

Background: Protein-disulfide isomerase (PDI) has previously been identified to bind bisphenol A (BPA), an endocrine disrupter.
Results: BPA inhibited Ero1α-PDI-mediated disulfide bond formation.
Conclusion: BPA significantly inhibited the Ero1α and PDI oxidative cycle, probably through closure of the substrate- and Ero1α-binding pocket in the PDI b′ domain.
Significance: BPA may have inhibitory effects on oxidative folding of secretory and membrane proteins.
Keywords: Endoplasmic Reticulum (ER), Enzyme Inhibitor, Protein Conformation, Protein Folding, Protein Isomerase
Abstract
Bisphenol A (BPA) is an endocrine disruptor that may have adverse effects on human health. We recently isolated protein-disulfide isomerase (PDI) as a BPA-binding protein from rat brain homogenates and found that BPA markedly inhibited PDI activity. To elucidate mechanisms of this inhibition, detailed structural, biophysical, and functional analyses of PDI were performed in the presence of BPA. BPA binding to PDI induced significant rearrangement of the N-terminal thioredoxin domain of PDI, resulting in more compact overall structure. This conformational change led to closure of the substrate-binding pocket in b′ domain, preventing PDI from binding to unfolded proteins. The b′ domain also plays an essential role in the interplay between PDI and ER oxidoreduclin 1α (Ero1α), a flavoenzyme responsible for reoxidation of PDI. We show that BPA inhibited Ero1α-catalyzed PDI oxidation presumably by inhibiting the interaction between the b′ domain of PDI and Ero1α; the phenol groups of BPA probably compete with a highly conserved tryptophan residue, located in the protruding β-hairpin of Ero1α, for binding to PDI. Consistently, BPA slowed down the reoxidation of PDI and caused the reduction of PDI in HeLa cells, indicating that BPA has a great impact on the redox homeostasis of PDI within cells. However, BPA had no effect on the interaction between PDI and peroxiredoxin-4 (Prx4), another PDI family oxidase, suggesting that the interaction between Prx4 and PDI is different from that of Ero1α and PDI. These results indicate that BPA, a widely distributed and potentially harmful chemical, inhibits Ero1-PDI-mediated disulfide bond formation.
Introduction
Bisphenol A (BPA4; 2,2′-bis(4-hydroxyphenyl) propane) is an estrogenic endocrine-disrupting chemical widely used in the manufacture of plastics (1). BPA competes with several steroid ligands for binding to steroid receptors due to its structural similarity to steroids, and hence it inhibits or sometimes stimulates physiological functions, such as tumor cell proliferation (2, 3). BPA is a metabolically recalcitrant organic compound (Fig. 1A) and accumulates in tissues once taken up by living organisms. In addition to its antagonist/agonist effects on steroid hormone receptors, BPA induces a number of detrimental effects on the central nervous system, including induction of abnormal behavior in mice and morphogenic malformation in Xenopus laevis (4). Several lines of evidence demonstrate that BPA exposure causes adverse effects on human health (5).
FIGURE 1.

Inhibition of Ero1α by BPA. A, chemical structure of BPA. B, domain organization of PDIfull-length. The four thioredoxin domains, which are designated a, b, b′, and a′, near the N terminus and the C-terminal α-helical domain c′ are shown in boxes. Active site sequences (CGHC) and tryptophan residues (W) are also indicated. C, left, oxidative activity of Ero1α (4 μm) toward PDI (10 μm) in the presence of various concentrations of BPA was assessed by measuring the decrease in A340. This is due to NADPH consumption mediated by glutathione reductase in a buffer containing GSH and PDI. Right, plot of percentage inhibition as a function of BPA concentration determined from the initial velocity of the decrease in A340. D, detection of BPA binding to Ero1α and PDI by SPR. E, chaperone activity of PDI in the presence of BPA (20 or 40 μm). The chaperone activity of PDI was analyzed using luciferase as a model substrate.
Recently, we isolated a BPA-binding protein from rat brain extracts, which was identified as the canonical protein-disulfide isomerase (PDI) (6). PDI, an oxidoreductase abundantly present in the endoplasmic reticulum (ER), plays a central role in the folding of secretory proteins by catalyzing disulfide bond formation and preventing aggregation of unfolded or misfolded proteins (7, 8). PDI consists of four thioredoxin domains, termed a, b, b′, and a′, from the N terminus and an additional ill defined α-helical c domain located at the C terminus (9, 10) (Fig. 1B). The a and a′ domains contain a catalytic active site composed of a CGHC sequence, whereas the central domains, b and b′, possess hydrophobic surfaces responsible for the binding of incompletely folded protein substrates (11–15). Additionally, PDI contains an x-linker region between the b′ and a′ domains, which may function as a gate for access to the substrate-binding site of the b′ domain (10, 16, 17).
During disulfide bond formation (18, 19), Ero1 enzymes generate a protein disulfide bond de novo consuming an oxygen molecule as an electron acceptor and transfer it to PDI, resulting in oxidation of PDI (20, 21). To react specifically with PDI, a protruding β-hairpin loop of Ero1α binds a hydrophobic pocket in the PDI b′ domain (22, 23). Because Ero1 family enzymes yield hydrogen peroxide, a source of reactive oxygen species, as a result of the generation of protein disulfide bonds, their PDI oxidation activity must be tightly regulated in response to the redox environment of the ER. Regulatory mechanisms for the Ero1α-PDI oxidative cycle have been studied in detail and are outlined in several review articles (19–21, 24).
We previously found that BPA binds the PDI b′ domain and thereby inhibits the disulfide bond formation activity of PDI (25, 26). Because both Ero1α and BPA bind the PDI b′ domain, we hypothesized that BPA may interfere with Ero1α-mediated oxidation of PDI. To gain further insights into the structural and functional effects of BPA on PDI, detailed biochemical and biophysical analyses of PDI were performed in the presence of BPA. Consequently, BPA binding significantly inhibited oxidative folding of bovine pancreas trypsin inhibitor (BPTI) catalyzed by Ero1α and PDI. BPA induced significant spatial rearrangement of the a and b′ domains of PDI, leading to a more compact overall structure. Thus, BPA probably inhibits the Ero1-PDI disulfide bond formation pathway of the ER, an essential organelle engaged in the synthesis of secretory and membrane proteins.
EXPERIMENTAL PROCEDURES
Materials
BPA was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). All other chemicals and solvents used were of the highest grade available.
Expression and Purification of PDIfull-length, PDI(120–353), PDI(354–509), ERp46, Prx4, and Ero1α
Rat PDI cDNA was cloned as described previously (6). Plasmids used for expression of PDIfull-length, PDI(120–353), and PDI(354–509) fragments were constructed as described previously (25). Plasmids for ERp46, Prx4, and Ero1α have been described previously (23, 27, 28). Each construct was overexpressed in Escherichia coli. Cells were harvested and homogenized. Recombinant proteins were then purified by a combination of several types of chromatography. The final preparation was subjected to SDS-PAGE, and the purity was confirmed to be over 90% for all recombinant proteins (data not shown) (29).
Circular Dichroism (CD) Spectroscopy
CD measurements were performed on a JASCO J-600 and J-720 spectropolarimeter (JASCO Corp., Tokyo, Japan). The CD spectra of PDI(120–353) and PDI(354–509) were measured in 20 mm phosphate buffer (pH 7.4) at room temperature, using a quartz cell with a 1.0-mm path length for far-UV (260–200 nm) and a 10-mm path-length for near-UV spectra (340–250 nm). Protein concentrations were as follows: for PDIfull-length, 0.95 mg/ml in near-UV spectra and 0.24 mg/ml in far-UV spectra; for PDI(120–353), 0.69 mg/ml in near-UV spectra and 0.31 mg/ml in far-UV spectra; for PDI(354–509), 0.93 mg/ml in near-UV spectra and 0.31 mg/ml in far-UV spectra. All samples were centrifuged at 15,000 × g for 15 min, and then supernatants were used for measurements.
Differential Scanning Calorimetry (DSC)
DSC measurements were performed using a Microcal VP-capillary DSC platform (GE Healthcare) at a scan rate of 60 °C h−1, 120 °C h−1, and 200 °C h−1 for PDI(120–353) and 200 °C h−1 for PDI(354–509). Protein solutions were dialyzed against 20 mm phosphate buffer (pH 7.0) and thereafter centrifuged at 15,000 × g for 15 min. Supernatants were filtered through a 0.22-μm pore size membrane before measurement. Protein concentration of PDI(120–353) was 0.32 mg/ml for 200 °C h−1 and 0.97 mg/ml for 60 and 120 °C h−1. Protein concentration of PDI(354–509) was 0.32 mg/ml. Reversibility of heat denaturation for PDI(120–353) was performed at a scan rate of 60 °C h−1 in 20 mm phosphate buffer (pH 7.0) with a protein concentration of 0.72 mg/ml. Thermograms were integrated using ORIGIN software (Microcal Software, Northampton, MA) to estimate binding constants.
Dynamic Light Scattering (DLS)
The hydrodynamic sizes of PDI(120–353) and PDI(354–509) in solution were measured with a Zetasizer NanoZS (Malvern Instruments, Malvern, UK), based on the principle of dynamic light scattering (30). Briefly, hydrodynamic diameter was converted by the Stokes-Einstein equation. Polydispersity, or heterogeneity of Brownian motion, was calculated from the S.D. value of the distribution at each BPA concentration. All samples were centrifuged at 15,000 × g for 10 min, and the supernatants were filtered through a 0.22-μm pore size membrane before measurement. The protein concentration was 0.89 mg/ml for PDI(120–353) and 1.18 mg/ml for PDI(354–509).
Homology Modeling of PDI(120–353) and PDI(354–509)
To predict the structure of PDI(120–353) and PDI(354–509), a protein homology/analogy recognition engine (Phyre) was used (31).
Small Angle X-ray Scattering (SAXS)
SAXS data were collected on the BL45XU beamline at the RIKEN SPring-8 Center (Hyogo, Japan) (32). The x-ray wavelength was 1.0 Å, and a PILATUS 300K-W system (DECTRIS, Baden, Switzerland) was employed as a detector with a camera distance of ∼2,000 mm. Scattering intensity was recorded at 293.2 K with 18 continuous images, each with an exposure time of 10 s. Images were checked for radiation damage, and data thus selected were averaged. All protein solutions were prepared in 20 mm phosphate buffer (pH 7.0), and SAXS profiles for PDI(120–353) in the presence or absence of BPA (with a 1:1 molar ratio of PDI(120–353)/BPA) were collected in a concentration range of 1.5–4.0 mg/ml. SAXS profiles for bovine serum albumin (Mr of 66,400; Sigma-Aldrich) were also collected as a reference for determining the relative molecular weight of PDI(120–353). All samples were centrifuged at 15,000 × g for 15 min, and supernatants were used for measurements.
Small angle scattering profiles were fitted by Guinier approximation with the equation,
where I(0) and Rg are the forward scattering intensity at Q = 0 and the radius of gyration, respectively (33). The I(0) value is proportional to the averaged molecular weight and concentration. The pair distribution function, P(r), was calculated by indirect Fourier transformation using GNOM (34).
NADPH Consumption Assay
Glutathione reductase (1 unit) was incubated with NADPH (200 μm), reduced glutathione (300 μm), human PDI (10 μm), the hyperactive form, including mutations of C104A and C131A, of human Ero1α (4 μm) (27), and various concentrations of BPA (5–2000 μm). The oxidation of NADPH by glutathione reductase led to a decrease in absorbance at 340 nm (A340). A molar extinction coefficient of 6,200 m−1 cm−1 for NADPH was used for the calculations. The reaction was initiated by adding Ero1α. Changes in absorbance at 340 nm were monitored with a Hitachi U-3310 spectrophotometer at 30 °C in 50 mm Tris-HCl, pH 7.5, 0.3 m NaCl. IC50 values were calculated by curve fitting using ImageJ.
BPTI Folding Assay
Reduction and denaturation of BPTI (Takara Bio Inc.) were performed as described previously (27, 35–37). Fully denatured and reduced BPTI (30 μm) was refolded at 30 °C in oxygen-saturated buffer containing 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, 1 μm PDI, a 1 μm concentration of the hyperactive form of Ero1α, and 40 μm BPA. For the Prx4-PDI- or glutathione-PDI-mediated folding of BPTI, the same procedure was used, but the buffer was degassed by flushing N2 gas. Reactions were performed at 30 °C in buffer containing 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, 1 μm PDI, 0.1 μm Prx4, 200 μm H2O2, and 40 μm BPA (27, 28) or buffer containing 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, 1 μm PDI, 2 mm GSH, 0.2 mm GSSG, and 40 μm BPA (38, 39). The resulting sample was analyzed by HPLC using a TSKgel Protein C4-300 column (4.6 × 150 mm; Tosoh Bioscience) at 229 nm, and identities of the peaks obtained were confirmed by MALDI-TOF/MS (Bruker), as described previously (28).
Chaperone Activity of PDI
The chaperone activity of PDI was measured using luciferase as a model substrate (40). To monitor effect of PDI on the thermal aggregation of luciferase, the substrate protein was diluted to a final concentration of 2 μm in buffer containing 40 mm HEPES-KOH, pH 7.5, PDI (4 μm) and BPA (20 or 40 μm) under constant stirring and incubated at 45 °C during the measurement. Thermal aggregation was monitored by measuring light scattering using a Hitachi Spectrofluorometer F2500. The wavelength of light entered and monitored was 350 nm.
Analyzing the Oxidation Status of PDI in HeLa Cells
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Nacalai Tesque) supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. Where indicated, cells were treated with 10 mm dithiothreitol (DTT), 1 mm diamide, or the indicated concentrations of BPA for the indicated times before preparing samples. To analyze the redox state of PDI in cells, cells were washed twice with phosphate-buffered saline (PBS) and treated with ice-cold 10% trichloroacetic acid. After a 20-min incubation on ice, cellular proteins were collected by centrifugation, washed once with ice-cold acetone to remove acid, and dissolved in alkylation buffer (100 mm Tris-HCl (pH 6.8), 2% SDS) containing 5.5 mm PEG-maleimide 2000 (SUNBRIGHT ME-020MA, NOF) and protease inhibitors (10 μg/ml pepstatin A, 1 mm benzamide, and 1 mm phenylmethylsulfonyl fluoride) (41). The samples were then agitated for 20 min and incubated for another 30 min at 37 °C to obtain PEG-maleimide 2000-treated cell lysates. Proteins from the resulting cell lysates were separated by 10% SDS-polyacrylamide gel electrophoresis, blotted onto an Immobilon-P membrane (Millipore), incubated with rabbit α-PDI antiserum (27) and with an appropriate secondary antibody, and detected with ECL Select Western blotting Detection Reagent (GE Healthcare) and LAS-3000 mini (Fujifilm) to visualize the oxidation status of PDI in the cells.
DTT Pulse Experiment
To study the effect of BPA on the reoxidation of PDI in HeLa cells, the culture of HeLa cells was treated with 10 mm DTT for 10 min to reduce PDI (pulse) (42). DTT was then removed by washing the cells twice with fresh medium containing the indicated concentrations of BPA. The cultures of the resulting washed cells were then grown in the fresh medium for 10 min to allow the reoxidation of PDI (chase). The chase was performed in the presence of BPA to study the effect of BPA on the reoxidation of PDI. The cultures were then subjected to alkylation with PEG-maleimide 2000 and subsequent immunoblotting with anti-PDI antiserum.
RESULTS
Inhibition of the Ero1α-PDI Oxidative Cycle by BPA in Vitro
To determine the rate of Ero1α-mediated PDI oxidation, we previously developed an NADPH consumption assay, in which oxidation of NADPH is coupled to Ero1α-mediated catalysis of PDI oxidation through the intermediary actions of glutathione reductase and glutathione (27). In this assay, the rate of Ero1α-mediated PDI oxidation was compared by measuring the initial velocity of the decrease in A340 due to the consumption of NADPH (Fig. 1C, left). BPA markedly inhibited Ero1α-mediated catalysis of PDI oxidation in a dose-dependent manner, with an IC50 of 41 μm (Fig. 1C, right). To examine whether BPA acts on PDI or Ero1α in exerting this inhibitory effect, we analyzed the binding of BPA to PDI or Ero1α by surface plasmon resonance (SPR). We observed that BPA directly associated with PDI but not with Ero1α (Fig. 1D). Hiroi et al. (6) indicated that the affinity of BPA for PDI was 22.6 μm, which is almost comparable with the IC50 value estimated by the PDI oxidation assay (Fig. 1C). It is conceivable that BPA inhibits Ero1α-mediated catalysis of PDI oxidation by binding a site on PDI critical for functional interplay with Ero1α. We next investigated the effect of BPA on the chaperone activity of PDI using luciferase as a model substrate (40). Luciferase aggregated, taking hundreds of seconds at 45.0 °C (Fig. 1E). Although the presence of PDI prevented the luciferase aggregation to a significant extent, the addition of BPA again caused the aggregation in a dose-dependent manner. Thus, BPA has an inhibitory effect on the chaperone activity of PDI.
Inhibitory Effect of BPA on Oxidative Protein Folding Catalyzed by the Ero1α-PDI Pathway
The results shown above suggest that BPA may inhibit oxidative protein folding catalyzed by Ero1α and PDI in combination. To test this possibility, we measured the oxidative folding of BPTI, as a model substrate, in the presence of Ero1α, PDI, saturated levels of oxygen, and BPA. BPTI contains six cysteine residues that participate in three native intramolecular disulfide bonds (Cys5–Cys55, Cys14–Cys38, and Cys30–Cys51). Glutathione-mediated oxidative folding pathways of BPTI have been well characterized by HPLC (Fig. 2A) (43, 44). The species R, species (5–55) and (30–51), des species (des(30–51), des(5–55), and des(14–38)), and N represent no disulfide bond, one disulfide bond, two disulfide bonds, and native disulfide bonds, respectively. As predicted, BPA substantially slowed down the generation of the N species (Fig. 2, B and C). Notably, whereas BPA inhibited both the early and late steps of BPTI folding, the effect was even more pronounced in the conversion of the des species to the N species than in the initial disulfide introduction during the early stage of folding. Previous in depth studies indicate that PDI plays a critical role in the formation of native disulfide bonds within partially structured and kinetically trapped folding intermediates, especially des species, of BPTI during the late steps of folding (7, 28). Likewise, the present glutathione-PDI-mediated BPTI refolding assay demonstrated that BPA inhibited slightly but significantly the conversion of kinetically trapped intermediates to a native state, probably due to the reduced disulfide rearrangement ability of BPA-bound PDI (Fig. 3).
FIGURE 2.

Ero1α-PDI-driven oxidative folding of BPTI in the presence of BPA. A, schematic representation of the oxidative folding pathways of BPTI in the presence of glutathione. R and N represent reduced and native form of BPTI (5–55, 14–38, and 30–51). The disulfide parings of folding intermediates at each step are shown in parentheses, and the missing disulfide is indicated with the prefix “des.” B, HPLC profiles indicating the time course of oxidative folding of BPTI catalyzed by Ero1α-PDI in the absence (left) and presence (right) of BPA. Folding was initiated by the addition of Ero1α and PDI to reduced BPTI (30 μm) in a buffer saturated with molecular oxygen (no thiol agents, such as glutathione, were present). At the indicated time points, the reaction was quenched by HCl and analyzed by HPLC. C, quantitative analysis of each folding species based on the HPLC profiles shown in B. The occupancy of each species was plotted as a function of refolding time.
FIGURE 3.

Glutathione-PDI-mediated oxidative folding of BPTI in the presence of BPA. A, HPLC profiles indicating the time course of oxidative folding of BPTI catalyzed by glutathione-PDI in the absence (left) and presence (right) of BPA. Folding was initiated by the addition of PDI to reduced BPTI (30 μm) in a degassed buffer containing 2 mm GSH and 0.2 mm GSSG (39). At the indicated time points, the reaction was quenched by HCl and analyzed by HPLC. B, quantitative analysis of each folding species based on the HPLC profiles shown in A. The occupancy of each species was plotted as a function of refolding time.
Meanwhile, BPA had no effect on the Prx4-catalyzed PDI oxidation (Fig. 4A). In line with this, the early stage of BPTI folding catalyzed by Prx4-PDI pathway was little affected by the addition of BPA (Fig. 4B). However, the late steps of BPTI folding catalyzed by this pathway were significantly inhibited (Fig. 4C), as was the case with glutathione-PDI-mediated folding (Fig. 3B). We thus infer that BPA compromises the ability of PDI to recognize folding intermediates as well as the functional interplay between PDI and Ero1α.
FIGURE 4.

Oxidative activity of Prx4 toward PDI in the presence of BPA. A, oxidative activity of Prx4 (0.4 μm) toward PDI (10 μm) in the presence of various concentrations of BPA was assessed by measuring the decrease in A340. B, Prx4-PDI-mediated oxidative folding of BPTI. HPLC profiles indicate the time course of oxidative folding of BPTI catalyzed by Prx4-PDI in the absence (left) and presence (right) of BPA (40 μm). Folding was initiated by the addition of Prx4 (0.1 μm) and PDI (1 μm) to reduced BPTI (30 μm) in a buffer containing H2O2 (200 μm). At the indicated time points, reactions were quenched with HCl and analyzed by HPLC. GSH/GSSG redox reagents were not included in this assay. C, quantitative analysis of each folding species based on the HPLC profiles shown in B. The occupancy of each species was plotted as a function of refolding time.
BPA Retards the Reoxidation of PDI in HeLa Cells
Because BPA inhibited the ability of Ero1α to oxidize PDI in vitro, we envisaged that BPA might slow down the reoxidation of PDI in cells. To test this possibility, we first treated the cultures of HeLa cells with 10 mm DTT for 10 min to reduce PDI and washed the cells to remove DTT from the culture (42). We then cultivated the cells in a medium without DTT for 10 min to allow the reoxidation of PDI. This step was performed in the presence of various concentrations of BPA to study the effect of BPA on the reoxidation of PDI within the cells.
To determine the oxidation (disulfide-bonded) status of PDI in the cells, we treated the cells directly with acid to prevent post-harvest oxidation of cysteines and then alkylated free cysteines with PEG-maleimide 2000 (average molecular mass, 2,300 Da). This modification stabilizes disulfide-linked complexes and retards the migration of the reduced form of proteins on a gel (41). The oxidation status of PDI in the cells was then visualized by immunoblotting using antibody to PDI (Fig. 5A). In addition to the oxidized and reduced forms of PDI, we detected a number of bands with different apparent molecular masses that reacted with antibody to PDI (Fig. 5A). We suggest that the latter bands represent disulfide-linked complexes between PDI and its partners for two reasons. First, they ran slower than the monomeric forms of PDI. Second, when the samples were treated with reducing agent before electrophoresis, these bands disappeared, giving rise to bands corresponding to the monomeric forms of this protein (Fig. 5B). Remarkably, the treatment of the culture with BPA caused the accumulation of the reduced form of PDI with concomitant decreases in the levels of the disulfide-linked complexes (Fig. 5A). This effect was enhanced as the BPA concentration increased, suggesting that BPA slowed down the reoxidation of PDI in the cells.
FIGURE 5.

BPA affects the redox state of PDI in HeLa cells. A and B, the effect of BPA on the reoxidation of PDI was studied by cultivating DTT-pretreated HeLa cells in fresh medium without DTT in the presence of the indicated concentrations of BPA for 10 min (lanes 3–6). To visualize the oxidation status of PDI, cellular proteins were subjected to alkylation with PEG-maleimide 2000 and separation on SDS-PAGE under non-reducing conditions in A and reducing conditions in B, followed by immunoblotting with antibody to PDI. Each lane contains 5 μg of proteins from each lysate. The positions of the disulfide-linked complexes that involve PDI are indicated on the right in A. The band assignable to PDI-Ero1α complex, which was also recognized by antibody to Ero1α (not shown), is marked as PDI-Ero1α. To know the positions of the fully oxidized (oxi) and reduced (red) forms of PDI, cells were treated with 1 mm diamide and 10 mm DTT for 10 min, respectively, before alkylation of free cysteines (lanes 1 and 2). Note that some disulfide-linked complexes are refractory to these treatments (see lanes 1 and 2 in A). The numbers of PEG-maleimide 2000 alkylation on the active site cysteines of PDI are indicated on the right in B. Note that the presence of PEG-maleimide 2000 in the sample caused slower migration of even the oxidized form of PDI on the gels when compared with PDI that was not treated with the alkylation reagent (not shown). We suggest that this could have been caused by the alkylation of two free non-catalytic cysteines on PDI (Cys312 and Cys343). Bands marked with an asterisk may represent some degradation products of PDI or nonspecific bands that appeared when the cultures were treated with diamide. C and D, to examine the effect of BPA on the redox state of PDI in cells, HeLa cells that had not been treated with DTT were incubated with or without 400 μm BPA for the indicated times. In lanes 3, 5, 7, and 9, dimethyl sulfoxide (a solvent of BPA) was added to the culture to a final concentration of 0.2%. To visualize the redox state of PDI, the cellular proteins were alkylated with PEG-maleimide 2000 as described in the legend to A. In D, proteins were reduced with 50 mm DTT before electrophoresis. Note that BPA caused the accumulation of a large portion of PDI in its reduced form, in agreement with the in vitro finding that BPA inhibited the Ero1α-catalyzed oxidation of PDI.
Because disulfide bonds that form within a protein or between proteins can alter the mobility of the protein on a gel, we treated the cell lysates with a reducing agent to observe the oxidation status of PDI itself more directly. Indeed, separation of the PEG-maleimide-treated cell lysate on a reducing gel allowed us to identify five distinct bands whose changes in the mobility should reflect changes in the numbers of alkylation on free cysteines in PDI (Fig. 5B). As observed clearly in Fig. 5B, lanes 3–6, BPA treatment increased the level of alkylation on PDI in a dose-dependent manner, indicating again that BPA can slow down the reoxidation of PDI in the cells.
BPA Affects the Oxidation Status of PDI in HeLa Cells
We next proceeded to examine whether BPA has an impact on the oxidation status of PDI in the cells. For this purpose, we cultured HeLa cells without DTT treatment in the presence of 400 μm BPA for various times and looked at the oxidation status of PDI in the cells. As observed in Fig. 5, C and D, BPA caused the reduction of PDI in HeLa cells in a time-dependent manner, indicating that BPA can have a great impact on the redox homeostasis of PDI in the cells. These findings are consistent with the observation that BPA can inhibit the ability of Ero1α to oxidize PDI in vitro (see above).
BPA Has No Effect on Oxidative Protein Folding Catalyzed by the Prx4-ERp46 Pathway
To further examine the possible role of BPA as an inhibitor of oxidative pathways other than Ero1α-PDI, we analyzed Prx4-catalyzed ERp46 oxidation in the presence of BPA. A BPA titration assay showed little or no effect on Prx4 oxidative activity toward ERp46 (Fig. 6A) as well as toward PDI (Fig. 4A). In line with this, the addition of BPA had little effect on the HPLC profiles, indicating the time course of BPTI oxidative folding (Fig. 6B).
FIGURE 6.

Oxidative activity of Prx4 and Ero1α toward ERp46 in the presence of BPA. A, oxidative activity of Prx4 (0.4 μm) toward ERp46 in the presence of various concentrations of BPA was assessed by measuring the decrease in A340, as above. B, Prx4-ERp46-driven oxidative folding of BPTI. HPLC profiles indicate the time course of oxidative folding of BPTI catalyzed by Prx4-ERp46 in the absence (left) and presence (right) of BPA (40 μm). Folding was initiated by the addition of Prx4 (0.1 μm) and ERp46 (1 μm) to reduced BPTI (30 μm) in a buffer containing H2O2 (200 μm). At the indicated time points, reactions were quenched with HCl and analyzed by HPLC. GSH/GSSG redox reagents were not used in this assay. C, left, oxidative activity of Ero1α (4 μm) toward ERp46 in the presence of various concentrations of BPA was assessed by measuring the decrease in A340. Right, plot of percentage inhibition as a function of BPA concentration determined from the initial velocity of the decrease in A340.
By contrast, BPA had weak but significant inhibitory effect on Ero1α-mediated ERp46 oxidation. The IC50 value was 538 μm, more than 10-fold higher than that of Ero1α-mediated PDI oxidation in the presence of BPA (IC50 = 41 μm) (Fig. 6C). Thus, BPA had the ability to more or less inhibit the oxidative pathways constituted by Ero1α and PDI family members (also see “Discussion”).
BPA-induced Conformational Change Revealed by CD
The functional defects caused by BPA suggest that BPA may induce conformational changes in PDI. To examine this possibility, we measured near-UV and far-UV CD spectra of PDI in the presence or absence of BPA. Although there was no significant change in the far-UV region (Fig. 7A, left), a slight but significant difference was observed in the near-UV region; a CD signal at ∼290 nm increased upon titration of BPA (Fig. 7A, right). The results suggest that although BPA had little effect on secondary structure, BPA induced a slight but significant change in the chemical environment around the tryptophan residues of PDI.
FIGURE 7.

BPA-induced conformational changes in PDI. A, far-UV (left) and near-UV (right) CD spectra of PDIfull-length. The CD curves at BPA/PDIfull-length molar ratios of 0 (blue), 1 (green), and 10 (red) are displayed. B, structural model of PDI(120–353) and PDI(354–509) predicted using Phyre (31). Tryptophan residues contained in these two fragments are indicated by W below the schematic structure. C, far-UV (left) and near-UV (right) CD spectra of PDI(354–509). The CD curves at BPA/PDI(354–509) molar ratios of 0 (blue), 0.25 (green), 0.5 (orange), and 1 (red) are displayed. D, far-UV (left) and near-UV (right) CD spectra of PDI(120–353). The CD curves at BPA/PDI(120–353) molar ratios of 0 (blue), 0.25 (green), 0.5 (orange), and 1 (red) are displayed.
To explore which region of PDI undergoes BPA-induced conformational changes, we measured near-UV CD spectra of PDI(120–353), which consists of the C-terminal helix of domain a and intact domains b and b′ (Fig. 7B, left), and that of PDI(354–509), which consists of the x-linker and domains a′ and c (Fig. 7B, right). The former fragment contains only one tryptophan residue (Trp130) in the C-terminal region, whereas the latter contains one tryptophan residue in the x-linker region and two in domain a′.
CD spectra of PDI(354–509) in the near- and far-UV regions were almost identical regardless of the presence or absence of BPA (Fig. 7C), indicating that the secondary and tertiary structure of PDI(354–509) were unaltered upon the addition of BPA. By contrast, PDI(120–353) exhibited a marked increase in CD signal at ∼290 nm in proportion to the concentration of BPA added (Fig. 7D, right), whereas far-UV CD spectra were insensitive to BPA (Fig. 7D, left). These results strongly suggest that the location and surrounding environment of Trp130 contained in domain a of PDI(120–353) were greatly affected by BPA despite the lack of large conformational changes in secondary structure.
BPA Binds to PDI(120–353) Fragment
To investigate possible changes in the thermal stability of PDI(120–353) upon the addition of BPA, we performed DSC analysis. Fig. 8A shows that the DSC curves for PDI(120–353) were shifted to higher melting temperature (Tm) values in accordance with the BPA/PDI(120–353) molar ratio; the Tm of PDI(120–353) increased by 4.4 °C in the presence of a 10-molar equivalent excess of BPA at a scan rate of 60 °C h−1 (Table 1). Similar phenomena were observed at different scan rates of 120 and 200 °C h−1 (Table 1 and Fig. 8A). Precise analysis of thermal denaturation requires confirmation of its reversibility. To this end, a second run was performed by reducing the temperature after the first run. Successive rescans of PDI(120–353) showed a superimposable curve (89.3%) at a scan rate of 60 °C h−1 at pH 7.0 (Fig. 8B), indicating that the observed unfolding transition was highly reversible. Changes in unfolding enthalpy by BPA binding are listed in Table 1, suggesting that they are within experimental errors at three different scan rates. Thus, unfolding enthalpy is not drastically affected by BPA binding to PDI.
FIGURE 8.

DSC measurements for PDI(120–353) and PDI(354–509) in the presence of BPA. A, DSC curves of PDI(120–353) at BPA/PDI(120–353) molar ratios of 0 (blue), 1 (green), 5 (orange), and 10 (red) are displayed. The DSC curves were measured at a scan rate of 60, 120, and 200 °C h−1, respectively. B, DSC curve of the second run (red curve) was measured for PDI(120–353) at a scan rate of 60 °C h−1 immediately after cooling of the first run (blue curve). The recovery area of excess heat capacity was 89.3%. C, DSC curves of PDI(354–509) at BPA/PDI(354–509) molar ratios of 0 (blue), 1 (green), and 5 (orange) are displayed. The DSC curves were measured at a scan rate of 200 °C h−1.
TABLE 1.
Melting temperature (Tm) and the enthalpy of unfolding in accordance with the BPA/PDI(120–353) molar ratio at a scan rate of 60 °C h−1 (left), 120 °C h−1 (middle), and 200 °C h−1 (right)
The ΔCp value of 1.66 × 10 kJmol−1 K−1 was used for calculation of ΔH at 56.6 °C.
| BPA/PDI(120–353) | Scan rate of 60 °C h−1 |
Scan rate of 120 °C h−1 |
Scan rate of 200 °C h−1 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm | ΔH(Tm) | ΔH at 56.6 °C | Tm | ΔH(Tm) | ΔH at 57.1 °C | Tm | ΔH(Tm) | ΔH at 57.5 °C | |
| °C | kJ/mol | kJ/mol | °C | kJ/mol | kJ/mol | °C | kJ/mol | kJ/mol | |
| 0 | 56.6 ± 0.2 | 318 ± 19 | 319 ± 20 | 57.1 ± 0.1 | 313 ± 4 | 313 ± 3 | 57.5 ± 0.1 | 293 ± 25 | 294 ± 26 |
| 1 | 59.0 ± 0.5 | 382 ± 22 | 342 ± 16 | 59.4 ± 0.3 | 375 ± 19 | 337 ± 14 | 58.9 ± 0.6 | 315 ± 27 | 293 ± 17 |
| 5 | 60.5 ± 0.1 | 399 ± 21 | 334 ± 20 | 60.8 ± 0.2 | 397 ± 18 | 336 ± 15 | 60.5 ± 0.4 | 354 ± 45 | 305 ± 52 |
| 10 | 61.0 ± 0.3 | 376 ± 4 | 302 ± 2 | 61.2 ± 0.1 | 412 ± 10 | 344 ± 112 | |||
The equation for the relationship between the binding constant of a ligand to a protein and the melting temperature of a protein can be described as follows (45, 46),
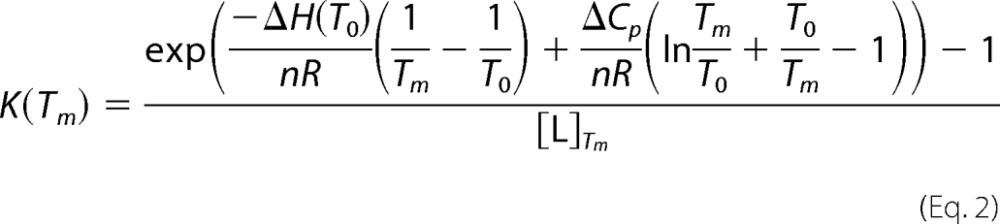 |
where Tm and T0 are melting temperatures in the presence and the absence of BPA, respectively, [L]Tm is the concentration of unbound BPA at Tm, n is the difference in the number of bound molecules of BPA in the unfolded and folded states, K is a binding constant, R is the gas constant, and ΔCp is a heat capacity change due to heat denaturation of a protein. The ΔCp value upon PDI(120–353)-BPA complex formation was estimated to be 1.66 × 10 kJ mol−1 K−1, using the water-accessible nonpolar surface area and polar surface area of PDI(120–353) calculated from structure prediction with Phyre (45, 46). This equation is applicable only when the enthalpy change of binding is negligible compared with the enthalpy change of protein denaturation (47). In the case of BPA binding, the enthalpy change was negligible within experimental errors as described.
The dissociation constant Kd (1/Ka) between PDI(120–353) and BPA was calculated to be 1.17 × 10−5, 1.36 × 10−5, and 1.09 × 10−5 m−1 at a scan rate of 60, 120, and 200 °C h−1, respectively (Table 2). The dissociation constants obtained by DSC experiments were similar to that obtained by previous reports (22.6 μm) (6). In sharp contrast, the DSC curves of PDI(354–509) were unchanged even upon the addition of a 5-molar equivalent excess of BPA; DSC curves showed peaks with a Tm value of 51.3 °C, irrespective of the presence or absence of BPA (Fig. 8C). These results suggest that only PDI(120–353) enhances the thermal stability due to BPA binding, but BPA does not have any effect on PDI(354–509).
TABLE 2.
Determination of the dissociation constant between PDI(120–353) and BPA
| BPA/PDI(120–353) |
Kd |
||
|---|---|---|---|
| Scan rate of 60 °C h−1 | Scan rate of 120 °C h−1 | Scan rate of 200 °C h−1 | |
| m−1 | |||
| 0 | 1.17 × 10−5 | 1.36 × 10−5 | 1.09 × 10−5 |
| 1 | 1.23 × 10−5 ± 0.37 × 10−5 | 1.39 × 10−5 ± 0.28 × 10−5 | 1.55 × 10−5 ± 0.79 × 10−5 |
| 5 | 4.29 × 10−5 ± 0.33 × 10−5 | 5.35 × 10−5 ± 0.46 × 10−5 | 4.48 × 10−5 ± 1.03 × 10−5 |
| 10 | 7.02 × 10−5 ± 0.84 × 10−5 | 9.14 × 10−5 ± 0.41 × 10−5 | |
Molecular Diameter and Geometrical Shape of PDI(120–353) in the Presence of BPA
To gain further structural insights into the BPA-bound form of PDI(120–353), we performed DLS and SAXS analyses. DLS is often employed to measure the size of target particles (48). Fig. 9A shows the molecular diameters of PDI(120–353) and PDI(354–509) measured at different molar ratios of BPA/PDI (ranging from 0 to 10). No significant change was observed in the mean diameter of PDI(354–509) in this range of BPA/PDI molar ratios (Fig. 9A, right); however, the molecular size of PDI(120–353) markedly decreased upon titration of BPA up to a BPA/PDI molar ratio of ∼5 (Fig. 9A, left).
FIGURE 9.

Size and molecular shape of BPA-bound PDI(120–353) in solution. A, molecular diameters of PDI(120–353) (left) and PDI(354–509) (right) at various molecular ratios of BPA/PDI. Concentrations of PDI(120–353) and PDI(354–509) were 0.89 and 1.18 mg/ml, respectively. B, SAXS curves of PDI(120–353) in the presence (red) or absence (blue) of BPA. The natural logarithm of zero-extrapolated scattering intensity, lnI(Q), is shown as a function of scattering angular momentum, Q. C, protein concentration dependence of the apparent radius of gyration of PDI(120–353) in the presence (red) or absence (blue) of BPA. The continuous lines denote the least-squares fit of a linear equation to the data. D, protein concentration dependence of normalized forward intensity, I(0)/c, for PDI(120–353) in the presence (red) or absence (blue) of BPA. The continuous lines denote the least squares fit of a linear equation to the data. E, pair distribution functions of PDI(120–353) in the presence (red) or absence (blue) of BPA, extracted from SAXS curves shown in B.
To determine the structural parameters and the molecular shape of BPA-bound and -unbound forms of PDI(120–353), SAXS analysis was performed in the presence or absence of BPA. Notably, there was a striking difference between the scattering curves of the PDI(120–353)-BPA complex and PDI(120–353) alone around the q value of 0.10 Å−1 (Fig. 9B), suggesting a significant conformational change upon BPA binding. The apparent radius of gyration, Rg(c), and the normalized forward intensity, I(0)/c, showed only marginal protein concentration dependence in the range of 0–5 mg/ml (Fig. 9, C and D). The extrapolated Rg value at c = 0 was 26.7 Å for the PDI(120–353)-BPA complex and 28.0 Å for PDI(120–353) alone. Thus, PDI(120–353) assumes a more compact structure when it binds BPA. This is consistent with the DLS observations, where the molecular diameter of PDI(120–353) markedly decreased upon the addition of BPA.
To extract information on the geometrical shape of PDI(120–353), we calculated a pair distribution function, P(r) (Fig. 9E). Because P(r) indicates the distribution of linear distances between every pair of atoms in a particle, the most frequent r value and the largest r value (Dmax) can be determined from this function. The Dmax values were thus estimated to be 82.0 Å for PDI(120–353) alone and 79.5 Å for the PDI(120–353)-BPA complex, corroborating the present finding that BPA binding results in a more compact overall shape for PDI(120–353).
DISCUSSION
The present study determined that BPA, an endocrine disruptor, markedly inhibited oxidative protein folding catalyzed by the Ero1α-PDI combination in vitro. Consistent with the findings, BPA slowed down the reoxidation of PDI in HeLa cells, leading to the accumulation of a large amount of PDI in the reduced form. Thus, BPA can have an enormous influence on the properties of PDI both in vitro and in cells.
One of the major effects of BPA on this oxidative system was the rearrangement of the thioredoxin domain of PDI through inward movement of the N-terminal a domain, resulting in more compact overall shape. This conformational change probably led to inhibition of the capture of partially folded or misfolded proteins by PDI. Accordingly, the BPTI refolding assay clearly demonstrated that BPA inhibited the late stages of protein folding (and early stages to a lesser extent) from kinetically trapped intermediates to a native state (Figs. 2B, 3A, and 4B). In this context, several lines of evidence suggest the significant plasticity of the bb′ segment of PDI. Crystal structures of reduced and oxidized forms of human PDI showed a redox-dependent domain arrangement of the b domain relative to b′ (15). In addition, human PDI bb′ cannot be superimposed onto yeast PDI bb′ (13, 15). Our study represents the first report that significant rearrangement of the N-terminal a domain can also occur when PDI is complexed with BPA. The intrinsically flexible nature of PDI is likely to be advantageous during oxidative folding by enabling PDI to adapt to the size, shape, and folded states of substrate proteins.
Another major effect of BPA on the Ero1α-PDI oxidative cycle could be that BPA prevents Ero1α from binding to the hydrophobic pocket of the PDI b′ domain and hence inhibits the Ero1α-mediated catalysis of PDI oxidation. We previously revealed that Trp272, a highly conserved residue in the protruding β-hairpin of Ero1α, plays a pivotal role during the functional interplay of Ero1α and PDI (22). It is conceivable that phenol groups of BPA compete with this tryptophan residue for binding to the same site in PDI b′ (25). In support of this, our previous mutational study suggested that His258, Gln245, and Asn300, residues located near the hydrophobic pocket of PDI, were closely involved in BPA binding (25).
Ero1 and PDI have long been known to constitute a major disulfide bond formation pathway, although more recently more than 20 members of the PDI family and more than five enzymes with significant oxidative activity toward PDI family members have been identified in the mammalian ER (18, 19). Thus, the oxidative folding network in mammalian cells is more complicated and diverse than previously thought. Presumably, multiple PDI family members and their oxidation enzymes work in a distinct, cooperative, and sometimes complementary manner to ensure efficient production of multidisulfide-containing proteins (17, 18, 27, 28, 49).
We recently revealed that although Prx4 can oxidize a broad range of PDI family members (27, 50), ERp46 and P5 are the primary partners of Prx4 in cells. The ERp46-Prx4 and P5-Prx4 oxidative pathways are probably dedicated to rapid but promiscuous disulfide bond introduction during the early stages of oxidative protein folding (27, 28). The present study demonstrated that BPA had a marginal effect on Prx4 oxidative activity toward both ERp46 (Fig. 6A) and PDI (Fig. 4A). Meanwhile, BPA significantly inhibited Ero1α-catalyzed oxidation of ERp46 although with a lower potency (IC50 = 538 μm) than that of PDI (IC50 = 41 μm) (Figs. 1C and 6C). The large difference in the effects of BPA on Ero1α- and Prx4-driven oxidative pathways presumably reflects the completely different mode of interaction of these two oxidative enzymes toward PDI family members. Systematic studies indicate that Prx4 oxidizes the thioredoxin domains of PDI family members at almost the same rate regardless of whether they are present on an isolated peptide or as part of an intact protein (27, 28). Ero1α recognizes and oxidizes the tandemly positioned b′-a′ domains of PDI as a minimal element (22, 23). Whereas the BPA binding site of ERp46 remains to be specified, BPA may influence the function of a wide range of PDI family members and hence may alter the redox environment in the ER.
An increasing number of studies have investigated the physiological effects of BPA on mammalian tissues. In vivo, BPA induces heat shock protein 90α expression in mouse uterus (51) and induces oxidative stress in rat liver due to decreased antioxidant enzyme activity (52). Also, BPA down-regulates the production and secretion of adiponectin (53), which is widely known as an adipocyte-derived secretory protein that has potent insulin-sensitizing and anti-apoptotic properties (54). Of note, BPA induces ER stress as indicated by the increased expression level of GRP78/Bip, an ER stress marker (55, 56). One possible interpretation is that the inhibitory effect of BPA on PDI activity may cause the accumulation of misfolded proteins in the ER, inducing the unfolded protein response. Thus, BPA may interfere with the synthesis of secretory proteins and their secretion pathways in mammalian cells. In this context, it would be interesting to perform a global analysis of the effects of BPA on secretory protein production and secretion. The present finding of BPA-mediated inhibition of the functional interplay between Ero1α and PDI thus provides feasible insights into the mechanisms by which BPA causes adverse health effects.
Acknowledgments
We thank Dr. Daizo Hamada, Marina Nawata (Kobe University), Dr. Satoshi Takahashi (Tohoku University), Dr. Shinichi Segawa, Tomohisa Shibano, Atsushi Matsuoka, Satomi Uramoto (Kwansei Gakuin University), and Michiyo Takehira (RIKEN SPring-8 Center, Harima Institute) for considerable assistance and for discussion of this work. Synchrotron radiation experiments were performed on BL45XU in SPring-8 with the approval of RIKEN (Proposal 20110112).
This work was supported by funding from the Next Generation World-leading Researchers program (MEXT) (to K. I.), by a grant-in-aid for scientific research (B) (to S. I.), by a grant-in-aid for scientific research (C) (to H. Y. and H. K.), by a grant-in-aid for scientific research on innovative areas (to K. I. and H. K.), and by a grant-in-aid for Japan Society for the Promotion of Science fellows (to M. O., S. H., and S. M.).
- BPA
- bisphenol A
- DLS
- dynamic light scattering
- DSC
- differential scanning calorimetry
- ER
- endoplasmic reticulum
- Ero1
- ER oxidoreduclin 1
- PDI
- protein-disulfide isomerase
- Prx4
- peroxiredoxin-4
- SAXS
- small angle x-ray scattering
- BPTI
- bovine pancreas trypsin inhibitor
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Vandenberg L. N., Hauser R., Marcus M., Olea N., Welshons W. V. (2007) Human exposure to bisphenol A (BPA). Reprod. Toxicol. 24, 139–177 [DOI] [PubMed] [Google Scholar]
- 2. Obata T., Kubota S. (2000) Formation of hydroxy radicals by environmental estrogen-like chemicals in rat striatum. Neurosci. Lett. 296, 41–44 [DOI] [PubMed] [Google Scholar]
- 3. Jenkins S., Wang J., Eltoum I., Desmond R., Lamartiniere C. A. (2011) Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ. Health Perspect. 119, 1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Imaoka S., Mori T., Kinoshita T. (2007) Bisphenol A causes malformation of the head region in embryos of Xenopus laevis and decreases the expression of the ESR-1 gene mediated by Notch signaling. Biol. Pharm. Bull 30, 371–374 [DOI] [PubMed] [Google Scholar]
- 5. Fagin D. (2012) Toxicology: the learning curve. Nature 490, 462–465 [DOI] [PubMed] [Google Scholar]
- 6. Hiroi T., Okada K., Imaoka S., Osada M., Funae Y. (2006) Bisphenol A binds to protein disulfide isomerase and inhibits its enzymatic and hormone-binding activities. Endocrinology 147, 2773–2780 [DOI] [PubMed] [Google Scholar]
- 7. Weissman J. S., Kim P. S. (1993) Efficient catalysis of disulphide bond rearrangements by protein disulphide isomerase. Nature 365, 185–188 [DOI] [PubMed] [Google Scholar]
- 8. Narayan M. (2012) Disulfide bonds: protein folding and subcellular protein trafficking. FEBS J. 279, 2272–2282 [DOI] [PubMed] [Google Scholar]
- 9. Edman J. C., Ellis L., Blacher R. W., Roth R. A., Rutter W. J. (1985) Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature 317, 267–270 [DOI] [PubMed] [Google Scholar]
- 10. Nguyen V. D., Wallis K., Howard M. J., Haapalainen A. M., Salo K. E., Saaranen M. J., Sidhu A., Wierenga R. K., Freedman R. B., Ruddock L. W., Williamson R. A. (2008) Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b′ domain. J. Mol. Biol. 383, 1144–1155 [DOI] [PubMed] [Google Scholar]
- 11. Klappa P., Ruddock L. W., Darby N. J., Freedman R. B. (1998) The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pirneskoski A., Ruddock L. W., Klappa P., Freedman R. B., Kivirikko K. I., Koivunen P. (2001) Domains b′ and a′ of protein disulfide isomerase fulfill the minimum requirement for function as a subunit of prolyl 4-hydroxylase. The N-terminal domains a and b enhance this function and can be substituted in part by those of ERp57. J. Biol. Chem. 276, 11287–11293 [DOI] [PubMed] [Google Scholar]
- 13. Tian G., Xiang S., Noiva R., Lennarz W. J., Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73 [DOI] [PubMed] [Google Scholar]
- 14. Tian G., Kober F. X., Lewandrowski U., Sickmann A., Lennarz W. J., Schindelin H. (2008) The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. J. Biol. Chem. 283, 33630–33640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang C., Li W., Ren J., Fang J., Ke H., Gong W., Feng W., Wang C. C. (2013) Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 19, 36–45 [DOI] [PubMed] [Google Scholar]
- 16. Byrne L. J., Sidhu A., Wallis A. K., Ruddock L. W., Freedman R. B., Howard M. J., Williamson R. A. (2009) Mapping of the ligand-binding site on the b′ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 423, 209–217 [DOI] [PubMed] [Google Scholar]
- 17. Kojima R., Okumura M., Inaba K. (2013) Structural basis of disulfide bond formation in the bacterial periplasm and mammalian ER. eLS 10.1002/9780470015902.a0024169 [DOI] [Google Scholar]
- 18. Sato Y., Inaba K. (2012) Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J. 279, 2262–2271 [DOI] [PubMed] [Google Scholar]
- 19. Bulleid N. J., Ellgaard L. (2011) Multiple ways to make disulfides. Trends Biochem. Sci. 36, 485–492 [DOI] [PubMed] [Google Scholar]
- 20. Araki K., Inaba K. (2012) Structure, mechanism, and evolution of Ero1 family enzymes. Antioxid. Redox Signal. 16, 790–799 [DOI] [PubMed] [Google Scholar]
- 21. Tavender T. J., Bulleid N. J. (2010) Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox Signal. 13, 1177–1187 [DOI] [PubMed] [Google Scholar]
- 22. Masui S., Vavassori S., Fagioli C., Sitia R., Inaba K. (2011) Molecular bases of cyclic and specific disulfide interchange between human ERO1α protein and protein-disulfide isomerase (PDI). J. Biol. Chem. 286, 16261–16271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inaba K., Masui S., Iida H., Vavassori S., Sitia R., Suzuki M. (2010) Crystal structures of human Ero1α reveal the mechanisms of regulated and targeted oxidation of PDI. EMBO J. 29, 3330–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramming T., Appenzeller-Herzog C. (2012) The physiological functions of mammalian endoplasmic oxidoreductin 1: on disulfides and more. Antioxid. Redox Signal. 16, 1109–1118 [DOI] [PubMed] [Google Scholar]
- 25. Hashimoto S., Shiomoto K., Okada K., Imaoka S. (2012) The binding site of bisphenol A to protein disulphide isomerase. J. Biochem. 151, 35–45 [DOI] [PubMed] [Google Scholar]
- 26. Hashimoto S., Yoshimura H., Okada K., Uramaru N., Sugihara K., Kitamura S., Imaoka S. (2012) Effects of polybrominated diphenyl ethers (PBDEs) and their derivatives on protein disulfide isomerase activity and growth hormone release of GH3 cells. Chem. Res. Toxicol. 25, 656–663 [DOI] [PubMed] [Google Scholar]
- 27. Sato Y., Kojima R., Okumura M., Hagiwara M., Masui S., Maegawa K., Saiki M., Horibe T., Suzuki M., Inaba K. (2013) Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 3, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kojima R., Okumura M., Masui S., Kanemura S., Inoue M., Saiki M., Yamaguchi H., Hikima T., Suzuki M., Akiyama S., Inaba K. (2014) Radically different thioredoxin domain arrangement of ERp46, an efficient disulfide bond introducer of the mammalian PDI family. Structure 22, 431–443 [DOI] [PubMed] [Google Scholar]
- 29. Hashimoto S., Ito L., Okumura M., Shibano T., Nawata M., Kumasaka T., Yamaguchi H., Imaoka S. (2012) Crystallization and preliminary crystallographic analysis of the complex between triiodothyronine and the bb′ fragment of rat protein disulfide isomerase. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 68, 476–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Privalov P. L., Khechinashvili N. N. (1974) A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J. Mol. Biol. 86, 665–684 [DOI] [PubMed] [Google Scholar]
- 31. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 32. Fujisawa T., Inoue K., Oka T., Iwamoto H., Uruga T., Kumasaka T., Inoko Y., Yagi N., Yamamoto M., Ueki T. (2000) Small-angle x-ray scattering station at the SPring-8 RIKEN beamline. J. Appl. Crystallogr. 33, 797–800 [Google Scholar]
- 33. Rice S. A. (1956) Small angle scattering of x-rays. A. Guinier and G. Fournet. Translated by C. B. Wilson and with a bibliographical appendix by K. L. Yudowitch. Wiley, New York, 1955. 268 pp. J. Polym. Sci. 10.1002/pol.1956.120199326 [DOI] [Google Scholar]
- 34. Svergun D. (1991) Mathematical methods in small-angle scattering data analysis. J. Appl. Crystallogr. 24, 485–492 [Google Scholar]
- 35. Okumura M., Saiki M., Yamaguchi H., Hidaka Y. (2011) Acceleration of disulfide-coupled protein folding using glutathione derivatives. FEBS J. 278, 1137–1144 [DOI] [PubMed] [Google Scholar]
- 36. Okumura M., Shimamoto S., Hidaka Y. (2012) A chemical method for investigating disulfide-coupled peptide and protein folding. FEBS J. 279, 2283–2295 [DOI] [PubMed] [Google Scholar]
- 37. Okumura M., Shimamoto S., Nakanishi T., Yoshida Y., Konogami T., Maeda S., Hidaka Y. (2012) Effects of positively charged redox molecules on disulfide-coupled protein folding. FEBS Lett. 586, 3926–3930 [DOI] [PubMed] [Google Scholar]
- 38. Okumura M., Shimamoto S., Hidaka Y. (2014) Chemical methods for producing disulfide bonds in peptides and proteins to study folding regulation. Curr. Protoc. Protein Sci. 76, 28.27.21–28.27.13 [DOI] [PubMed] [Google Scholar]
- 39. Walker K. W., Gilbert H. F. (1995) Oxidation of kinetically trapped thiols by protein disulfide isomerase. Biochemistry 34, 13642–13650 [DOI] [PubMed] [Google Scholar]
- 40. Shao F., Bader M. W., Jakob U., Bardwell J. C. (2000) DsbG, a protein disulfide isomerase with chaperone activity. J. Biol. Chem. 275, 13349–13352 [DOI] [PubMed] [Google Scholar]
- 41. Kadokura H., Saito M., Tsuru A., Hosoda A., Iwawaki T., Inaba K., Kohno K. (2013) Identification of the redox partners of ERdj5/JPDI, a PDI family member, from an animal tissue. Biochem. Biophys. Res. Commun. 440, 245–250 [DOI] [PubMed] [Google Scholar]
- 42. Cao Z., Subramaniam S., Bulleid N. J. (2014) Lack of an efficient endoplasmic reticulum-localized recycling system protects peroxiredoxin IV from hyperoxidation. J. Biol. Chem. 289, 5490–5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weissman J. S., Kim P. S. (1991) Reexamination of the folding of BPTI: predominance of native intermediates. Science 253, 1386–1393 [DOI] [PubMed] [Google Scholar]
- 44. Weissman J. S., Kim P. S. (1995) A kinetic explanation for the rearrangement pathway of BPTI folding. Nat. Struct. Biol. 2, 1123–1130 [DOI] [PubMed] [Google Scholar]
- 45. Plotnikov V., Rochalski A., Brandts M., Brandts J. F., Williston S., Frasca V., Lin L. N. (2002) An autosampling differential scanning calorimeter instrument for studying molecular interactions. Assay Drug Dev. Technol. 1, 83–90 [DOI] [PubMed] [Google Scholar]
- 46. Schellman J. A. (1975) Macromolecular binding. Biopolymers 14, 999–1018 [Google Scholar]
- 47. Schellman J. A. (1976) The effect of binding on the melting temperature of biopolymers. Biopolymers 15, 999–1000 [Google Scholar]
- 48. Shiba K., Niidome T., Katoh E., Xiang H., Han L., Mori T., Katayama Y. (2010) Polydispersity as a parameter for indicating the thermal stability of proteins by dynamic light scattering. Anal. Sci. 26, 659–663 [DOI] [PubMed] [Google Scholar]
- 49. Araki K, Iemura S, Kamiya Y, Ron D, Kato K, Natsume T, Nagata K. (2013) Ero1α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 202, 861–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tavender T. J., Springate J. J., Bulleid N. J. (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Papaconstantinou A. D., Fisher B. R., Umbreit T. H., Goering P. L., Lappas N. T., Brown K. M. (2001) Effects of β-estradiol and bisphenol A on heat shock protein levels and localization in the mouse uterus are antagonized by the antiestrogen ICI 182,780. Toxicol. Sci. 63, 173–180 [DOI] [PubMed] [Google Scholar]
- 52. Bindhumol V., Chitra K. C., Mathur P. P. (2003) Bisphenol A induces reactive oxygen species generation in the liver of male rats. Toxicology 188, 117–124 [DOI] [PubMed] [Google Scholar]
- 53. Kidani T., Kamei S., Miyawaki J., Aizawa J., Sakayama K., Masuno H. (2010) Bisphenol A downregulates Akt signaling and inhibits adiponectin production and secretion in 3T3-L1 adipocytes. J. Atheroscler. Thromb. 17, 834–843 [DOI] [PubMed] [Google Scholar]
- 54. Wijesekara N., Krishnamurthy M., Bhattacharjee A., Suhail A., Sweeney G., Wheeler M. B. (2010) Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J. Biol. Chem. 285, 33623–33631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Asahi J., Kamo H., Baba R., Doi Y., Yamashita A., Murakami D., Hanada A., Hirano T. (2010) Bisphenol A induces endoplasmic reticulum stress-associated apoptosis in mouse non-parenchymal hepatocytes. Life Sci. 87, 431–438 [DOI] [PubMed] [Google Scholar]
- 56. Tabuchi Y., Takasaki I., Kondo T. (2006) Identification of genetic networks involved in the cell injury accompanying endoplasmic reticulum stress induced by bisphenol A in testicular Sertoli cells. Biochem. Biophys. Res. Commun. 345, 1044–1050 [DOI] [PubMed] [Google Scholar]