP (Affinity Bioreagents, Golden, CO), and goat anti-C-terminal GFP (Santa Cruz Biotechnology, Inc., Dallas, TX) antibodies. After washing, cells were treated with relevant Alexa fluor 405-, 488-, and 594-conjugated anti-mouse, anti-rabbit, and anti-goat secondary antibodies (Invitrogen). After staining, cells were mounted and imaged using the Zeiss LSM 700 confocal microscope, Zen2009 software (Carl Zeiss AG, Oberkochen, Germany), 63× or 100× Plan-Apo oil immersion objectives, and appropriate filter sets for each dye.
FRET Quantification by Flow Cytometry
FRET measurements were performed as described earlier (15). Briefly, cells were transfected with homomeric FRET enzyme constructs (mCer and mVen constructs). For competition assays, cells were also co-transfected with an HA-tagged homomeric or heteromeric interacting or non-interacting enzyme construct. After 24 h the cells were detached from the plates with 2× trypsin-EDTA (Sigma), collected by centrifugation, and resuspended in 1.5 ml of fresh culture media before analyses with CyFlowTM flow cytometer (Partec GmbH, Münster, Germany) equipped with the appropriate filter sets for mCer (excitation, 405 nm; emission, 425–475 nm) and mVen (excitation, 488 nm; emission, 515–540 nm). In each measurement, 2–5 × 103 cells expressing both mCer and mVen were selected by gating and used for quantification of FRET-positive cells. The FRET filter set (excitation, 405 nm; emission, 515–540 nm) and FloMAXTM software were used for quantification. Three separate experiments were used for the quantification. The results are presented as percentages (mean ± S.D.) of control cells expressing only the two FRET constructs.
FRET Microscopy
FRET microscopy measurements were performed using the Zeiss LSM700 confocal microscope, mVen and mChe variants as the donor/acceptor FRET pair, and the acceptor bleaching protocol with appropriate filter sets for mChe. Samples were fixed before analysis as described for immunofluorescence (see above). The samples were subjected to iterative bleaching (30 cycles, 20 iterations, 555 nm, 70% laser intensity) during which the intensity values of the mVen were recorded. Background values were subtracted from the measured intensity values. FRET % was calculated from the acceptor-corrected intensities (a macro package from Zeiss) using the formula
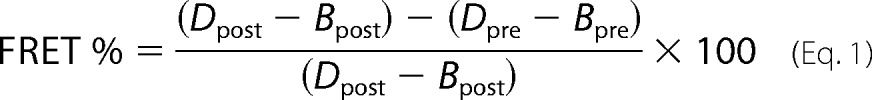 |
where the D = donor intensity and B = background intensity.
FRAP Microscopy and Mobility Assays
COS-7 cells were grown on Ibidi μ-dishes (Ibidi GmbH, Martinsried, Germany), transfected with the appropriate plasmids for 20–24 h, and treated with cycloheximide (100 μm) for 1 h before the analyses to prevent the synthesis of new fluorescent constructs during the measurements. Cells were placed on a microscope chamber under 5% CO2 at 37 °C and imaged using the LSM700 confocal microscope. The mobility of the complexes was measured using the FRAP approach (36). Selected regions of interest were photobleached using 100% 488-laser intensity with 50 iterations. Fluorescence intensities were recorded and averaged pixel-by-pixel to recovery rates of the bleached regions of interest with time. Single exponential protein kinetics was used for the calculations. To determine the size of the mobile fractions in each case, fluorescent intensities were determined also in a similar-sized area adjacent to the bleached regions of interest. The values for mobility fractions were then calculated as percentages of the intensity values at the end of each measurement in the bleached and non-bleached regions of interest areas.
Immunoprecipitation, SDS-PAGE, and Immunoblotting
One μg of plasmids encoding HA- and myc-tagged enzyme constructs was used to transfect COS-7 cells as described above. 24 h after transfection, cells were subjected to immunoprecipitation according to Flügel et al. (37). Briefly, cells were solubilized by scraping and agitation at 4 °C for 20 min either at pH 6.5 or pH 7.5 using Tris/HCl buffer (50 mm Tris/HCl, 150 mm NaCl, 1% TX-100, 2 mm EDTA, 2 mm EGTA, 10 mm Na2PO7) that was supplemented with a protease inhibitor cocktail (Complete Mini, Roche Applied Science). Lysates were cleared by centrifugation at 12 000 × g at 4 °C for 15 min, and aliquots (200 μg of protein) were incubated in the presence of 2 μg of monoclonal anti-HA antibody (Sigma) or polyclonal anti-GalT-I antibody (Sigma) for 1 h before the addition of protein G- or A-Sepharose beads (30 μl of pre-made slurry/reaction, GE Healthcare). The beads were collected by centrifugation, and the supernatant fractions with the remaining enzymes were precipitated with 10% TCA, collected by centrifugation and washed with acetone. Both fractions were suspended into 2× Laemmli sample loading buffer (pH 6.5 or 7.5) and separated by using a 7.5% SDS-PAGE gel. After transfer to a nitrocellulose filter and immunoblotting, a polyclonal anti-myc antibody (Abcam) was used to detect myc-tagged enzymes on the blots. Quantification of protein bands from the blots was done with ImageLab software (Bio-Rad).
Statistical Analyses
Statistical analyses were performed using Student's t test for the comparison of small normally distributed samples.
RESULTS
N-Glycosyltransferase Homomers Are Assembled in the ER
Previously, we have shown that all the main Golgi N-glycosyltransferases exist either as homomeric or heteromeric complexes in the cells (15), the former consisting of the enzyme itself and the latter of interacting (i.e. sequentially acting) medial-Golgi enzymes GnT-I and GnT-II or the trans-Golgi enzymes GalT-I and ST6Gal-I or GalT-I and ST3Gal-III. To examine whether the formation of enzyme homomers and heteromers are competitive and interrelated processes, we utilized a FRET-based competition assay in which the formation of enzyme homomers was inhibited by the enzyme itself or by heteromeric enzyme constructs (Fig. 1, A–F). In each case, FRET measurements between C-terminal-tagged monomeric Cerulean (donor) or Venus (acceptor) constructs showed that FRET signal was markedly (by 51–87%) inhibited by the enzyme itself and also by an interacting heteromeric enzyme construct (by 70–96%). In contrast, non-interacting enzyme constructs did not significantly inhibit the formation of enzyme homomers (8–24% inhibition). These results show that enzyme homomers and heteromers are competitive and thus interrelated enzyme species that use the same interaction surfaces for binding.
FIGURE 1.

Formation of glycosyltransferase homomers in the presence of homomeric and heteromeric enzyme constructs. A, schematic representation of the plasmid constructs used for the transfections. B–F, cells were transfected with the two depicted FRET enzyme constructs of each enzyme (control, left columns) or together with a HA-tagged homomeric or heteromeric interacting (middle columns) or non-interacting (right columns) enzyme constructs. An equivalent amount of each plasmid cDNA (500 ng) was used for the transfections. The two sialyltransferase constructs in D are both known to interact with GalT-I. Quantification of the FRET signals were performed by flow cytometry (15) as described under “Experimental Procedures.” Note that in each case only the enzyme itself and an interacting heteromeric enzyme are able to inhibit the formation of enzyme homomers, whereas non-interacting enzyme constructs are not. These data indicate that the formation of enzyme homomers and heteromers are competitive processes and are thus interrelated phenomena. The results are presented as percentages of the control values (mean ± S.D., n = 3).
To examine whether the enzyme complexes form shortly after their synthesis in the ER and before their transport to the Golgi, we utilized the BiFC approach. For the BiFC measurements, we tagged each enzyme C-terminally with both VN and VC fragments (VN, amino acids 1–173; VC, amino acids 155–238) of the monomeric Venus (mVenus) fluorescent protein variant (35, 38). To prevent their exit from the ER, we also tagged each enzyme construct N-terminally with either the FLAG or HA epitope tag (the pCMV plasmids in Fig. 2A). These N-terminal tags apparently prevent ER exit by masking ER export signals in the cytoplasmic tails of these enzymes. These signals are normally needed for Sar1/Sec23p/Sec24p-mediated cargo selection into COP II carriers and transport to the Golgi (39, 40). The tags also enabled us to demonstrate that the enzyme constructs accumulate in the ER. This was shown by co-localization of the constructs with the ER marker protein disulfide isomerase (data not shown). Co-transfection of the homomeric BiFC enzyme constructs gave also a typical ER-like staining pattern (Fig. 2B). BiFC measurements (using the YFP filter set) in the same cells also revealed a clear BiFC signal in the ER, indicating the formation of enzyme homomers in the organelle (Fig. 2B, bottom row). In contrast, co-transfection of the cells with interacting heteromeric BiFC enzyme constructs (Fig. 2C) did not produce any signal despite the presence of the enzyme constructs in the ER. These results demonstrate that the enzyme homomers are assembled in the ER, whereas the heteromers are not.
FIGURE 2.

Formation of homomeric N-glycosyltransferase complexes in the ER. A, schematic representation of the ER-localized BiFC (top), FRET (middle), and epitope-tagged (bottom) glycosyltransferase constructs used for the transfections. B, localization of the homomeric BiFC enzyme constructs and the measurement of the BiFC signal using the YFP filter set. The expressed proteins were stained by using antibodies against their N-terminal tags. The pictures show the typical ER-like staining pattern of the expressed enzyme proteins and also the clear BiFC signal, indicating the presence of homomeric enzyme complexes in the ER. Scale bar, 10 μm. C, localization of the heteromeric BiFC constructs in the ER and the BiFC measurement. Note the absence of BiFC signal in the ER between the depicted heteromeric enzyme constructs. D, FRET microscopy measurements of the ER-localized glycosyltransferase homomers in the presence of a homomeric or heteromeric enzyme construct. Cells were co-transfected with the two FRET constructs (mVen and mChe) of each enzyme (depicted on top) with or without a myc-tagged homomeric or heteromeric enzyme construct (depicted below). Equivalent amounts (500 ng) of each plasmid cDNA were used for the transfections. Left, fluorescence microscopy shows co-localization of all three constructs in the ER. Right, inhibition of the FRET signal by homomeric or heteromeric enzyme construct. FRET signals were measured by using confocal microscopy and the acceptor bleaching method. Note that only the myc-tagged enzyme itself (dark columns) is able to inhibit FRET signal between mVen- and mChe-tagged enzyme constructs in the ER, whereas a heteromeric enzyme construct (light columns) was without effect. The results are presented as percentages (mean ± S.D., n = 8) of the controls in which only the two FRET enzyme constructs were used for the transfections. (**, p < 0.01; ***, p < 0.001). E, co-immunoprecipitation (Co-IP) and Western blotting (WB) of the depicted GalT-I homomers and GalT-I/ST6Gal-I heteromers. Immunoprecipitation was performed at pH 7.5 using the anti-HA antibody. Immunoblotting was done using the anti-myc antibody (left). Note that the myc-tagged homomeric enzyme co-immunoprecipitates with the HA-tagged construct and is recovered in the pellet (p), whereas >90% of the heteromeric myc-ST6Gal-I enzyme construct (right) is recovered in the supernatant fraction (s).
To provide further proof for the absence of enzyme heteromers in the ER, we next tested whether an interacting heteromeric enzyme construct can inhibit homomer formation in the ER. FRET microscopy-based competition assays were carried out with the ER-localized homomeric enzyme constructs that carry C-terminal mVen (donor) or mCherry (acceptor) tags (for the plasmid constructs, see Fig. 1A). Co-transfection of COS-7 cells with equivalent amounts (500 ng/each plasmid) of homomeric enzyme constructs (Fig. 2D) revealed that the FRET signal between each enzyme homomer was inhibited by 43–55%, whereas the interacting heteromeric constructs did not (3–12%). Moreover, we also performed conventional pulldown experiments (Fig. 2, A and E) with COS-7 cells expressing N-terminal-tagged homomeric (GalT-I) or heteromeric (GalT-I/ST6Gal-I) enzyme constructs. Anti-HA antibody immunoprecipitates of the cleared cell lysates and Western blotting with anti-myc antibody showed that roughly one-third (37%) of the myc-tagged GalT-I immunoprecipitated with the HA-tagged GalT-I at pH 7.5 (the pH close to that of the ER). This value means that practically all of the expressed GalT-I exist as homomers, as only one (HA/myc) out of two other potential (myc/myc, HA/HA) enzyme combinations can be detected by this approach. In contrast, immunoprecipitation experiments with cells expressing heteromeric GalT-I/ST6Gal-I constructs showed that almost all (93%) of the expressed ST6Gal-I was recovered in the supernatant.
Assembly of Heteromers Takes Place in the Golgi Apparatus
Next, to clarify whether the enzyme heteromers are assembled at the ER exit sites or in adjacent nocodazole-induced Golgi mini-stacks, we utilized Golgi-localized BiFC enzyme constructs. Nocodazole, a microtubule disrupting drug, induces the formation of such Golgi mini-stacks by blocking anterograde ER-to-Golgi transport and coalescence of Golgi membranes into the compact juxtanuclear structure typically seen e.g. in COS-7 cells (41, 42). This was also confirmed by using antibodies against the GM130 and the ER exit site marker protein, Sec23 (data not shown). We transfected COS-7 cells with the Golgi-localized (i.e. without any N-terminal tags; Fig. 3A) homomeric or heteromeric BiFC enzyme constructs and treated the cells with nocodazole for 16 h (added 8 h post-transfection) before staining with anti-GFPVN- and anti-GFPVC-specific antibodies (Fig. 3A). Consistent with the formation of enzyme homomers in the ER, we detected a clear BiFC signal in nearly all scattered Golgi mini-stacks. In contrast, only a small number (∼15%) of the Golgi mini-stacks showed a BiFC signal when the cells were transfected with the heteromeric enzyme constructs (GalT-I/ST6Gal-I). However, removal of the drug (4 h recovery; Fig. 3B) resulted in the coalescence of nearly all Golgi elements into a compact Golgi structure with a clear BiFC signal. These data indicate that the enzyme heteromers are not assembled at the ER exit sites nor in the nocodazole-induced Golgi mini-stacks but, rather, in the compact, juxtanuclear Golgi structures typically seen in COS-7 cells.
FIGURE 3.

Formation of glycosyltransferase heteromers in the Golgi after recovery from nocodazole treatment as detected by the BiFC approach. A, immunofluorescence microscopy of the cells transfected with the depicted homomeric and heteromeric GalT-I BiFC constructs. For transfections the plasmids without the N-terminal HA or FLAG tags were used. 8 h post-transfection cells were treated with 1 μg/ml nocodazole (NDZ) for 16 h and fixed before staining with the anti-GFP-VN- and anti-VC-specific antibodies. Note the localization of the enzyme constructs in the Golgi mini-stacks and the clear BiFC signal in the Golgi mini-stacks with the homomeric GalT-I constructs (top). In contrast, no BiFC signal was detected with the heteromeric GalT-I/ST6Gal-I constructs (bottom row). Scale bar, 10 μm. B, formation of GalT-I/ST6Gal-I heteromers after recovery from the nocodazole treatment. Cells treated as above were allowed to recover from nocodazole treatment for 4 h by removing the drug. Note the clear BiFC signal in the compact juxtanuclear Golgi structure. C, inhibition of homomer formation in the Golgi by the heteromeric enzyme constructs. In each case cells were transfected with the depicted homomeric FRET enzyme constructs (top) together with interacting (dark columns) or non-interacting (light columns) heteromeric enzyme constructs. Note that in each case only an interacting heteromeric enzyme construct significantly decreased the amount of homomers in the Golgi. The results are presented as percentages (the mean ± S.D., n = 10) of controls in which only the FRET constructs were used for the transfections. Fluorescence microscopy (right) shows the localization of the enzyme constructs in the Golgi of the transfected cells (***, p < 0.001).
Further proof for the formation of enzyme heteromers in the Golgi was obtained from FRET-based competition assays (Fig. 3C) carried out with the Golgi-localized (i.e. without the N-terminal tags) homomeric FRET enzyme constructs. Co-transfection of the cells with interacting and non-interacting heteromeric enzyme constructs showed that in each case an interacting heteromeric construct markedly inhibited (by 71 ± 8 to 81 ± 6%, p < 0.0001) the amount of enzyme homomers in the Golgi. Non-interacting heteromeric FRET enzyme constructs, however, did not cause such an inhibition (<8 ± 12% inhibition, p = 0.266). These data are in contrast to the ER (Fig. 2D), where co-transfection of an interacting heteromeric enzyme did not inhibit the formation of enzyme homomers and thus confirm that enzyme heteromers form upon or after arrival in the Golgi.
Assembly of Enzyme Heteromers Is Golgi pH-dependent and Accompanied by the Disassembly of Homomers
The observed pH sensitivity of the enzyme heteromers, but not of homomers (15), suggests that the acidic luminal pH is the driving force for the assembly of enzyme heteromers in the Golgi. To assess whether this is indeed the case, we performed pulldown experiments either at pH 6.5 (the pH of medial-Golgi) or at pH 7.5 (to mimic the pH of the ER; Fig. 2). Transfection of COS-7 cells with Golgi-localized homomeric (GalT-I) or heteromeric (GalT-I/ST6Gal-I) enzyme constructs and immunoblotting of the precipitates (Fig. 4A) revealed that 41% of the total GalT-I was recovered in the pellet as homomeric GalT-I complexes at pH 6.5. We also found that the majority (69%) of the expressed ST6Gal-I was recovered in the pellet when the cells were co-transfected with the heteromeric enzyme constructs (GalT-I/ST6Gal-I). Thus, enzyme heteromers are the preferred species under normal pH conditions of the Golgi lumen. We also found that 88% of the endogenous GalT-I was also immunoprecipitated upon transfection of the cells with the ST6Gal-I enzyme construct. However, due to the lack of suitable antibodies against endogenous ST6Gal-I, we could not confirm whether endogenous ST6Gal-I and GalT-I also interact and form a complex with each other. Nevertheless, our co-immunoprecipitation experiments at pH 7.5 with the same enzyme constructs (Fig. 4B) also showed that the amount of GalT-I homomers was almost equal to that detected at pH 6.5. In contrast, the amount of ST6Gal-I heteromers with either overexpressed or endogenous GalT-I was only 7.1 and 8.6%, respectively, that of the amount of expressed ST6Gal-I in the cells.
FIGURE 4.

The pH dependence of the Golgi-localized glycosyltransferase homomers and heteromers. A and B, co-immunoprecipitation (Co-IP) and Western blotting (WB) of GalT-I homomers or GalT-I/ST6Gal-I heteromers at pH 6.5 and 7.5. Cells were transfected with the depicted HA- and/or myc-tagged enzyme constructs, subjected to immunoprecipitation as described under “Experimental Procedures,” and immunoblotted from both the pellet (p) and supernatant (s) fractions with anti-myc or anti-GalT-I (for endogenous GalT-I) antibodies at both pH 6.5 (A) and pH 7.5 (B). At pH 6.5, note that both enzyme homomers and heteromers were recovered mainly in the pellet fractions, whereas at pH 7.5 >90% of the interacting enzymes were detected in the supernatant fractions. C and D, FRET microscopy quantification of the Golgi-localized enzyme heteromers (C) and homomers (D) in control and chloroquine-treated cells. Briefly, cells were transfected with the depicted (bottom) mVenus-, mCherry-, and HA-tagged enzyme constructs for 24 h before 40 μm CQ treatment. Note that the 0.4 pH unit increase in Golgi luminal pH induced by CQ markedly reduces the amount of enzyme heteromers in the Golgi (C), whereas it increases that of the enzyme homomers (D). The results are presented as percentages of non-treated controls (mean ± S.D., n = 10; *, p < 0.05; **, p < 0.01).
Having confirmed the pH sensitivity of the enzyme heteromers, we next examined whether the formation of heteromers in the Golgi is coupled with the concomitant disassembly of the homomers and vice versa in a case when Golgi pH is elevated e.g. by using CQ. To measure this we utilized FRET microscopy (Fig. 4, C and D) and quantified the relative levels of heteromers (Fig. 4C) and homomers (Fig. 4D) in the Golgi both at normal and at slightly elevated Golgi pH. For this purpose we utilized 40 μm CQ, which raises Golgi pH by only 0.4 pH units without causing any major structural changes in the Golgi (43). We transfected cells with the two highly pH-sensitive heteromeric FRET enzyme constructs (GalT-I/ST3Gal-III and ppGalNAcT-6/C3GnT-1) together with an HA-tagged homomeric construct. The latter was included to allow equal possibility to form both enzyme homomers and FRET-positive heteromers in the Golgi. We found that CQ decreased the relative amount of the Golgi-localized heteromers by 49 ± 24% (p = 0.001) and 39 ± 19% (p = 0.006), respectively (Fig. 4C). A similar decrease takes place also in the absence of the HA-tagged construct (15). In contrast, when cells were transfected with the two homomeric FRET enzyme constructs (Fig. 4D) together with a FRET-incompatible (HA-tagged) heteromeric enzyme construct, we anticipated that the relative amount of homomers should increase upon CQ treatment. Quantification of the FRET signal revealed that CQ indeed increased the relative amount of enzyme homomers in the Golgi by 135 ± 45 and 66 ± 25%, respectively. These values correspond to a 2.4 (p = 0.0001)- and 1.7 (p = 0.0002)-fold increase relative to control cells that were not treated with CQ, respectively. Taken together, these data indicate that the formation of enzyme heteromers involves concomitant disassembly of the homomers at normal luminal Golgi pH, whereas at elevated Golgi pH, this transition between enzyme homomers and heteromers is markedly inhibited. Thus, the acidic Golgi microenvironment seems to be crucial for the formation and maintenance of enzyme heteromers in the Golgi.
Enzyme Heteromers Are Disassembled upon Their Brefeldin A- and Nocodazole-induced Redistribution to the ER
The preferred Golgi trafficking models entail that Golgi residents move laterally and along the cis-trans axis of the Golgi stack. To assess whether the enzyme homomers and heteromers are compatible with these models, we measured their mobilities in the Golgi membranes of living cells by using the FRAP approach (36). Due to the nearly irreversible nature of the bimolecular fluorescence complementation of the Venus fragments (38), transfection of the cells with the Golgi-localized BiFC constructs ensures that the mobility of the enzyme homomers or heteromers is measured. Based on FRAP measurements, the calculated mobility fractions of both enzyme homomers and heteromers varied between 63 ± 27 and 91 ± 19%, showing that the majority of the complexes are mobile Golgi membrane constituents. Their measured diffusion coefficients (D) were also roughly similar between different enzymes and also between homomers and heteromers. The calculated diffusion coefficients (D) ranged from 0.14 to 0.32 μm2/s (Table 1). These values are only slightly lower than the previously measured values for GFP-tagged ManII and GalT-I constructs (0.2–0.5 μm2/s) (36). Thus, the complexes are compatible with the cisternal maturation or rapid partitioning models for Golgi trafficking.
TABLE 1.
The measured recovery rates, diffusion coefficients, and the mobility fractions of the listed glycosyltransferase homomers and heteromers in live COS-7 cells
| D | Mobile fraction | |
|---|---|---|
| μm2/s | % | |
| Homomers | ||
| GnT-I | 0.14 ± 0.01 | 91.3 ± 18.5 |
| GnT-II | 0.14 ± 0.01 | 63.0 ± 27.1 |
| GalT-I | 0.16 ± 0.01 | 81.1 ± 10.4 |
| ST6Gal-I | 0.21 ± 0.04 | 87.5 ± 24.0 |
| ST3Gal-III | 0.32 ± 0.04 | 90.8 ± 10.9 |
| Heteromers | ||
| GnT-II/GnT-I | 0.13 ± 0.02 | 76.8 ± 14.1 |
| GalT-I/ST6Gal-I | 0.15 ± 0.01 | 78.6 ± 33.1 |
| GalT-I/ST3Gal-III | 0.27 ± 0.06 | 71.7 ± 28.2 |
To examine whether the Golgi-localized N-glycosyltransferases are also able to recycle as complexes, we treated cells expressing selected FRET enzyme constructs with brefeldin A for 1 h (BFA added 16 h post-transfection) to induce coalescence of the Golgi membranes with the ER (44). Their re-localization into the ER was confirmed by co-staining of the BFA-treated cells with an anti-protein disulfide isomerase antibody (Fig. 5A). FRET measurements revealed that N-glycosyltransferase homomers were present in the ER in nearly equal amounts to those present in the Golgi region of untreated cells. The relative FRET signals ranged from 98 ± 24 to 74 ± 23% that of the Golgi-localized homomers (Fig. 5B). In contrast, the amount of ER localized GnT-I/GnT-II heteromers was reduced to 54 ± 23% (p = 0.002) of their original levels in the Golgi (Fig. 5B). This decrease was even more pronounced with the trans-Golgi- localized GalT-I/ST6Gal-I (to 27 ± 14%) or GalT-I/ST3Gal-III (31 ± 16%, p < 0.0001) heteromers. These results indicated that a substantial fraction of enzyme heteromers are disassembled during their BFA-induced relocation back to the ER, whereas enzyme homomers remain mostly intact.
FIGURE 5.

The amount of enzyme homomers and heteromers after their BFA- or nocodazole-induced re-localization into the ER or to the Golgi mini-stacks, respectively. A, immunofluorescence micrograph showing the accumulation of the depicted FRET enzyme constructs in the ER in BFA-treated cells as determined by their co-localization with the ER marker protein (PDI). Scale bar, 10 μm. B, FRET microscopy quantification of the amount of enzyme homomers and heteromers in the ER after BFA treatment. Note that the drug does not markedly reduce the amount of enzyme homomers in the ER (left, dark columns) relative to the Golgi-localized homomers in untreated control cells (light columns). In contrast, the amount of heteromers is significantly (by 50–70%) reduced in the drug-treated cells. The results are presented as percentages of untreated controls (mean ± S.D., n = 8). C, relocalization of the depicted FRET enzyme constructs upon nocodazole (NDZ, 1 μg/ml) treatment or after a 4-h recovery period. Note that the enzymes accumulate in the Golgi mini-stacks after nocodazole treatment and relocalize back to the juxtanuclear Golgi structure after removal of the drug. D–F, FRET microscopy measurements in cells expressing the depicted enzyme homomers or heteromers either before or after nocodazole treatment or after the 4-h recovery period. The results are presented as percentages of untreated controls (mean ± S.D., n = 10; *, p < 0.05; **, p < 0.01; ***, p < 0.001). D, enzyme homomers. Note that the drug-induced relocalization into the ER does not affect the amount of enzyme homomers when compared with that of the Golgi-localized homomers in untreated cells. E, enzyme heteromers. Note the marked reduction in the amount of enzyme heteromers after their accumulation in the Golgi mini-stacks in nocodazole-treated cells and their reappearance after the 4-h recovery period. F, FRET measurements with the GnT-I or GalT-I homomers in the presence of an interacting HA-tagged heteromeric enzyme construct. Note the marked increase in the amount of enzyme homomers in nocodazole-treated cells and the decrease after the 4-h recovery period.
Further proof for the disassembly of enzyme heteromers during their suggested recycling between the ER and the Golgi was obtained by inducing their relocalization via the ER to the Golgi mini-stacks with nocodazole. Cells transfected with homomeric or heteromeric FRET enzyme constructs were, therefore, treated 20 h post-transfection with nocodazole for 4 h (Fig. 5C). FRET microscopy measurements showed that drug treatment did not markedly reduce the amount of enzyme homomers in the cells (Fig. 5D). In contrast, the amount of enzyme heteromers was reduced by 73 ± 7 and 74 ± 6% of the original levels, respectively (middle columns in Fig. 5E). Removal of the drug (4 h recovery) resulted in the relocalization of the enzymes close to the nucleus as part of the formation of the compact juxtanuclear Golgi (right columns) and a concomitant recovery of the FRET signal nearly to the original levels detected in the Golgi without drug treatment (to 85 ± 12 to 70 ± 11%). Moreover, we measured whether the nocodazole treatment induces a concomitant increase in the amount of enzyme homomers by transfecting cells with the two homomeric FRET constructs together with an interacting HA-tagged heteromeric enzyme construct (Fig. 5F). We found that nocodazole treatment increased by 3.7 (±0.5)- and 2.8 (±0.4)-fold the amount of both GnT-I and GalT-I homomers relative to Golgi-localized homomers in untreated cells. After the 4-h recovery, the amount of homomers significantly decreased close to the original levels. These data indicate that recycling of Golgi N-glycosyltransferases involves organelle microenvironment-dependent transitions from enzyme heteromers back to homomers and vice versa when the homomers are transported back to the more acidic Golgi luminal milieu.
DISCUSSION
In this study we investigated the assembly and organizational interplay of the Golgi N-glycosyltransferase complexes by utilizing various microscopic approaches as well as pulldown experiments with both endogenous GalT-I and overexpressed enzyme constructs. We showed that enzyme homomers constitute the majority of the enzyme species in the ER. Upon their transport to the Golgi they undergo transition to form enzyme heteromers given that a relevant heteromeric interaction partner is present. This transition is highly dependent on the acidic Golgi microenvironment, as chloroquine, a pH gradient dissipating drug, markedly inhibited heteromer assembly in live cells. By utilizing BFA and nocodazole treatments, we also showed that enzyme heteromers undergo a reverse transition to form homomers upon their transport back to the ER or to adjacent Golgi mini-stacks (for a summary diagram, see Fig. 6).
FIGURE 6.
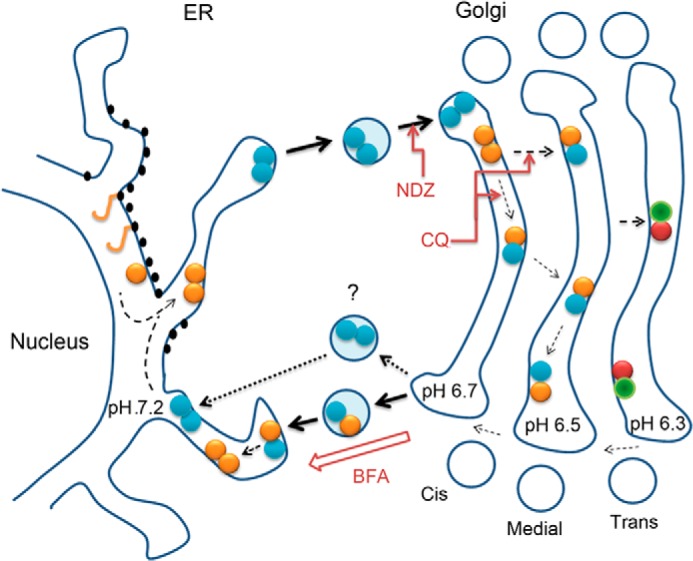
Schematic diagram of the organizational interplay between N-glycosyltransferase homo- and heteromers in live cells. After de novo synthesis, glycosyltransferase polypeptides (yellow sticks) form homodimers (double yellow or blue dots) during their folding and/or before their transport to the Golgi. In the Golgi acidic luminal milieu favors the formation of heteromers between sequentially acting medial-Golgi (yellow-blue dots) or trans-Golgi (red-green dots) enzymes at the expense of enzyme homomers. Nocodazole (NDZ) inhibits this process due to its ability to block anterograde transport and coalescence of transport vesicles into a compact Golgi structure. CQ, a pH gradient dissipating drug that impairs heteromer formation, was used to show that this transition is a pH-dependent process. Heteromers remain mobile in the Golgi membranes and are able to recycle to the ER. This was evidenced by using nocodazole-induced anterograde transport block that resulted in accumulation of the complexes in a more alkaline ER and a reverse transition from heteromers to enzyme homomers. Similar observations were obtained by using BFA that forces coalescence of Golgi membranes in the ER membranes. The question mark denotes a possibility that heteromer disassembly may already take place before the fusion of retrograde transport vesicles with the ER.
These findings are important as they provide new insights into glycosylation, glycosylation-associated diseases, and general Golgi functioning in mammalian cells. First of all, they show that glycosyltransferase homomers and heteromers are spatially and temporally distinct yet are interdependent membrane constituents that undergo dynamic transitions between each other during their trafficking between the ER and the Golgi. Second, they emphasize that glycosyltransferase homomers and heteromers are not competing enzyme species that utilize the same substrates and/or acceptors but, rather, have distinct functional roles in glycosylation. Based on their predominant localization in the ER (Fig. 1), enzyme homomers are very likely to have a role in helping proper folding and/or transport of glycosyltransferases to the Golgi. In contrast, enzyme heteromers must be the most important enzyme species for efficient glycosylation. In strong support of this, our previous studies have shown that the heteromers are enzymatically more active than the corresponding enzyme homomers (15). Here we also showed that the heteromers are both assembled (Fig. 3) and can exist (Fig. 5) only in the acidic Golgi microenvironment. Heteromer formation can thus be regarded as to provide a simple means to localize the most active enzyme species in the Golgi and thereby to increase its glycosylation potential. The observations that elevated pH inhibited heteromer formation both in vitro and in vivo (Fig. 4) are consistent with this view.
The strict pH dependence of enzyme heteromers (but not of homomers) also provides an explanation for their absence in acidification/glycosylation-defective cancer cells (15). In addition, we showed that their absence in non-cancerous cells at elevated Golgi pH correlated with altered cell surface glycosylation. Only a 0.2 pH unit increase was found to be sufficient to induce expression of the oncofetal Tn and T-antigens, both of which are known to contribute to the invasive and metastatic behavior of cancer cells (43). Thus, the observed absence of the enzyme homomers in cancer cells is very likely one reason for their tendency to invade and metastasize into adjacent organs. An important issue in this respect is also to unravel the molecular defects that are responsible for altered Golgi pH regulation in cancer cells. In this context, however, it should be also emphasized that an alternative way to reduce the amount of enzyme heteromers in cancer cells and their glycosylation potential is the down-regulation of any single glycosyltransferase (45, 46) that normally forms a complex with a relevant enzyme partner. Collectively, these observations highlight the functional importance of Golgi acidity not only in providing an optimal pH for each individual glycosyltransferase but also in regulating the organizational interplay between Golgi glycosyltransferase homomers and heteromers.
Our findings also provide new insights into the existing Golgi trafficking and retention phenomena. It is well known that sequences in the cytoplasmic tails, transmembrane regions, and luminal domains of various Golgi proteins are required for their Golgi localization (34, 47). Yet it is still unclear how these sequences confer Golgi residency. Given that both enzyme homomers and heteromers localize almost exclusively in the Golgi membranes even in transfected cells (see e.g. Fig. 3C), we do not expect any marked differences in their Golgi targeting mechanisms. Yet the signals are slightly different between enzyme homomers and heteromers. The possibility that such subtle differences may influence sub-compartmental localization in the Golgi or restrict/enhance their trafficking through the organelle cannot be ruled out. Whether such differences indeed exist between enzyme homomers and heteromers requires more detailed studies preferentially at the single molecule level.
One still prevailing view holds that oligomerization catalyzed by the cisternal environmental factors (31, 48) facilitates the formation of complexes that are large enough to prevent their exit into post-Golgi compartments. The general tendency of glycosyltransferases to exist both as homomers and heteromers is consistent with this view (49). However, based on our size exclusion chromatography results (15), the size of the GalT-I and ST6Gal-I homomers and heteromers corresponds to enzyme tetramers/dimers, making it unlikely that tetramerization would be enough to cause that effect. The observed transport of the ER-localized enzyme homomers to the Golgi and the mobility of both complexes in the Golgi membranes provide strong support for this view. Thus, complex formation itself appears to neither restrict intra-Golgi transport nor their recycling between the Golgi and the ER. Therefore, the formation of glycosyltransferase complexes is fully compatible with the “cisternal maturation” or the “rapid partitioning” models, which both entail that the cisternal contents change both laterally and along the cis-trans axis of the Golgi stack (29, 30, 50, 51). The data are also compatible with the suggested recycling of the Golgi glycosyltransferases between the Golgi and the ER (42). However, continuous recycling of heteromers does not seem to be beneficial to glycosylation, at least if not compensated by the transport of homomers and the formation of new heteromers in the Golgi. Otherwise, recycling would result eventually in loss of Golgi localized heteromers and, thus, to lowered glycosylation potential. This is not the case with enzyme homomers and their recycling, as their main role seems to be in helping folding and/or transport from the ER to the Golgi. In fact, it may even be advantageous, as reappearance of the homomers in the Golgi would allow another round of trials to find a relevant heteromeric interaction partner for glycan synthesis. This may be important, considering the fact that Golgi cisternae possess different sets of enzymes along the cis-trans axis of the Golgi stack, each with unique luminal milieu. Finally, although these issues are only speculative at this point, our results emphasize that the existence and dynamic organizational interplay between Golgi glycosyltransferase homomers and heteromers need be taken into account when addressing Golgi targeting/retention-related phenomena.
This work was supported by the Finnish Glycoscience Graduate School, The University of Oulu, Magnus Ehrnrooth Foundation, Emil Aaltonen Foundation, and the Finnish Cultural Foundation.
- GalT
- galactosyltransferase
- GnT
- N-acetylglucosaminyltransferase
- ER
- endoplasmic reticulum
- BiFC
- bimolecular fluorescence complementation
- FRAP
- fluorescence recovery after photobleaching
- mVen
- monomeric Venus
- mCer
- mCerulean
- mChe
- mCherry
- CQ
- chloroquine
- BFA
- brefeldin A
- D
- diffusion coefficient.
REFERENCES
- 1. Hart G. W. (1992) Glycosylation. Curr. Opin. Cell Biol. 4, 1017–1023 [DOI] [PubMed] [Google Scholar]
- 2. Apweiler R., Hermjakob H., Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 [DOI] [PubMed] [Google Scholar]
- 3. Varki A. (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. Glycobiology 3, 97–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Freeze H. H. (2006) Genetic defects in the human glycome. Nat. Rev. Genet. 7, 537–551 [DOI] [PubMed] [Google Scholar]
- 5. Varki A. (2008) Sialic acids in human health and disease. Trends Mol. Med. 14, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ungar D. (2009) Golgi linked protein glycosylation and associated diseases. Semin. Cell Dev. Biol. 20, 762–769 [DOI] [PubMed] [Google Scholar]
- 7. Ohtsubo K., Chen M. Z., Olefsky J. M., Marth J. D. (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 17, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kornfeld R., Kornfeld S. (1985) Assembly of Asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 9. Dunphy W. G., Brands R., Rothman J. E. (1985) Attachment of terminal N-acetylglucosamine to asparagine-linked oligosaccharides occurs in central cisternae of the Golgi stack. Cell 40, 463–472 [DOI] [PubMed] [Google Scholar]
- 10. Cummings R.D., Esko J.D. (2009) Principles of Glycan Recognition, Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) 2nd Ed., pp. 387–402, The Consortium of Glycobiology Editors, La Jolla, CA, Cold Spring Harbor, NY [Google Scholar]
- 11. Rabouille C., Hui N., Hunte F., Kieckbusch R., Berger E. G., Warren G., Nilsson T. (1995) Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J. Cell Sci. 108, 1617–1627 [DOI] [PubMed] [Google Scholar]
- 12. Nilsson T., Hoe M. H., Slusarewicz P., Rabouille C., Watson R., Hunte F., Watzele G., Berger E. G., Warren G. (1994) Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J. 13, 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jungmann J., Munro S. (1998) Multi-protein complexes in the cis Golgi of Saccharomyces cerevisiae with α-1, 6-mannosyltransferase activity. EMBO J. 17, 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCormick C., Duncan G., Goutsos K. T., Tufaro F. (2000) The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. U.S.A. 97, 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hassinen A., Pujol F. M., Kokkonen N., Pieters C., Kihlström M., Korhonen K., Kellokumpu S. (2011) Functional organization of the Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J. Biol. Chem. 286, 38329–38340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maccioni H. J., Quiroga R., Ferrari M. L. (2011) Cellular and molecular biology of glycosphingolipid glycosylation. J. Neurochem. 117, 589–602 [DOI] [PubMed] [Google Scholar]
- 17. Schoberer J., Liebminger E., Botchway S. W., Strasser R., Hawes C. (2013) Time-resolved fluorescence imaging reveals differential interactions of N-glycan processing enzymes across the Golgi stack in planta. Plant Physiol. 161, 1737–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamaguchi N., Fukuda M. N. (1995) Golgi retention mechanism of β-1,4-galactosyltransferase. Membrane-spanning domain-dependent homodimerization and association with α- and β-tubulins. J. Biol. Chem. 270, 12170–12176 [DOI] [PubMed] [Google Scholar]
- 19. Opat A. S., Houghton F., Gleeson P. A. (2000) Medial Golgi but not late Golgi glycosyltransferases exist as high molecular weight complexes. Role of luminal domain in complex formation and localization. J. Biol. Chem. 275, 11836–11845 [DOI] [PubMed] [Google Scholar]
- 20. Ouzzine M., Gulberti S., Netter P., Magdalou J., Fournel-Gigleux S. (2000) Structure/function of the human Galβ1, 3-glucuronosyltransferase dimerization and functional activity are mediated by two crucial cysteine residues. J. Biol. Chem. 275, 28254–28260 [DOI] [PubMed] [Google Scholar]
- 21. Sasai K., Ikeda Y., Tsuda T., Ihara H., Korekane H., Shiota K., Taniguchi N. (2001) The critical role of the stem region as a functional domain responsible for the oligomerization and Golgi localization of N-acetylglucosaminyltransferase V. The involvement of a domain homophilic interaction. J. Biol. Chem. 276, 759–765 [DOI] [PubMed] [Google Scholar]
- 22. Yen T. Y., Macher B. A., Bryson S., Chang X., Tvaroska I., Tse R., Takeshita S., Lew A. M., Datti A. (2003) Highly conserved cysteines of mouse core 2 β1, 6-N-acetylglucosaminyltransferase I form a network of disulfide bonds and include a thiol that affects enzyme activity. J. Biol. Chem. 278, 45864–45881 [DOI] [PubMed] [Google Scholar]
- 23. Giraudo C. G., Maccioni H. J. (2003) Ganglioside glycosyltransferases organize in distinct multienzyme complexes in CHO-K1 cells. J. Biol. Chem. 278, 40262–40271 [DOI] [PubMed] [Google Scholar]
- 24. Dayhoff J. E., Shoemaker B. A., Bryant S. H., Panchenko A. R. (2010) Evolution of protein binding modes in homooligomers. J. Mol. Biol. 395, 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hashimoto K., Madej T., Bryant S. H., Panchenko A. R. (2010) Functional states of homooligomers: insights from the evolution of glycosyltransferases. J. Mol. Biol. 399, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Losev E., Reinke C. A., Jellen J., Strongin D. E., Bevis B. J., Glick B. S. (2006) Golgi maturation visualized in living yeast. Nature 441, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 27. Matsuura-Tokita K., Takeuchi M., Ichihara A., Mikuriya K., Nakano A. (2006) Live imaging of yeast Golgi cisternal maturation. Nature 441, 1007–1010 [DOI] [PubMed] [Google Scholar]
- 28. Emr S., Glick B. S., Linstedt A. D., Lippincott-Schwartz J., Luini A., Malhotra V., Marsh B. J., Nakano A., Pfeffer S. R., Rabouille C., Rothman J. E., Warren G., Wieland F. T. (2009) Journeys through the Golgi: taking stock in a new era. J. Cell Biol. 187, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackson C. L. (2009) Mechanisms of transport through the Golgi complex. J. Cell Sci. 122, 443–452 [DOI] [PubMed] [Google Scholar]
- 30. Glick B. S., Luini A. (2011) Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect. Biol. 3, a005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weisz O. A., Swift A. M., Machamer C. E. (1993) Oligomerization of a membrane protein correlates with its retention in the Golgi complex. J. Cell Biol. 122, 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gleeson P. A. (1998) Targeting of proteins to the Golgi apparatus. Histochem. Cell Biol. 109, 517–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tu L., Banfield D. K. (2010) Localization of Golgi-resident glycosyltransferases. Cell. Mol. Life Sci. 67, 29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Banfield D. K. (2011) Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 3, a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hassinen A., Rivinoja A., Kauppila A., Kellokumpu S. (2010) Golgi N-glycosyltransferases form both homo- and heterodimeric enzyme complexes in live cells. J. Biol. Chem. 285, 17771–17777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cole N. B., Smith C. L., Sciaky N., Terasaki M., Edidin M., Lippincott-Schwartz J. (1996) Diffusional mobility of Golgi proteins in membranes of living cells. Science 273, 797–801 [DOI] [PubMed] [Google Scholar]
- 37. Flügel D., Görlach A., Kietzmann T. (2012) GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood 119, 1292–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kerppola T. K., (2008) Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 37, 465–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang W., Storrie B. (1997) Expression of a cytoplasmically epitope-tagged, human Golgi glycosyltransferase in homologous cells results in mislocalization of multiple Golgi proteins. Cell Biol. Int. 21, 223–228 [DOI] [PubMed] [Google Scholar]
- 40. Giraudo C. G., Maccioni H. J. (2003) Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol. Biol. Cell 14, 3753–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cole N. B., Sciaky N., Marotta A., Song J., Lippincott-Schwartz J. (1996) Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 7, 631–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Storrie B., White J., Röttger S., Stelzer E. H., Suganuma T., Nilsson T. (1998) Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143, 1505–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rivinoja A., Kokkonen N., Kellokumpu I., Kellokumpu S. (2006) Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J. Cell Physiol. 208, 167–174 [DOI] [PubMed] [Google Scholar]
- 44. Lippincott-Schwartz J., Yuan L., Tipper C., Amherdt M., Orci L., Klausner R. D. (1991) Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67, 601–616 [DOI] [PubMed] [Google Scholar]
- 45. Brockhausen I. (1999) Pathways of O-glycan biosynthesis in cancer cells. Biochim. Biophys. Acta 1473, 67–95 [DOI] [PubMed] [Google Scholar]
- 46. Burchell J., Poulsom R., Hanby A., Whitehouse C., Cooper L., Clausen H., Miles D., Taylor-Papadimitriou J. (1999) An α2,3 sialyltransferase (ST3Gal I) is elevated in primary breast carcinomas. Glycobiology 9, 1307–1311 [DOI] [PubMed] [Google Scholar]
- 47. Nilsson T., Au C. E., Bergeron J. J. (2009) Sorting out glycosylation enzymes in the Golgi apparatus. FEBS Lett. 583, 3764–3769 [DOI] [PubMed] [Google Scholar]
- 48. Nilsson T., Slusarewicz P., Hoe M. H., Warren G. (1993) Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 330, 1–4 [DOI] [PubMed] [Google Scholar]
- 49. de Graffenried C. L., Bertozzi C. R. (2004) The roles of enzyme localisation and complex formation in glycan assembly within the Golgi apparatus. Curr. Opin. Cell Biol. 16, 356–363 [DOI] [PubMed] [Google Scholar]
- 50. Rabouille C., Klumperman J. (2005) The maturing role of COPI vesicles in intra-Golgi transport. Nat. Rev. Mol. Cell Biol. 6, 812–817 [DOI] [PubMed] [Google Scholar]
- 51. Patterson G. H., Hirschberg K., Polishchuk R. S., Gerlich D., Phair R. D., Lippincott-Schwartz J. (2008) Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell 133, 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]