

Background: The amyloid-β peptide (Aβ) is crucially involved in the onset and progression of Alzheimer disease (AD).
Results: A designed ankyrin repeat protein (DARPin) was selected to bind and neutralize Aβ.
Conclusion: DARPins can prevent amyloid formation and associated neurotoxic effects of Aβ.
Significance: DARPins provide a therapeutic potential in the treatment of AD.
Keywords: Alzheimer Disease, Amyloid, Immunotherapy, Neurodegenerative Disease, Protein Design
Abstract
Passive immunization with anti-amyloid-β peptide (Aβ) antibodies is effective in animal models of Alzheimer disease. With the advent of efficient in vitro selection technologies, the novel class of designed ankyrin repeat proteins (DARPins) presents an attractive alternative to the immunoglobulin scaffold. DARPins are small and highly stable proteins with a compact modular architecture ideal for high affinity protein-protein interactions. In this report, we describe the selection, binding profile, and epitope analysis of Aβ-specific DARPins. We further showed their ability to delay Aβ aggregation and prevent Aβ-mediated neurotoxicity in vitro. To demonstrate their therapeutic potential in vivo, mono- and trivalent Aβ-specific DARPins (D23 and 3×D23) were infused intracerebroventricularly into the brains of 11-month-old Tg2576 mice over 4 weeks. Both D23 and 3×D23 treatments were shown to result in improved cognitive performance and reduced soluble Aβ levels. These findings demonstrate the therapeutic potential of Aβ-specific DARPins for the treatment of Alzheimer disease.
Introduction
The amyloid-β peptide (Aβ)2 is considered a central component in the onset and progression of Alzheimer disease (AD) (1). The following three major pathways have been identified as potential therapeutic channels to alleviate the clinical progression and cognitive loss: (i) the inhibition of Aβ production from amyloid precursor protein (APP); (ii) interference with the formation of toxic aggregation intermediates, and (iii) the accelerated elimination of Aβ from the brain into the periphery (2, 3). From a clinical point of view, these concepts strongly support the use of therapeutic strategies with proteins that can specifically bind, neutralize, and prevent the aggregation and propagation of misfolded proteins throughout the brain.
Based on the effective clearance of β-amyloid from the brains of mouse models of brain amyloidosis upon repeated Aβ immunization (4, 5), numerous immunotherapy clinical trials in humans have subsequently been initiated (6–8). To date, more than one dozen anti-amyloid immunotherapy clinical trials, testing both passive immunotherapy and active vaccination strategies, are currently underway for the treatment of AD (9).
Designed ankyrin repeat proteins (DARPins) can be effectively selected in vitro for binding to a broad variety of target proteins with high specificity and affinity, and they have been shown to provide new therapeutic avenues (10). DARPins are built from several ankyrin repeat modules that are tightly packed and capped by terminating repeats that shield the hydrophobic core, resulting in high stability and solubility with a low aggregation tendency (11, 12). Additionally, the absence of redox-sensitive disulfide bonds has enabled DARPins to be used for both intracellular and extracellular applications (13, 14) and, combined with the lack of endogenous receptors, allows an improved fine-tuning of tissue distribution and clearance (15) as compared with conventional immunoglobulin-based (IgG) scaffolds. At one-tenth the molecular weight of IgGs, DARPins might cross the blood-brain barrier more efficiently than antibodies upon peripheral administration (16, 17). Unlike IgG-Aβ complexes (18), DARPins bound to Aβ will probably be removed quickly and efficiently from the body, making them ideal amyloid-lowering therapeutics with a low risk for immunogenicity and production of neutralizing antibodies following repeated administration (19). Here, we describe the selection of a novel class of potential Aβ-specific therapeutics based on the ankyrin fold, their affinity determination, as well as their ability to prevent Aβ aggregation, reduce Aβ-mediated neurotoxicity in a cell culture model, and show its therapeutic potential in APP transgenic mice (Tg2576) in vivo, an animal model of Alzheimer disease.
EXPERIMENTAL PROCEDURES
Preparation of the Amyloid-β Peptides
Recombinant Aβ(1–42)-peptide was purchased as a hexafluoroisopropanol film from rPeptide (Bogart, GA). All biotinylated variants (Aβ(1–28), Aβ(1–40), and Aβ(1–42)) were obtained from Anaspec (Fremont, CA) and processed as described previously (20).
Ribosome Display Selection of DARPins
The enrichment of anti-Aβ DARPins was performed using previously described N2C and N3C DARPin libraries (12). Ribosome display selections using decreasing concentrations of biotinylated Aβ(1–28) (400 to 100 nm) as target peptide were performed in analogy to previous reports (21). The library was subjected to a total of four selection cycles, the first of which was performed on biotinylated peptide bound to a NeutrAvidin-coated microtiter plate, and the three subsequent rounds were carried out in solution using streptavidin-coated magnetic particles for pulldown of the ternary DARPin-ribosome-mRNA complexes.
Screening and Titration ELISA
DARPin expression and cell lysis were performed as described previously (22). For analysis of individual DARPin clones, microtiter plates (Corning Glass) were coated with 66 nm NeutrAvidin followed by immobilization of 250 nm of the target peptides for 1 h at 4 °C. Bound DARPins were detected via an anti-RGS-His6 antibody (Qiagen, dilution 1:1000) followed by an anti-mouse-HRP conjugate (GE Healthcare, dilution 1:2000). The assay was quantified colorimetrically (tetramethylbenzidine in 30 mm citric acid (1:20)) and stopped with 1 m H2SO4. The absorbance at 450 nm was recorded against a reference wavelength of 690 nm with a standard ELISA reader SunriseTM (Tecan, Männedorf, Switzerland). The data were fitted to a sigmoid dose-response equation using Graph Pad Prism 5 (GraphPad Software) to calculate the concentration of half-maximal binding (EC50).
Competition ELISA
Competition studies were performed using a constant concentration of 10 nm DARPin with increasing quantities of soluble Aβ(1–42) (0–5000 nm in TBS150 for 1 h at 4 °C). Target peptide was immobilized at 250 nm. Signal detection was performed as described for titration ELISA.
Epitope Mapping by Monoclonal Antibodies
Aβ(1–42) at 200 nm was immobilized onto a DARPin D23-coated microtiter plate and probed with 1 ng μl−1 of the monoclonal antibodies 6E10, 4G8 (Covance, Basel, Switzerland), and 22C4 (in-house). Bound antibodies were detected as described for titration ELISA.
Epitope Mapping by Peptide Competition
Aβ(1–28)-biotin (250 nm solution) was surface-immobilized on a NeutrAvidin layer of a microtiter plate. DARPin D23 at 50 nm was preincubated with a 50-fold molar excess of the peptide fragments Aβ(−4–6), Aβ(−3–7), Aβ(−2–8), Aβ(−1–9), Aβ(1–8), Aβ(2–11), Aβ(3–12), Aβ(4–13), Aβ(5–14), Aβ(6–15) (peptides&elephants, Potsdam, Germany), Aβ(1–11), Aβ(1–16), Aβ(12–28), Aβ(29–40), Aβ(31–35), Aβ(33–42), Aβ((1–28), Aβ(1–38) (Bachem, Bubendorf, Switzerland) and scrambled Aβ(1–42) (from N to C terminus: AIAEGDSHVLKEGAYMEIFDVQGHVFGGKIFRVVDLGSHNVA) (rPeptide). The fraction of surface-bound D23 was detected as described for titration ELISA.
Aβ Aggregation Assays
The inhibitory effect of equimolar concentrations of D23 on Aβ(1–42) aggregation (both at 5 μm) was assessed in real time by thioflavin T (ThT) fluorescence as described previously (20). The concentrations of the Aβ stock solutions were determined spectrophotometrically (ϵ = 1730 m−1 cm−1) and, for reasons of reproducibility, used within 24 h of preparation (23). The elongation rates were mathematically deduced via linear regression from the aggregation curves using signals from 20 to 80% of the steady state for all three experimental conditions.
Neurotoxicity Assays
The neuroprotective effect of D23 on Aβ-mediated neurotoxicity (both at 5 μm) was investigated in a cell culture model of primary cortical neurons from rat embryos (day E18) and performed identically to previously published protocols (20). Three wells per treatment were measured, and the assay was repeated two more times. Data are given as means ± S.E.
Cloning Strategy for Multivalent DARPins
The coding sequence of D23 was excised from the expression plasmid pQE30 (Qiagen, Hildesheim, Germany) using the restriction enzymes BamHI and HindIII (New England Biolabs, Ipswich, MA) and subsequently inserted into the recipient plasmid (pQI-bi-1_1) via a BamHI/HindIII (cassette 1) and BglII and BsaI (cassette 2) restriction digestion. Trivalent D23 was constructed from bivalent D23 via PCR using the primers MVD3_forward (5′-AAAGAGGAGAAATTAACTATGAGAGGATC-3′) and QiBi_MVD_reverse (5′-AAGGATAGGTCTCAAGCTAGAGAGTCATTACCCCAGGCGTTTAAGG-3′), and the resulting amplification was digested with BamHI/BsaI and inserted into the pQI-bi-1_1 recipient vector via its restriction sites for BglII and BsaI. The vector had previously been modified to already contain a DARPin in the 5′-expression cassette. DARPins were genetically engineered as head-to-tail fusions and expressed in a single open reading frame (ORF) as described previously (15).
Surface Plasmon Resonance
SPR was recorded with a BIAcore T100 instrument (GE Healthcare) similarly to previous descriptions using a streptavidin-coated sensor chip and Aβ(1–42)-biotin as immobilized target (12). Association and dissociation events were measured at a constant flow rate of 30 μl min−1 with analyte concentrations doubling from 0.5 to 512 nm. Kinetic data were globally fitted to a 1:1 binding model (D23) or a bivalent binding model (3×D23) as part of the BIAevaluation software 2.0.3 (GE Healthcare), but the complicated kinetics of 3×D23 preclude a numeric evaluation.
Animals
This study used Tg2576 transgenic mice expressing human APP carrying the Swedish mutation K670N/M671L (24). A 12-h light-dark cycle was maintained in the housing room, and except for the time of testing, water and food were provided ad libitum.
Cognitive Behavioral Testing
At the time of testing, mice were weighed and examined for general health indicators to ensure that the mice were physically able to conduct the cognitive-behavioral test and to rule out any adverse side effects due to the surgical procedure or DARPin therapy. Spatial working memory was assessed in mice using the Y-maze (Y-shaped plastic maze, with 40 × 20 × 10-cm arm sizes). During a 5-min trial, the sequence of arm entries was recorded using the ANY-maze Video Tracking System (Stoelting Co.). The percent alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries −2) × 100%.
DARPin Intracerebroventricular Administration via Surgical Implantation of Alzet® Osmotic Minipumps
Twelve-month-old Tg2576 mice were deeply anesthetized; a small midline incision was made to expose the skull, and a subcutaneous pocket was prepared in the midscapular area of the back of the mouse so that a sterile Alzet® minipump (model 2004) filled with DARPin solution or vehicle could be inserted. Subsequently, an Alzet Brain infusion kit 3 cannula was lowered into the left lateral ventricle (coordinates according to bregma: anterior-posterior, −0.2 mm; medial-lateral, 0.9 mm; and dorsal-ventral, 2.5 mm) and two small screws were placed within the skull and dental cement applied to firmly attach to the skull. Groups of 19–23 mice received intracerebroventricular infusion over 28 days (equimolar concentration of 50 μm) of either D23, 3×D23, E2_5, or PBS control.
Biochemical and Histochemical Analyses
Brains from treated mice were obtained as described previously (25). The right hemibrain was homogenized using a sequential extraction protocol using a glass-Teflon homogenizer (20 strokes, 315 rpm) in RIPA buffer containing complete protease inhibitor tablets (Sigma). The brain extract was ultracentrifuged (100,000 × g at 4 °C for 1 h), supernatant-extracted, and stored at −80 °C for later biochemical analysis. The remaining pellet was frozen on dry ice, resuspended in 70% formic acid, sonicated for 30 s at 30% power, and ultracentrifuged (30 min). The supernatant was extracted, lyophilized, reconstituted in RIPA buffer, and stored at −80 °C for later analysis.
β-Amyloid Analysis
Aβ fragments were measured in plasma and brain homogenates using a MSD 3plex multi-SPOT Aβ human kit (MesoScale Discovery, Rockville, MD), based on electrochemiluminescence detection, with capture antibodies specific for Aβ(x-38), Aβ(x-40), and Aβ(x-42), in accordance to the manufacturer's instructions. The MSD SECTOR Imager 6000 reader was used for analysis, and the MSD DISCOVERY WORKBENCH software (Version 3.0.17) with the Data Analysis Toolbox was used to calculate sample concentrations by comparing them against a standard curve (five-parameter logistic curve).
Histochemistry
Thioflavine S staining was done according to a previously published protocol (25). All chemicals were obtained from Sigma. Antibody 4G8 (Covance, Basel, Switzerland) was used at 1 ng μl−1 to detect amyloid deposits.
The effect of Aβ addition on neuronal morphology was observed in a cell culture model of primary cortical neurons from rat embryos (day E18) and performed as described previously (20). Shortly, cells were washed in PBS, fixated by 4% paraformaldehyde (in PBS) for 15 min, and subsequently washed by TBS + 0.05% Triton X-100. Cells were blocked with a mixture of 5% goat serum and 5% horse serum (in TBS + 0.05% Triton X-100) for 1 h at 4 °C. Aβ deposits were visualized by a polyclonal anti-Aβ antibody (Zymed Laboratories Inc.) at 1:500, and neurons were stained by an anti-MAP2 antibody (Sigma) at 1:1000, followed by secondary Cy2-/Cy3-conjugated antibodies.
Statistical Analysis
Data analysis was performed using GraphPad Prism 4.03 software. Tests for normal distribution were performed before statistical testing, according to the results of the Shapiro-Wilk and the Kolmogorov-Smirnov Test for normality. Either Student's t test or Mann-Whitney U test for two sample groups or analysis of variance for multiple comparisons was performed (followed by post hoc Tukey's or Mann-Whitney U test). A p value < 0.05 was considered statistically significant. Error bars are S.E. if not indicated differently.
Approval Animal Studies
All animal experiments were approved by the veterinary office of the Cantonal Health Department Zurich.
RESULTS
DARPin D23 Specifically Binds to Soluble Monomeric Aβ
Ribosome display was used to enrich specific DARPins against the Aβ peptide. Libraries in the N2C and N3C format (with 2 or 3 randomized repeats between the N- and C-capping repeats) were subjected to surface-immobilized C-terminally biotinylated Aβ(1–28). This truncated Aβ variant was chosen over Aβ(1–42) for its reduced propensity to form aggregates. In every round, the selection pressure was increased through a decrease in the amount of target peptide, increased washing stringency, and a reduced number of PCR cycles to re-amplify the selected sub-pool. From cycle 2 onward, selection was performed in solution to drive the selection of DARPins toward recognizing soluble Aβ species (Fig. 1a). After four cycles, a screening ELISA revealed 30 out of 96 clones as specific for Aβ(1–28) (31%) with negligible reactivity to control proteins. These controls include NeutrAvidin, bovine serum albumin (BSA), and directly (= hydrophobically) coated Aβ(1–42). All specific binders came from the N2C library. DNA sequence analysis of 20 randomly selected Aβ binders confirmed differences mainly in the variable interaction residues and with lower frequency also in the N-terminal cap. Because of best performance in a series of binding experiments, the DARPin termed D23 was selected as a lead candidate for further characterization. A titration ELISA (Fig. 1b) demonstrated that the selected clone also recognized the pathologically relevant variant Aβ(1–42) with a similar EC50 (17 nm for Aβ(1–28) and 31 nm for Aβ(1–42)). The binding of DARPin D23 to surface-immobilized Aβ was specifically competed using a molar excess of soluble Aβ(1–42) with a 50% drop in binding signal at a competitor concentration of <50 nm, indicating a low nanomolar affinity of D23 for soluble Aβ(1–42) (Fig. 1c).
FIGURE 1.

Selection of anti-Aβ DARPins through ribosome display in vitro. a, DARPin libraries were selected against decreasing amounts of the biotinylated Aβ(1–28)-peptide via ribosome display in four selection rounds. Biotinylated peptides were presented via NeutrAvidin immobilized on a microtiter plate (round 1) or recovered from solution through streptavidin-coated magnetic particles in all subsequent rounds. DNA was re-amplified at the end of each round with decreasing numbers of PCR cycles (arrow numbers). After four rounds, Escherichia coli was transformed (transf), and individual clones were analyzed for Aβ binding activity through ELISA. b, ELISA titration with increasing amounts of DARPin D23 binding to immobilized Aβ(1–28) and Aβ(1–42). The half-maximal intensity (EC50) is determined at 17 nm (Aβ(1–28)) and 31 nm (Aβ(1–42)). A nonselected library member, E2_5, did not bind Aβ(1–42) at any concentration. c, inhibition of DARPin D23 binding to immobilized Aβ(1–28)-biotin by increasing concentrations of the soluble nonbiotinylated peptide Aβ(1–42). The competition showed half-maximal inhibition at ∼50 nm Aβ(1–42). Data in b and c are means from three independent experiments, represented as means ± S.D.
D23 Binds a Conformational Aβ Epitope Involving the Free N Terminus
D23 bound three C-terminally biotinylated variants (Aβ(1–28), Aβ(1–40), and Aβ(1–42)) with similar affinities but did not recognize N-terminally biotinylated Aβ(1–42) in ELISA (data not shown). We utilized the three monoclonal antibodies 6E10 (recognizing the N terminus), 4G8 (central domain), and 22C4 (C terminus) with known epitopes on the Aβ peptide to analyze the DARPin-specific epitope by competition experiments (26, 27). The addition of these antibodies to immobilized DARPin-Aβ complexes revealed that the DARPin-Aβ interaction strongly interfered with peptide recognition by antibody 6E10 and to a lesser extent by 4G8, suggesting that the N-terminal Aβ stretch was already bound by DARPin D23 (Fig. 2a). The use of truncated Aβ(1–28) during ribosome display selections focused selection on the N-terminal region. Thus, DARPin D23 does not interfere with the C-terminal antibody 22C4.
FIGURE 2.
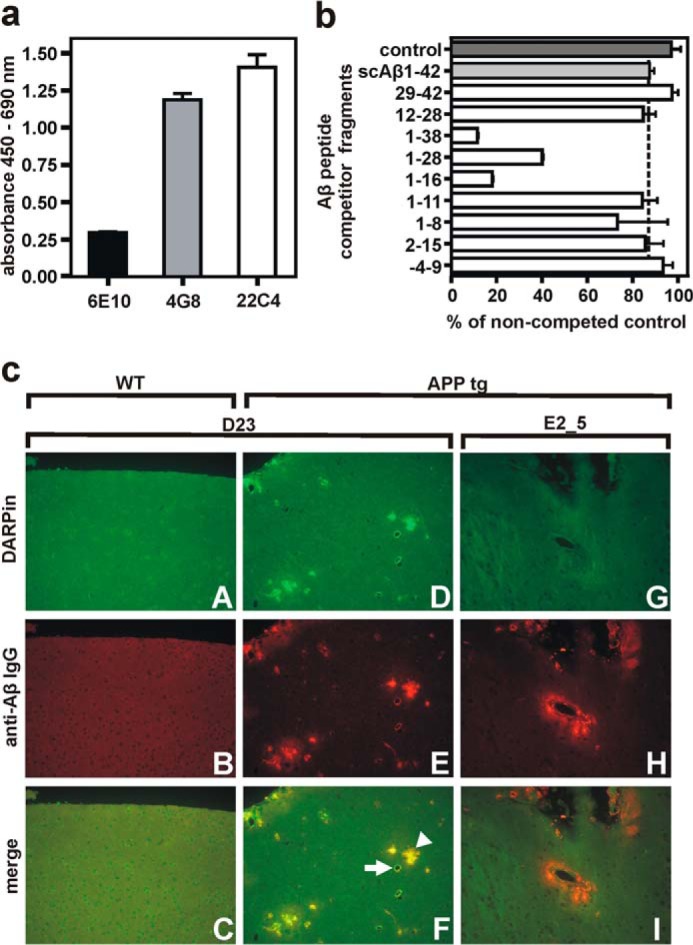
Determination of the DARPin D23 epitope and binding to amyloid deposits in ex vivo murine brain tissue. a, in a direct ELISA, Aβ(1–42) was bound to immobilized DARPin D23, and the accessibility of different parts of the peptide was probed with monoclonal anti-Aβ antibodies directed against the N terminus (6E10), central part (4G8), or C terminus (22c4) of the peptide. The lowest binding signal was observed for antibody 6E10, specific for the N terminus of Aβ. b, determination of the Aβ epitope recognized by D23 by competition with a 50-fold molar excess of Aβ-derived peptide fragments. Biotinylated Aβ(1–28)-peptide was immobilized via streptavidin, and ELISA signals were normalized to a noncompeted control. Inhibition was considered specific if signals were below that of scrambled Aβ(1–42) control (dotted line). c, GFP-fused DARPin D23 specifically recognized amyloid-β plaques in brain slices from transgenic ArcAβ mice. The specificity of plaque binding by the selected DARPin D23 was confirmed by applying DARPins C-terminally fused to superfolder GFP (sfGFP) onto brain sections from wild-type mice (panels A–C) and ArcAβ (panels D–F) transgenic mice. The control DARPin E2_5 did not show any binding activity (panels G–I). D23 recognized vascular (arrow) and parenchymal (arrowhead) amyloid deposits without cross-reactivity to WT brain structures. Top row, DARPin binding (GFP channel); middle row, anti-Aβ antibody binding (antibody 4G8, Cy3 channel); bottom row, merge of GFP and Cy3 channel. Data in a and b are means from three independent experiments; represented as means ± S.D.
To dissect the DARPin-Aβ interaction more closely, a peptide scanning approach with soluble short Aβ fragments was used to compete the DARPin-Aβ(1–28) interaction (Fig. 2b). A 50-fold molar excess of partially overlapping peptide fragments was used for competition, and only signals below those of scrambled Aβ(1–42) were regarded as specific inhibition. Not only was the importance of the N-terminal aspartate residue confirmed for high affinity binding, but we also observed that longer peptide fragments (Aβ(1–16) and Aβ(1–38)) with a free N terminus inhibited the interaction more effectively than shorter ones, suggesting that the N-terminal stretch is part of a conformational rather than a linear epitope. In an immunohistochemical analysis of brain sections from APP transgenic mice, we observed the specific staining of parenchymal and vascular amyloid deposits through D23 but not by the unselected control DARPin E2_5 (Fig. 2c). Importantly, D23 does not cross-react with other brain structures in wild-type (WT) mice.
D23 Prevents Aβ(1–42) Aggregation and Mediated Neurotoxicity
To study the DARPin's functional effect on Aβ aggregation, equimolar concentrations of Aβ(1–42) were incubated under constant agitation either alone or with DARPins D23 and the control DARPin E2_5, and thioflavin T (ThT) fluorescence signals were recorded over time (Fig. 3a). Although a 5 μm preparation of monomeric Aβ(1–42) readily aggregated into fibrils, the addition of equimolar amounts of D23, but not E2_5, was able to significantly prolong the lag time and reduce the elongation rate of Aβ oligomers thereby delaying the formation of higher molecular weight aggregation products. The addition of D23 to the aggregation assay resulted in a 7-fold prolonged elongation rate (elongation rates: 8.58 ± 0.13 min−1 (Aβ only), 6.28 ± 0.09 min−1 (Aβ + E2_5), and 1.17 ± 0.01 min−1 (Aβ + D23)), whereas E2_5 only had a minor effect on the formation of Aβ fibrils. We hypothesize that D23 stabilizes the monomeric form of Aβ peptide and sequesters it from the dynamic aggregation equilibrium. This idea is underscored by the observation that D23 does not interfere with the Aβ aggregation process if added during the log phase or after the plateau has been reached, but it delays the aggregation process if added in substoichiometric amounts at the beginning of the assay (Fig. 3b).
FIGURE 3.

Inhibition of Aβ aggregation and Aβ-mediated neurotoxicity through D23 addition. a, inhibitory effect of DARPin D23 on Aβ(1–42) aggregation was monitored by ThT fluorescence. Equimolar concentrations of Aβ(1–42) and DARPins D23 or E2_5 (5 μm each) were co-incubated, and ThT fluorescence was monitored for 3 h at 489 nm. The assays were performed in triplicate in stirred cuvettes. Data are presented as means ± S.D. b, half-equimolar concentrations of D23 were added at different time points during the exponential growth phase of the Aβ fibrils. The addition occurred at t1 = 20 min (red curve) and t2 = 37 min (gray curve). c, effects of Aβ(1–42) and DARPin addition on neuronal morphology. Upper panels, neurons were incubated either in the absence (−) or in the presence (+) of 5 μm Aβ(1–42). Aβ deposits were stained orange (polyclonal anti-Aβ antibody) and neurons green (antibody to MAP2). Lower panels, co-incubation of Aβ(1–42) with Aβ-specific DARPin D23 (left, lower panel) or control DARPin E2_5 (right, lower panel). Insets to all four micrographs show magnifications of anti-MAP2-stained neurons from the aforementioned treatment conditions. Scale bars correspond to 20 μm. d, primary rat neurons (day 5 in vitro) were co-incubated with equimolar amounts of recombinant Aβ(1–42) and DARPins D23 or E2_5 (all components 5 μm). After 48 h, the level of Aβ(1–42)-mediated toxicity was assessed by the activity of neuronal proteases (cleaving luciferin off a coupled substrate). D23 is able to significantly decrease Aβ(1–42)-mediated cytotoxicity as compared with controls (mean ± S.E.; *, p < 0.05; one-way analysis of variance, followed by Tukey's test).
Aβ-mediated neurotoxicity, as measured by the release of cytoplasmic proteases from rat primary cortical neurons, was quantified either alone or in equimolar combination of Aβ with DARPin D23 or the control DARPin E2_5. The anti-Aβ DARPin could reduce toxicity by about 40%, although E2_5 did not show any effect on neurotoxicity (Fig. 3d). We previously determined a 48-h incubation of 5 μm Aβ as most toxic to a culture of primary neurons, although an equimolar preparation of scrambled Aβ(1–42) did not induce any toxicity. In microscopy experiments, we observed that the addition of D23 had a pronounced effect not only on the size of the Aβ aggregates, but it also prevented dendrite retraction thereby contributing to a preserved neuronal morphology (Fig. 3c).
Multivalent DARPins Show Higher Avidity on Immobilized Aβ(1–42)
To further optimize Aβ binding, we constructed multivalent DARPins that were connected by flexible amino acid linkers. All constructs eluted as stable monomers from a size exclusion column with apparent molecular masses of 12 kDa (D23), 30 kDa (2×D23), and 44 kDa (3×D23) (Fig. 4a). In ELISAs with coated Aβ(1–28), the concentration of half-maximal signal (EC50 values) of the multivalent DARPins was only slightly improved over the monomeric DARPin, consistent with only a partial multivalent engagement, presumably because head-to-tail fusions interfere with the unidirectional display of surface-immobilized Aβ(1–28) (data not shown).
FIGURE 4.

Stability and binding of multivalent DARPin constructs to immobilized Aβ(1–42). a, multivalent DARPin variants of D23 were constructed by fusing monovalent units by (Gly4Ser)2 linkers. These constructs were well expressed and eluted as monomeric peaks from size exclusion chromatography with apparent molecular masses of 12 kDa (VE = 1.86 ml, D23), 30.4 kDa (VE = 1.57 ml, 2×D23), and 44.3 kDa (VE = 1.45 ml, 3×D23). The elution volumes of molecular mass standards are shown above the graph. mAU, arbitrary units. b, surface plasmon resonance kinetics of monovalent DARPin D23 on surface-immobilized Aβ(1–42). The C-terminally biotinylated peptide Aβ(1–42) was immobilized on a streptavidin sensor chip, and monovalent DARPin D23 was injected in duplicate at increasing concentrations of 1–128 nm. Association and dissociation phases were recorded, and the sensorgrams were globally fitted (red dotted lines) to a 1:1 binding model. c, equilibrium binding constant (KD) of monovalent D23 was determined as 1.59 × 10−7 m from evaluation of the plateau heights as a function of D23 concentration. d, SPR kinetics of trivalent DARPin on surface-immobilized Aβ(1–42). Trivalent 3×D23 was attempted to be fitted with a bivalent binding model (red dotted lines), but the more complicated situations with several modes of bivalent and possibly trivalent binding are not described by the fits well enough and preclude a numerical evaluation.
An analysis of the association and dissociation phases via SPR revealed substantial differences in the association and dissociation phases of the individual constructs (Fig. 4, b and d). Although the monovalent DARPin D23 (Fig. 4b) showed very fast equilibration precluding a kinetic evaluation, the trivalent construct gave rise to slower and multiphasic dissociation kinetics, indicating multivalent binding to the immobilized Aβ(1–42) on the sensor chip. The monovalent construct was evaluated by measuring the plateau levels, indicating a KD of 1.589 × 10−7 m (Fig. 4c). Because the observed association phase is the sum of the association and the dissociation process, they appear slower for the trivalent construct, simply because koff gets slower. Because of the various modes of multivalent binding, the sensorgrams of 3×D23 cannot be satisfactorily fit, and thus no kinetic constants are reported (Fig. 4d).
Encouraged by the long term stability and preservation of Aβ binding activity of D23 under physiological conditions of at least 3 weeks (data not shown), in accordance with previous descriptions (15), we tested the effect of intracerebroventricular infusions of mono- and trivalent DARPin constructs in Tg2576 mice, which express the human APP695 isoform with the double mutation K670N/M671L (24).
Intracerebroventricular DARPin Infusion Improves Cognition and Lowers the Soluble Aβ Pool in a Mouse Model of Brain Amyloidosis
Two weeks after intracerebroventricular implantation of osmotic pumps, all Tg2576 mice underwent Y-maze behavioral testing, a paradigm for testing working memory in APP transgenic mice (28, 29). Cognitive assessment in the Y-maze revealed a significant improvement in percentage alternation, as a measure for memory function upon treatment with both mono- and trivalent anti-Aβ DARPins, as compared with E2_5 and PBS-treated control mice (p < 0.05, Fig. 5a). The number of total arm entries and distance traveled did not differ between groups (data not shown), excluding a confounding effect due to altered activity of the mice. No differences in general health measures or any treatment-induced side effects were observed across all experimental groups.
FIGURE 5.

Restoration of memory deficits in APPSwe transgenic mice by DARPin treatment and influence on soluble brain Aβ levels. a, 2 weeks after continuous intracerebroventricular infusion of monovalent (hatched), trivalent (black) DARPins, nonspecific DARPin (E2_5, gray), or PBS (white), Tg2576 mice were cognitively assessed in a Y-maze experiment by recording the number of alternating arm entries. b, after 4 weeks of anti-Aβ therapy, animals were sacrificed, and the brain cortical area covered by amyloid deposits determined. DARPin treatment appeared to influence amyloid deposition but did not reach significance. c–h, total levels of both soluble (RIPA dissolved, c–e) and insoluble (formic acid dissolved (f–h)) brain Aβ(1–38), Aβ(1–40), and Aβ(1–42) levels were measured with a human Aβ triplex kit. Animals per group, n = 10–12. Data are shown as mean ± S.E. Statistical analysis by analysis of variance was followed by Mann-Whitney U test; *, p < 0.05; # trend p < 0.10.
After 4 weeks of infusion, animals were perfused and a compact plaque load in the cortex was quantified by thioflavine S histochemical analysis of brain sections. Already after 4 weeks of treatment a reduction of plaque area by 23% in both D23 and 3×D23 mice was revealed, as compared with E2_5 and PBS-treated mice, but did not reach statistical significance (Fig. 5b). As a next step, we determined Aβ levels biochemically in two sequentially extracted protein fractions from hemi-brain tissue (Fig. 5, c–h). In agreement with our histological findings, D23 and 3×D23-treated mice showed a significant reduction of Aβ(1–40) (by 21 and 16%), Aβ(1–38) (by 21 and 16%), and Aβ(1–42) (by 31 and 25%), respectively, in the detergent-soluble (RIPA-soluble, Fig. 5, c–e) protein fraction, as compared with E2_5 and PBS control mice. A similar trend was observed in the detergent-insoluble (formic acid soluble, Fig. 5, f–h) Aβ pool, although the effect was not significant due to the initiation phase of amyloid deposition and concomitantly higher variability of plaque load in these animals. These findings provide the first in vivo evidence that DARPins may successfully reverse Aβ-mediated cognitive deficits in a transgenic mouse model of brain amyloidosis and that the continuous infusion of DARPins can have a modulatory effect on brain Aβ levels.
DISCUSSION
Ribosome display of combinatorial DARPin libraries has been utilized to select for DARPin binders against a diverse set of folded target proteins (10), but selection against peptide antigens without defined structure has not been reported. In this study, we were able to select a DARPin binder, termed D23, which showed nanomolar affinities for the Aβ peptide in both direct and competition ELISAs. Therefore, D23 was selected as the lead candidate for further characterization and to test the therapeutic potential of DARPins administered in vivo for the treatment of brain amyloidosis in APP transgenic mice.
The binding of D23 to surface-immobilized Aβ(1–28) and Aβ(1–42) was shown to be in the low nanomolar range; however, the physiologically more relevant specific binding to Aβ in solution (as it principally occurs in prodromal phases of AD in vivo) is difficult to address and needs to be clarified. Aβ can occur in solution in various quaternary structures, ranging from soluble monomers (30), oligomeric intermediates (31), to fibrillar states (32). Because these forms differ in conformation, accessibility of epitopes and the possibility of bi- or even multivalent interactions, it is not surprising that Aβ-binding macromolecules can display varying specificities for Aβ, depending on its state.
A competition assay using freshly dissolved monomeric Aβ(1–42) as competitor confirmed the observations made with surface-immobilized Aβ and suggested a KI value of ∼50 nm. This inhibition was specific and not due to general hydrophobic interactions, as the use of scrambled Aβ(1–42) did not significantly reduce D23 binding to the surface-immobilized target (Fig. 2b). We had noticed that C-terminal amino acid extensions (to result in a peptide longer than the 28 residues used for selection) resulted in slightly decreased apparent affinities, possibly because some molecules had formed aggregates and were not able to bind.
We demonstrated that the free N terminus of the peptide must be involved in D23 binding. Furthermore, we could confirm the N-terminal Aβ domain to be involved in DARPin recognition, and of the several monoclonal antibodies with comparable affinities tested (33), 6E10 was most impaired in binding the complex of D23 and Aβ(1–42) and thus competes for the epitope. A peptide scanning approach enlarged this binding region to the central Aβ domain, suggesting the binding of a discontinuous rather than linear epitope, as generally described for DARPins binding to different targets (10, 12, 34). Aβ is thought to contain a largely unstructured N terminus (residues 1–11) and two domains spanning residues 12–21 and 24–42, respectively, with varying contacts among each other, whereas the connecting residues (22 and 23) face the solvent (35). The particular binding mode of D23 is thus different from that of previously reported monoclonal antibodies that recognize a linear stretch of the Aβ peptide and also cross-react to APP (at least 6E10 and 4G8) (36).
The targeting of conformational Aβ epitopes is highly advantageous for the specificity of anti-Aβ therapeutics. As the conformation of the free monomeric Aβ peptide is most likely different from the one inside APP, D23 cross-reactivity to APP is limited (as indeed observed in cell culture binding experiments, data not shown) and could enable repeated dosing without the risk of peripheral or central APP to either act as off-target scavenger or to create an immune reaction against APP-carrying cells. To determine the binding characteristics of D23 ex vivo, we performed immunohistochemical analyses with GFP-fused DARPins on brain tissue from an ArcAβ mouse model of AD (Fig. 2c). The staining of Aβ deposits in this model with an increased proportion of vascular amyloid demonstrated D23 to preferentially bind the cross-β-pleated dense central cores of the amyloid deposits, in contrast to more diffuse anti-Aβ antibody stainings. Such findings underscore D23's recognition of discontinuous amino acid stretches on both the N-terminal and central domain of the Aβ peptide that still exist in both the monomeric and aggregated form. Nonspecific adherence of the DARPin scaffold to the “sticky” amyloid plaques could be excluded, as no tissue reactivity was observed with the nonspecific E2_5 control DARPin.
As a first test of the DARPin's in vitro activity, we were able to demonstrate that D23 delays Aβ aggregation (Fig. 3a), using ThT aggregation assays. In contrast, the addition of D23 during the lag phase or after complete fibrillization did not lead to any decrease in ThT signal. This likely indicates that D23 binds to and stabilizes a monomeric form of Aβ and excludes it from the aggregation process. We were further able to demonstrate that D23, but not the control DARPin E2_5, when administered into the medium of rat primary cortical neuron cultures, could reduce Aβ-mediated neurotoxicity and preserve neuronal morphology (Fig. 3, b and c).
To optimize D23 binding behavior for potential therapeutic use, a trivalent DARPin was engineered by fusing three monovalent units of D23 into one continuous open reading frame. Although these constructs showed similar stability and resistance against degradation and bound the Aβ peptide with nanomolar affinity, surface plasmon resonance kinetic analysis revealed differences in on- and off-rates for each individual construct (Fig. 4, b–d). The increased molecular weight of multivalent DARPins reduces diffusion and potentially limits their tissue penetration, although their multivalency prolongs the half-life of DARPin-Aβ complexes in case of polymeric Aβ surfaces such as oligomers and fibrils.
Finally, we were able to demonstrate the in vivo therapeutic potential of D23 and 3×D23 in Tg2576 mice. Following the intracerebroventricular infusion of mono- and trivalent DARPin constructs over a 28-day treatment, D23- and 3×D23-treated mice showed improved cognitive performance, compared with mice treated with the control DARPin E2_5 or PBS. These results were supported by histochemical analyses, showing a trend for reduction of plaque deposition in both D23- and 3×D23-treated mice (Fig. 5b). However, as we infused anti-Aβ DARPins in Tg2576 mice already at 11 months of age, the majority of the amyloid burden is still present in soluble Aβ forms, explaining the limited number of amyloid deposits found in the cortices of treated animals.
This preventive treatment paradigm was accompanied by a biochemical analysis that showed significant reductions of brain Aβ levels for both D23- and 3×D23-treated mice with the most pronounced modulatory effects on the soluble Aβ pool (Fig. 5, c–e). Interestingly, despite anti-Aβ multivalency, 3×D23 did not perform better than its monovalent counterpart D23 in vivo. There are several possible explanations for this finding. If the action of D23 predominantly involves binding of monomeric Aβ, then a trimer would have, at most, the same activity as three monomers, but avidity would play no role. It is also possible that the increased molecular weight of 3×D23 could have restricted its rapid tissue penetration such that it could not sufficiently benefit from the additional interaction surfaces (15, 37).
DARPins lack the Fc domain intrinsic to the immunoglobulin G molecule, which has been shown to activate resident microglia in the vicinity of antibody-decorated amyloid deposits (29, 38, 39). Activated microglia can degrade fibrillar Aβ by lysosomal proteolysis, but local inflammatory reactions might, at worst, result in microhemorrhages (29, 40, 41). The biological activity of DARPins could be based on their association with these deposits, and they could sterically block recruitment of Aβ monomers to the amyloid plaque by binding to monomers, or they could clear amyloid deposits independent of microglia in analogy to other molecular scaffolds without a functional Fc domain (42–44). DARPin-mediated Aβ removal would have to rely on the capture and elimination of these species from the central nervous system via the bulk flow of interstitial fluid (45).
Our findings demonstrate the therapeutic potential of Aβ-specific DARPins for the treatment of Alzheimer disease. With the advent of recombinant protein expression and efficient in vitro selection technologies, this novel class of engineered protein scaffolds presents attractive opportunities for both diagnostic and therapeutic use. If longer serum half-lives are required, this could be achieved by site-specific PEGylation (15) or by hijacking an abundant serum protein with a long half-life, like albumin or immunoglobulin G through peptide tags that bind to them to escape glomerular filtration (46, 47). Knowing the recent set backs of human anti-Aβ therapies, this study opens new avenues away from antibodies toward DARPins to fight brain amyloidosis effectively.
Acknowledgments
We thank K. Wollenick, W. Buck, N. Lucke, and A. Jeske for excellent technical support.
This work was supported in part by the Swiss National Science Foundation and a Forschungskredit grant of the University of Zurich. S. Cattepoel is now an employee of CSL Behring AG, but declares no competing interests. M. Maier is now an employee of Neurimmune Holding AG, but declares no competing interests. A. Plückthun is a shareholder of Molecular Partners, commercializing the DARPin technology.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- APP
- amyloid precursor protein
- SPR
- surface plasmon resonance
- DARPin
- designed ankyrin repeat protein
- ThT
- thioflavin T.
REFERENCES
- 1. Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 2. Citron M. (2010) Alzheimer's disease: strategies for disease modification. Nat. Rev. Drug Discov. 9, 387–398 [DOI] [PubMed] [Google Scholar]
- 3. Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug. Discov. 10, 698–712 [DOI] [PubMed] [Google Scholar]
- 4. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 [DOI] [PubMed] [Google Scholar]
- 5. Morgan D., Diamond D. M., Gottschall P. E., Ugen K. E., Dickey C., Hardy J., Duff K., Jantzen P., DiCarlo G., Wilcock D., Connor K., Hatcher J., Hope C., Gordon M., Arendash G. W. (2000) Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408, 982–985 [DOI] [PubMed] [Google Scholar]
- 6. Rinne J. O., Brooks D. J., Rossor M. N., Fox N. C., Bullock R., Klunk W. E., Mathis C. A., Blennow K., Barakos J., Okello A. A., Rodriguez Martinez de Liano S., Liu E., Koller M., Gregg K. M., Schenk D., Black R., Grundman M. (2010) 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 9, 363–372 [DOI] [PubMed] [Google Scholar]
- 7. Siemers E. R., Friedrich S., Dean R. A., Gonzales C. R., Farlow M. R., Paul S. M., Demattos R. B. (2010) Safety and changes in plasma and cerebrospinal fluid amyloid β after a single administration of an amyloid β monoclonal antibody in subjects with Alzheimer disease. Clin. Neuropharmacol. 33, 67–73 [DOI] [PubMed] [Google Scholar]
- 8. Relkin N. R., Szabo P., Adamiak B., Burgut T., Monthe C., Lent R. W., Younkin S., Younkin L., Schiff R., Weksler M. E. (2009) 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol. Aging 30, 1728–1736 [DOI] [PubMed] [Google Scholar]
- 9. Delrieu J., Ousset P. J., Caillaud C., Vellas B. (2012) “Clinical trials in Alzheimer's disease”: immunotherapy approaches. J. Neurochem. 120, 186–193 [DOI] [PubMed] [Google Scholar]
- 10. Boersma Y. L., Plückthun A. (2011) DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr. Opin. Biotechnol. 22, 849–857 [DOI] [PubMed] [Google Scholar]
- 11. Binz H. K., Stumpp M. T., Forrer P., Amstutz P., Plückthun A. (2003) Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332, 489–503 [DOI] [PubMed] [Google Scholar]
- 12. Binz H. K., Amstutz P., Kohl A., Stumpp M. T., Briand C., Forrer P., Grütter M. G., Plückthun A. (2004) High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22, 575–582 [DOI] [PubMed] [Google Scholar]
- 13. Amstutz P., Binz H. K., Parizek P., Stumpp M. T., Kohl A., Grütter M. G., Forrer P., Plückthun A. (2005) Intracellular kinase inhibitors selected from combinatorial libraries of designed ankyrin repeat proteins. J. Biol. Chem. 280, 24715–24722 [DOI] [PubMed] [Google Scholar]
- 14. Kohl A., Amstutz P., Parizek P., Binz H. K., Briand C., Capitani G., Forrer P., Plückthun A., Grütter M. G. (2005) Allosteric inhibition of aminoglycoside phosphotransferase by a designed ankyrin repeat protein. Structure 13, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 15. Zahnd C., Kawe M., Stumpp M. T., de Pasquale C., Tamaskovic R., Nagy-Davidescu G., Dreier B., Schibli R., Binz H. K., Waibel R., Plückthun A. (2010) Efficient tumor targeting with high affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 70, 1595–1605 [DOI] [PubMed] [Google Scholar]
- 16. Banks W. A., Terrell B., Farr S. A., Robinson S. M., Nonaka N., Morley J. E. (2002) Passage of amyloid β protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides 23, 2223–2226 [DOI] [PubMed] [Google Scholar]
- 17. Bard F., Cannon C., Barbour R., Burke R. L., Games D., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Lieberburg I., Motter R., Nguyen M., Soriano F., Vasquez N., Weiss K., Welch B., Seubert P., Schenk D., Yednock T. (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919 [DOI] [PubMed] [Google Scholar]
- 18. Levites Y., Smithson L. A., Price R. W., Dakin R. S., Yuan B., Sierks M. R., Kim J., McGowan E., Reed D. K., Rosenberry T. L., Das P., Golde T. E. (2006) Insights into the mechanisms of action of anti-Aβ antibodies in Alzheimer's disease mouse models. FASEB J. 20, 2576–2578 [DOI] [PubMed] [Google Scholar]
- 19. Stumpp M. T., Binz H. K., Amstutz P. (2008) DARPins: a new generation of protein therapeutics. Drug Discov. Today 13, 695–701 [DOI] [PubMed] [Google Scholar]
- 20. Cattepoel S., Hanenberg M., Kulic L., Nitsch R. M. (2011) Chronic intranasal treatment with an anti-Aβ30–42 scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer's disease. PLoS ONE 6, e18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zahnd C., Amstutz P., Plückthun A. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4, 269–279 [DOI] [PubMed] [Google Scholar]
- 22. Wetzel S. K., Settanni G., Kenig M., Binz H. K., Plückthun A. (2008) Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J. Mol. Biol. 376, 241–257 [DOI] [PubMed] [Google Scholar]
- 23. Finder V. H., Vodopivec I., Nitsch R. M., Glockshuber R. (2010) The recombinant amyloid-β peptide Aβ 1–42 aggregates faster and is more neurotoxic than synthetic Aβ 1–42. J. Mol. Biol. 396, 9–18 [DOI] [PubMed] [Google Scholar]
- 24. Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 25. Biscaro B., Lindvall O., Hock C., Ekdahl C. T., Nitsch R. M. (2009) Aβ immunotherapy protects morphology and survival of adult-born neurons in doubly transgenic APP/PS1 mice. J. Neurosci. 29, 14108–14119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim K. S., Miller D. L., Sapienza V. J., Chen C. M., Bai C., Gundke-Iqbal I., Currie J. R., Wisniewski H. M. (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci. Res. Commun. 2, 121–130 [Google Scholar]
- 27. Mohajeri M. H., Saini K., Schultz J. G., Wollmer M. A., Hock C., Nitsch R. M. (2002) Passive immunization against β-amyloid peptide protects central nervous system (CNS) neurons from increased vulnerability associated with an Alzheimer's disease-causing mutation. J. Biol. Chem. 277, 33012–33017 [DOI] [PubMed] [Google Scholar]
- 28. Arendash G. W., Gordon M. N., Diamond D. M., Austin L. A., Hatcher J. M., Jantzen P., DiCarlo G., Wilcock D., Morgan D. (2001) Behavioral assessment of Alzheimer's transgenic mice following long-term Aβ vaccination: task specificity and correlations between Aβ deposition and spatial memory. DNA Cell Biol. 20, 737–744 [DOI] [PubMed] [Google Scholar]
- 29. Wilcock D. M., Rojiani A., Rosenthal A., Levkowitz G., Subbarao S., Alamed J., Wilson D., Wilson N., Freeman M. J., Gordon M. N., Morgan D. (2004) Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. 24, 6144–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeMattos R. B., Bales K. R., Cummins D. J., Dodart J.-C., Paul S. M., Holtzman D. M. (2001) Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 98, 8850–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 32. O'Nuallain B., Wetzel R. (2002) Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. U.S.A. 99, 1485–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramakrishnan M., Kandimalla K. K., Wengenack T. M., Howell K. G., Poduslo J. F. (2009) Surface plasmon resonance binding kinetics of Alzheimer's disease amyloid β peptide-capturing and plaque-binding monoclonal antibodies. Biochemistry 48, 10405–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schweizer A., Roschitzki-Voser H., Amstutz P., Briand C., Gulotti-Georgieva M., Prenosil E., Binz H. K., Capitani G., Baici A., Plückthun A., Grütter M. G. (2007) Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure 15, 625–636 [DOI] [PubMed] [Google Scholar]
- 35. Vitalis A., Caflisch A. (2010) Micelle-like architecture of the monomer ensemble of Alzheimer's amyloid-β peptide in aqueous solution and its implications for Aβ aggregation. J. Mol. Biol. 403, 148–165 [DOI] [PubMed] [Google Scholar]
- 36. Tampellini D., Magrané J., Takahashi R. H., Li F., Lin M. T., Almeida C. G., Gouras G. K. (2007) Internalized antibodies to the Aβ domain of APP reduce neuronal Aβ and protect against synaptic alterations. J. Biol. Chem. 282, 18895–18906 [DOI] [PubMed] [Google Scholar]
- 37. Colcher D., Pavlinkova G., Beresford G., Booth B. J., Choudhury A., Batra S. K. (1998) Pharmacokinetics and biodistribution of genetically-engineered antibodies. Q. J. Nucl. Med. 42, 225–241 [PubMed] [Google Scholar]
- 38. Koenigsknecht-Talboo J., Meyer-Luehmann M., Parsadanian M., Garcia-Alloza M., Finn M. B., Hyman B. T., Bacskai B. J., Holtzman D. M. (2008) Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. J. Neurosci. 28, 14156–14164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang A., Das P., Switzer R. C., 3rd, Golde T. E., Jankowsky J. L. (2011) Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-β immunotherapy. J. Neurosci. 31, 4124–4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Racke M. M., Boone L. I., Hepburn D. L., Parsadainian M., Bryan M. T., Ness D. K., Piroozi K. S., Jordan W. H., Brown D. D., Hoffman W. P., Holtzman D. M., Bales K. R., Gitter B. D., May P. C., Paul S. M., DeMattos R. B. (2005) Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J. Neurosci. 25, 629–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfeifer M., Boncristiano S., Bondolfi L., Stalder A., Deller T., Staufenbiel M., Mathews P. M., Jucker M. (2002) Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science 298, 1379. [DOI] [PubMed] [Google Scholar]
- 42. Bacskai B. J., Kajdasz S. T., McLellan M. E., Games D., Seubert P., Schenk D., Hyman B. T. (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J. Neurosci. 22, 7873–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilcock D. M., DiCarlo G., Henderson D., Jackson J., Clarke K., Ugen K. E., Gordon M. N., Morgan D. (2003) Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J. Neurosci. 23, 3745–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilcock D. M., Alamed J., Gottschall P. E., Grimm J., Rosenthal A., Pons J., Ronan V., Symmonds K., Gordon M. N., Morgan D. (2006) Deglycosylated anti-amyloid-β antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J. Neurosci. 26, 5340–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shibata M., Yamada S., Kumar S. R., Calero M., Bading J., Frangione B., Holtzman D. M., Miller C. A., Strickland D. K., Ghiso J., Zlokovic B. V. (2000) Clearance of Alzheimer's amyloid-β1–40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Invest. 106, 1489–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dennis M. S., Zhang M., Meng Y. G., Kadkhodayan M., Kirchhofer D., Combs D., Damico L. A. (2002) Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 277, 35035–35043 [DOI] [PubMed] [Google Scholar]
- 47. Nguyen A., Reyes A. E., 2nd, Zhang M., McDonald P., Wong W. L., Damico L. A., Dennis M. S. (2006) The pharmacokinetics of an albumin-binding Fab (AB. Fab) can be modulated as a function of affinity for albumin. Protein Eng. Des. Sel. 19, 291–297 [DOI] [PubMed] [Google Scholar]