

Background: Signaling pathways underlying beneficial effects of extracorporeal shock wave treatment (ESWT) remain to be completely elucidated.
Results: ESWT enhances cell proliferation in vitro and wound healing in vivo.
Conclusion: ESWT-induced ATP release and subsequent extracellular signal-regulated kinase (ERK) activation are prerequisites for enhanced cell proliferation and wound healing.
Significance: Deciphering the involved signaling cascades provides the basis for ESWT as clinical wound healing treatment.
Keywords: ATP, Cell Proliferation, Extracellular Signal-regulated Kinase (ERK), Signal Transduction, Wound Healing, Shock Wave Treatment
Abstract
Shock wave treatment accelerates impaired wound healing in diverse clinical situations. However, the mechanisms underlying the beneficial effects of shock waves have not yet been fully revealed. Because cell proliferation is a major requirement in the wound healing cascade, we used in vitro studies and an in vivo wound healing model to study whether shock wave treatment influences proliferation by altering major extracellular factors and signaling pathways involved in cell proliferation. We identified extracellular ATP, released in an energy- and pulse number-dependent manner, as a trigger of the biological effects of shock wave treatment. Shock wave treatment induced ATP release, increased Erk1/2 and p38 MAPK activation, and enhanced proliferation in three different cell types (C3H10T1/2 murine mesenchymal progenitor cells, primary human adipose tissue-derived stem cells, and a human Jurkat T cell line) in vitro. Purinergic signaling-induced Erk1/2 activation was found to be essential for this proliferative effect, which was further confirmed by in vivo studies in a rat wound healing model where shock wave treatment induced proliferation and increased wound healing in an Erk1/2-dependent fashion. In summary, this report demonstrates that shock wave treatment triggers release of cellular ATP, which subsequently activates purinergic receptors and finally enhances proliferation in vitro and in vivo via downstream Erk1/2 signaling. In conclusion, our findings shed further light on the molecular mechanisms by which shock wave treatment exerts its beneficial effects. These findings could help to improve the clinical use of shock wave treatment for wound healing.
Introduction
The application of extracorporeal shock wave treatment (ESWT)3 is a novel and effective therapeutic strategy to treat musculoskeletal disorders as well as chronic soft tissue wounds including bedsores, burn wounds, and diabetic and vascular ulcers (1, 2). In addition to the accelerated healing rates in patients treated with ESWT, this therapeutic approach is non-invasive and cost-effective. Despite the growing evidence of the beneficial clinical effects of ESWT in treating musculoskeletal disorders and soft tissue wounds in animal models and clinical studies (3–10), the underlying mechanisms of how ESWT exerts these beneficial effects have remained unclear.
The process of wound healing consists of four major phases, namely hemostasis, inflammation, proliferation, and tissue remodeling that involve complex cellular processes such as chemotaxis, phagocytosis, angiogenesis, collagen synthesis, and epithelialization (11). In recent years, various research groups have focused on the angiogenic impact of shock waves (4, 5, 12, 13). They have shown that ESWT up-regulates angiogenesis via mediators such as vascular endothelial growth factor (VEGF), endothelial nitric-oxide synthase, hypoxia-inducible factor 1a, and CD31 (4, 5, 14, 15). ESWT was shown to augment the expression of proliferating cell nuclear antigen and the recruitment of fibroblasts and to down-regulate production of proinflammatory cytokines (16). Other studies have shown that the expression of certain cytokines, chemokines, and matrix metalloproteinases with proangiogenic roles is enhanced during ESWT-promoted wound healing (12).
Shock waves generate an impulse that elicits mechanical stimulation via pressure changes that elicit a biological response such as cell differentiation and proliferation (17, 18). Shock waves are capable of initiating such cellular responses by triggering various intracellular signaling events that involve integrins, calcium channels, phospholipase C, and mitogen-activated protein kinases (MAPKs). For example, in osteoblasts, one of the earliest intracellular signaling responses caused by mechanical stimulation is the phosphorylation of extracellular signal-regulated kinase 1/2 (Erk1/2) (18). In a similar fashion, ESWT has been shown to induce Erk1/2 or p38 MAPK activation in mesenchymal stem cells, immune cells, and osteoblasts, resulting in distinct cellular effects including osteogenic differentiation of mesenchymal stem cells, angiogenesis, and T cell proliferation (19–24). MAPK signaling pathways are known to play a central role in cell proliferation, differentiation, apoptosis, inflammation, and development (25–28). Moreover, Erk1/2 can induce hypoxia-inducible factor 1a activation and angiogenesis via VEGFA expression (20).
Recent evidence suggests that shock waves trigger adenosine triphosphate (ATP) release by activating purinergic signaling and p38 MAPK activation (22, 23). The crucial role of purinergic signaling in the regulation of numerous physiological processes has only recently been appreciated (29, 30). In addition to the regulation of neurotransmission and -modulation, it is involved in differentiation, motility, apoptosis, regeneration, and proliferation. ATP that is released from stimulated cells can bind to P2X and P2Y purinergic receptors, which are ion channels and G-protein coupled receptors, respectively (29, 31). Purinergic signaling also contributes to wound healing (29). Signaling in wound repair is to some extent dependent on the local release of ATP from the wound that subsequently causes the activation of Erk1/2 effector pathways to induce wound healing (32). In our current study, we show that shock wave treatment triggers the release of cellular ATP, which in turn elicits Erk1/2 signaling pathway activation and increases cell proliferation in vitro and wound healing in vivo.
EXPERIMENTAL PROCEDURES
Reagents
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich. Antibodies for phospho-p38 MAPK (Thr-180/Tyr-182), total p38 MAPK, phospho-Akt (Ser-473), total Akt, phospho-p44/42 MAPK (Thr-202/Tyr-204) (phospho-Erk1/2), total p44/42 MAPK (total Erk1/2), phospho-Mek1/2 (Ser-217/221), total Mek1/2, phospho-S6 ribosomal protein (Ser-240/244), and total S6 ribosomal protein were obtained from Cell Signaling Technology. The secondary antibodies IRDye® 680LT donkey anti-rabbit IgG, IRDye 800CW goat anti-rabbit IgG, and IRDye 800CW goat anti-mouse IgG were obtained from LI-COR Biosciences.
Cell Culture
C3H10T1/2 is a stable multipotent mesenchymal cell line derived from mouse embryos with a finite lifetime (33). Cells were grown in basal medium Eagle supplemented with 10% FBS, 1% glutamine, and 1% penicillin-streptomycin. Cells used in experiments were between passages 7 and 11. Adipose tissue-derived stem cells (ASCs) were cultured in EGM-2 medium (Lonza, Walkersville, MD) supplemented with 5% FBS. Cells were used between passages 2 and 7. For proliferation experiments, ASCs were grown in DMEM/F-12 medium. Human Jurkat T cells were maintained in RPMI 1640 medium supplemented with 10% FBS, 1% glutamine, and 1% penicillin-streptomycin. To assess cell cycle progression and proliferation, cells were first deprived of serum for 24 h in medium containing 0.2% FBS (starvation medium) to obtain synchronization in G0/G1. Cells were then stimulated to re-enter the cell cycle by adding 10% FBS. The shock wave group additionally received shock wave treatment at this time point.
Shock Wave Treatment
In this study, shock wave treatment was performed with the electrohydraulic device DermaGold®100 and an OP155 applicator (Tissue Regeneration Technologies, LLC, manufactured by MTS Europe GmbH). To allow the unhampered physical propagation and reproducible application of shock waves to the sample in vitro, shock wave treatment was performed using a water bath setup (34) where 8 × 105 cells suspended in 400 μl of medium in a 15-ml polypropylene tube are exposed to shock waves under uniform and reproducible treatment conditions in terms of temperature and distance to the shock wave applicator. In vitro, 10–300 pulses of shock waves using energy flux densities between 0.03 and 0.19 mJ/mm2 at 3 Hz were applied. According to previous experiments and in vivo studies (35), 100 pulses at 0.13 mJ/mm2 and 3 Hz were used for the shock wave treatment in a rodent ischemic excision wound healing model. Control animals were treated identically but received no shock wave treatment.
Metabolic Activity
To exclude possible adverse effects of shock wave treatment on the metabolic activity of cells, the effect of 100 shock wave pulses at 0.07 and 0.19 mJ/mm2 was analyzed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. After shock wave treatment, cells were seeded to 96-well plates and incubated for the indicated time frames. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reagent was added at a final concentration of 650 μg/ml, and cells were incubated for 1 h at 37 °C in a 5% CO2 environment. Medium was discarded, precipitated formazan was dissolved in DMSO by mechanical shaking in the dark for 20 min, and absorbance was measured immediately at 540 nm.
Cell Proliferation
Propidium iodide DNA staining was used to specifically determine the amount of cells undergoing S phase. Cells were harvested by trypsinization and fixed by rapid submersion in ice-cold 85% EtOH. Samples were stored at −20 °C for at least overnight or longer. For cell cycle analysis, DNA was stained with 0.25 mg/ml propidium iodide, 0.05 mg/ml RNase A, and 0.01% Triton X-100 in citrate buffer, pH 7.8. Cells were analyzed on a BD FACSCanto II using BD FACSDiva software, and data were further processed using FlowJo software.
5-Bromo-2-deoxyuridine (BrdU) incorporation into newly synthesized DNA of cells treated with/without shock waves was used as an indicator for actively proliferating cells. The BrdU enzyme-linked immunosorbent assay (Roche Applied Science) was performed according to the manufacturer's instructions. In brief, cells were deprived of serum for growth arrest and restimulated by serum addition combined with/without shock wave treatment. Cells were then seeded into 96-well plates and incubated with media containing 100 μm BrdU for 3 h at the indicated time points. FixDenat® solution was added for 30 min followed by incubation with anti-BrdU peroxidase antibody for 1 h at room temperature. After three washing steps with PBS, tetramethyl benzidine was added as a substrate for 30 min. By adding 1 m H2SO4, the reaction was terminated, and absorbance was measured at 450 nm.
ATP Release
The amount of ATP release of C3H10T1/2 cells, Jurkat T cells, and adipose tissue-derived stem cells into the supernatant was determined with the CellTiter-Glo assay (Promega). Cells were adjusted to 8 × 105/400 μl and allowed to rest for 1 h at 37 °C in a humidified incubator before shock wave treatment was applied. Afterward cells were centrifuged at 1000 × g for 5 min at 4 °C, and 100 μl of supernatant was transferred to a 96-well plate. After an equal amount of CellTiter-Glo reagent was added, the plate was horizontally shaken for 2 min, and after incubation for 10 min at room temperature, the luminescence was measured. The calibration of measured luminescence to ATP concentrations was performed by using ATP standard solutions of known concentrations.
Immunoblotting
Total protein of cells was extracted by repeated freeze and thaw cycles. In brief, cells were harvested by trypsinization, and cell pellets were washed three times with PBS and lysed in Nonidet P-40 buffer containing 40 mm HEPES, pH 7.9, 120 mm NaCl, 1 mm EDTA, pH 8.0, 10 mm 2-glycerol phosphate, 50 mm NaF, 0.5 mm Na3VSO4, 1% Nonidet P-40 substitute, and 1 mm PMSF supplemented with 2 μg/ml aprotinin, 2 μg/ml leupeptin, 0.3 μg/ml benzamidine chloride, and 10 μg/ml trypsin inhibitor. Samples were incubated on ice for 20 min and then centrifuged at 15,000 rpm for 20 min at 4 °C. Supernatants were collected, and protein concentrations were determined (Protein Assay kit II, Bio-Rad).
Equal amounts of protein (up to 20 μg/lane) were resolved on an SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane. Membranes were blocked with 5% milk in TBS buffer with 0.1% Tween (TBS/T), and primary antibodies in 5% BSA in TBS/T were incubated at 4 °C overnight. Membranes were incubated in 5% milk-TBS/T containing secondary antibody, and signals were detected using the Odyssey Fc infrared imaging system (LI-COR Biosciences). After membranes were incubated in 1× Stripping Buffer (LI-COR Biosciences) on a shaker at room temperature for 5 min and washed three times in PBS, membranes were reprobed with total antibodies. Ratios of phosphorylated/total proteins were analyzed using densitometry (Image Studio Lite, LI-COR Biosciences).
Erk1/2 Inhibition
The Mek1/2 inhibitor U0126 (Cell Signaling Technology) was solubilized in DMSO and used in vitro to block Erk1/2 activation. Cells were serum-deprived using starvation medium for 24 h with 10 μm U0126 present for the last 2.5 h. Cells were detached, adjusted to 8 × 105/400 μl, and allowed to rest for 1 h followed by shock wave treatment or the addition of ATP to the supernatant of untreated cells at the indicated concentrations for 10 min, respectively. Cells were then either seeded on 96-well plates for cell proliferation assays or washed with PBS and processed for total protein isolation.
Purinergic Signaling
Cells were adjusted to 8 × 105/400 μl and allowed to rest at 37 °C in a humidified incubator for 30 min. Purinergic receptor antagonists suramin, pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate, and theophylline were added at a concentration of 100 μm, and cells were incubated for a further 30 min. Cells received either shock wave treatment at the indicated energies or ATP at the indicated concentrations added for 10 min. Directly after treatment, protein was isolated for immunoblotting. Purinergic receptor agonists adenosine, uridine triphosphate (UTP), ATP, and 2-MeSATP were added at the indicated concentrations (5, 50, or 100 μm) after cells were allowed to rest at 37 °C in a humidified incubator for 1 h. Cells were incubated with agonists at 37 °C for 10 min and immediately processed for protein isolation. For hydrolysis of released ATP in the supernatant of shock wave-treated cells, 20 units/ml apyrase was added prior to treatment.
Erk1/2 Inhibition in Vivo
Sprague-Dawley rats were randomly assigned to one of the following four groups: control group, control group receiving inhibitor, ESWT group, and ESWT group receiving inhibitor. Rats in the inhibitor groups were treated with GSK1120212 (AbMole BioScience), a potent inhibitor of Mek1/2 activity (36). GSK1120212 was dissolved in DMSO, diluted in hydroxyethylcellulose (1%, w/v), and administered orally once a day. Treatment with the inhibitor started 4 days before surgery with a singular dose of 0.3 mg/kg followed by 0.1 mg/kg GSK1120212 until day 10 after surgery. Control animals received vehicle (DMSO) in hydroxyethylcellulose via the same administration route.
Rodent Ischemic Excision Wound Healing Model
Forty male Sprague-Dawley rats, weighing between 350 and 450 g, were used in this study. A standard rodent ischemic epigastric flap model was modified to study the effect of shock wave treatment on ischemia-impaired wound healing (37).4 In brief, rats were anesthetized in an inhalation box using isoflurane (2.5 volume %). Anesthesia was maintained by intraperitoneal injection of a mixture of ketamine (110 mg/kg; Pharmacia and Upjohn, Germany) and xylazine (12 mg/kg; Bayer, Leverkusen, Germany).
The abdomen of the rats was shaved and depilated, and an epigastric adipocutaneous flap was created as described in detail elsewhere (35). Ligation of a unilateral inferior epigastric neurovascular bundle induces ischemia in the corresponding abdominal area, whereas the contralateral side remains adequately perfused by the intact neurovascular bundle. After the flap was sutured back, the ischemic side of the flap was treated with 100 shock wave pulses at 0.13 mJ/mm2. Thereafter, circular excision wounds with a diameter of 1.3 cm were created on both the ischemic and normal perfused sides, leaving the fascia intact. Thus, the wound healing progress in an ischemia-disturbed excision wound can be compared with the regular perfused internal counterpart. Control animals were treated identically without receiving shock wave treatment.
To determine the rate of wound healing, wounds were traced on a transparent acrylic foil, which was captured by digital imaging and further analyzed using a planimetric software program. Wound size obtained immediately after surgery represents baseline values, and wound size recordings from days 1, 5, and 10 after surgery were referred to this baseline. Wounds from the ischemic side reflect impaired healing and were compared with wound healing in the contralateral normal perfused area. Animals were euthanized on day 10, and tissue biopsies from all wounds with adjacent unaffected tissue were harvested for histological and immunohistochemical analysis.
Histology and Immunohistochemistry
Full-thickness biopsies from the excision wounds were processed according to standard procedures. In brief, samples were fixed in formalin for 24 h, dehydrated in a gradient series of alcohol, and embedded in paraffin. Deparaffinized sections were rehydrated in graded alcohols and stained with hematoxylin and eosin (H&E) for standard histology. Immunohistochemical staining for proliferation and activation of Erk1/2 was performed on formalin-fixed, paraffin-embedded sections using a Ki-67 (RM-9106, Thermo Scientific) and phospho-Erk1/2 antibody, respectively. Quantification of positively stained cells in the area of the wound was performed in a double-blinded fashion with the HistoQuest analysis software (TissueGnostics GmbH, Vienna, Austria).
Statistical Analysis
All data are presented as mean + S.D. except when indicated otherwise. Normal distribution of data was tested with the Kolmogorov-Smirnov test. Comparisons between groups were calculated using either Student's t test (unpaired, two-tailed), one-way ANOVA with Tukey's multiple comparison test, or Kruskal-Wallis test with Dunn's multiple comparison test, and p values ≤0.05 were considered as statistically significant. All calculations were performed using GraphPad software (GraphPad Software, Inc., San Diego, CA).
Study Approval
The in vivo study was conducted in accordance with protocols approved by the local Committee on Animal Experiments of Vienna, Austria. All experiments were performed according to the policies, procedures, and responsibilities for the care and use of laboratory animals at the Ludwig Boltzmann Institute for Experimental and Clinical Traumatology, Vienna, Austria. For human ASCs, liposuction material of male and female patients between 31 and 40 years of age was used. The collection of human adipose tissue was approved by the local ethical review board, and informed, written consent was obtained from each patient prior to the use of the collected material for this research project.
RESULTS
Shock Wave Treatment Increases in Vitro Proliferation of Mesenchymal Progenitor Cells in a Dose- and Time-dependent Fashion
To date, no routine standard protocol exists for the application of in vitro shock wave treatment. Therefore we began our studies by assessing the influence of shock wave treatment on the metabolic activity of C3H10T1/2 mesenchymal progenitor cells in vitro (Fig. 1A). Cells were treated with 100 shock wave pulses at an energy level of 0.07 or 0.19 mJ/mm2, and metabolic activity was measured with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay after 15, 24, and 36 h. Shock wave treatment significantly increased metabolic activity 15 h after treatment but not at later time points. These findings indicate that shock waves can enhance the metabolic activity of C3H10T1/2 cells and that energy levels of 0.07 or 0.19 mJ/mm2 do not adversely affect cell viability under our experimental conditions.
FIGURE 1.

Dose- and time-dependent effect of shock wave treatment on metabolic activity and proliferation of mesenchymal progenitor cells in vitro. A, C3H10T1/2 cells were treated with 100 shock wave pulses at 0.07 and 0.19 mJ/mm2, and metabolic activity was assessed with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay 15, 24, and 36 h after treatment (n = 4 in triplicates; mean + S.D. (error bars); Kruskal-Wallis test with Dunn's multiple comparison test). B, for determination of proliferation, C3H10T1/2 cells were synchronized in G0/G1 phase using 0.2% serum for 24 h and restimulated using 10% serum. Then cells were treated with 100 shock wave pulses at 0.03, 0.07, or 0.19 mJ/mm2. Untreated cells were treated identically except they did not receive shock wave treatment. Cells were harvested at the indicated time points and stained with propidium iodide, and DNA content was assessed by flow cytometry (2N and 4N, diploid and tetraploid DNA content, respectively). Characteristic cell cycle profiles of untreated cells (gray area) compared with shock wave-treated cells (overlaying black line) are shown 15 (upper panel) and 21 h (lower panel) after shock wave treatment. The corresponding percentages of proliferating cells in S phase 15 (C) and 21 h (D) after shock wave treatment (n = 6; *, p < 0.05; **, p < 0.01; one-way ANOVA with Tukey's multiple comparison test) are shown.
Next, we studied whether shock wave treatment affects cell proliferation. C3H10T1/2 cells were subjected to shock wave treatment using 100 pulses at 0.03, 0.07, or 0.19 mJ/mm2. Along with respective controls that did not receive shock wave treatment, cells were harvested and fixed at the indicated time points and stained with propidium iodide, and cell cycle characteristics were assessed using flow cytometric analysis (Fig. 1B). The cell cycle profiles revealed more cells in the S phase in the samples treated with shock waves compared with control cells without shock waves. This effect increased with energy and was particularly clear 21 h after treatment with 100 pulses at 0.19 mJ/mm2, revealing a significant increase in proliferating cells in the S phase (Fig. 1D). However, shock wave treatment with 100 pulses using 0.19 mJ/mm2 increased the cell numbers in S phase by up to 48% compared with untreated control already 15 h after treatment (Fig. 1C). These results indicate a dose- and time-dependent proproliferative effect of shock wave treatment in vitro.
Shock Wave Treatment Triggers ATP Release and Activates MAPK Signaling Pathways
Shock wave treatment exerts a mechanical stimulus on cells, which can release cellular ATP into the extracellular space (23). We exposed C3H10T1/2 cells to shock wave treatment and measured ATP release immediately and activation of Erk1/2, its upstream effector Mek1/2, and p38 MAPK 10 min after shock wave treatment. To assess the dose-dependent effects of shock waves, the cells were treated at a constant energy level (0.19 mJ/mm2) with different numbers of shock wave pulses (0–300; Fig. 2, A, B, and E) or with 100 shock wave pulses at different energy levels (0–0.19 mJ/mm2; Fig. 2, C, D, and F).
FIGURE 2.

Shock wave treatment releases cellular ATP and activates intracellular signaling in a pulse number- and energy-dependent fashion. Immediately after shock wave treatment with the indicated pulse numbers at a constant energy of 0.19 mJ/mm2 (A, B, and E) or varying energies at a constant number of 100 shock wave pulses (C, D, and F), cells were placed on ice, and ATP concentrations in supernatants were determined (E and F, n = 2 in triplicates; **, p < 0.01; ***, p < 0.001; Kruskal-Wallis test with Dunn's multiple comparison test). Cells were lysed, and Akt, Erk1/2, Mek1/2, p38 MAPK, and S6 protein activation was determined by immunoblot analysis. Representative blots of three different experiments are shown (A and C; # denotes unspecific band), and intensity ratios depicted in corresponding bar graphs were calculated using phosphorylated (p-) and total protein expression and normalized to the untreated controls (B and D, n = 3; mean + S.D. (error bars); *, p < 0.05; **, p < 0.01; ***, p < 0.001; one-way ANOVA with Tukey's multiple comparison test compared with untreated controls).
The application of 50, 100, or 300 shock wave pulses at 0.19 mJ/mm2 caused a significant release of ATP into the supernatant that increased with the number of shock wave pulses administered (Fig. 2E). Western blot analysis 10 min after shock wave treatment revealed significant and dose-dependent activation of Erk1/2, Mek1/2, and p38 MAPK signaling reaching levels up to 10-fold higher than in unstimulated controls (Fig. 2B). Activation of Akt and ribosomal S6 protein, a downstream target of the mammalian target of rapamycin, did not follow this dose-dependent pattern, which can be appreciated in the representative Western blots (Fig. 2A).
Shock wave treatment elicited ATP release in an energy level-dependent fashion, reaching levels significantly higher than in control samples at ≥0.07 mJ/mm2 (Fig. 2F). Similarly, Erk1/2, Mek1/2, and p38 MAPK activation was increased by up to 8-fold in response to shock waves at 0.19 mJ/mm2, whereas the phosphorylation of Akt and ribosomal S6 remained unchanged (Fig. 2, C and D). Thus, shock wave treatment results in a pulse- and energy-dependent increase in ATP release and Erk1/2 and p38 MAPK activation.
Shock Wave Treatment Promotes Proliferation and MAPK Signaling Pathway Activation via Purinergic Signaling
Shock wave treatment has been shown to have proproliferative effects on T cells where ATP release plays an essential role (22). BrdU incorporation assays showed that shock wave treatment increased proliferation of C3H10T1/2 cells (Fig. 3A). Shock wave treatment with energy levels of 0.07 or 0.19 mJ/mm2 significantly increased proliferation by up to 1.5 times the value of untreated control cells. To test the hypothesis that ATP release contributes to the observed proproliferative effect of shock wave treatment, we treated cells with shock waves in the presence of apyrase (20 units/ml) to rapidly hydrolyze extracellular ATP. Scavenging ATP from the supernatant of shock wave-treated cells abolished the enhancing effect of shock waves on proliferation (Fig. 3A). Analysis of Erk1/2 phosphorylation showed that apyrase (20 units/ml) and the nonspecific P2 receptor antagonist suramin (100 μm) reversed the stimulatory effects of shock wave treatment on Erk1/2 activation (Fig. 3, B and C). Apyrase abolished p38 MAPK phosphorylation in control cells and cells treated with ESWT, whereas suramin did not reduce p38 MAPK phosphorylation, suggesting the involvement of suramin-insensitive mechanisms in the p38 MAPK signaling of C3H10T1/2 cells (Fig. 3C). These results indicate that purinergic signaling is responsible for the enhancement of Erk1/2 activation and proliferation of shock wave-treated C3H10T1/2 cells.
FIGURE 3.

Shock wave treatment promotes proliferation and Erk1/2 and p38 MAPK pathway activation via purinergic signaling. A, C3H10T1/2 cells were growth-arrested for 24 h, restimulated by addition of 10% serum, and treated with 100 shock wave pulses at 0.07 or 0.19 mJ/mm2 in the absence or presence of 20 units/ml apyrase during shock wave treatment. BrdU incorporation at 21 h post-treatment was used as a measure of cell proliferation (n = 3 in triplicates; *, p < 0.05; **, p < 0.001; ***, p < 0.001; one-way ANOVA with Tukey's multiple comparison test compared with unstimulated controls). B and C, representative Western blots are shown of three independent experiments with C3H10T1/2 cells subjected to 100 shock wave pulses at 0.19 mJ/mm2 in the presence or absence of apyrase (20 units/ml) or suramin (100 μm). Protein lysates were prepared immediately after shock wave treatment for determination of MAPK activation. Intensity ratios of Erk1/2 and p38 MAPK were calculated using activated and total protein expression and normalized to corresponding untreated controls (n ≥ 3; mean + S.D. (error bars); two-tailed unpaired Student's t test compared with unstimulated controls; n.d., not detectable). p-Erk1/2, phospho-Erk1/2; p-p38, phospho-p38.
Erk1/2 Signaling Is Crucial for the Proliferative Effects of Shock Wave Treatment
To further test the notion that shock wave treatment accelerates proliferation by Erk1/2 activation, we used the Mek1/2 inhibitor U0126. C3H10T1/2 cells were treated with U0126 (10 μm) for 2.5 h before shock wave treatment at 0.19 mJ/mm2, and Western blot analysis was performed to assess Erk1/2 activation. In some cases, exogenous ATP (500 nm) was added for 10 min prior to analysis of Erk1/2 activation. The significant increase in Erk1/2 phosphorylation induced by shock wave treatment was reduced 6-fold by inhibition of Mek1/2, the upstream kinases of Erk1/2 (Fig. 4A). Addition of extracellular ATP had no significant effect on Erk1/2 phosphorylation compared with control cells without shock wave treatment (Fig. 4A). Under control conditions, shock wave treatment with 100 pulses at 0.07 and 0.19 mJ/mm2 increased proliferation by up to 1.6-fold as measured by BrdU incorporation 21 h after treatment (Fig. 4B). Pretreatment with U0126 reduced the enhancing effect of shock wave treatment on proliferation (Fig. 4B). These findings support the conclusion that Erk1/2 signaling is essential for the proliferative effects of shock wave treatment.
FIGURE 4.
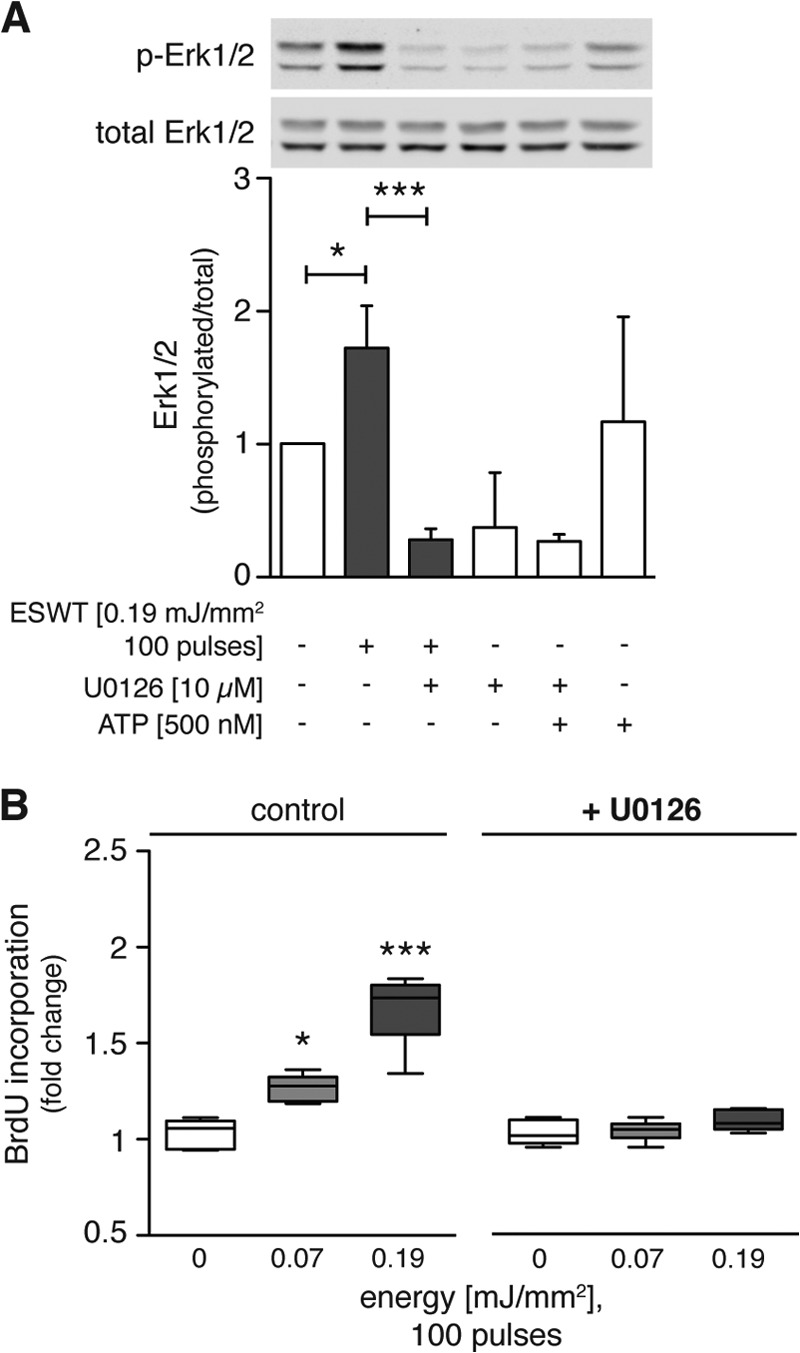
Erk1/2 signaling is crucial for the proliferative effects of shock wave treatment. A, C3H10T1/2 cells were serum-deprived for 24 h and incubated with the Mek1/2 inhibitor U0126 (10 μm) for 2.5 h before restimulation and treatment with 100 shock wave pulses at 0.19 mJ/mm2. Extracellular ATP (500 nm) was incubated with cells at 37 °C for 10 min. Directly after treatment, samples were placed on ice, and cells were processed for protein extraction and Western blot analysis. A representative Western blot of three independent experiments is shown. Ratios between activated and total Erk1/2 were calculated, and data were normalized to the non-treated control group (n = 3; mean + S.D. (error bars); *, p < 0.05; ***, p < 0.001; one-way ANOVA with Tukey's multiple comparison test). B, BrdU incorporation was assessed 21 h after shock wave treatment of C3HT101/2 cells pretreated with or without U0126 (10 μm) (n = 3 in duplicates; *, p < 0.05; ***, p < 0.001; data are normalized to corresponding controls; one-way ANOVA with Tukey's multiple comparison test). p-Erk1/2, phospho-Erk1/2.
Shock Wave Treatment-activated Purinergic Signaling Stimulates Erk1/2 Phosphorylation
Extracellular ATP is rapidly broken down by ectonucleotidases that are found on the cell surfaces of many cell types. This results in the formation of adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine and the potential stimulation of P2X, P2Y, and P1 (adenosine) receptors (38). To determine whether these different purinergic receptor classes contribute to Erk1/2 activation in response to shock wave treatment, we used agonists and antagonists of these different receptors. P2X purinergic receptors are solely activated by ATP, whereas ATP and UTP are both agonists for P2Y receptors (39). We found that extracellular ATP and UTP increased Erk1/2 activation (Fig. 5A). Treatment with 2-MeSATP, an agonist of P2X receptors, and adenosine, a P1 agonist (39), did not significantly increase Erk1/2 activation. Taken together, these findings suggest that P2Y receptors are primarily responsible for the stimulatory effect of purinergic signaling on C3H10T1/2 cells.
FIGURE 5.

Shock wave treatment-activated purinergic signaling stimulates Erk1/2 activation. A, C3H10T1/2 cells were treated with the indicated concentrations of the P2 receptor agonists ATP, UTP, and 2-MeSATP or with the P1 receptor agonist adenosine for 10 min at 37 °C. Then cells were placed on ice, and protein was extracted for Western blotting. The bar graph shows -fold stimulation of the Erk1/2 protein level normalized to control. A representative Western blot of four independent experiments is shown (n = 4; mean + S.D. (error bars); *, p < 0.05; **, p < 0.001; Kruskal-Wallis test with Dunn's multiple comparison test). B and C, C3H10T1/2 cells were pretreated with 100 μm P2 receptor antagonists pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate (PPADS) or suramin or P1 receptor antagonist theophylline. Cells were shock wave-treated using 100 pulses at 0.19 mJ/mm2 (B), or 5 μm ATP was added and incubated for 10 min (C) before protein was extracted. Bar graphs depict -fold change of the Erk1/2 protein level normalized to untreated controls (B, n = 4; C, n = 5; mean + S.D. (error bars); *, p < 0.05; **, p < 0.001; one-way ANOVA with Tukey's multiple comparison test; lanes were run on the same gel but were noncontiguous). p-Erk1/2, phospho-Erk1/2.
To investigate the roles of P2 and P1 receptors in Erk1/2 activation by shock wave treatment, we used pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate, an antagonist of P2X receptors (40); suramin, which is a nonselective antagonist of P2X and P2Y receptors (30); and theophylline, which is an antagonist of P1 receptors (30). Cells were treated with these antagonists for 30 min before shock wave treatment (Fig. 5B) or addition of extracellular ATP (Fig. 5C), and Erk1/2 activation was assessed. We found that suramin but neither pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate nor theophylline reduced Erk1/2 activation by shock wave or ATP treatment (Fig. 5, B and C). These findings suggest that P2Y receptors seem to be predominantly responsible for Erk1/2 activation in response to shock wave treatment.
Shock Wave Treatment Enhances Proliferation by Inducing ATP Release and Subsequent Erk1/2 Activation in Human Cells
Shock wave treatment has been shown previously to beneficially influence wound healing in preclinical and clinical trials. Therefore, we studied the effect of shock wave treatment on human cell types that may be involved in wound healing, namely human stem cells and T lymphocytes. Primary, adherent adipose tissue-derived stem cells were obtained from three different donors, and Jurkat T lymphocytes were cultured in vitro. First we tested whether shock wave treatment has an effect on the proliferation of these cells similar to that described above for C3H10T1/2 cells. Cell cycle time course experiments using flow cytometry suggested the accumulation of both cell types in the S phase after shock wave treatment. ASCs and Jurkat T cells showed significantly higher percentages of cells in S phase at 21 and 18 h after shock wave treatment, respectively (Figs. 6A and 7A). Similar to C3H10T1/2 cells, both human cell types showed dose-dependent increases in ATP release and Erk1/2, Mek1/2, and p38 MAPK activation in response to shock wave treatment (Figs. 6, B, C, F, G, H, and J, and 7, B, C, F, G, H, and J). To test whether the activation of Erk1/2 and p38 MAPKs is dependent upon ATP released in response to shock wave treatment, ASCs and Jurkat T cells were treated with either apyrase or suramin, which hydrolyzes ATP or blocks purinergic P2 receptors, respectively. As with C3H10T1/2 cells, both apyrase and suramin treatment abolished the activation of Erk1/2 and p38 MAPK in both human cell types (Figs. 6, D and E, and 7, D and E). Finally, we found that shock wave treatment induced proliferation in both ASCs and Jurkat T cells as assessed by BrdU incorporation. This proliferative effect was reduced by apyrase and U0126, indicating that proliferation in response to shock wave treatment depends on ATP release and activation of Erk1/2 signaling (Figs. 6I and Fig. 7I). Taken together with the results in C3H10T1/2 cells shown above, these findings indicate that ATP release, P2 receptor activation, and Erk1/2 signaling are universal mechanisms by which shock wave treatment influences the activation and function of different cell types.
FIGURE 6.

Purinergic signaling and Erk1/2 activation are essential in the shock wave treatment-induced proliferation in human adipose tissue-derived stem cells. A, for assessment of cell cycle progression, ASCs were serum-deprived for 24 h. Cells were then restimulated with 10% serum and shock wave-treated with 0.19 mJ/mm2 and 100 pulses. Cell were harvested and fixed at the indicated time points, and DNA content was assessed by flow cytometry. Corresponding percentages of cells in S phase at various time points (0, 15, 18, 21, and 24 h) after shock wave treatment are shown (n = 3 in duplicates; *, p < 0.05; two-tailed unpaired Student's t test for pairwise comparison at different time points). Immediately after shock wave treatment with varying energies at a constant number of 100 pulses (B, C, and F) or with the indicated pulse numbers at a constant energy of 0.19 mJ/mm2 (G, H, and J), ATP concentrations in supernatants were determined (F and J; n = 3 in triplicates; *, p < 0.05; ***, p < 0.001; Kruskal-Wallis test with Dunn's multiple comparison test). Cells were lysed, and Erk1/2, Mek1/2, and p38 MAPK activation was determined by Western blot analysis. Representative blots of three different experiments are shown (B and G; # denotes unspecific band). Intensity ratios depicted in corresponding bar graphs were calculated using phosphorylated (p-) and total protein expression and normalized to untreated controls (C and H; n = 3; mean + S.D. (error bars); *, p < 0.05; **, p < 0.01; ***, p < 0.001; one-way ANOVA with Tukey's multiple comparison test). D and E, representative Western blots are shown of five independent experiments with ASCs subjected to 100 pulses at 0.19 mJ/mm2 in the presence or absence of apyrase (20 units/ml) or suramin (100 μm). Intensity ratios of Erk1/2 and p38 MAPK were calculated and normalized to corresponding controls (n = 5; mean + S.D. (error bars); two-tailed unpaired Student's t test). I, ASCs were serum-deprived for 24 h and incubated either with the Mek1/2 inhibitor U0126 (10 μm) for 2.5 h or with 20 units/ml apyrase for 30 min before restimulation with 10% serum and shock wave treatment with 100 pulses at 0.19 mJ/mm2. BrdU incorporation was assessed 18 and 21 h after shock wave treatment as a measure of cell proliferation (n = 3 in triplicates; *, p < 0.05; two-tailed unpaired Student's t test compared with corresponding controls; n.s., not significant).
FIGURE 7.

Purinergic signaling and Erk1/2 activation are essential in the shock wave treatment-induced proliferation in human Jurkat T cells. A, for assessment of cell cycle progression, Jurkat T cells were serum-deprived for 48 h. Cells were restimulated with 10% serum and treated with 100 shock wave pulses at 0.19 mJ/mm2. Then cells were harvested and fixed at the indicated time points, and DNA content was assessed by flow cytometry. Corresponding percentages of cells in S phase at various time points (0, 15, 18, 21, and 24 h) after shock wave treatment are shown (n = 3; *, p < 0.05; two-tailed unpaired Student's t test for pairwise comparison at different time points). Immediately after shock wave treatment with varying energies at a constant number of 100 pulses (B, C, and F) or with the indicated pulse numbers at a constant energy of 0.19 mJ/mm2 (G, H, and J), ATP concentrations in supernatants were determined (F and J; n = 3 in triplicates; *, p < 0.05; ***, p < 0.001; Kruskal-Wallis test with Dunn's multiple comparison test). Cells were lysed, and Erk1/2, Mek1/2, and p38 MAPK activation was determined by Western blot analysis. Representative blots of three different experiments are shown (B and G). Intensity ratios depicted in corresponding bar graphs were calculated using phosphorylated (p-) and total protein expression and normalized to untreated controls (C and H; n = 4; mean + S.D. (error bars); *, p < 0.05; **, p < 0.01; ***, p < 0.001; one-way ANOVA with Tukey's multiple comparison test). D and E, representative Western blots are shown of two independent experiments with Jurkat T cells subjected to 100 pulses at 0.19 mJ/mm2 in the presence or absence of apyrase (20 units/ml) or suramin (100 μm). Intensity ratios of Erk1/2 and p38 MAPK were calculated and normalized to corresponding controls (n = 8; mean + S.D. (error bars); two-tailed unpaired Student's t test). I, Jurkat T cells were serum-deprived for 48 h and incubated either with the Mek1/2 inhibitor U0126 (10 μm) for 2 h or with 20 units/ml apyrase for 30 min before restimulation with 10% serum and shock wave treatment with 100 pulses at 0.19 mJ/mm2. BrdU incorporation was assessed 18 and 21 h after shock wave treatment as a measure of cell proliferation (n = 4 in triplicates; *, p < 0.05; **, p < 0.01; two-tailed unpaired Student's t test compared with corresponding controls; n.s., not significant).
Shock Wave Treatment Enhances Wound Healing in a Rat Model by Promoting Proliferation via Erk1/2 Signaling
Shock wave treatment of ischemic skin flaps has been shown to improve wound healing in a standard rodent ischemic epigastric flap model (35). We used this model and the Mek1/2 inhibitor GSK1120212 to test the hypothesis that Erk1/2 signaling is crucial for improved wound healing in response to shock wave treatment. Planimetric analysis of the wound size area on the ischemic side of the epigastric flap was performed immediately after surgery (day 0) and on days 1, 5, and 10 after surgery (Fig. 8, C and D). Shock wave treatment of the ischemic side of the epigastric flap (100 pulses at 0.13 mJ/mm2) significantly decreased the wound size compared with control animals (Fig. 8A). In animals receiving the Mek1/2 inhibitor GSK1120212 (0.1 mg/kg daily), wound sizes in the control and shock wave groups did not differ (Fig. 8B). These results suggest that shock wave treatment exerts its beneficial effects on wound healing by induction of Erk1/2 signaling.
FIGURE 8.

In vivo shock wave treatment-enhanced wound healing is dependent on Erk1/2 signaling. Sprague-Dawley rats did not receive (A and C) or received GSK1120212 (B and D) on a daily basis, starting 4 days before surgery and until day 10 post surgery. After detachment of the epigastric adipocutaneous flap, the unilateral inferior epigastric neurovascular bundle was ligated according to a randomization protocol to induce an ischemic area. This area was then shock wave-treated using 100 pulses at 0.13 mJ/mm2 followed by the creation of 1.3-cm2-diameter wounds on the ischemic as well as on the adequately perfused side (vital) of the flap. A and B, wound size on the ischemic side was monitored at days 1, 5, and 10 postsurgery and is depicted as percentage of postoperative wound area (day 0) for each animal (n = 9; mean ± S.E. (error bars); *, p < 0.05; two-tailed unpaired Student's t test or Mann-Whitney U test (GSK1120212 day 5) for pairwise comparison at different time points).
Quantitative immunohistochemical analysis of the effects of shock wave treatment on proliferation and Erk1/2 phosphorylation in ischemic wounds was performed by staining with the proliferation marker Ki-67 and a phospho-Erk1/2 antibody, respectively. Regions of interest in the granulation tissue area close to the remaining wound were analyzed under the microscope, and representative pictures of each study group showing sections of the ischemic wound harvested 10 days after surgery are depicted in Fig. 9A. Wounds of animals treated with 100 shock wave pulses at 0.13 mJ/mm2 exhibited a significant increase in Ki-67- and phospho-Erk1/2-positive cells in the granulation tissue that was 6- and ∼5-fold higher than in untreated controls, respectively. In animals receiving GSK1120212, shock wave treatment did not significantly change the percentage of Ki-67- or phospho-Erk1/2-positive cells (Fig. 9, B and C). These data support our findings that ischemic wound healing in shock wave-treated animals is significantly enhanced by shock wave treatment and is dependent on Erk1/2 pathways.
FIGURE 9.

Erk1/2 pathway inhibition impedes the proliferative effect of shock wave treatment in vivo. A, representative IHC sections of each study group at day 10 post surgery showing H&E (HE), Ki-67, and phospho-Erk1/2 (p-Erk1/2) staining in the control group and the group receiving GSK1120212. Bar graphs depict percentages of Ki-67-positive (B) or phospho-Erk1/2-positive (C) cells of all cells assessed in regions of interest (ROI) (n = 7; mean + S.D. (error bars); **, p < 0.01; ***, p < 0.001; two-tailed unpaired Student's t test comparing the shock wave-treated group of control animals/animals receiving GSK1120212 inhibitor with the corresponding untreated group; scale bars, 100 μm; ns, not significant).
DISCUSSION
Despite the clinical evidence that ESWT can benefit patients with musculoskeletal disorders, non-union bone fractures, and chronic soft tissue wounds (2, 8, 9), the molecular mechanisms underlying these beneficial effects have remained unclear. Here, we sought to fill this gap in knowledge by exploring these mechanisms using several cell types that contribute to wound healing. Our results indicate that shock wave treatment affects target cells and tissues by eliciting ATP release, which in turn promotes P2 receptor-mediated Erk1/2 signaling and thereby enhances cell proliferation (Fig. 10). Through this mechanism, ESWT improved ischemic wound healing in a rodent model that recapitulates one of the clinically most remarkable features of ESWT, namely its ability to induce the healing of chronic ulcers and non-healing wounds (1–5).
FIGURE 10.
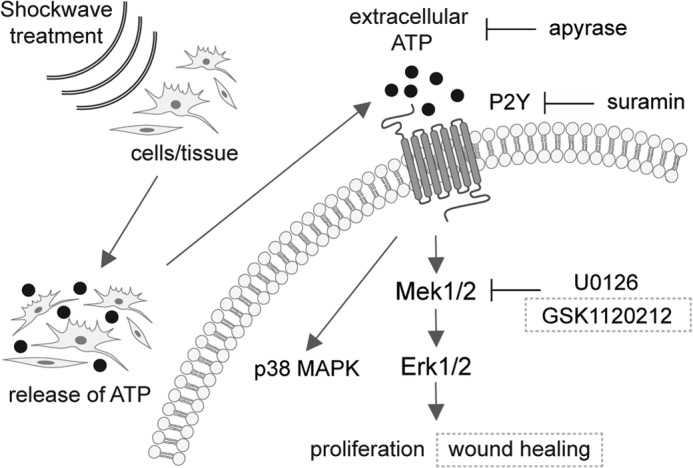
Shock wave treatment triggers purinergic signaling and regulates cell function. Shown is a schematic illustration of how shock wave treatment induces intracellular signaling pathways in cells and tissue, finally leading to enhanced proliferation and wound healing. Shock waves exert a mechanical stimulus on cells and tissue, possibly leading to changes in membrane integrity or even actively opening channels to release ATP. The released ATP then binds to purinergic receptors, in this model predominantly P2Y receptors, leading to downstream signaling events. Uptake of ATP consequently activates p38 MAPK as well as Mek1/2-Erk1/2 signaling pathways, ultimately leading to increased cellular proliferation in vitro and enhanced wound healing in vivo. If extracellular released ATP is either degraded by apyrase or P2Y receptors are blocked by suramin, the transduction of the stimulus into the cell to activate Erk1/2 is inhibited, and the proliferative effect of shock wave treatment is abolished. Inhibition of Mek1/2 by using U0126 in vitro or GSK1120212 in vivo to block the activation of Erk1/2 results in the loss of the proliferative and wound healing effect of shock wave treatment. Together, these signaling events transduce the primary mechanical stimulus of shock waves, resulting in proliferation and wound healing.
Shock wave treatment has been shown to influence the function of a number of different cells including mesenchymal stem cells (24, 41), osteoblasts (20), tenocytes (42), and T cells (22). In mouse mesenchymal progenitor cells, we could show that the proliferative effects of shock wave treatment are mediated by the release of cellular ATP in an energy- and dose-dependent manner and through P2 receptors. P2 receptor stimulation has been shown to induce proliferation of many different cell types, suggesting that purinergic signaling plays an important, perhaps universal, role in processes involved in wound healing (29, 32, 43–45). We found that ATP is released from shock wave-treated cells. However, the mechanisms of release are unclear. ATP can be released by damaged and dying cells or in response to physiological stimuli and mechanical stretch (30). Mechanical stimulation has been shown to mediate the release of cellular ATP of various cells such as astrocytes, osteoblasts, chondrocytes, and epithelial cells (46–49). Studies by Sun et al. (23) and Yu et al. (22) support our finding that ESWT at our chosen settings releases ATP without negatively affecting cell viability as they showed that subjection of human mesenchymal stem cells to <200 shock wave pulses at 0.18 mJ/mm2 resulted in >95% viability directly or 24 h after ESWT.
We found that the released ATP activated P2 receptors and increased Erk1/2 and p38 MAPK signaling via mechanisms similar to those reported in osteoblasts, mesenchymal stem cells, and T cells (20, 22–24). In addition to Erk1/2 and p38 MAPK, the downstream signaling mechanisms triggered by shock wave-induced P2 receptor activation may also involve additional signaling intermediates such as Ras and hypoxia-inducible factor 1a transactivation (20, 23). A study dealing with stretch-induced injury reported the activation of Erk1/2 to be dependent on the rate and amplitude of mechanical impact, involving released ATP and activated P2 receptors (46). Akt and mammalian target of rapamycin signaling pathways are known as essential regulators of cell proliferation (50–52). However, we did not observe a change in the phosphorylation of Akt and ribosomal S6 protein in our study, suggesting that these pathways do not play a major role in the proproliferative effects of shock wave treatment of C3H10T1/2 cells.
Once released into the extracellular space, ATP cannot only stimulate P2 receptors, but it can also be degraded by ectonucleotidases found on cell surfaces to ADP, AMP, and adenosine, which in turn can activate different sets of P2X, P2Y, and P1 (adenosine) receptors (30, 53). We found that exogenous ATP and UTP could both replicate the stimulatory effect of shock wave treatment on Erk1/2 signaling in C3H10T1/2 cells, implying the involvement of P2Y receptors in this response (39, 47, 54). Moreover, in epithelial cells, the P2Y receptor antagonist reactive blue 2 inhibited wound healing in vitro (49) and released ATP after injury-induced Erk1/2 phosphorylation through activation of P2Y receptors (55).
We could demonstrate that shock wave treatment caused Erk1/2 activation in the mouse mesenchymal progenitor cell line (C3H10T1/2) as well as in two different human cell types that may be involved in wound healing, namely primary adherent adipose tissue-derived stem cells and the Jurkat T cell line. ESWT has been shown to trigger inflammatory processes by activating immune cells including T cells (3, 22, 34, 56). Our findings with these different cell types suggest that ESWT may aid tissue repair by influencing the inflammatory and regenerative aspects of wound healing. The effects of shock waves on primary human stem cells suggest that clinical ESWT may promote the growth and perhaps the differentiation of stem or progenitor cells during tissue regeneration (1, 57–59). In all three cell types we tested in vitro, we found a uniform scheme of shock wave-induced ATP release, subsequent stimulation of purinergic receptors, and the activation of Erk1/2 signaling, finally resulting in enhanced cell proliferation. We therefore conclude that this mechanism plays a central role in the response of mammalian cells to shock wave treatment. Our in vivo experiments demonstrated that the biological responses triggered by ESWT improved tissue repair. It remains to be seen to what extent the here proposed mechanism induced by ESWT also influences other aspects of wound healing such as neovascularization, angiogenesis, stem cell differentiation, and the inflammatory response involved in wound repair as shock wave treatment already has been reported to favor these phases of wound healing (2, 10, 12, 19, 60). Our in vivo studies further revealed that ESWT improves wound healing in a rat ischemic skin flap model and that Erk1/2 signaling is involved in this process. Taken together with previous reports (5, 16) and with our in vitro and in vivo findings, this suggests that ESWT promotes wound healing by local ATP release and P2 receptor activation that foster cell proliferation and tissue remodeling via Erk1/2 activation.
Patients with diabetes often suffer from chronic ulcers, resulting in significant discomfort, risk of infection, and impaired quality of life. The treatment of such chronic wounds is difficult, and success is uncertain. ESWT is a novel treatment approach with a remarkable success rate (1, 2, 9). Schaden et al. (1) reported that a single session of ESWT can induce wound healing in patients suffering from different wound disorders. The clinical observation that a single ESWT session can affect outcome is echoed by our in vitro findings where shock wave treatment had prolonged effects on subsequent cellular functions. Furthermore, the advantage of using ESWT in an outpatient setting and its cost effectiveness (compared with other clinical procedures) underline the clinical relevance and the need for the integration of shock wave treatment into future clinical routine. Additionally, our results might help to explain the non-responsiveness to shock wave treatment in some patients suffering wound healing disorders. Besides well known factors contributing to wound healing such as age and lifestyle, the Erk1/2 signaling pathway has been described as crucial in the wound healing process (61, 62). We therefore hypothesize that the non-responding patients might have an impaired Erk1/2 signaling pathway resistant to shock wave treatment.
In conclusion, our findings shed new light on the underlying mechanisms by which ESWT may exert its clinical effects, namely by causing the release of ATP, the subsequent stimulation of P2 receptors, and activation of Erk1/2 signaling, which initiate processes involved in wound healing. Although future studies are required to further define these processes, we think that our findings may help to refine the use of shock wave treatment to further improve its clinical efficacy.
Acknowledgments
We thank B. Grabner and M. Schlederer from the Ludwig Boltzmann Institute for Cancer Research, Vienna, Austria and R. Szoeloesi from the Ludwig Boltzmann Institute for Experimental and Clinical Traumatology for sharing their knowledge, time, and equipment for immunohistochemical stainings. We also thank the whole animal facility team from the Ludwig Boltzmann Institute for Experimental and Clinical Traumatology for support of the in vivo study. We are particularly thankful for receiving human adipose tissue-derived stem cells from E. Oberbauer, Ludwig Boltzmann Institute for Experimental and Clinical Traumatology, in cooperation with the Red Cross Blood Transfusion Service of Upper Austria, Linz, Austria. We are especially grateful to W. Schaden from the Unfallkrankenhaus Meidling, Vienna, Austria for continuous scientific comments and invaluable discussions and thank C. Rupp from the Max F. Perutz Laboratories, Vienna, Austria for input on Mek1/2 inhibitors.
This work was supported by the NewTissue Project (Österreichische Forschungsförderungsgesellschaft Grant 818412 and City of Vienna MA27 Grant 06-06) and the City of Vienna Competence Team reacTissue Project (MA27 Grant 12-06).
A. Hoffmann, J. Hartinger, S. Pfeifer, H. Redl, and R. Mittermayr, submitted manuscript.
- ESWT
- extracorporeal shock wave therapy
- ASC
- adipose tissue-derived stem cell
- 2-MeSATP
- 2-methylthio-ATP
- ANOVA
- analysis of variance.
REFERENCES
- 1. Schaden W., Thiele R., Kölpl C., Pusch M., Nissan A., Attinger C. E., Maniscalco-Theberge M. E., Peoples G. E., Elster E. A., Stojadinovic A. (2007) Shock wave therapy for acute and chronic soft tissue wounds: a feasibility study. J. Surg. Res. 143, 1–12 [DOI] [PubMed] [Google Scholar]
- 2. Mittermayr R., Antonic V., Hartinger J., Kaufmann H., Redl H., Téot L., Stojadinovic A., Schaden W. (2012) Extracorporeal shock wave therapy (ESWT) for wound healing: technology, mechanisms, and clinical efficacy. Wound Repair Regen. 20, 456–465 [DOI] [PubMed] [Google Scholar]
- 3. Davis T. A., Stojadinovic A., Anam K., Amare M., Naik S., Peoples G. E., Tadaki D., Elster E. A. (2009) Extracorporeal shock wave therapy suppresses the early proinflammatory immune response to a severe cutaneous burn injury. Int. Wound J. 6, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayashi D., Kawakami K., Ito K., Ishii K., Tanno H., Imai Y., Kanno E., Maruyama R., Shimokawa H., Tachi M. (2012) Low-energy extracorporeal shock wave therapy enhances skin wound healing in diabetic mice: a critical role of endothelial nitric oxide synthase. Wound Repair Regen. 20, 887–895 [DOI] [PubMed] [Google Scholar]
- 5. Kuo Y. R., Wang C. T., Wang F. S., Chiang Y. C., Wang C. J. (2009) Extracorporeal shock-wave therapy enhanced wound healing via increasing topical blood perfusion and tissue regeneration in a rat model of STZ-induced diabetes. Wound Repair Regen. 17, 522–530 [DOI] [PubMed] [Google Scholar]
- 6. Moretti B., Notarnicola A., Maggio G., Moretti L., Pascone M., Tafuri S., Patella V. (2009) The management of neuropathic ulcers of the foot in diabetes by shock wave therapy. BMC Musculoskelet. Disord. 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saggini R., Figus A., Troccola A., Cocco V., Saggini A., Scuderi N. (2008) Extracorporeal shock wave therapy for management of chronic ulcers in the lower extremities. Ultrasound Med. Biol. 34, 1261–1271 [DOI] [PubMed] [Google Scholar]
- 8. Thiel M. (2001) Application of shock waves in medicine. Clin. Orthop. Relat. Res. 18–21 [DOI] [PubMed] [Google Scholar]
- 9. Wang C. J. (2003) An overview of shock wave therapy in musculoskeletal disorders. Chang Gung Med. J. 26, 220–232 [PubMed] [Google Scholar]
- 10. Yang G., Luo C., Yan X., Cheng L., Chai Y. (2011) Extracorporeal shock wave treatment improves incisional wound healing in diabetic rats. Tohoku J. Exp. Med. 225, 285–292 [DOI] [PubMed] [Google Scholar]
- 11. Reinke J. M., Sorg H. (2012) Wound repair and regeneration. Eur. Surg. Res. 49, 35–43 [DOI] [PubMed] [Google Scholar]
- 12. Stojadinovic A., Elster E. A., Anam K., Tadaki D., Amare M., Zins S., Davis T. A. (2008) Angiogenic response to extracorporeal shock wave treatment in murine skin isografts. Angiogenesis 11, 369–380 [DOI] [PubMed] [Google Scholar]
- 13. Yan X., Zeng B., Chai Y., Luo C., Li X. (2008) Improvement of blood flow, expression of nitric oxide, and vascular endothelial growth factor by low-energy shockwave therapy in random-pattern skin flap model. Ann. Plast. Surg. 61, 646–653 [DOI] [PubMed] [Google Scholar]
- 14. Dumfarth J., Zimpfer D., Vögele-Kadletz M., Holfeld J., Sihorsch F., Schaden W., Czerny M., Aharinejad S., Wolner E., Grimm M. (2008) Prophylactic low-energy shock wave therapy improves wound healing after vein harvesting for coronary artery bypass graft surgery: a prospective, randomized trial. Ann. Thorac. Surg. 86, 1909–1913 [DOI] [PubMed] [Google Scholar]
- 15. Meirer R., Brunner A., Deibl M., Oehlbauer M., Piza-Katzer H., Kamelger F. S. (2007) Shock wave therapy reduces necrotic flap zones and induces VEGF expression in animal epigastric skin flap model. J. Reconstr. Microsurg. 23, 231–236 [DOI] [PubMed] [Google Scholar]
- 16. Kuo Y. R., Wang C. T., Wang F. S., Yang K. D., Chiang Y. C., Wang C. J. (2009) Extracorporeal shock wave treatment modulates skin fibroblast recruitment and leukocyte infiltration for enhancing extended skin-flap survival. Wound Repair Regen. 17, 80–87 [DOI] [PubMed] [Google Scholar]
- 17. Higuera G. A., van Boxtel A., van Blitterswijk C. A., Moroni L. (2012) The physics of tissue formation with mesenchymal stem cells. Trends Biotechnol. 30, 583–590 [DOI] [PubMed] [Google Scholar]
- 18. Hughes-Fulford M. (2004) Signal transduction and mechanical stress. Sci. STKE 2004, RE12. [DOI] [PubMed] [Google Scholar]
- 19. Chen Y. J., Kuo Y. R., Yang K. D., Wang C. J., Sheen Chen S. M., Huang H. C., Yang Y. J., Yi-Chih S., Wang F. S. (2004) Activation of extracellular signal-regulated kinase (ERK) and p38 kinase in shock wave-promoted bone formation of segmental defect in rats. Bone 34, 466–477 [DOI] [PubMed] [Google Scholar]
- 20. Wang F. S., Wang C. J., Chen Y. J., Chang P. R., Huang Y. T., Sun Y. C., Huang H. C., Yang Y. J., Yang K. D. (2004) Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1α and VEGF-A expression in shock wave-stimulated osteoblasts. J. Biol. Chem. 279, 10331–10337 [DOI] [PubMed] [Google Scholar]
- 21. Wang F. S., Wang C. J., Sheen-Chen S. M., Kuo Y. R., Chen R. F., Yang K. D. (2002) Superoxide mediates shock wave induction of ERK-dependent osteogenic transcription factor (CBFA1) and mesenchymal cell differentiation toward osteoprogenitors. J. Biol. Chem. 277, 10931–10937 [DOI] [PubMed] [Google Scholar]
- 22. Yu T., Junger W. G., Yuan C., Jin A., Zhao Y., Zheng X., Zeng Y., Liu J. (2010) Shockwaves increase T-cell proliferation and IL-2 expression through ATP release, P2X7 receptors, and FAK activation. Am. J. Physiol. Cell Physiol. 298, C457–C464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun D., Junger W. G., Yuan C., Zhang W., Bao Y., Qin D., Wang C., Tan L., Qi B., Zhu D., Zhang X., Yu T. (2013) Shockwaves induce osteogenic differentiation of human mesenchymal stem cells through ATP release and activation of P2X7 receptors. Stem Cells 31, 1170–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raabe O., Shell K., Goessl A., Crispens C., Delhasse Y., Eva A., Scheiner-Bobis G., Wenisch S., Arnhold S. (2013) Effect of extracorporeal shock wave on proliferation and differentiation of equine adipose tissue-derived mesenchymal stem cells in vitro. Am. J. Stem Cells 2, 62–73 [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang W., Liu H. T. (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 [DOI] [PubMed] [Google Scholar]
- 26. Meloche S., Pouysségur J. (2007) The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239 [DOI] [PubMed] [Google Scholar]
- 27. Aouadi M., Binetruy B., Caron L., Le Marchand-Brustel Y., Bost F. (2006) Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie 88, 1091–1098 [DOI] [PubMed] [Google Scholar]
- 28. Mebratu Y., Tesfaigzi Y. (2009) How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 8, 1168–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burnstock G., Verkhratsky A. (2010) Long-term (trophic) purinergic signalling: purinoceptors control cell proliferation, differentiation and death. Cell Death Dis. 1, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burnstock G., Knight G. E., Greig A. V. (2012) Purinergic signaling in healthy and diseased skin. J. Invest. Dermatol. 132, 526–546 [DOI] [PubMed] [Google Scholar]
- 31. Baroja-Mazo A., Barberà-Cremades M., Pelegrín P. (2013) The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 1828, 79–93 [DOI] [PubMed] [Google Scholar]
- 32. Chi C., Trinkaus-Randall V. (2013) New insights in wound response and repair of epithelium. J. Cell. Physiol. 228, 925–929 [DOI] [PubMed] [Google Scholar]
- 33. Reznikoff C. A., Brankow D. W., Heidelberger C. (1973) Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res. 33, 3231–3238 [PubMed] [Google Scholar]
- 34. Holfeld J., Tepeköylü C., Kozaryn R., Urbschat A., Zacharowski K., Grimm M., Paulus P. (2014) Shockwave therapy differentially stimulates endothelial cells: implications on the control of inflammation via toll-like receptor 3. Inflammation 37, 65–70 [DOI] [PubMed] [Google Scholar]
- 35. Mittermayr R., Hartinger J., Antonic V., Meinl A., Pfeifer S., Stojadinovic A., Schaden W., Redl H. (2011) Extracorporeal shock wave therapy (ESWT) minimizes ischemic tissue necrosis irrespective of application time and promotes tissue revascularization by stimulating angiogenesis. Ann. Surg. 253, 1024–1032 [DOI] [PubMed] [Google Scholar]
- 36. Gilmartin A. G., Bleam M. R., Groy A., Moss K. G., Minthorn E. A., Kulkarni S. G., Rominger C. M., Erskine S., Fisher K. E., Yang J., Zappacosta F., Annan R., Sutton D., Laquerre S. G. (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 17, 989–1000 [DOI] [PubMed] [Google Scholar]
- 37. Sacchi V., Mittermayr R., Hartinger J., Martino M. M., Lorentz K. M., Wolbank S., Hofmann A., Largo R. A., Marschall J. S., Groppa E., Gianni-Barrera R., Ehrbar M., Hubbell J. A., Redl H., Banfi A. (2014) Long-lasting fibrin matrices ensure stable and functional angiogenesis by highly tunable, sustained delivery of recombinant VEGF164. Proc. Natl. Acad. Sci. U.S.A. 111, 6952–6957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmermann H. (2000) Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch. Pharmacol. 362, 299–309 [DOI] [PubMed] [Google Scholar]
- 39. Ralevic V., Burnstock G. (1998) Receptors for purines and pyrimidines. Pharmacol. Rev. 50, 413–492 [PubMed] [Google Scholar]
- 40. Lambrecht G., Friebe T., Grimm U., Windscheif U., Bungardt E., Hildebrandt C., Bäumert H. G., Spatz-Kümbel G., Mutschler E. (1992) PPADS, a novel functionally selective antagonist of P2 purinoceptor-mediated responses. Eur. J. Pharmacol. 217, 217–219 [DOI] [PubMed] [Google Scholar]
- 41. Zhao Y., Wang J., Wang M., Sun P., Chen J., Jin X., Zhang H. (2013) Activation of bone marrow-derived mesenchymal stromal cells-a new mechanism of defocused low-energy shock wave in regenerative medicine. Cytotherapy 15, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 42. Leone L., Vetrano M., Ranieri D., Raffa S., Vulpiani M. C., Ferretti A., Torrisi M. R., Visco V. (2012) Extracorporeal shock wave treatment (ESWT) improves in vitro functional activities of ruptured human tendon-derived tenocytes. PLoS One 7, e49759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heo J. S., Han H. J. (2006) ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathways. Stem Cells 24, 2637–2648 [DOI] [PubMed] [Google Scholar]
- 44. Chen J. B., Liu W. J., Che H., Liu J., Sun H. Y., Li G. R. (2012) Adenosine-5′-triphosphate up-regulates proliferation of human cardiac fibroblasts. Br. J. Pharmacol. 166, 1140–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hill L. M., Gavala M. L., Lenertz L. Y., Bertics P. J. (2010) Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J. Immunol. 185, 3028–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neary J. T., Kang Y., Willoughby K. A., Ellis E. F. (2003) Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J. Neurosci. 23, 2348–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Katz S., Boland R., Santillán G. (2006) Modulation of ERK 1/2 and p38 MAPK signaling pathways by ATP in osteoblasts: involvement of mechanical stress-activated calcium influx, PKC and Src activation. Int. J. Biochem. Cell Biol. 38, 2082–2091 [DOI] [PubMed] [Google Scholar]
- 48. Garcia M., Knight M. M. (2010) Cyclic loading opens hemichannels to release ATP as part of a chondrocyte mechanotransduction pathway. J. Orthop. Res. 28, 510–515 [DOI] [PubMed] [Google Scholar]
- 49. Yin J., Xu K., Zhang J., Kumar A., Yu F. S. (2007) Wound-induced ATP release and EGF receptor activation in epithelial cells. J. Cell Sci. 120, 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hall M. N. (2008) mTOR—what does it do? Transplant. Proc. 40, S5–S8 [DOI] [PubMed] [Google Scholar]
- 51. Corradetti M. N., Guan K. L. (2006) Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene 25, 6347–6360 [DOI] [PubMed] [Google Scholar]
- 52. Mendoza M. C., Er E. E., Blenis J. (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36, 320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Junger W. G. (2011) Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 11, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu D., Soleymani S., Madakshire R., Insel P. A. (2012) ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 26, 2580–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang L., Cranson D., Trinkaus-Randall V. (2004) Cellular injury induces activation of MAPK via P2Y receptors. J. Cell. Biochem. 91, 938–950 [DOI] [PubMed] [Google Scholar]
- 56. Schwiebert E. M. (2010) Underlying purinergic signaling contributes to T lymphocyte activation in tissue repair. Focus on “shockwaves increase the T-cell proliferation and IL-2 expression through ATP release, P2X7 receptors, and FAK activation.” Am. J. Physiol. Cell Physiol. 298, C446–C447 [DOI] [PubMed] [Google Scholar]
- 57. Aicher A., Heeschen C., Sasaki K., Urbich C., Zeiher A. M., Dimmeler S. (2006) Low-energy shock wave for enhancing recruitment of endothelial progenitor cells: a new modality to increase efficacy of cell therapy in chronic hind limb ischemia. Circulation 114, 2823–2830 [DOI] [PubMed] [Google Scholar]
- 58. Chen Y. J., Wurtz T., Wang C. J., Kuo Y. R., Yang K. D., Huang H. C., Wang F. S. (2004) Recruitment of mesenchymal stem cells and expression of TGF-β1 and VEGF in the early stage of shock wave-promoted bone regeneration of segmental defect in rats. J. Orthop. Res. 22, 526–534 [DOI] [PubMed] [Google Scholar]
- 59. Tepeköylü C., Wang F. S., Kozaryn R., Albrecht-Schgoer K., Theurl M., Schaden W., Ke H. J., Yang Y., Kirchmair R., Grimm M., Wang C. J., Holfeld J. (2013) Shock wave treatment induces angiogenesis and mobilizes endogenous CD31/CD34-positive endothelial cells in a hindlimb ischemia model: implications for angiogenesis and vasculogenesis. J. Thorac. Cardiovasc. Surg. 146, 971–978 [DOI] [PubMed] [Google Scholar]
- 60. Wang C. J., Wang F. S., Yang K. D., Weng L. H., Hsu C. C., Huang C. S., Yang L. C. (2003) Shock wave therapy induces neovascularization at the tendon-bone junction. A study in rabbits. J. Orthop. Res. 21, 984–989 [DOI] [PubMed] [Google Scholar]
- 61. Lima M. H., Caricilli A. M., de Abreu L. L., Araújo E. P., Pelegrinelli F. F., Thirone A. C., Tsukumo D. M., Pessoa A. F., dos Santos M. F., de Moraes M. A., Carvalheira J. B., Velloso L. A., Saad M. J. (2012) Topical insulin accelerates wound healing in diabetes by enhancing the AKT and ERK pathways: a double-blind placebo-controlled clinical trial. PLoS One 7, e36974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schreml S., Szeimies R. M., Prantl L., Landthaler M., Babilas P. (2010) Wound healing in the 21st century. J. Am. Acad. Dermatol. 63, 866–881 [DOI] [PubMed] [Google Scholar]