

Background: How IκB kinaseβ K171E and K171T mutations mediate lymphomagenesis is not known.
Results: We performed biochemical, molecular modeling and TALEN-based knockin studies on wild-type and mutant IKKβ.
Conclusion: Mutant IKKβ molecules are constitutively active in an activation-loop phosphorylation-independent manner.
Significance: These results have broad implications for the function of positively charged residues in all activation loop-dependent kinases.
Keywords: Lymphoma, Molecular Modeling, Mutant, NF-kappa B (NF-κB), Protein Kinase, Serine/Threonine Protein Kinase, Signaling, Transcription Activator-like Effector Nuclease (TALEN), I Kappa B Kinase Beta
Abstract
Somatic mutations altering lysine 171 of the IKBKB gene that encodes (IKKβ), the critical activating kinase in canonical (NFκB) signaling, have been described in splenic marginal zone lymphomas and multiple myeloma. Lysine 171 forms part of a cationic pocket that interacts with the activation loop phosphate in the activated wild type kinase. We show here that K171E IKKβ and K171T IKKβ represent kinases that are constitutively active even in the absence of activation loop phosphorylation. Predictive modeling and biochemical studies establish why mutations in a positively charged residue in the cationic pocket of an activation loop phosphorylation-dependent kinase result in constitutive activation. Transcription activator-like effector nuclease-based knock-in mutagenesis provides evidence from a B lymphoid context that K171E IKKβ contributes to lymphomagenesis.
Introduction
The canonical nuclear factor κB (NFκB)3 pathway is required for normal marginal zone (MZ) B cell development (1) as is non-canonical NFκB activation (2–5). Another crucial pathway in MZ B cell development involves the activation of Notch signaling (6–8). Canonical NFκB signaling and Notch activation function synergistically during normal MZ B cell development (9, 10), so it is not surprising that mutations in the NFκB and Notch pathways have been found in 40 of 117 cases of splenic marginal zone lymphoma (11), a B cell lymphoma that arises from marginal zone B cells.
Mutations in the IKBKB gene encoding the IκB kinase β (IKKβ) protein have been observed frequently in splenic marginal zone lymphoma (11, 12). IKKβ is a catalytic subunit of the IκB kinase (IKK) complex that drives canonical NFκB signaling by phosphorylating inhibitor of NFκB α (IκBα) and targeting it for degradation. In 8/117 splenic marginal zone lymphoma recurrent mutations in IKBKB have been observed that convert lysine 171 to either a glutamate (K171E IKKβ) or a threonine (K171T IKKβ) (11, 12). The somatic variant encoding K171E IKKβ has also been documented in a single subject with multiple myeloma (13). Lysine 171 lies in a cationic pocket that interacts with a critical phosphorylated serine residue in the activation loop of IKKβ. Serine 181 in the activation loop of IKKβ is the crucial residue that needs to be phosphorylated by an upstream kinase for it to switch to its active conformation (13). A phospho-mimetic mutation in serine 181 can lead to constitutive activation of IKKβ, and constitutively activated IKKβ can contribute to lymphoma generation in transgenic mice (14). Lysine 171, however, is situated parallel to serine 181 in the activation loop, and no structural models or experimental data exist to indicate whether the cancer-associated alterations found in this residue of IKKβ activate or inactivate this critically important kinase, or if these changes represent functionally irrelevant “passenger” mutations.
Activating mutations in the NFκB pathway have been functionally identified in diffuse large B cell lymphomas (15–17). Mutated genes include CARD11 (caspase recruitment domain 11), TRAF2 (TNF receptor-associated factor 2) and Myd88 (myeloid differentiation primary response 88). Inactivating mutations in A20 can also contribute to constitutive NFκB activation (18). All of these genes are also mutated in splenic marginal zone lymphoma, but IKKβ lysine 171 alterations have not been reported in diffuse large B cell lymphomas. Other mutations in IKKβ have been described in the Catalogue of Somatic Mutations in Cancer (COSMIC) database, many of these may well be “passengers,” and no direct evidence exists for any activating mutations in IKKβ in any human tumor.
Next Generation Sequencing has revealed numerous mutations in tumors, including lymphomas. One of the most specific definitions of a “driver” mutation is one that “directly or indirectly confers a growth advantage to the cell in which it occurs” (19). Commonly used mutation predictive methods such as Polyphen 2 (20) or SIFT (Sorting Intolerant From Tolerant) (21), can suggest whether a mutation is “potentially damaging,” but this categorization cannot distinguish between activating and inactivating mutations. Most mutations in lymphomas have been functionally analyzed in overexpression studies and, short of generating knock-in mice, methods to readily interrogate mutations in a B lymphoid context to distinguish between potential passengers and drivers of lymphomagenesis have not been described. Transcription activator-like effector nuclease (TALEN) and CRISPR (clustered regularly interspersed short palindromic repeats) technology have been used to generate mutations in human-induced pluripotent cells (22–24), but this kind of knock-in approach has not been utilized in the study of cancer mutations to identify drivers.
We show here that the K171E and K171T IKKβ proteins result in the constitutive activation of NFκB in an activation loop phosphorylation-independent manner. Molecular modeling was used to predict the mechanism of constitutive activation by these two distinct mutations, and the predictions were confirmed biochemically in overexpression studies. Confirmation of the potential contribution of activating mutations to lymphomagenesis was obtained in a B lymphoid cell line and non-overexpression context by TALEN-based knock-in mutagenesis of the tumor-specific K171E IKKβ alteration in human BJAB B cells.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
Xpress-tagged pcDNA3.1/His C-IKKβ was kindly provided by S. Ghosh and used as a template to generate IKKβ mutants. A QuikChange site-directed mutagenesis kit (Stratagene) was used to create the plasmids for all mutants according to the manufacturer's instructions. The mutagenesis and sequencing primers are described in Tables 1 and 2.
TABLE 1.
Primers for mutagenesis
| Construct | Sequence |
|---|---|
| IKKβ-K44M | 5′-GAGCAGATTGCCATCATGCAGTGCCGGCAGGAG-3′ |
| IKKβ-R57A | 5′-CCCCGGAACCGAGAGGCGTGGTGCCTGGAGATC-3′ |
| IKKβ-R144A | 5′-AACAGAATCATCCATGCGGATCTAAAGCCAGAA-3′ |
| IKKβ-K171T | 5′-ACCTAGGATATGCCACGGAGCTGGATCAGGG-3′ |
| IKKβ-K171E | 5′-GACCTAGGATATGCCGAGGAGCTGGATCAGGGC-3′ |
| IKKβ-S177A | 5′-GAATGATGTGCAAAGAGCGCCCTGATCCAGCTC-3′ |
| IKKβ-S181A | 5′-GGCAGTCTTTGCACAGCATCGTGGGGACCCTG-3′ |
| IKKβ-SSEE | 5′-GAGCTGGATCAGGGCGAGCTTTGCACAGAATTCGTGGGGACCCT-3′ |
| IKKβ-A360S | 5′-CAGGAAGCGGGCCTGTCGTTGATCCCCGATA-3′ |
| IKKβ-D639N | 5′-AGCTTAATGAATGAGAATGAGAAGACTGTTG-3′ |
| IKKβ-G667C | 5′-TAGAGCAAGGTCCGTTGTCCTGTCAGTGGAA-3′ |
| IKKβ-N677D | 5′-AGCCCGGATAGCATGGATGCCTCTCGACTTA-3′ |
TABLE 2.
Primers for sequence verification
| Primer description/Name | Primer orientation | Sequence |
|---|---|---|
| Sequencing IKKβ 1–150 aaa | ||
| IKKβ-Seq-FP1 | Sense | 5′-CGAGAGCGGTGGTGCCTGGAG-3′ |
| IKKβ-Seq-RP1 | Antisense | 5′-GTCAATAATTTTGTGTATTAACCTC-3′ |
| Sequencing IKKβ 100–250 aa | ||
| IKKβ-Seq-FP2 | Sense | 5′-TCCGGAAGTACCTGAACCAG-3′ |
| IKKβ-Seq-RP2 | Antisense | 5′-CTTCTGCCGCACTTTTGAAT-3′ |
| Sequencing IKKβ 250–400 aa | ||
| IKKβ-Seq-FP3 | Sense | 5′-GTGCGGCAGAAGAGTGAGGTG-3′ |
| IKKβ-Seq-RP3 | Antisense | 5′-AGGCGAGATTCCTCTTGGGCT-3′ |
| Sequencing IKKβ 580–730 aa | ||
| IKKβ-Seq-FP4 | Sense | 5′-GAACGCTGGACGACCTAGAGG-3′ |
| IKKβ-Seq-RP4 | Antisense | 5′-CTTCTTCCGTCTGTAACCAGC-3′ |
a aa, amino acids.
Lymphoid Cell Lines
The ABC-DLBCL (active B-cell diffuse large B-cell lymphoma) cell line, NU-DUL-1, the Burkitt's lymphoma cell lines, BJAB, Ramos, and Raji and the GCB-DLBCL (germinal center B-cell diffuse large B-cell lymphoma) cell line OCI-Ly17 were obtained from ATCC.
Antibodies
Rabbit monoclonal GST, phospho-IKKβ (Ser-177), phospho-IKKβ (Ser-177/181), p65, phopho-p65 (Ser-536), β-actin, mouse monoclonal phospho-IκBα (Ser-32/36), and IκBα antibodies were obtained from Cell Signaling Technology. Mouse Anti-Xpress monoclonal antibody was obtained from Invitrogen. An anti-β-tubulin mouse monoclonal antibody was from Upstate Biotechnology.
Immunofluorescence
Hela cells were seeded on coverslips in six-well culture plates and transiently transfected with WT-IKKβ, K171E-IKKβ, and K171T-IKKβ DNAs. Cells were fixed for 20 min with 4% paraformaldehyde, and permeabilized for 5 min with PBS containing 1% Nonidet P-40. After washing three times with PBS containing 10 mm glycine, cell were blocked with PBS containing 3% BSA for 30 min. Immunostaining was performed using antibodies as indicated in Fig. 1C, and images were acquired using a Olympus IX81 spinning disk confocal microscope.
FIGURE 1.

Recurrent mutations encoding K171E and K171T IKKβ activate NFκB signaling. A, wild type IKKβ (WT), lymphoma-derived K171E and K171T IKKβ, and A360S, D639N, G667C, and N677D IKKβ proteins from non-lymphoid cancers were co-transfected with an NFκB-driven firefly luciferase reporter construct along with a Renilla luciferase reporter construct into HeLa cells. The upper panel depicts a typical Dual-Luciferase assay. In the lower panel, an aliquot from each of the cell lysates depicted above was loaded on a 10% SDS-PAGE gel and subjected to anti-Xpress quantitative Western blots (WB) to determine protein levels. Luciferase levels were normalized to protein levels, and the WT value was set at 1. Bars represent the means of two individual experiments, and the error bars represent ± S.D. ***, p < 0.001. B and C, kinase activity was determined using GST-IκBα as a specific substrate. IKKβ, phosphorylated GST-IκBα, unphosphorylated GST-IκBα, IκBα, and phosphorylated IκBα bands are indicated in B. The protein levels of IκBα and phosphorylated IκBα were quantified in C. This Western blot is representative of three individual experiments, and the error bars represent ± S.D. *, p < 0.01. D, HeLa cells were transfected with IKKβ WT, K171T, and K171E encoding mutants for 24 h and visualized by spinning disk confocal microscopy following immunostaining for IKKβ (green), p65 (red), and DAPI (blue). Based on the distribution of p65 in transfected and untransfected cells, they were labeled as nuclear (nuc), nuclear and cytoplasmic (nuc-cyt), or cytoplasmic (cyt), respectively. E, the cell numbers of each group in un-transfected and transfected cells for each construct were counted as described under “Experimental Procedures.”
Quantification of Fluorescence Images
Immunofluorescent pictures were analyzed using ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD) as reported previously (25, 26). To more objectively quantify the nuclear-cytoplasmic distribution, confocal images were acquired for at least 100 cells from untransfected cells and cells transfected with each construct, respectively. The representative confocal cross-sections (8-bit grayscale or 24- bit RGB) were converted to TIFF files and used to determine the ratio of fluorescence in the cytoplasm relative to the nucleus. Mean pixel density was assessed for manually cropped areas of uniform intensity in both nucleus and cytoplasm, and ratios were determined after subtraction from background fluorescence. According to the ratios, cells were categorized as follows: <0.75 (predominantly cytoplasmic), 0.75–1.25 (nuclear and cytoplasmic), and >1.25 (predominantly nuclear).
NFκB Luciferase Reporter Gene Assays
Hela cells were transiently transfected with the pBII×Luc NFκB reporter construct containing firefly luciferase under the control of four NFκB binding sites. A Renilla luciferase reporter construct, pRL-TK (Promega) was used as an internal control to normalize for transfection. Cells were assessed after 48 h post-transfection with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.
Kinase Assays
Hela cells were collected in lysis buffer (50 mm HEPES (pH 8.0), 150 mm NaCl, 25 mm EGTA, 1 mm EDTA, 0.1% Tween 20, 10% glycerol, 50 mm NaF, and 0.1 mm Na3VO4) containing a mixture of protease inhibitors (Calbiochem). Xpress-tagged WT and mutated IKKβ proteins in HeLa cells were immunoprecipitated with anti-Xpress antibody, and then incubated in a 20-μl kinase buffer containing 50 mm HEPES (pH 8.0). 10 mm MgCl2, 2.5 mm EGTA, 1 mm DTT, 10 μm β-glycerophosphate, 1 mm NaF, 0.1 mm Na3VO4, 0.1 mm PMSF, and 10 μm ATP. After adding 1 mm GST-IκBα as a substrate and 0.2 mm ATP (10 mCi/ml), the reaction mixture was incubated for 30 min at 30 °C, the reaction was stopped by addition of 20 μl 4× SDS-loading buffer and then subjected to Western blotting using anti-GST, anti-IκBα, anti- phospho-IκBα (Ser-32/36), and anti-Xpress antibodies.
Molecular Modeling
The structures of K171E and K171T IKKβ kinase domains were modeled based on the recently published structure of human IKKβ (27) using the one-to-one threading algorithm of Phyre2 (28). The figures for the structural models were generated in PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4 Schrödinger, LLC).
Construction of TALE Nucleases
TALENs were designed using the ZiFiT Targeter web server, such that the nucleotide to be altered, to create the K171E and K171T changes, lay within the “spacer” region that is flanked by the two TALEN binding sites. The TALEN monomers were designed to bind to 18 base pair target sites that are 16 base pairs apart. This configuration was used because it has been shown to function robustly (29). The DNA fragments encoding the repeat arrays of the TALEN monomers were built on a liquid handling system using the previously described fast ligation-based automatable solid-phase high-throughput system (29). These fragments were then cloned into expression vectors that provide the required Δ152 amino acid N terminus, the final 0.5 repeat domain, the +63 amino acid C terminus, and the fokI endonuclease domain. These constructs were then sequences verified as described (30) and used to create the required genomic alternations. All of the expression vectors are available from Addgene.
Construction of Donor Template
The 1.5-kb DNA fragment the IKKβ K171E mutation was amplified out of genomic DNA by nested PCR (Table 3). 3′ Adenosine overhangs were added to nested PCR products by adding 1 μl of Taq polymerase and incubating at 72 °C for 9 min. PCR products were then cloned into TOPO TA vector according to the TOPO TA Cloning protocol from Invitrogen. Site-direct mutagenesis was performed as mentioned before on 1.5-kb genomic DNA inserts in TOPO-TA. Colonies with successful ligations were grown in large volumes, and donor template DNA was extracted using Qiagen Plasmid Maxi Prep Kit.
TABLE 3.
Primers for TALEN donor template construction
FP, forward primer; RP, reverse primer; mut, mutagenesis; seq, sequencing.
| Primer name | Primer orientation | Sequence |
|---|---|---|
| K171E nested FP | Sense | TTGTTGCAGATTGAGGCAAG |
| K171E nested RP | Antisense | GCAGGTCTTCCTGTGTGTGA |
| K171E 1.5 kb F BamH1 | Sense | AAAAAGGATCCCGCCTCTGACTCTAAATGG |
| K171E 1.5 kb R AgeI | Antisense | AAAAAACCGGTGCCCTTTTCACCTCCTTACC |
| K171E mut. FP | Sense | GACCTAGGATATGCCGAGGAGCTGGATCAGG |
| K171E mut. RP | Antisense | AGCTCCTCGGATATCCTAGGTCCCCTGATC |
| K171E seq. FP | Sense | CCCTCAGCTTTCTCCTTCCT |
| K171E seq. RP | Antisense | CAAGGCCCACTTCTTACCAG |
Nucleofection
Nucleofector and cell line nucleofector kits were obtained from Amaxa (Cologne, Germany). 2 × 106 BJAB cells were resuspended in 100 μl of Nucleofector solution V containing 500 ng each of TALEN, 1 μg of pUC with a mutagenized insert, and 200 ng of tdTomato as a marker for transfected cells. Nucleofection of cells was carried out using Program E-23, and cells were diluted into 2 ml of complete RPMI 1640 medium containing 0.1% β-mercaptoethanol and placed in a humidified incubator at 37 °C for 48 h. FACS sorting was performed by gating on the red fluorescent protein-positive cell population. Cells were grown in 10 ml complete RPMI 1640 medium containing 0.1% β-mercaptoethanol until red fluorescence was lost, at which point they were diluted to 0.1 cell/well and 0.2 cell/well dilutions in 96-well plates. Single cell colonies were screened by sequencing, and gene modification of endogenous IKKβ was demonstrated using Mutation Surveyor DNA variant analysis software (Softgenetics).
Apoptosis Assay
WT BJAB, K171E/WT BJAB, and K171E/K171E BJAB cells were seeded in 12-well plates at a density of 5 × 105 per well and were either left untreated or treated with 1 μm staurosporine for 16 h. The cells were washed in phosphate-buffered saline, resuspended in annexin V-binding buffer (Biolegend), and stained with pacific blue-conjugated annexin V (Biolegend) and propidium iodide (Invitrogen). The cells were incubated for 10 min and then sorted by flow cytometry using a MACSQuant analyzer (Miltenyi Biotech). Annexin V- positive, propidium iodide-negative cells are early apoptotic cells. Cells that were propidium iodide-positive as well as annexin V-positive are late apoptotic or dead cells.
RNA Extraction and Quantitative RT-PCR
Total RNA was extracted with the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. For reverse transcriptase PCR (RT-PCR), 1 μg RNA sample were reverse transcribed with Retro-Script MMLV reverse transcriptase kit (Invitrogen). The cDNA stock was diluted into 200 μl, and 2 μl was used for PCR using primers (Table 4) and SYBR Green Master Mix (ABI). β-Actin gene expression was determined for normalization. Real-time PCR was performed in duplicate using the ABI 7500 Real-Time PCR System (Applied Biosystem) as follows: hold 50 °C for 1 min; hold 95 °C 15 min; 40 cycles (95 °C for 15 s; 60 °C for 1 min). Results were collected with 7500 software (Applied Biosystems), and relative quantification was performed using the comparative threshold (CT) method. Transcript levels were normalized to β-actin mRNA levels (ΔCT), and the normalized data were used to determined changes in gene expression (2−ΔΔCT). To analyze the difference between WT and mutants, WT samples were set as a calibrator (control).
TABLE 4.
Primers for quantitative real-time RT-PCR and ChIP quantitative real-time PCR
FP, forward primer; RP, reverse primer.
| Primer names | Primer orientation | Sequence |
|---|---|---|
| Quantitative real-time RT-PCR | ||
| BCL2-RT-PCR-FP | Sense | 5′-TCCGCATCAGGAAGGCTAGA-3′ |
| BCL2-RT-PCR-RP | Antisense | 5′-AGGACCAGGCCTCCAAGCT-3′ |
| BCL2L1-RT-PCR-FP | Sense | 5′-CCCAGGGACAGCATATCAGA-3′ |
| BCL2L1-RT-PCR-RP | Antisense | 5′-CTCCTTGTCTACGCTTTCCAC-3′ |
| BCL2A1-RT-PCR-FP | Sense | 5′-AGGCTGGCTCAGGACTATCT-3′ |
| BCL2A1-RT-PCR-RP | Antisense | 5′-TGTTCTGGCAGTGTCTACGG-3′ |
| CFLAR-RT-PCR-FP | Sense | 5′-TCAAGGAGCAGGGACAAGTTA-3′ |
| CFLAR-RT-PCR-RP | Antisense | 5′-GACAATGGGCATAGGGTGTTATC-3′ |
| BIRC5-RT-PCR-FP | Sense | 5′-GCACCACTTCCAGGGTTTAT-3′ |
| BIRC5-RT-PCR-RP | Antisense | 5′-GCCACTGTTACCAGCAGCAC-3′ |
| XIAP-RT-PCR-FP | Sense | 5′-CCTTGTGATCGTGCCTGGTC-3′ |
| XIAP-RT-PCR-RP | Antisense | 5′-CCCTCCTCCACAGTGAAAGC-3′ |
| ACTIN-RT-PCR-FP | Sense | 5′-CATGTACGTTGCTATCCAGGC-3′ |
| ACTIN-RT-PCR-RP | Antisense | 5′-CTCCTTAATGTCACGCACGAT-3′ |
| ChIP quantitative real-time PCR | ||
| B-actin-ChIP-FP | Sense | 5′-CTCAGGCCTTGGGGACATTC-3′ |
| B-actin-ChIP-RP | Antisense | 5′-GTGCTCAAGTCCCCAAATGC-3′ |
| BCL2-ChIP-FP | Sense | 5′-TGCATCTCATGCCAAGGG-3′ |
| BCL2-ChIP-RP | Antisense | 5′-CCCCAGAGAAAGAAGAGGAGTT-3′ |
| BCL2L1-ChIP-FP | Sense | 5′-CTTTAGGGTTTCGGACGCCT-3′ |
| BCL2L1-ChIP-RP | Antisense | 5′-TGGGAGCCAGGAGTACTCTC-3′ |
Chromatin Immunoprecipitation (ChIP)
The Magna ChIPTM A kit (Millipore) was used as described by the manufacturer. Briefly, cellular proteins and DNA were cross-linked with 1% formaldehyde at room temperature for 10 min, and glycine was added at a final concentration of 0.125 m to neutralize formaldehyde. Cells were washed three times with 10 ml of ice-cold PBS containing a protease inhibitor mixture and collected by centrifugation. Cells were then resuspended in SDS lysis buffer, incubated at 4 °C for 10 min, and sonicated using a Q800R sonicator (Qsonica). Total sonication time was 30 min with a sonication pulse rate of 30 s on and 30 s off. Supernatants were recovered by centrifugation at 12,000 rpm in an Eppendorf microfuge for 10 min at 4 °C before being diluted 10 fold with ChIP dilution buffer. Immunoprecipitation was performed overnight at 4 °C with anti-NFκB p65 (RelA) mouse monoclonal antibody (Millipore) and Magnetic protein A beads. Antibody-bound protein/DNA complexes were washed, eluted, and treated with proteinase K to digest proteins. DNA was then purified using QIAquick PCR purification Kit (Qiagen). An aliquot of chromatin that was not incubated with an antibody was used as the input control sample.
Immunoprecipitated and input DNA were analyzed by real-time PCR using the ABI SYBR Green Master Mix and an ABI 7500 real time PCR system. The specific primers used are listed in Table 4. All values were calculated relative to input DNA level.
Cell Growth Curves
Cells were placed in triplicate in six-well dishes at 1 × 105 cell per well. For growth curves, the cells were counted using an ADAM-MC automated cell counter (Digital Bio) at different time points as described.
Cell Proliferation Assay and Cell Cycle Analysis
Nucleotide incorporation was detected using a Click-iT EdU flow cytometry assay kit (Invitrogen) according to the manufacturer's instructions. Cell cycle distribution was monitored by analysis of propidium iodide-stained cells on a MACSQuant analyzer (Miltenyi Biotech).
Statistical Analysis
Data are presented as mean ± S.D. and were analyzed by Student's t test using GraphPad Prism (version 5, GraphPad Software, Inc.). Statistical significance was defined with the following: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
The Lymphoma and Myeloma-associated K171E and K171T IKKβ Variants Exhibit Enhanced Activation of NFκB
A number of somatic variants of the IKBKB gene and of other genes in the NFκB pathway have been identified in a range of tumors. Although K171E IKKβ was described in 6/117 marginal zone lymphomas (11) and in one multiple myeloma sample (13), K171T IKKβ was identified in 2/117 marginal zone lymphomas (11). Mutations involving other IKKβ residues have also been identified in a number of other non-lymphoid tumors reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) database, which stores and displays a large compendium of somatic mutations in different genes in a variety of cancers. The A360S, D639N, G667C, and N677D variants of IKKβ were observed respectively in breast, ovarian, lung, and stomach cancers. Wild type and mutant forms of IKKβ were transfected into HeLa cells, and luciferase assays were performed using an NFκB-driven luciferase reporter construct. As seen in Fig. 1A, the K171E and K171T IKKβ variants exhibit a markedly increased ability to activate NFκB as compared with wild type IKKβ. The non-lymphoid tumor associated variants did not activate NFκB above wild type levels, suggesting that they represent passenger mutations. The kinase activity of the K171E and K171T IKKβ variants were considerably increased above wild type levels as determined by radioactive and non-radioactive kinase assays (Fig. 1B and data not shown), and these mutant kinases induced more nuclear p65 accumulation in transfected HeLa cells as seen in Fig. 1, C and D.
Modeling Predicts the Mechanisms That Contribute to the Enhanced Activity of K171E and K171T IKKβ Proteins
The activation of IKKβ requires phosphorylation on serine 181 (and to a lesser extent on serine 177) by an upstream activating kinase such as TAK1 (31). Comparison of the inactive (Figs. 2, A and B) and active (Figs. 2, C and D) structures of this kinase based on the recently published structure of human IKKβ (27) shows striking differences between the two states. In the active, phosphorylated IKKβ, the negatively charged phosphate moiety on serine 181 makes electrostatic interactions with cationic pocket residues Arg-57, Arg-144, and Lys-171, located, respectively, in the C-terminal helix, catalytic loop, and activation loop (Fig. 2, C and D). In the absence of activation loop phosphorylation, the positive charge on these residues induces repulsion between these three critical regions, preventing the kinase domain from adopting a catalytically competent conformation. The interaction between phosphoserine 181 (Ser(P)-181) and both Lys-171 and Arg-144 neutralizes the repulsion between the activation and catalytic loops, facilitating the formation of a β6-β9 β-sheet structure. Formation of this β-sheet is a hallmark of kinase activation and has been proposed to set off a network of hydrogen bonds that result in the proper positioning of residues involved in ATP binding as well as phosphoryl transfer (32). The interaction between phosphoserine 181 and Arg-57 is suggested to promote a closer interaction of the N-terminal lobe of the kinase with the C-terminal lobe, thus facilitating proper positioning of the γ-phosphate of kinase bound ATP for catalytic transfer. When Lys-171 is replaced by a glutamate, it can be predicted that the Glu-171 residue forms an electrostatic interaction with Arg-57 and a salt bridge with Arg-144 (Fig. 3A), thus mimicking the effect of a phosphorylation event on serine 181, bridging the cationic pocket residues. Such a kinase should already be maximally active and constitutively “on” even in the absence of any ligand or activating upstream kinase. However, the polar yet uncharged nature of a threonine in position 171 would only allow this Thr-171 residue to engage in a hydrogen bond with Arg-144, while making it incapable of forming salt bridges or electrostatic interactions (Fig. 3B).
FIGURE 2.

Models of the inactive and active conformation of IKKβ kinase domain. A, the inactive conformation of the IKKβ kinase domain (PDB 4KIK) with ATP from the active PKA structure (PDB 1ATP) superimposed onto its active site. Important components of the kinase active site such as the αC-helix, activation loop, catalytic loop, and p + 1 loop are labeled. Residues that make up part of the cationic pocket are labeled and shown as sticks. In the absence of phosphorylation, the activation loop is disordered, and its approximate location is shown as a magenta dotted line. B, an enlarged, side view of the disordered activation loop in inactive, unphosphorylated IKKβ. C, the active conformation of the IKKβ kinase domain (PDB 4KIK) represented as described in A. The activation loop is ordered in the Ser(P)-181-phosphorylated form except for three residues (residues 174–176), which are missing due to disorder. A network of electrostatic interactions (dashed black lines) between the phosphate moiety of Ser(P)-181 and cationic pocket residues Arg-57, Arg-144, and Lys-171 stabilizes the active conformation of the activation loop. D, an enlarged view of the interactions between phosphoserine 181 (pS181) and cationic pocket residues in active, phosphorylated IKKβ. Formation of a beta sheet between beta strands β6 and β9 is observed. C-lobe, C-terminal lobe; N-lobe, N-terminal lobe.
FIGURE 3.
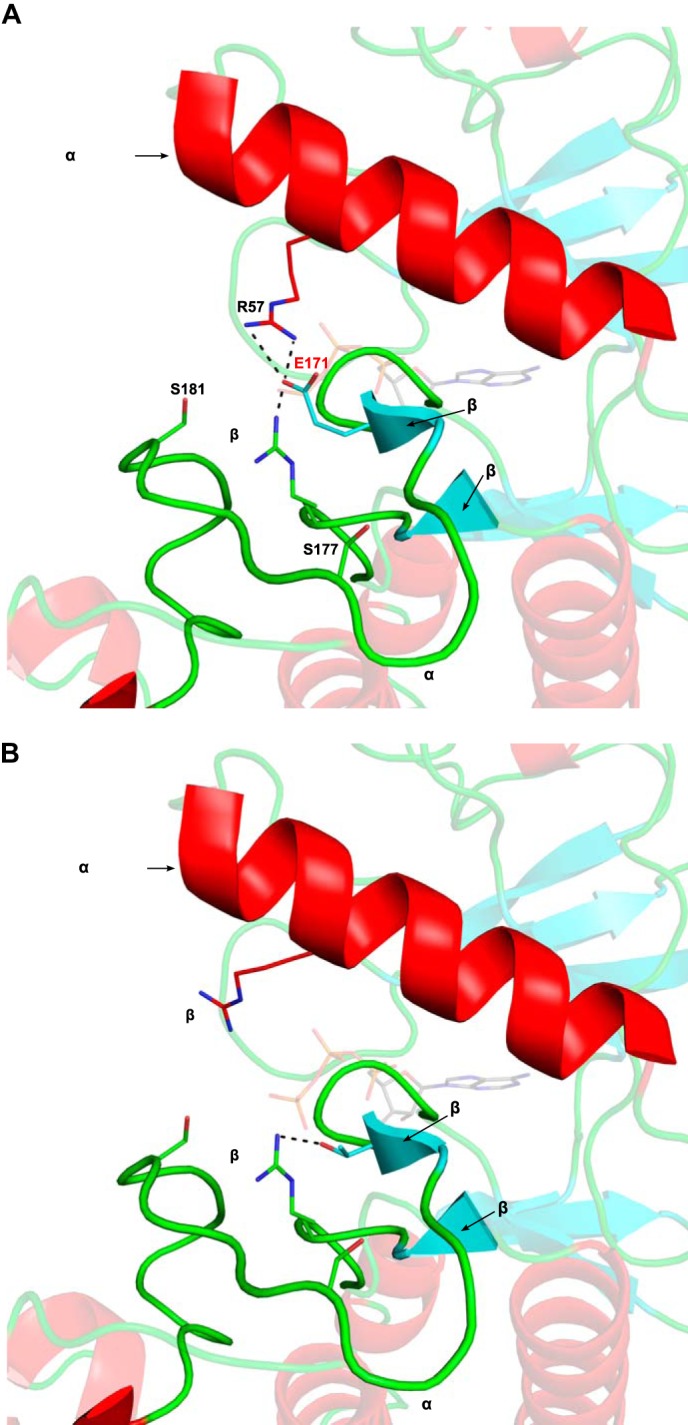
Predicted structures of K171E IKKβ and K171T IKKβ. A, the kinase domain of K171E IKKβ modeled on the recently published structure of human IKKβ (PDB 4KIK). ATP from the active PKA structure (PDB 1ATP) was superimposed onto its active site. Important components of the kinase active site such as the αC-helix and activation loop are labeled. Residues that make up part of the cationic pocket are labeled and shown as sticks. Glu-171 is labeled in red. The activation loop is ordered, and Glu-171 is able to engage in the same electrostatic interactions that the phosphate on Ser(P)-181 makes in the active conformation. The interactions between Glu-171 and cationic pocket residues Arg-57 and Arg-144 IKKβ are labeled. Formation of a β-sheet between β-strands β6 and β9 is observed. B, the kinase domain of K171T IKKβ modeled on the recently published IKKβ structure (PDB 4KIK) and represented as described in A. The uncharged, polar side chain of Thr-171 is able to make a hydrogen bond to Arg-144 and induce formation of a beta sheet between β-strands β6 and β9, yet it is too far from Arg-57 in the αC helix to interact with it.
K171E and K171T IKKβ Are Constitutively Active in an Activation Loop Phosphorylation-independent Manner
Based on the predictive modeling, K171E and Thr mutations should be active independent of activation loop phosphorylation. Mutation of both activation loop serines to alanines (SSAA) results in a loss of NFκB activating potential and a specific loss of IKKβ kinase activity (Fig. 4, A and B) as reported previously (33). The K171E and Thr mutations were introduced into the SSAA background. Luciferase and kinase assays reveal that both the IKKβ K171E and the K171T mutant proteins are constitutively active on the SSAA background, indicating that they are both active in a ligand-independent manner (Fig. 4, A and B).
FIGURE 4.

Kinase activity of K171E and K171T IKKβ is independent of activation loop phosphorylation. A, luciferase activity of NFκB reporter genes in HeLa cells co-transfected with empty vector (Mock), WT IKKβ, SSAA IKKβ, and K171T/K171E IKKβ in the SSAA background are represented as means derived from three individual experiments. Error bars represent ± S.D. ***, p < 0.001. B and C, kinase activity of mock, WT IKKβ, K171E/K171T IKKβ, SSAA IKKβ, and K171E/K171T IKKβ in the SSAA background were determined using GST-IκBα as a specific substrate. IKKβ, phosphorylated GST-IκBα, unphosphorylated GST-IκBα, IκBα, and phosphorylated IκBα bands are indicated in B. The protein levels of IκBα and phosphorylated IκBα were quantified in C. This Western blot is representative of three individual experiments, and the error bars represent ± S.D. *, p < 0.01; **, p < 0.005. D, luciferase activity of NFκB reporter genes in HeLa cells co-transfected with WT IKKβ, K171E IKKβ, and K171E IKKβ in the R57A, R144A, or R57A/R144A backgrounds were determined as described in Fig. 1A. Bars represent the means of three individual experiments, and the error bars represent ± S.D. ***, p < 0.001. E, kinase activity of WT IKKβ, K171E IKKβ, and K171E mutants in R57A, R144A, or R57A/R144A background were determined as described in Fig. 1B. This Western blot (WB) is representative of three individual experiments.
The predictive modeling in Fig. 3 suggested that the activity of K171E IKKβ depends on the interactions of Glu-171 with the two remaining residues in the cationic pocket, Arg-57 and Arg-144. Mutagenesis of individual arginine residues to alanines resulted in compromised K171E IKKβ activity, and mutagenesis of both Arg-57 and Arg-144 completely abrogated K171E IKKβ activity (Fig. 4, C–E).
Responsiveness to TNF Suggests Maximal Activation of K171E IKKβ but Not of K171T IKKβ
Thr-171 would induce formation of the β6-β9 sheet but would not be able to provide a link between the N- and C-terminal lobes because Arg-57 would be too distant from it to engage in a hydrogen bond. Such a kinase would be predicted to be constitutively, but not maximally, active. It would therefore be expected to remain responsive to activation by an upstream kinase, whereas Glu-171 IKKβ would not. We tested this prediction by examining NFκB activation before and after TNF stimulation of transfected cells using a luciferase assay for NFκB-dependent transcription (Fig. 5A) as well as a kinase assay (Fig. 5, B and C). As hypothesized, TNF did not enhance the activation of K171E IKKβ but did contribute to further activation of K171T IKKβ.
FIGURE 5.

K171T IKKβ is responsive to TNF treatment but K171E IKKβ is not. A, NFκB activity of WT IKKβ and K171T IKKβ and K171E IKKβ both without stimulation or following stimulation with TNF-α (20 ng/ml) for 6 h were detected by a Dual-Luciferase assay as described in Fig. 1A. Bars represent the means of four individual experiments, and error bars represent ± S.D. ***, p < 0.001. B and C, kinase activity of WT IKKβ, K171T IKKβ, and K171E IKKβ with or without TNF-α treatment for 15 min was detected by an in vitro kinase assay as described in Fig. 1B. This Western blot (WB) is representative of four individual experiments. N.S., not significant.
Overexpressed IKKβ can form higher order multimers and may be activated by trans-autophosphorylation even in the absence of an upstream kinase (34). We examined the phosphorylation of this kinase in transfected HeLa cells using phospho-specific antibodies. Both K171T and K171E IKKβ exhibited enhanced phosphorylation on residues Ser-177 and Ser-181 (data not shown).
TALEN-based Mutagenesis Demonstrates Constitutive Activation of NFκB in a K171E IKBKB Knock-in B Cell Line
Most of our understanding of the effects of mutations in cancer has depended on the study of biochemical alterations observed following the overexpression of mutant cDNAs in transfected cells. Ideally, non-overexpression-based studies in lymphoid lines should be undertaken to distinguish potential driver mutations of lymphomas from passengers. TALEN-based mutagenesis offers the possibility of generating heterozygous or homozygous knock-ins in cell lines, although such studies have so far never been reported in a cancer context. To choose lymphoma cell lines with less active NFκB signaling, we assayed phosphorylation of p65 in six lymphoma cell lines and demonstrated that BJAB has relative low phosphorylation of p65 (Fig. 6, A and B). We reasoned that cell lines in which existing mutations already generate constitutively active NFκB would be unsuitable for studies of this type. We used TALENs to create knock-in mutations that are both heterozygous and homozygous for K171E IKKβ into human BJAB cells (Fig. 6, C and D). As seen in Fig. 6, E–G, K171E IKKβ in both heterozygous and homozygous forms of BJAB cells resulted in constitutive activation of NFκB as analyzed by the phosphorylation of p65 and by a reporter-based assay for transcriptional activity.
FIGURE 6.

Heterozygous and homozygous K171E IKBKB variants created by TALEN mutagenesis constitutively activate NFκB signaling. A, the protein levels of phospho-p65 (Ser-536), p65, and β-actin were measured using indicated antibody in HeLa, three Burkitt's lymphoma cell lines BJAB, Ramos, and Raji, a GCB-DLBCL cell line OCI-Ly17, and a ABC-DLBCL cell line NU-DUL-1. B, the ratio of phosphorylated p65 to p65:actin was quantified among six cancer cell lines. The bars represent the means ± S.D. of three independent experiments. C, a schematic overview depicting the targeting strategy for IKBKB K171E variant generation using TALEN mutagenesis. D, illustration of sequencing results. Homozygous and heterozygous K171E IKBKB variants introduced into the BJAB B cell line by TALEN-based mutagenesis were detected by Sanger sequencing. E and F, Western blot analysis showed phospho-p65 (Ser-536), p65, and β-actin protein expression in WT BJAB B cells, a homozygous K171E IKBKB (K171E/K171E) knock-in BJAB cell line, a heterozygous K171E IKBKB (K171E/WT) knock-in BJAB B cell line in C. Protein levels were also quantified in terms of the ratio of P-p65 (Ser-536)/(p65:β-actin) in D based on three separate experiments. Error bars represent ± S.D. *, p < 0.01. G, a firefly luciferase NFκB reporter construct and a Renilla luciferase reporter construct were nucleofected into BJAB WT, BJAB K171E/K171E, and BJAB K171E/WT cells for 16 h. Luciferase firefly activity was determined and normalized for transfection efficiency on the basis of Renilla activity. Bars represent the means of three individual experiments, and the error bars represent ± S.D. ***, p < 0.001.
K171E IKBKB Knock-in BJAB Cells Are More Resistant to Apoptosis
Aberrantly active NFκB signals can contribute to tumorigenesis by regulating genes that promote the growth and survival of cancer cells. Examination of the cell cycle and of proliferation revealed no differences between wild type and mutant IKKβ expressing BJAB cells (data not shown). To investigate whether K171E and K171T IKKβ affect cell apoptosis, wild type, heterozygous K171E/WT IKKβ, and homozygous K171E/K171E IKKβ harboring BJAB cells were exposed to staurosporine, a non-selective protein kinase inhibitor that can induce apoptosis. A significant inhibition of apoptosis was noted in knock-in cell lines expressing K171E IKKβ after 16 h of treatment (Fig. 7, A and B). The mRNA levels of six established anti-apoptotic NFκB target genes, including BCL2, BCL2L1, BCL2A1, CFLAR, BIRC5, and XIAP, were analyzed by quantitative real-time RT-PCR. Interestingly, only the expression of BCL2 was significantly enhanced in knock-in BJAB cells harboring both heterozygous and homozygous IKBKB K171E mutations (Fig. 7C), which indicates that enhanced NF-κB signaling in these may selectively regulate target genes. ChIP studies were then performed to detect the recruitment of p65 to BCL2, BCL2L1, and β-actin promoters. Analysis of these ChIP samples using quantitative PCR only showed occupancy of the BCL2 promoter by p65 in K171E/WT BJAB cells and not the promoters of BCL2L1 or β-actin (Fig. 7D).
FIGURE 7.

Heterozygous and homozygous K171E IKBKB BJAB variants created by TALEN mutagenesis are more resistant to apoptosis. A and B, control (WT) BJAB cells and K171E IKBKB heterozygous (K171E/WT) or homozygous (K171E/K171E) BJAB cells were treated with1 μm staurosporine for 16 h and stained with annexin V and propidium iodide (PI) followed by analysis by flow cytometry. Representative contour plots show the total percent of early (annexin V+PI−) and late (annexin V+PI−) apoptotic cells. The bars represent the means of four individual experiments, and the error bars represent ± S.D. ***, p < 0.001 for control BJAB versus K171E IKBKB heterozygous or homozygous BJAB cells. C, quantitative RT-PCR analysis was performed using primers specific for BCL2, BCL2L1, BCL2A1, CFLAR, BIRC5, XIAP, and β-actin control, using total RNA prepared from WT, K171E/WT, and K171E/K171E BJAB cells. The bars represent the means ± S.D. of three independent experiments. ***, p < 0.001. D, ChIP assays were performed using anti-p65 antibody on nuclear extracts from WT and K171E/WT BJAB cells. Immunoprecipitated DNA was subjected to real-time PCR using primers specific for the indicated promoters. The graph represents the means ± S.D. of three independent experiments. ***, p < 0.001.
DISCUSSION
Tumor-specific somatic mutations of a gene encoding a kinase may often not alter the function of the kinase as noted for the mutations in IKBKB found in a number of cancers. Those are clearly passenger mutations. Lysine 171 of IKKβ is a residue that is located parallel to the phosphorylation site in the activation loop. Examination of the structure of phosphorylated versus non-phosphorylated (27) IKKβ revealed that this residue is part of a cationic pocket, also made up of Arg-57 in the C-terminal helix and Arg-144 in the catalytic loop. The recently resolved structure of human IKKβ suggests that in its active conformation, the activation loop phosphoserine 181 interacts with these cationic pocket residues, including lysine 171, to switch the kinase domain to a catalytically active conformation. The absence of activation loop phosphorylation results in a disordered activation loop, as can be noted in the missing region in the inactive IKKβ structure. It could have been argued that the drastic mutations in this interacting lysine residue might result in a less active kinase, but clearly its association with lymphomagenesis suggested, purely from a theoretical context, that the K171E and K171T IKKβ proteins might represent activated kinases.
Our overexpression studies of K171E IKKβ as well as of K171T IKKβ indicated that these two proteins, found originally in splenic marginal zone lymphomas, are constitutively active in an activation loop phosphorylation-independent manner, essentially even in the absence of an upstream kinase. Why are these kinases constitutively active? Predictive modeling indicates that conversion of lysine 171 of IKKβ into a glutamate would allow this residue to interact with the other two cationic pocket residues and thus contribute to kinase activation. Conversion of this residue into a threonine would not support an electrostatic interaction with Arg-57 but would permit hydrogen bond formation with Arg-144, inducing formation of the β6-β9 sheet, thus allowing constitutive activation of this kinase as well. The Thr-171 change, however, was predicted to result in submaximal activation, thereby being permissive for more complete activation if serine 181 were to be phosphorylated following cellular activation. These predictions were all borne out in experimental biochemical studies.
One approach that has been widely used to get around potential artifacts of overexpression is to generate knock-in mice. It is already clear that dramatically increased numbers of marginal zone B cells in knock-in mice constitutively expressing IKKβ (35) and phospho-mimetic activation loop IKKβ knock-in mice cause lymphomagenesis (14). Although TALEN- or CRISPR-based mutagenesis has not yet been reported in genetic studies of cancer, we establish here that this is a powerful approach that can be employed in the study of tumorigenesis. Nevertheless, knock-in mice remain the gold standard.
Our data suggest that in BJAB B cells one of the major consequences of constitutive NFκB activation is the induction of Bcl-2 expression. More broad analyses using RNA-seq or microarray technology may reveal a more detailed picture of how constitutively active IKKβ drives lymphomagenesis.
This work was supported, in whole or in part, by Grants AI 064930 and AI 076505 from the National Institutes of Health (to S. P.) and National Institutes of Health Director's Pioneer Award DP1 GM105378 (to J. K. J.). J. K. J. has a financial interest in Editas Medicine and Transposagen Biopharmaceuticals. J. K. J.'s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
- NFκB
- nuclear factor κB
- IKKβ
- IκB kinase β
- ΙκΒα
- inhibitor of NFκB α
- TALEN
- transcription activator-like effector nuclease
- MZ
- marginal zone
- PDB
- Protein Data Bank.
REFERENCES
- 1. Cariappa A., Liou H. C., Horwitz B. H., Pillai S. (2000) Nuclear factor κ B is required for the development of marginal zone B lymphocytes. J. Exp. Med. 192, 1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xie P., Stunz L. L., Larison K. D., Yang B., Bishop G. A. (2007) Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27, 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pappu B. P., Lin X. (2006) Potential role of CARMA1 in CD40-induced splenic B cell proliferation and marginal zone B cell maturation. Eur. J. Immunol. 36, 3033–3043 [DOI] [PubMed] [Google Scholar]
- 4. Conze D. B., Zhao Y., Ashwell J. D. (2010) Non-canonical NF-kappaB activation and abnormal B cell accumulation in mice expressing ubiquitin protein ligase-inactive c-IAP2. PLoS biology 8, e1000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sasaki Y., Calado D. P., Derudder E., Zhang B., Shimizu Y., Mackay F., Nishikawa S., Rajewsky K., Schmidt-Supprian M. (2008) NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc. Natl. Acad. Sci. U.S.A. 105, 10883–10888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuroda K., Han H., Tani S., Tanigaki K., Tun T., Furukawa T., Taniguchi Y., Kurooka H., Hamada Y., Toyokuni S., Honjo T. (2003) Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity 18, 301–312 [DOI] [PubMed] [Google Scholar]
- 7. Saito T., Chiba S., Ichikawa M., Kunisato A., Asai T., Shimizu K., Yamaguchi T., Yamamoto G., Seo S., Kumano K., Nakagami-Yamaguchi E., Hamada Y., Aizawa S., Hirai H. (2003) Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 18, 675–685 [DOI] [PubMed] [Google Scholar]
- 8. Santos M. A., Sarmento L. M., Rebelo M., Doce A. A., Maillard I., Dumortier A., Neves H., Radtke F., Pear W. S., Parreira L., Demengeot J. (2007) Notch1 engagement by Delta-like-1 promotes differentiation of B lymphocytes to antibody-secreting cells. Proc. Natl. Acad. Sci. U.S.A. 104, 15454–15459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moran S. T., Cariappa A., Liu H., Muir B., Sgroi D., Boboila C., Pillai S. (2007) Synergism between NF-κ B1/p50 and Notch2 during the development of marginal zone B lymphocytes. J. Immunol. 179, 195–200 [DOI] [PubMed] [Google Scholar]
- 10. Pillai S., Cariappa A. (2009) The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 9, 767–777 [DOI] [PubMed] [Google Scholar]
- 11. Rossi D., Trifonov V., Fangazio M., Bruscaggin A., Rasi S., Spina V., Monti S., Vaisitti T., Arruga F., Famà R., Ciardullo C., Greco M., Cresta S., Piranda D., Holmes A., Fabbri G., Messina M., Rinaldi A., Wang J., Agostinelli C., Piccaluga P. P., Lucioni M., Tabbò F., Serra R., Franceschetti S., Deambrogi C., Daniele G., Gattei V., Marasca R., Facchetti F., Arcaini L., Inghirami G., Bertoni F., Pileri S. A., Deaglio S., Foà R., Dalla-Favera R., Pasqualucci L., Rabadan R., Gaidano G. (2012) The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 209, 1537–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rossi D., Deaglio S., Dominguez-Sola D., Rasi S., Vaisitti T., Agostinelli C., Spina V., Bruscaggin A., Monti S., Cerri M., Cresta S., Fangazio M., Arcaini L., Lucioni M., Marasca R., Thieblemont C., Capello D., Facchetti F., Kwee I., Pileri S. A., Foà R., Bertoni F., Dalla-Favera R., Pasqualucci L., Gaidano G. (2011) Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood 118, 4930–4934 [DOI] [PubMed] [Google Scholar]
- 13. Chapman M. A., Lawrence M. S., Keats J. J., Cibulskis K., Sougnez C., Schinzel A. C., Harview C. L., Brunet J. P., Ahmann G. J., Adli M., Anderson K. C., Ardlie K. G., Auclair D., Baker A., Bergsagel P. L., Bernstein B. E., Drier Y., Fonseca R., Gabriel S. B., Hofmeister C. C., Jagannath S., Jakubowiak A. J., Krishnan A., Levy J., Liefeld T., Lonial S., Mahan S., Mfuko B., Monti S., Perkins L. M., Onofrio R., Pugh T. J., Rajkumar S. V., Ramos A. H., Siegel D. S., Sivachenko A., Stewart A. K., Trudel S., Vij R., Voet D., Winckler W., Zimmerman T., Carpten J., Trent J., Hahn W. C., Garraway L. A., Meyerson M., Lander E. S., Getz G., Golub T. R. (2011) Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calado D. P., Zhang B., Srinivasan L., Sasaki Y., Seagal J., Unitt C., Rodig S., Kutok J., Tarakhovsky A., Schmidt-Supprian M., Rajewsky K. (2010) Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 18, 580–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ngo V. N., Young R. M., Schmitz R., Jhavar S., Xiao W., Lim K. H., Kohlhammer H., Xu W., Yang Y., Zhao H., Shaffer A. L., Romesser P., Wright G., Powell J., Rosenwald A., Muller-Hermelink H. K., Ott G., Gascoyne R. D., Connors J. M., Rimsza L. M., Campo E., Jaffe E. S., Delabie J., Smeland E. B., Fisher R. I., Braziel R. M., Tubbs R. R., Cook J. R., Weisenburger D. D., Chan W. C., Staudt L. M. (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis R. E., Ngo V. N., Lenz G., Tolar P., Young R. M., Romesser P. B., Kohlhammer H., Lamy L., Zhao H., Yang Y., Xu W., Shaffer A. L., Wright G., Xiao W., Powell J., Jiang J. K., Thomas C. J., Rosenwald A., Ott G., Muller-Hermelink H. K., Gascoyne R. D., Connors J. M., Johnson N. A., Rimsza L. M., Campo E., Jaffe E. S., Wilson W. H., Delabie J., Smeland E. B., Fisher R. I., Braziel R. M., Tubbs R. R., Cook J. R., Weisenburger D. D., Chan W. C., Pierce S. K., Staudt L. M. (2010) Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lenz G., Davis R. E., Ngo V. N., Lam L., George T. C., Wright G. W., Dave S. S., Zhao H., Xu W., Rosenwald A., Ott G., Muller-Hermelink H. K., Gascoyne R. D., Connors J. M., Rimsza L. M., Campo E., Jaffe E. S., Delabie J., Smeland E. B., Fisher R. I., Chan W. C., Staudt L. M. (2008) Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679 [DOI] [PubMed] [Google Scholar]
- 18. Novak U., Rinaldi A., Kwee I., Nandula S. V., Rancoita P. M., Compagno M., Cerri M., Rossi D., Murty V. V., Zucca E., Gaidano G., Dalla-Favera R., Pasqualucci L., Bhagat G., Bertoni F. (2009) The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 113, 4918–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vogelstein B., Papadopoulos N., Velculescu V. E., Zhou S., Diaz L. A., Jr., Kinzler K. W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumar P., Henikoff S., Ng P. C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 22. Ding Q., Lee Y. K., Schaefer E. A., Peters D. T., Veres A., Kim K., Kuperwasser N., Motola D. L., Meissner T. B., Hendriks W. T., Trevisan M., Gupta R. M., Moisan A., Banks E., Friesen M., Schinzel R. T., Xia F., Tang A., Xia Y., Figueroa E., Wann A., Ahfeldt T., Daheron L., Zhang F., Rubin L. L., Peng L. F., Chung R. T., Musunuru K., Cowan C. A. (2013) A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 12, 238–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding Q., Regan S. N., Xia Y., Oostrom L. A., Cowan C. A., Musunuru K. (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henderson B. R. (2000) Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat. Cell Biol. 2, 653–660 [DOI] [PubMed] [Google Scholar]
- 26. Gupta M., Korol A., West-Mays J. A. (2013) Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor β-induced epithelial to mesenchymal transition of lens epithelial cells. Mol. Vis. 19, 1017–1028 [PMC free article] [PubMed] [Google Scholar]
- 27. Liu S., Misquitta Y. R., Olland A., Johnson M. A., Kelleher K. S., Kriz R., Lin L. L., Stahl M., Mosyak L. (2013) Crystal structure of a human IkappaB kinase β asymmetric dimer. J. Biol. Chem. 288, 22758–22767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 29. Reyon D., Tsai S. Q., Khayter C., Foden J. A., Sander J. D., Joung J. K. (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 30, 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reyon D., Maeder M. L., Khayter C., Tsai S. Q., Foley J. E., Sander J. D., Joung J. K. (2013) Engineering customized TALE nucleases (TALENs) and TALE transcription factors by fast ligation-based automatable solid-phase high-throughput (FLASH) assembly. Curr. Protoc. Mol. Biol. 10.1002/0471142727.mb1216s103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 32. Nolen B., Taylor S., Ghosh G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 33. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., Rao A. (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278, 860–866 [DOI] [PubMed] [Google Scholar]
- 34. Polley S., Huang D. B., Hauenstein A. V., Fusco A. J., Zhong X., Vu D., Schröfelbauer B., Kim Y., Hoffmann A., Verma I. M., Ghosh G., Huxford T. (2013) A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 11, e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sasaki Y., Derudder E., Hobeika E., Pelanda R., Reth M., Rajewsky K., Schmidt-Supprian M. (2006) Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 24, 729–739 [DOI] [PubMed] [Google Scholar]