

Background: Auxiliary proteins bind to viral capsid surfaces, forming symmetric arrays of polypeptides.
Results: Collagen-like gp12 binds cooperatively to multiple sites of the bacteriophage SPP1 capsid in a reversible fashion.
Conclusion: The collagen fold and interaction with the capsid determine gp12 thermostability and folding/association properties.
Significance: Gp12 represents a novel type of viral capsid binders characterized by thermoswitchable properties.
Keywords: Bacteriophage, Collagen, Protein Folding, Virus Assembly, Virus Structure, Auxiliary Protein, Thermal Denaturation, Viral Capsid
Abstract
Icosahedral capsids of viruses are lattices of defined geometry and homogeneous size. The (quasi-)equivalent organization of their protein building blocks provides, in numerous systems, the binding sites to assemble arrays of viral polypeptides organized with nanometer precision that protrude from the capsid surface. The capsid of bacterial virus (bacteriophage) SPP1 exposes, at its surface, the 6.6-kDa viral polypeptide gp12 that binds to the center of hexamers of the major capsid protein. Gp12 forms an elongated trimer with collagen-like properties. This is consistent with the fold of eight internal GXY repeats of gp12 to build a stable intersubunit triple helix in a prokaryotic setting. The trimer dissociates and unfolds at near physiological temperatures, as reported for eukaryotic collagen. Its structural organization is reacquired within seconds upon cooling. Interaction with the SPP1 capsid hexamers strongly stabilizes gp12, increasing its Tm to 54 °C. Above this temperature, gp12 dissociates from its binding sites and unfolds reversibly. Multivalent binding of gp12 trimers to the capsid is highly cooperative. The capsid lattice also provides a platform to assist folding and association of unfolded gp12 polypeptides. The original physicochemical properties of gp12 offer a thermoswitchable system for multivalent binding of the polypeptide to the SPP1 capsid surface.
Introduction
Viruses are infectious agents characterized by an extracellular state, the virus particle or virion, which protects the viral genome from environmental aggression and ensures its highly efficient delivery to host cells for virus multiplication. The viral particle is a protein nanocage, sometimes combined with a lipid membrane, surrounding the nucleic acid molecule(s) that code(s) for the hereditary genetic information of the virus. A large number of prokaryotic and eukaryotic virions have an icosahedral protein shell of homogeneous size, termed the capsid. Its self-assembly exploits (quasi-)equivalent interactions between a large number of identical protein subunits (1–3). Viruses with long dsDNA genomes, like tailed bacterial viruses (bacteriophages or phages) and the eukaryotic pathogen herpesvirus, first assemble an icosahedral protein lattice, the procapsid (4, 5) (Fig. 1). This structure is formed by major capsid protein subunit hexamers found at the planar faces of the icosahedron and by pentamers that define its angular vertices (2, 3). Viral DNA is then translocated to the procapsid interior through a specialized portal vertex by a powerful nanomotor, leading to tight packing of dsDNA in the capsid interior. During DNA packaging, the capsid undergoes a major conformational change called expansion. It leads to a gain in volume, stability, and, in numerous viral systems, to the creation of capsid auxiliary protein binding sites (3, 6–9). Those proteins cement structurally weak capsid points by establishing additional interhexamer bonding or attach to the center of hexamers (9–16). In both cases, they establish a symmetrically organized array of polypeptides at the capsid surface. In contrast to the conserved fold of the major capsid protein of the tailed bacteriophage-herpesviruses lineage (3, 5), their auxiliary proteins can have diverse length, structure, and biochemical properties (9–16).
FIGURE 1.
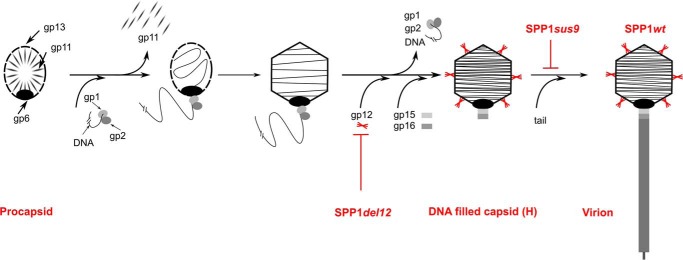
Schematic of the SPP1 virion assembly pathway. Phage proteins involved in capsid assembly are labeled as follows: gp1-gp2, terminase complex; gp6, portal protein; gp11, procapsid scaffolding protein; gp12, capsid auxiliary protein; gp13, major capsid protein; gp15, connector adaptor protein; gp16, connector stopper protein (25). Virus strains impaired in production of gp12 (SPP1del12) and of tail structures (SPP1sus9) are shown in red, and the step of assembly they affect is identified. SPP1del12 was used to produce SPP1 infectious particles lacking gp12, whereas SPP1sus9 and SPP1sus9del12 were used to produce tailless DNA-filled capsids with (capsid H) and without gp12 (capsid HΔ12). Gp12 trimers are also highlighted in red.
The high fidelity of viral capsids assembly yields a population of homogeneous, robust particles. Their symmetric elements are arranged accurately with nanometer precision, offering excellent systems to engineer versatile enzymatic or bioactive nanoparticles (17–21). This can be efficiently achieved by molecular biology and chemical approaches (22, 23) that rely on detailed knowledge of the molecular structure of the virion and of the biochemical behavior of its components.
SPP1, like all other tailed bacterial viruses and herpesviruses, assembles a procapsid that serves as a container for subsequent viral DNA packaging (Fig. 1). The structure is composed of 415 subunits of the major capsid protein gp13, organized following quasi-equivalent interaction rules to build an icosahedron with a triangulation number (T) of 7 (16, 24). DNA pumping to the procapsid interior through a specialized portal vertex is accompanied by a major rearrangement of the capsid lattice that acquires a clear icosahedral outfit, increasing in diameter more than 50 Å (24, 25). This expansion process creates the binding site for gp12 (Fig. 1) (26) at the center of each of the 60 gp13 hexamers in the icosahedral lattice (16). Viral DNA packaging is followed by binding of a tail to the DNA-filled capsid, yielding the infectious virion (Fig. 1) (27, 28). To uncover the molecular principles of how auxiliary proteins interact with the surfaces of viral particles, we investigated the properties of gp12. This 6.6-kDa polypeptide is shown to adopt a collagen-like fold with the remarkable property of binding reversibly in a temperature-dependent fashion to its 60 sites at the SPP1 capsid surface.
EXPERIMENTAL PROCEDURES
Cloning Procedures and Creation of the Gene 12 Knockout SPP1 Strain
Gene 12 was cloned into plasmid pRSET A (Invitrogen) for protein overproduction in Escherichia coli using the strategy described by Lurz et al. (29). The resulting plasmid, pBT453, codes for tag-gp12, in which the gp12 amino terminus is fused in-frame to a 36-amino acid-long tag that includes a hexahistidine sequence. To engineer a cleavage site for tobacco etch virus (TEV)3 protease (ENLYFQG) between the tag and the gp12 amino acid sequence, gene 12 was amplified from pBT453 DNA with oligonucleotides TGS (TAAGGTACCGGATCCGAGAATCTGTACTTCCAGGGCATGTCTAAGCGTATACCGCGTTTCTTGC; the BamHI site is underlined, the sequence coding for a TEV protease cleavage site is in italicized, and the beginning of gene 12 is shown in boldface) and TGA (ATACTCGAGTACCAGCTGCAGTTATTAAGTCGTTCC; the PstI site is underlined, the gene 12 complementary coding sequence is shown in boldface, and stop codons are double-underlined). The PCR fragment was then cleaved with BamHI-PstI and cloned into pRSET A, generating pMZ1.
Plasmid pBT450 was constructed in two steps. First a SfcI fragment bearing genes 15 and 16 of SPP1 (coordinates 8830–9787 of the SPP1 genome sequence, GenBankTM accession number X97918 (30)) was treated with a Klenow fragment to produce blunt ends and cloned in the SmaI site of pBluescript SK- (Stratagene). Secondly, the resulting plasmid was used to clone a NruI-EarI blunt-ended fragment (coordinates 6699–8778 of the SPP1 sequence), bearing genes 11-13, in the HincII site of the pBluescript SK- polylinker. The cloning strategy generated a polycistronic unit composed of SPP1 genes 11-13 and 15 and 16 under the control of a T7 promoter. A DNA fragment containing gene 11 and the beginning of gene 12 was produced by cleavage of pBT450 with BglI, treated with T4 polymerase to produce blunt ends, and digested with Asp718. This fragment was cloned into pBT450 previously digested with SmaI and KpnI to generate pBT451. In pBT451, gene 12 is disrupted by an out-of-frame deletion between its internal BglI and SmaI sites (coordinates 7618–7646 of the SPP1 sequence). The E. coli-Bacillus subtilis shuttle vector pHP13 (31) cut with PstI-SalI was used for cloning a PstI-XhoI fragment of pBT451 spanning genes 11 to 13 that flank the gene 12 knock-out deletion. The resulting plasmid was named pBT452.
The non-permissive strain B. subtilis YB886 (pBT452) was infected with a mutant phage carrying conditional lethal mutations in genes 11 and 13 (SPP1sus7sus31 (32)), forcing a double crossover that led to integration of the knockout mutation in gene 12 of the viral genome, as confirmed by DNA sequencing. The resulting phage, SPP1del12, was crossed with SPP1sus9, a mutant defective in a tail gene (33), to yield SPP1sus9del12 (16). SPP1del12 was used to produce SPP1 infectious particles lacking gp12, whereas SPP1sus9 and SPP1sus9del12 were used to produce tailless DNA-filled capsids with (capsid H) and without gp12 (capsid HΔ12), respectively.
Production and Purification of SPP1 Virions and DNA-filled Capsids
Procapsids, DNA-filled capsids, and viral particles were produced and purified as described previously (16, 33). Procapsids were kept in buffer R (50 mm potassium glutamate, 10 mm EDTA, 50 mm Hepes-KOH (pH 7.6), and 1 mm PMSF, added freshly (33)), whereas all other structures were stored stably and manipulated in TBT buffer (100 mm NaCl, 10 mm MgCl2, and 100 mm Tris-Cl (pH 7.5)). All interactions of tag-gp12/gp12 with viral structures were carried out in TBT buffer.
The concentration of capsid physical particles was estimated on the basis of their DNA content. Ultraviolet absorbance spectra of capsid suspensions were used to assess sample purity and the value at 260 nm to determine DNA concentration. This value was then used to calculate the concentration of capsid physical particles according the following equation:
where T is the concentration of physical particles/liter, c is the DNA concentration in grams/liter, NA is the Avogadro constant, and nbp is the average number of base pairs per SPP1 DNA molecule. The SPP1-packaged molecules were considered to have an average length of 45.9 kbp (16).
Production and Purification of Tag-gp12
Tag-gp12 was overproduced in E. coli BL21 (DE3) (pBT453). Cells were grown at 37 °C in Luria broth medium supplemented with 100 μg/ml ampicillin. An overnight culture was diluted 50-fold, grown to an optical density at 600 nm between 0.6 and 0.8, induced with isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm and shaken for 3 h. Cells were harvested (30,000 × g, 30 min, 4 °C), resuspended in buffer A (500 mm NaCl, 10 mm imidazole, and 50 mm NaH2PO4 (pH 8.0)) supplemented with protease inhibitor mixture (CompleteTM EDTA-free, Roche Applied Science) and disrupted by sonication on ice using three cycles of 2 min each spaced by 2-min pauses (Vibra Cell 72405, Fisher Bioblock, Illkirch, amplitude 60, pulse 3, 30–40 watt). The total soluble proteins extract obtained after centrifugation (30,000 × g, 1 h, 4 °C) was filtered through a 0.22-μm membrane. The filtrate was then loaded on a 5-ml HisTrapTM HP metal affinity column (GE Healthcare) coupled to an ÄKTA purification system (GE Healthcare). A three-step gradient was applied at 16 °C: 2% buffer B (500 mm NaCl, 500 mm imidazole, and 50 mm NaH2PO4 (pH 8.0)) for a first wash, 10% buffer B for a second wash, and 100% buffer B for elution. The tag-gp12 peak fractions were pooled and run through a preparative size exclusion chromatography column (HiLoad 26/60 SuperdexTM 200pg, GE Healthcare) pre-equilibrated in buffer C (500 mm NaCl and 50 mm Na2HPO4 (pH 8.0)) at 16 °C coupled to an ÄKTA purification system. Aggregates and contaminants were found mostly in the void volume, whereas tag-gp12 eluted as a single peak. Tag-gp12 was obtained at a yield of 3 mg/g wet cell weight and was more than 95% pure, as judged from SDS-PAGE analysis. Purified protein was stored in buffer C and dialyzed against other buffers immediately before use. Protein concentration was estimated using the Bio-Rad protein assay following the instructions of the manufacturer. Tag-gp12 was used to immunize rabbits following the protocols established for protein (pC)CAT (34) to obtain anti-tag-gp12 polyclonal serum.
TagTEV-gp12 was produced and purified according to the same protocol. The purified protein was then incubated at 16 °C for 4 h with TEV protease at a ratio of 1:20 (w/w). To remove the tag, the digestion product was loaded onto a 1-ml HisTrapTM HP metal affinity column. Gp12 was eluted with 80 mm imidazole. The tag and the TEV protease eluted at 500 mm imidazole. The purified gp12 carries an additional glycine at its amino terminus, preceding the initial methionine residue.
Mass Spectrometry
The collagenase digestion of tag-gp12 followed by trypsination was stopped by adding solid guanidine hydrochloride to a final concentration of 6 m, followed by incubation at 90 °C for 15 min. Peptides were precipitated at −20 °C over a weekend by adding 5 volumes of cold acetone. Peptides were recovered by centrifugation, dried, and resuspended in ammonium carbonate at 1 μg/μl. They were then analyzed by MALDI-TOF and nano-LC-MS/MS.
MALDI Peptide Mass Fingerprinting
Peptides (0.5 μl) were mixed with an equal volume of either α-cyano-4-hydroxycinnamic acid (10 mg/ml and 50% CH3CN, Sigma-Aldrich) or 2,5-dihydroxybenzoic acid (10 mg/ml and 20% CH3CN, Sigma-Aldrich). Peptide mixtures were analyzed by MALDI-TOF (Voyager-DESTR, Applied Biosystems) after external calibration. Crystals were obtained using the dried droplet method, and 500 MALDI mass spectra were averaged per spot. Mass spectrometry measurements were carried out at a maximum accelerating potential of 20 kV in positive reflectron mode. Peak lists were generated by Data Explorer software (Applied Biosystems), and processed data were submitted to the FindPept tool (available on the Expasy portal) using the following parameters: data bank gp12 protein; mass tolerance, 300 ppm; digest reagents, none.
Nano-LC-ESI-MS/MS Analyses
The peptide mixture was then analyzed with the Q/TOF Premier mass spectrometer (Waters) coupled to a nanoRSLC chromatography unit (Dionex) equipped with a trap column (Acclaim PepMap100 C18, 75 μm inner diameter × 2 cm, 3 μm, nanoViper) and an analytical column (Acclaim PepMapRSLC C18, 75 μm inner diameter × 15 cm, 2 μm, 100 Å, nanoViper). The loading buffer was H2O/CH3CN/TFA (98/2/0.05%). Buffer A and B were H2O/HCOOH (0.1%) and CH3CN/HCOOH (0.1%), respectively. A 2–50% B gradient was set for 40 min with a flow rate of 300 nl/min. Data-dependent scanning was applied to generate MS/MS spectra with a collision energy ramp of 15–40 volts. Standard MS/MS acquisitions were performed on the top of the three most intense parent ions of the previous MS scan. Raw data were processed with ProteinLynx Global Server (Waters). Peptide identification was achieved using the Mascot software with the following parameters: data bank gp12 protein; peptide tolerance, 15 ppm; fragment tolerance, 0.1 Da; digest reagent, none.
Digestion of Tag-gp12 with Collagenase
Collagenase VII from Clostridium histolyticum (8.8 units/mg) was purchased from Sigma-Aldrich. A stock solution was prepared at 1 mg/ml in buffer D (250 mm NaCl, 10 mm CaCl2, 10 mm 2-mercaptoethanol, and 20 mm HEPES-Na (pH 7.6)) and diluted 10-fold before use. Tag-gp12 (50 μg) was digested with 0.23 μg of collagenase for 4 h at 16 °C. The same result was obtained by digestion for 30 min at 37 °C. Digestion products were analyzed on SDS-PAGE gel stained with Coomassie Blue and by mass spectrometry as described above.
Analytical Size Exclusion Chromatography (SEC)
100 μl of purified tag-gp12 at 2 mg/ml was run at 16 °C using a flow of 0.5 ml/min on a SuperdexTM 200 10/300 GL (GE Healthcare) column equilibrated in buffer C (500 mm NaCl and 50 mm Na2HPO4 (pH 8.0)) and coupled to an ÄKTA purification system. Column calibration and Stokes radius estimation were carried out as described previously (35).
Analytical Ultracentrifugation
Analytical ultracentrifugation was carried out on a Beckman Optima XL-A ultracentrifuge (Beckman Coulter, Palo Alto, CA) equipped with 12-mm central cells on an ANTi-60 rotor. Runs were performed in buffer C at 16 °C and monitored by absorption at 280 nm. The tag-gp12 partial specific volume (0.7051 ml/g), buffer C solvent density (1.02647 g/ml), and solvent viscosity (1.1913 cP) were calculated using the SEDNTERP software (36).
Sedimentation velocity runs were carried out at a rotor speed of 220,000 × g using protein loading concentrations of 0.5, 1, and 2 mg/ml. Data points were recorded every 5 min and analyzed using the SEDFIT software, assuming a non-interacting species model.
Equilibrium sedimentation was performed at 16,300 × g using protein loading concentrations of 0.3, 0.5, and 0.8 mg/ml. Data were analyzed using SEDFAT for average mass determination.
CD Measurements
CD measurements were carried out on a Jasco J810 spectropolarimeter equipped with a Peltier temperature controller. The protein, at 2 mg/ml, was dialyzed against either buffer C or TBT buffer and loaded to a 0.1-mm path length quartz cell. Spectra at fixed temperatures were recorded at an equilibrium of between 190–260 nm every 0.2 nm using a bandwidth of 1 nm and a scanning speed of 20 nm/min. Each spectrum was an accumulation of five spectra after baseline correction using the buffer spectrum as blank. Thermal transition profiles were monitored at 200 nm with a 1 °C/min heating rate and a protein concentration of 2 mg/ml. One point was recorded for each 1 °C. The temperature was raised from 10 to 60 °C, kept at 60 °C for 30 min, and finally returned back to 10 °C using the same rate. Ellipticity was measured first and molar ellipticity was then calculated using the following equation:
where θ is the ellipticity in degrees, M is the molecular mass, c is the protein concentration in mg/ml, and l is the path length in centimeters. Tms was determined from data plots as the transition midpoint.
Fluorescence-based Thermal Shift Assay (FBTSA)
FBTSA experiments were performed as described previously (16). In brief, SPP1 virions or capsids (5.7 × 1010 particles) and/or purified tag-gp12 at different concentrations were mixed with diluted Sypro Orange dye (400-fold diluted from stock solution, Invitrogen) in TBT buffer to a final volume of 10 μl. Experiments were carried out in real-time PCR systems, and fluorescence was recorded in real time. Different heating-cooling cycles were applied to the samples as described in the figure legends and under “Results.” Experiments were carried out in ABI 7900HT and QuantStudio 12KFlex machines (Applied Biosystems) as detailed in the figure legends. The fluorescence profiles, derivatives, and Tms were determined using the analysis software of the manufacturer.
Gp12 Chimerization Experiments
A 2-fold molar excess of purified gp12 was mixed with tag-gp12 and kept either at 16 °C or heated for 15 min at 60 °C. Mixtures were then loaded onto a 1-ml metal affinity column (HisTrapTM HP metal affinity column, GE Healthcare), and proteins were eluted by applying a step gradient of imidazole concentration as used for tag-gp12 purification (see above).
Binding of Tag-gp12 to SPP1del12 Virions
100 μl of SPP1del12 virions at 5.6 × 1012 pfu/ml were mixed with a 10-fold molar excess of tag-gp12 protein (550 μl of a 2 mg/ml solution), considering 60 binding sites for tag-gp12 trimers per capsid, and incubated overnight at 16 °C. The mixture was then run through a discontinuous cesium chloride gradient to purify phage particles (37), and their protein composition was analyzed by Western blotting with rabbit polyclonal antibodies raised against purified tag-gp12 or against purified SPP1 virions.
Binding of Tag-gp12 to HΔ12 Capsids Analyzed by a Trypsin Protection Assay or FBTSA
Tag-gp12 was dialyzed against TBT. A range of tag-gp12 concentrations and purified SPP1 capsids lacking gp12 (HΔ12) were incubated separately for 1 h at 16 or 45 °C in a PCR machine (Biometra Tprofessional Trio thermocycler). A constant number of HΔ12 was then mixed with variable amounts of tag-gp12 to obtain different molar ratios and incubated at 16 or 45 °C for the desired reaction time according to the experimental schematic in Fig. 7, left panels. Samples were analyzed by FBTSA or incubated with 1 μg of trypsin at 45 °C for 30 min to proteolyse free tag-gp12 (not associated with capsids). Care was taken to avoid any cooling below 45 °C for samples whose mixtures were incubated at this temperature before FBTSA or trypsination. This was necessary to prevent rapid refolding/reassociation of free tag-gp12, which would facilitate assembly of free trimers and their binding to capsids. Capsids in trypsinated samples were separated on 0.8% agarose gels prepared in TAMg buffer (1 mm MgCl2 and 40 mm Tris acetate (pH 8.3)). The running buffer was TAMg, and the applied electric field intensity was 70 mA. Gels were stained with ethidium bromide in TAE buffer (40 mm Tris-acetate supplemented with 1 mm EDTA) in which EDTA led to disruption of capsids in situ, rendering viral DNA accessible to ethidium bromide binding, a more sensitive detection method than protein staining with Coomassie Blue.
FIGURE 7.

Capsid binding behavior of native and unfolded tag-gp12. Purified SPP1 tailless capsids lacking gp12 (capsid HΔ12) (blue characters and blue curves on the left and blue rectangles above the gels on the right) and tag-gp12 (red) were preincubated separately, mixed, and treated with trypsin (except in A) according to the different combinations of incubation conditions used in the experiments in A–E (see “Results” for details), as outlined on the left of each panel. Samples treated with trypsin in B–E are identified by yellow rectangles above the gel lanes on the right. Capsids were then resolved by agarose gel electrophoresis to assess their occupancy with tag-gp12. Wild-type SPP1 capsids with gp12 (H capsids) (green rectangles above the gels on the right) and HΔ12 were used as controls. The schematics in the center of A show the electrophoretic mobility of capsid H (with gp12 represented in red) and HΔ12. The experiment was repeated four times independently.
Bioinformatics
Protein secondary structure predictions were carried out using Jpred (38), and the three-dimensional structure was predicted using HHPred (39).
RESULTS
Gp12 Has a Collagen-like Sequence Motif
The SPP1 capsid auxiliary protein gp12 is a 64-amino acid-long polypeptide with a molecular mass of 6613 Da and a theoretical isoelectric point of 8.14. Its carboxyl terminus is predicted to form α-helices, whereas the central part features eight GXY repeats (Fig. 2A) (40). The repeated GXY motif is a sequence signature of collagen-like proteins in which three polypeptides are brought together to form an intermolecular, left-handed triple helix (41, 42).
FIGURE 2.

Properties of the SPP1 auxiliary protein gp12. A, Gp12 amino acid sequence showing position of the tag fused to its amino terminus. GXY triplets are underlined. The intermolecular collagen-like triple helix and α-helix predicted by bioinformatics are shown above the sequence. The collagenase cut of tag-gp12 inside the collagen-like sequence motif and peptides obtained from mass spectrometry analysis (E and F) are shown below. B, RH determination of native and unfolded-refolded tag-gp12. Native (dotted line) and tag-gp12 heated for 5 min at 90 °C and transferred directly to ice (continuous line) were analyzed by SEC at 16 °C as described under “Experimental Procedures.” The elution positions of thyroglobulin (RH = 85 Å), γ-globulin (RH = 48 Å), ovalbumin (RH = 30,5 Å), and myoglobin (RH = 20,7 Å) used to calibrate the column are indicated by arrows. Kav (partition coefficient) was calculated as described (35). mAU, milli absorbance units. C, sedimentation velocity of tag-gp12 at 220,000 × g (loading concentration of 1 mg/ml, 16 °C run). Gp12 has a sedimentation coefficient of 1.7 S (s20,w = 2.2 S). D, sedimentation equilibrium of tag-gp12 at 16,300 × g (loading concentration of 0.8 mg/ml, 16 °C run). The data (dots) were fit using a trimer model (continuous line). The best fit was obtained for a single species with an average mass of 31,270 ± 590 Da. The top panels in C and D show the deviation of experimental points from fitted curves. E and F, cleavage of tag-gp12 with collagenase VII analyzed by SDS-PAGE (inset in E) and mass spectrometry. The observed ion mass (4406.15 Da) in MALDI-TOF (E) is attributed to peptide 20–64 of the gp12 sequence, identifying the proteolysis site shown in A. The same peptide was detected by LC-MS/MS spectrometry, which also showed the presence of three other peptides, resulting from collagenase cleavage at the same position (F). The LS-MS/MS analysis had a tag-gp12 sequence coverage of 89%. For clarity, only the peptides in which one end was generated by the collagenase cleavage are listed in F. Those peptides were absent in the analysis of tag-gp12 not treated with collagenase.
Gp12 Is an Elongated Trimer in Solution
The gp12 amino terminus was fused to a 36-amino acid-long peptide including a hexahistidine tag to enhance protein production and allow easy purification. The 10.7-kDa recombinant protein (tag-gp12) eluted from a Superdex 200 analytical SEC column as a single symmetric peak (Fig. 2B). Its hydrodynamic radius (RH) on the basis of a protein calibration data set was 35 Å. The gp12 elongated shape observed in electron microscopy reconstructions of the bacteriophage SPP1 capsid (16) rendered SEC not suitable to estimate its native mass (43). The shape and oligomerization state of tag-gp12 were therefore investigated by analytical ultracentrifugation at 16 °C. Tag-gp12 behaved as a homogeneous species with a sedimentation coefficient of 1.7 S (s20,w = 2.2 S) (Fig. 2C) at all loading concentrations tested in sedimentation velocity experiments (0.5–2 mg/ml). Sedimentation equilibrium centrifugation was then used for shape-independent measurement of the tag-gp12 mass (Fig. 2D). The determined molecular mass (31,270 ± 590 Da) was only 3% lower than the theoretical mass of a tag-gp12 trimer. Using this experimental value and the sedimentation coefficient, we calculated a friction ratio (f/f0) of 1.79, showing that tag-gp12 is an elongated trimer in solution.
Gp12 Has a Collagen-like Fold
To probe that the (GXY)8 repeats of tag-gp12 form a collagen-like triple helix, the protein was challenged with collagenase VII, which cuts the triple helix at defined environments (44). The control SPP1 proteins gp6 and H16, a tagged form of gp16 (45), were insensitive to proteolysis (not shown), whereas tag-gp12 was cleaved (Fig. 2E, inset). MALDI-TOF (Fig. 2E) and nano-LC-MS/MS (Fig. 2F) identified the cut between Gln-19 and Gly-20 of tag-gp12 (Fig. 2A, arrow). This site, found at the beginning of the GXY repeat region, matches one of the expected cutting sites for collagenase VII (44).
Collagen left-handed triple helices are also characterized by a CD signature with a deep minimum of negative ellipticity at around 200 nm and a slightly positive ellipticity maximum at around 220 nm (46). The CD spectrum of native tag-gp12 had a strong minimum at 200 nm and a second minimum at 222 nm, where the ellipticity of α-helices masked the positive signal of the collagen helix (Fig. 3A). This profile strongly supports that tag-gp12 combines a collagen-like fold with α-helical regions.
FIGURE 3.
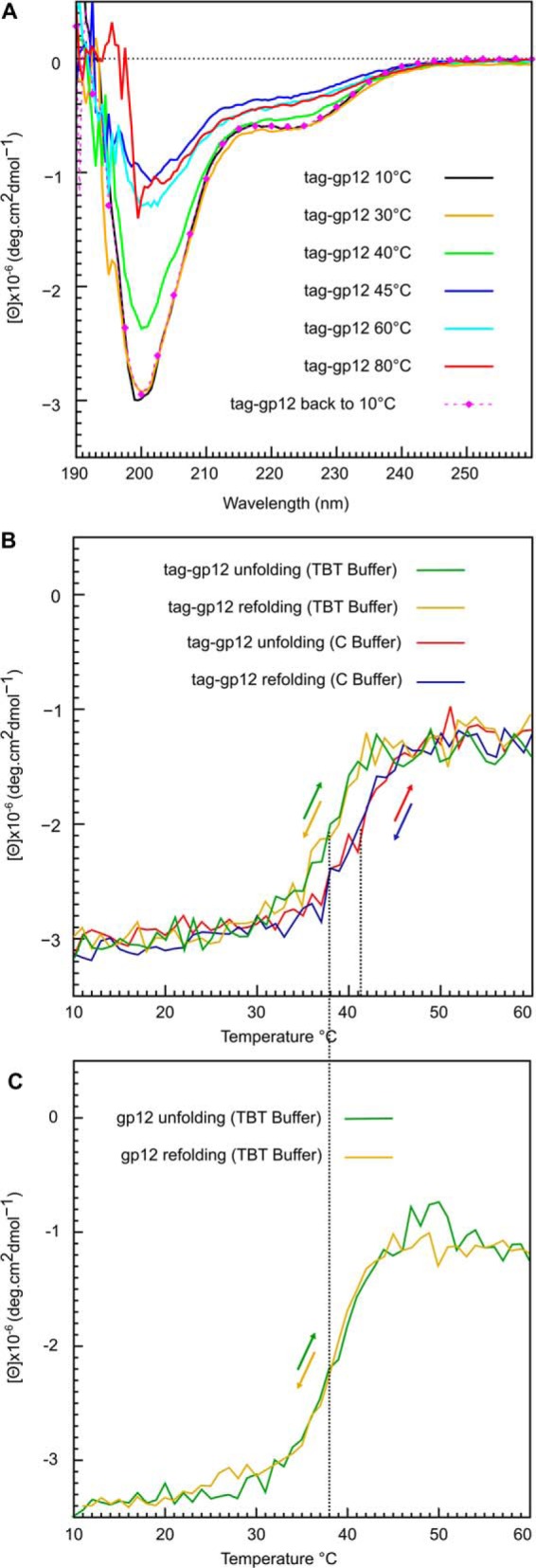
Reversibility of tag-gp12 trimer unfolding and dissociation. A, CD spectra of tag-gp12 (2 mg/ml) in buffer C were recorded using the same sample at different temperatures. Tag-gp12 was then maintained at 80 °C for 30 min and cooled back to 10 °C to record the spectra of the refolded protein (pink dotted line). B, tag-gp12 unfolding and refolding in buffer C and in TBT monitored by CD at 200 nm, the collagen-like triple helix local minimum signal, using a temperature gradient of 1 °C/min. The colored arrows show the direction of the temperature gradient (heating or cooling) for each individual color curve. C, gp12 unfolding and refolding in TBT monitored as in B. The dotted vertical lines in B and C are a visual aid to show the transition midpoints (Tm) in buffer C and TBT. The experiment was repeated twice independently.
Gp12 Dissociates and Unfolds Reversibly at Physiological Temperature
CD spectra showed a loss of tag-gp12 structure between 30 and 45 °C (Fig. 3A). The CD spectra from 45–80 °C were characteristic of an unfolded polypeptide chain. Fast (<1 min) or progressive cooling of the sample back to 10 °C led to complete recovery of the secondary and quaternary structure content, with a CD spectrum identical to the one of the native protein (Fig. 3A, pink dotted line).
To further analyze the dissociation-unfolding and refolding-reassociation transitions, a CD experiment was monitored at 200 nm (corresponding to the collagen-like helix minimum) by challenging the sample against a heating cycle from 10–60 °C (unfolding) and back to 10 °C (refolding) (Fig. 3B). Tag-gp12 showed a sharp transition with a Tm of 41 °C in the protein-high salt buffer and of 38 °C in a low monovalent salt solution with magnesium (TBT buffer that stabilizes SPP1 viral particles) (Fig. 3B). Unfolding and refolding followed the same kinetic profile upon heating and cooling (Fig. 3B). The thermal stability study of tag-gp12 by CD revealed a unique transition with complete loss of secondary structure and dissociation of the collagen-like triple helix (Fig. 3, A and B). The behavior of tag-free gp12 was identical to tag-gp12 (data not shown and Fig. 3C), revealing that the tag influenced neither the protein CD signature nor its dissociation/unfolding and refolding/reassociation properties. The SEC profiles of native and unfolded-refolded tag-gp12 were also indistinguishable, with a single symmetric peak of trimers and no detectable intermediate states (Fig. 2B). The complete population of refolded tag-gp12, therefore, retrieved its initial RH.
To define whether the tag-gp12 polypeptide chains physically separate upon thermal denaturation, we carried out a chimerization experiment between tag-gp12 (10.7 kDa subunit mass) and tag-free gp12 (6.6 kDa subunit mass). The hexahistidine-tagged tag-gp12 bound strongly to a metal affinity column and eluted only in the presence of 500 mm imidazole (Fig. 4, first panel). Gp12 also adsorbed to the column matrix but was completely released by a wash with 100 mm imidazole (Fig. 4, second panel). Loading of a tag-gp12:gp12 mixture kept at 16 °C led to differential elution of gp12 at 100 mm imidazole and of tag-gp12 at 500 mm imidazole (Fig. 4, third panel). When the tag-gp12:gp12 mixture was denatured at 60 °C and reassociated by cooling to 16 °C, there was a fraction of non-tagged gp12 that coeluted with tag-gp12 at 500 mm imidazole (Fig. 4, fourth panel). This behavior is explained by the presence of heterotrimers in which the tag-gp12-tagged subunit(s) led to retention of the non-tagged gp12 form present in the heterotrimer. The formation of chimeras showed that the gp12 and tag-gp12 trimers physically dissociated upon thermal denaturation and that reassociation led to the formation of heterotrimers, although homotrimerization appeared to be favored when comparing the intensity of bands in Fig. 4, fourth panel.
FIGURE 4.
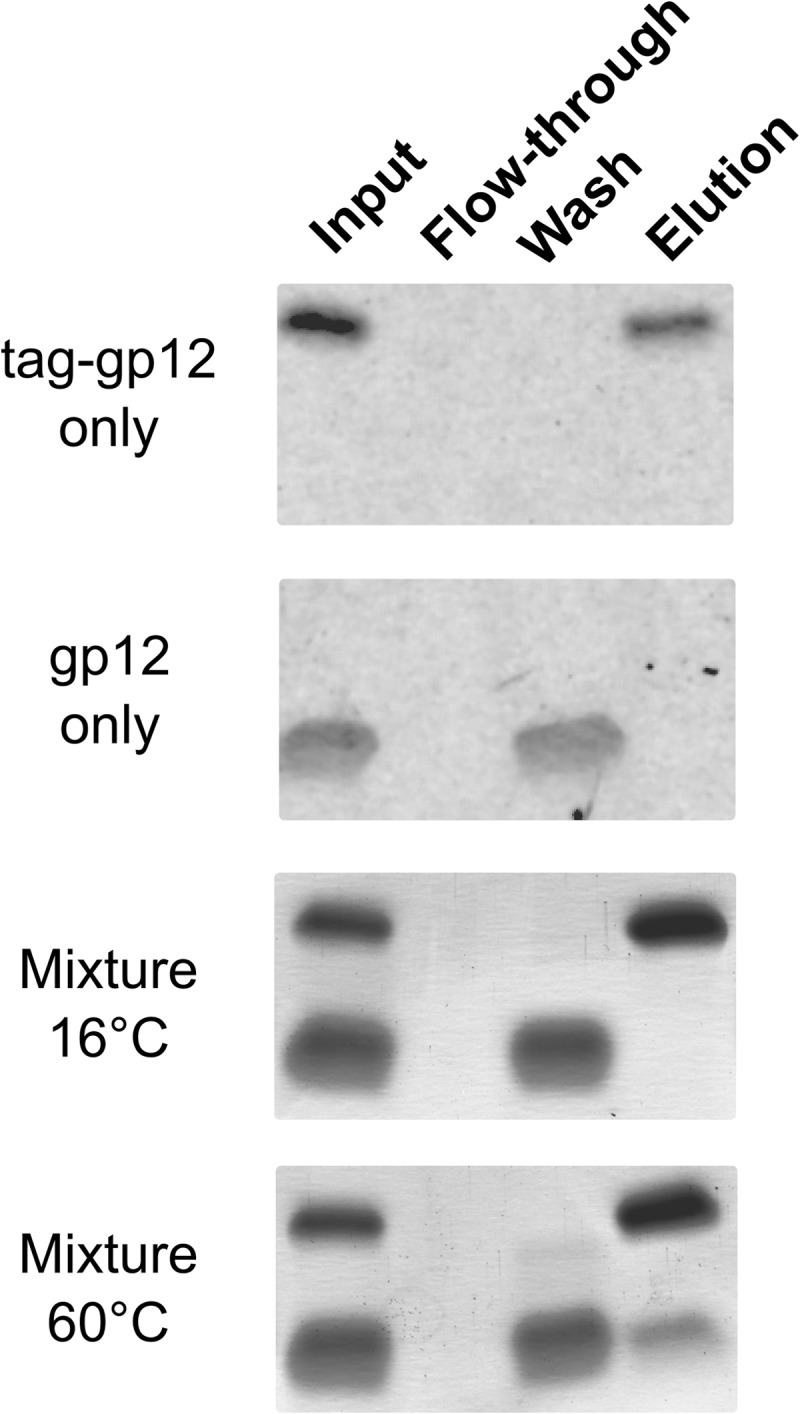
Tag-gp12/gp12 chimerization experiment. Isolated proteins and their mixture incubated at 16 or 60 °C (50 μl of 2 mg/ml in buffer C) were loaded onto a metal affinity column. Aliquots of the input proteins before chromatography, flow-through, washing with 100 mm imidazole, and elution with 500 mm imidazole were analyzed on a 12% Tris-N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine gel stained with Coomassie Blue. The experiment was repeated twice independently.
Binding of gp12 to the Capsid Lattice Is Reversible and Increases the Trimer Thermal Stability of 20 °C
To characterize the interaction of gp12 with SPP1 capsids, we generated viral particles (SPP1del12) and tailless expanded capsids (HΔ12) lacking gp12 by genetic engineering (Figs. 1 and 5A). These particles bound tag-gp12 in vitro, whereas wild-type virions whose capsid carries gp12 did not (Fig. 5B). Therefore, tag-gp12 interacts strongly and exclusively with specific sites in the SPP1 capsid lattice without any detectable exchange between free (tag-gp12) and capsid-bound (gp12) subunits.
FIGURE 5.

Binding of gp12 to SPP1 capsids. A, composition of SPP1 assembly intermediates (cf. Fig. 1) determined by SDS-PAGE gel stained with Coomassie Blue (left panel) and presence of gp12 in the structures detected with anti-tag-gp12 polyclonal antibodies (right panel). The major capsid protein gp13, the major tail tube protein gp17.1, and the procapsid internal scaffolding protein gp11 are also identified in the Coomassie Blue-stained gel. B, binding of tag-gp12 to wild-type SPP1 and to SPP1del12 particles. Virions incubated overnight at 16 °C with tag-gp12, as indicated above the Western blot analyses, were separated from free protein by isopycnic centrifugation in cesium chloride gradients, and the composition of particles was analyzed by Western blotting with polyclonal antibodies raised against purified SPP1 virions (left panel) and anti-tag-gp12 antibodies (right panel). Note that gp12 and the tail protein gp17.1* (28) are the most immunogenic proteins of the SPP1 particle despite of the fact that they are not the most abundant components of the virion (A, left panel) (Refs. 28, 40 and this work).
The FBTSA method allows monitoring independently of the thermal denaturation of gp12 and of the major capsid protein gp13 (16). The assay quantifies binding of the Sypro Orange dye to exposed hydrophobic regions of proteins challenged to a temperature gradient (47). In an aqueous environment, Sypro Orange has a low quantum yield, and in protein solutions, the dye access to non-polar environments is normally shielded by the protein fold. Protein thermal denaturation exposes hydrophobic regions where the dye binds, resulting in strong fluorescence emission. Isolated tag-gp12 exhibited the opposite behavior. An increase of temperature led to progressive loss of fluorescence, followed by a sharp transition at a Tm of 33.4 ± 0.7 °C in TBT buffer (Fig. 6A, red curve), which is 4.6 °C lower than the unfolding Tm determined by CD (Fig. 3B). Gp12 without a tag showed the same behavior. The profile of this transition revealed that Sypro Orange has one or several binding sites in native tag-gp12 that were destroyed when the protein started to lose its secondary and quaternary structure. Such a rare property provided a specific signature for tag-gp12 unfolding.
FIGURE 6.
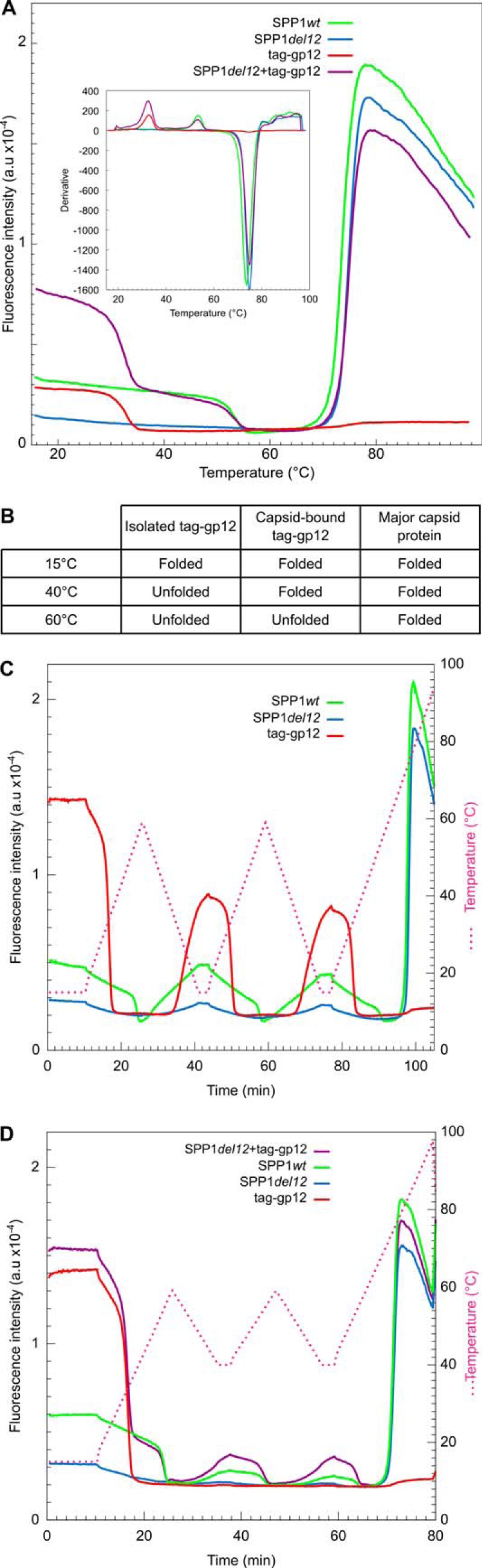
Cyclical gp12 association and dissociation from viral capsids. Sypro Orange was added to protein and viral particle samples that were submitted to different heating and cooling regimens at a rate of 3 °C/min and monitored by FBTSA in an ABI 7900HT machine. A, FBTSA of isolated tag-gp12 (red curve), wild-type SPP1 virions (green curve), SPP1del12 virions that lacked gp12 (blue curve), and SPP1del12 mixed with an excess of tag-gp12 (violet curve). The inset shows the opposite of the first derivative of the fluorescence signal. B, summary of the tag-gp12/gp12 and gp13 states at different temperatures. C, isolated tag-gp12, wild-type SPP1, and SPP1del12 virions submitted to cycles of heating to 60 °C and cooling to 15 °C. The experiment was finished with a denaturation step to 99 °C. The pink discontinuous line shows the temperature variation (coordinates are shown on the right). D, the same samples and a mix of SPP1del12 virions with a 5.5 molar excess of tag-gp12 (violet curve) challenged with two cycles of heating to 60 °C and cooling to 40 °C. Experiments were repeated at least twice independently.
The FBTSA profile of infectious SPP1 phage particles (Fig. 6A, green curve) was marked by two transitions similar to the ones found for the tailless capsids (data not shown) (16), showing that only the capsid proteins of phage particles gave a detectable signal under our experimental conditions. Both structures carry gp12 (Fig. 5A). The first transition, at 53.6 ± 0.2 °C, displayed the tag-gp12 signature and was absent from particles lacking gp12 (SPP1del12, Fig. 6A, blue curve). Mixing of SPP1del12 phages with tag-gp12 in vitro restored the signal at 53.6 °C, whereas the excess of free protein led to the typical Tm transition at 33.4 °C (Fig. 6A, violet curve). The identical Tm of gp12 and tag-gp12 was 20.2 °C higher than the one observed for isolated tag-gp12, showing that binding to the capsid lattice led to a major stabilization of the gp12 trimer. The second signal transition of viral particles with or without gp12 was characterized by a strong increase of fluorescence at 75 ± 0.3 °C because of cooperative denaturation of gp13.
The distinct melting temperatures of isolated tag-gp12 (33.4 °C), of capsid-bound tag-gp12 or gp12 (53.6 °C), and of major capsid protein (75 °C) allowed us to follow the behavior of the three species in gp12-capsid binding experiments. At 40 °C, capsid-bound gp12 was easily distinguished from isolated tag-gp12 because it was the only folded gp12 form at this temperature, whereas, at 60 °C, only the capsid protein was stable (Fig. 6B). Isolated tag-gp12, wild-type SPP1, and SPP1del12 particles were submitted to two heating-cooling cycles between 15 and 60 °C, followed by a final heating step from 15–99 °C (15–60-15–60-15–99 °C, 3 °C/min heating/cooling rate). Free tag-gp12 exhibited a loss of signal upon heating, and its partial reacquisition when cooling to 15 °C showed that the fluorophore binding site(s) was/were not completely restored in the tag-gp12 population (Fig. 6C, red curve) in spite of the fact that the protein fully reacquired its quaternary structure CD signature (Fig. 3). The temperatures of transition were remarkably reproducible, revealing that tag-gp12 underwent dissociation/unfolding and folding/reassociation cycles. Gp12 bound to SPP1 capsids exhibited a transition corresponding to a Tm of 53.6 °C (Fig. 6C, continuous green curve). The process was reversible upon cooling and reheating, apart from a slight loss of fluorescence from one cycle to another. Therefore, gp12 dissociated/unfolded reversibly from wild-type capsids and maintained its binding activity to the capsid.
To assess whether the capsid lattice influences gp12 refolding/reassociation, the cycling experiment was repeated with cooling steps to 40 °C (15–60-40–60-40–99 °C program, Fig. 6D), a temperature at which free tag-gp12 remained unfolded after the first heating step (Fig. 3D, red curve). Gp12 bound to phage capsids kept its signature (Tm of 53.6 °C) in heating cycles to 60 °C. Cooling to 40 °C led to recovery of some fluorescence signals (Fig. 6D, green curve) but significantly less than when the temperature was reduced to 15 °C (Fig. 6C). Therefore, at 40 °C, a subpopulation of gp12 rebound to phage capsids, yielding folded trimers that fixed Sypro Orange. Addition of a 5.5-fold molar excess of exogenous tag-gp12 to wild-type capsids restored most of the gp12 signal associated with capsids after each 60–40 °C cycle (Fig. 6D, violet curve), showing that tag-gp12 had efficiently replaced gp12, which left its capsid sites upon denaturation. Restoration of the tag-gp12 signal at 40 °C occurred exclusively in presence of the capsid lattice, showing that this structure promoted tag-gp12 refolding and reassociation.
Native and Unfolded gp12 Binds to SPP1 Capsids in a Distinct Way
The finding that both native and unfolded tag-gp12 bound to SPP1 phage capsids (Fig. 6, C and D) suggested two distinct types of interaction, prompting their characterization. Tailless capsids without gp12 (HΔ12) and purified tag-gp12 were preincubated separately at 16 or 45 °C, followed by mixing at different ratios for interaction at the two temperatures (Fig. 7). Reactions were then incubated at 45 °C with trypsin, which degraded free gp12/tag-gp12 (Fig. 8A). The tag of capsid-bound tag-gp12 was prone to trypsin attack, but the gp12 moiety attached to the capsid remained intact (Fig. 8B), explaining the lower electrophoretic mobility of capsids loaded with tag-gp12 not treated with trypsin when compared with those that were trypsinated (Fig. 7, A and B). This step prevented subsequent interactions of free tag-gp12 with capsids during downstream sample manipulation at room temperature and separation by gel agarose electrophoresis. SPP1 capsids with gp12 (H capsids) or HΔ12 loaded with tag-gp12 had a slower electrophoretic mobility than capsids lacking gp12 (Fig. 7), most likely because gp12/tag-gp12 reduces the capsid surface electronegative charge. In contrast, gp12 does not have a major effect on capsid diameter, which is almost identical in H and HΔ12 (∼610 Å (16)).
FIGURE 8.
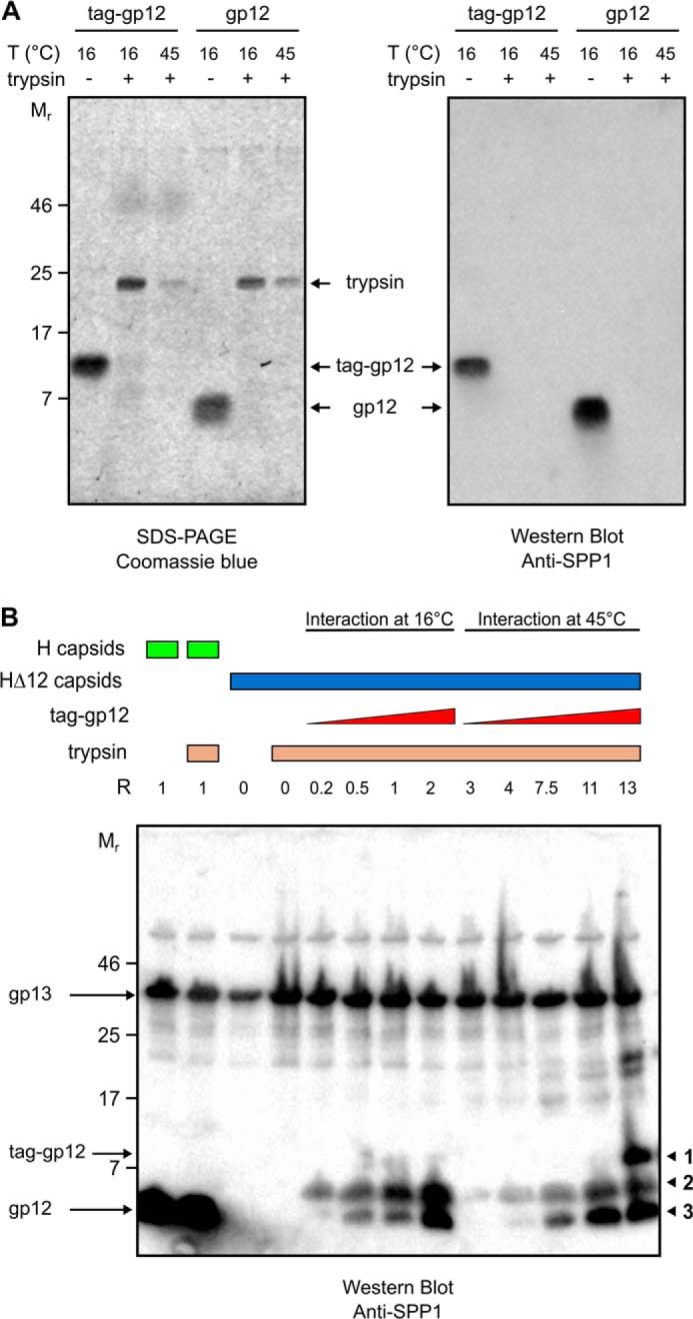
Trypsin sensitivity of free and capsid-bound gp12. A, purified tag-gp12 and gp12 were incubated with trypsin either at 16 or at 45 °C. Both proteins were completely digested by the protease at the tested temperatures, as assessed by Coomassie-stained SDS-PAGE (left panel) and Western blot analysis (right panel) with polyclonal anti-SPP1 antibodies that recognize gp12 (Fig. 5B). B, trypsin digestion of the binding reaction between capsids and tag-gp12 (labeled band 1 on the right of the figure) under the same conditions as in Fig. 7, B and C. Note that gp12 bound to H capsids is not sensitive to trypsin, whereas the tag of tag-gp12 is partially (band 2) or fully (band 3) digested by trypsin. The Western blot analysis was developed with anti-SPP1 antibodies that recognize gp12 but also, although giving a comparatively weak signal, the major capsid protein gp13, whose band was used to control the normalized input of capsids in the binding reaction.
When HΔ12 capsids were mixed at 16 °C with increasing amounts of tag-gp12 native trimers, the capsid species shifted from tag-gp12-free to capsids fully loaded with tag-gp12 (Fig. 7, A, B, and D). At a ratio (R = 60 tag-gp12 trimers/capsid) of 0.5, most capsids lacked tag-gp12, but a minority was already saturated with tag-gp12, whereas, at R = 1.5, almost all capsids were decorated with tag-gp12. Species with intermediate electrophoretic mobility were poorly detected, revealing that capsids partially occupied with tag-gp12 were a minor population, even at limiting amounts of tag-gp12 (e.g. R = 0.5). We attribute this behavior to high cooperative binding of tag-gp12 trimers to its 60 sites in the SPP1 capsid.
To characterize the interaction of unfolded tag-gp12 with the capsid, the two species were preheated individually at 45 °C and mixed at the same temperature (Fig. 7C). A significant excess of tag-gp12 per capsid (R between 4 and 13) was needed to promote a change of capsid electrophoretic mobility. Their discrete bands showed a migration pattern that progressed from the capsids lacking gp12 band behavior (R < 3) to the full tag-gp12-loaded capsid band (R ≥ 13) (Fig. 7C). Similar results were obtained when the interaction reaction was prolonged overnight at 45 °C (not shown). Furthermore, preheating of capsids at 16 °C or 45 °C showed that temperature did not affect their binding properties (Fig. 7). Stable interaction of unfolded tag-gp12 with HΔ12 capsids therefore required an excess of tag-gp12 that bound in an inefficient manner, leading to a population of capsids whose binding sites are only partially occupied by tag-gp12 at molar ratios as high as R = 11 (Fig. 7, C and E). The increase of occupancy with the rise of R correlated with an augmentation of the tag-gp12 signal in FBTSA experiments, consistent with the formation of tag-gp12 trimers in the capsid lattice (Fig. 9).
FIGURE 9.
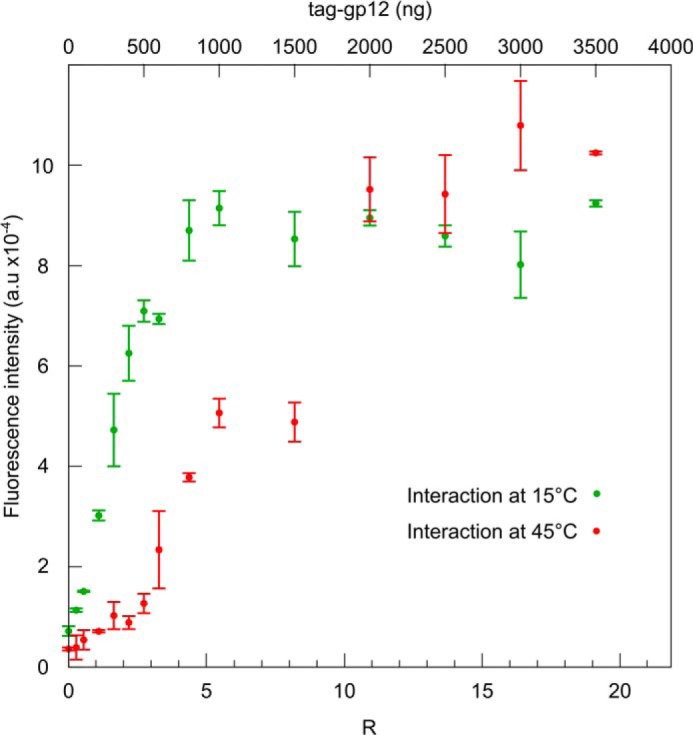
FBTSA of HΔ12 incubated with increasing amounts of tag-gp12. Capsids and tag-gp12 were preincubated separately at 15 (green) or 45 °C (red) and then mixed at the same temperature according to the experimental setup shown in the left panels of Fig. 7, A and C (not trypsinated), respectively. After coincubation, the samples were transferred to a QuantStudio 12Kflex machine for thermal denaturation at a heating rate of 3 °C/min in the presence of Sypro Orange. The amplitude of the gp12 signal with its characteristic transition at 53.6 °C (cf. Fig. 6A) was plotted against the R ratio of tag-gp12 relative to the input of HΔ12 capsids. The experimental points are averages of triplicates in two independent experiments.
DISCUSSION
The 6.6-kDa gp12 polypeptide of bacterial virus SPP1 was shown here to build an elongated trimer. Its properties indicate the presence of an intermolecular collagen-like triple helix that correlates with presence of eight GXY repeats at the center of the gp12 sequence, revealing a modular organization in which the collagen-like elongated segment connects two short amino and carboxyl terminus domains. Collagens are well studied protein components of the extracellular matrix of animals. They are characterized by the presence of 4-hydroxyproline at position Y of the GXY triplet, which is considered a major determinant of collagen stability (42). However, stable triple helices are built in synthetic model peptides by repeats longer than (GXY)9, showing that their association requires no amino acid posttranslational modifications (48). This property is consistent with the presence of collagen-like segments in prokaryote systems reported for streptococcal surface proteins (49, 50) and tail fibers of bacterial viruses (51), where several dozens of GXY triplets assemble long, flexible filaments. The remarkable feature of gp12 is that its short (GXY)8 stretch confers to the overall polypeptide a collagen-like behavior with a characteristic loss of quaternary structure, corresponding to a sharp transition at temperatures around 40 °C, like animal collagen (52, 53), that is rapidly and fully reversible upon cooling (Fig. 3). The process is accompanied by physical separation of the polypeptide chains that can reassociate into heterotrimers (Fig. 4). The unusual binding of Sypro Orange dye to folded gp12 resulting in fluorescence emission reveals the presence of an accessible hydrophobic binding environment in the trimer that is lost at the beginning of denaturation, correlating with a fast drop of fluorescence (Fig. 6). The gp12 native structure is, therefore, mainly stabilized by its intermolecular collagen-like triple helix rather than by a buried hydrophobic core that would become exposed for high-affinity binding of Sypro Orange upon unfolding, in contrast to the usual behavior of proteins (54).
The interaction of gp12 with SPP1 capsids does not change its capacity to bind Sypro Orange (Fig. 6A). However, it increases the protein gp12 thermal stability by 20.2 °C, to 53.6 °C. Such stability is not limited anymore by the collagen fold intrinsic stability, being strongly enhanced by gp12 binding to the capsid lattice. This stabilization mechanism ensures the perennial association of gp12 to viral particles that are liberated to the environment when infected cells lyse. Native trimers bind cooperatively to their 60 sites in the capsid, as best appraised when gp12 is provided in limiting concentrations to interact with HΔ12 capsids. A mixed population of capsids whose majority is either fully loaded with gp12 trimers or devoided of this auxiliary protein is observed under such conditions (Figs. 7, A, B, and D, and 10A). We hypothesize that initial binding of one trimer to a capsid hexamer creates a tectonic effect that spreads across the overall icosahedral shell, promoting a conformational change of other hexamers that strongly favors interaction with gp12 trimers. Such a maturation event uncovers a novel dynamic role of the expanded capsid surface that has previously been viewed as a rather passive lattice of independent binding sites for auxiliary viral polypeptides. The rearrangement resulting from the cross-talk between the 60 gp12 attachment sites is subtle, leading to no detectable difference when the structure of capsids before and after gp12 binding is compared at nanometer resolution (16).
FIGURE 10.
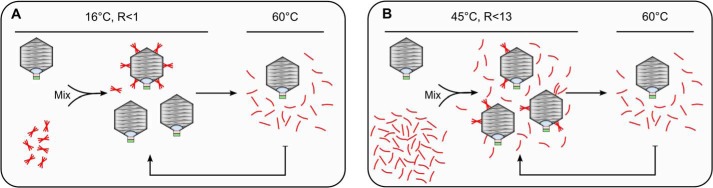
Models of native gp12 trimers (A) and unfolded gp12 polypeptides (B) binding to capsids and their dissociation from the capsid lattice in a temperature-dependent fashion.
Denatured gp12 also binds to capsid lattices (Fig. 7, C and E), leading to assembly of folded trimers when present in molar excess, as assessed in Sypro Orange binding experiments (Fig. 9). Therefore, the SPP1 capsid provides a platform for attachment of unfolded gp12 polypeptide chains. When three chains meet at a gp13 hexamer interaction site, their physical proximity likely provides a window of opportunity to twist together to trimerize at a temperature (45 °C) at which free gp12 chains remain fully unstructured (Fig. 10B). If unfolded gp12 is provided at less than a 13-fold excess relative to the number of its capsid binding sites, the reaction yields a relatively homogeneous population of capsids but those are only partially filled with gp12 (Fig. 7, C and E). Such behavior, resulting from the complexity of the interaction, contrasts with the very efficient cooperative binding of folded trimers.
CONCLUSIONS
The precise architecture of viral particles achieved by tightly regulated assembly of a few different polypeptides is an excellent system to understand how the polypeptides fold and how their physicochemical properties are exploited to build megadalton biomolecular assemblies of precise architecture with exquisite efficiency. The small capsid auxiliary protein gp12 of bacterial virus SPP1 exhibits novel and noteworthy properties. It uses a collagen-like fold to assemble an elongated trimer whose thermal stability properties render it a temperature dependent binder to the capsid multivalent icosahedral platform. Cooperative binding ensures very efficient full occupancy of gp12 sites in the capsid (Figs. 7, A, B, and D, and 10A), providing experimental evidence that an initial interaction of the viral auxiliary protein exerts long-range effects in the capsid lattice, favoring attachment to its other sites in the capsid. These properties of gp12, combined with its capacity to undergo fast reversible cycles of dissociation-unfolding and refolding-reassociation to capsids, offer a versatile system to engineer the SPP1 viral particle.
Acknowledgments
We thank Anja Dröge for generously communicating the initial identification of GXY repeats in the gp12 sequence. We also thank Manuela Argentini and David Cornu for mass spectrometry analyses (mass spectrometry platform of IMAGIF); Christophe Velours and Karine Madiona for CD and analytical ultracentrifugation analyses (biophysics platform of IMAGIF); and Isabelle Auzat, Sandrine Brasilès, Charlène Cornilleau, Stéphane Roche, and Stéphane Bressanelli for advice and experimental training (Unité de Virologie Moléculaire et Structurale).
This work was supported by institutional funding from the CNRS and from the Max Planck Institute for Molecular Genetics.
- TEV
- tobacco etch virus
- SEC
- size exclusion chromatography
- FBTSA
- fluorescence-based thermal shift assay.
REFERENCES
- 1. Caspar D. L., Klug A. (1962) Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27, 1–24 [DOI] [PubMed] [Google Scholar]
- 2. Baker T. S., Olson N. H., Fuller S. D. (1999) Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 63, 862–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prasad B. V., Schmid M. F. (2012) Principles of virus structural organization. Adv. Exp. Med. Biol. 726, 17–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casjens S., King J. (1975) Virus assembly. Annu. Rev. Biochem. 44, 555–611 [DOI] [PubMed] [Google Scholar]
- 5. Prevelige P. E., Fane B. A. (2012) Building the machines: scaffolding protein functions during bacteriophage morphogenesis. Adv. Exp. Med. Biol. 726, 325–350 [DOI] [PubMed] [Google Scholar]
- 6. Ren Z. J., Lewis G. K., Wingfield P. T., Locke E. G., Steven A. C., Black L. W. (1996) Phage display of intact domains at high copy number: a system based on SOC, the small outer capsid protein of bacteriophage T4. Protein Sci. 5, 1833–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang F., Forrer P., Dauter Z., Conway J. F., Cheng N., Cerritelli M. E., Steven A. C., Plückthun A., Wlodawer A. (2000) Novel fold and capsid-binding properties of the λ-phage display platform protein gpD. Nat. Struct. Biol. 7, 230–237 [DOI] [PubMed] [Google Scholar]
- 8. Roos W. H., Radtke K., Kniesmeijer E., Geertsema H., Sodeik B., Wuite G. J. (2009) Scaffold expulsion and genome packaging trigger stabilization of herpes simplex virus capsids. Proc. Natl. Acad. Sci. U.S.A. 106, 9673–9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parent K. N., Khayat R., Tu L. H., Suhanovsky M. M., Cortines J. R., Teschke C. M., Johnson J. E., Baker T. S. (2010) P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure 18, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tang L., Gilcrease E. B., Casjens S. R., Johnson J. E. (2006) Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 14, 837–845 [DOI] [PubMed] [Google Scholar]
- 11. Effantin G., Boulanger P., Neumann E., Letellier L., Conway J. F. (2006) Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361, 993–1002 [DOI] [PubMed] [Google Scholar]
- 12. Lander G. C., Evilevitch A., Jeembaeva M., Potter C. S., Carragher B., Johnson J. E. (2008) Bacteriophage λ stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryo-EM. Structure 16, 1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Q., Maluf N. K., Catalano C. E. (2008) Packaging of a unit-length viral genome: the role of nucleotides and the gpD decoration protein in stable nucleocapsid assembly in bacteriophage λ. J. Mol. Biol. 383, 1037–1048 [DOI] [PubMed] [Google Scholar]
- 14. Li Q., Shivachandra S. B., Zhang Z., Rao V. B. (2007) Assembly of the small outer capsid protein, Soc, on bacteriophage T4: a novel system for high density display of multiple large anthrax toxins and foreign proteins on phage capsid. J. Mol. Biol. 370, 1006–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin L., Fokine A., O'Donnell E., Rao V. B., Rossmann M. G. (2010) Structure of the small outer capsid protein, Soc: a clamp for stabilizing capsids of T4-like phages. J. Mol. Biol. 395, 728–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White H. E., Sherman M. B., Brasilès S., Jacquet E., Seavers P., Tavares P., Orlova E. V. (2012) Capsid structure and its stability at the late stages of bacteriophage SPP1 assembly. J. Virol. 86, 6768–6777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lucon J., Qazi S., Uchida M., Bedwell G. J., LaFrance B., Prevelige P. E., Jr., Douglas T. (2012) Use of the interior cavity of the P22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat. Chem. 4, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Neil A., Prevelige P. E., Basu G., Douglas T. (2012) Coconfinement of fluorescent proteins: spatially enforced communication of GFP and mCherry encapsulated within the P22 capsid. Biomacromolecules 13, 3902–3907 [DOI] [PubMed] [Google Scholar]
- 19. Clark J. R., March J. B. (2006) Bacteriophages and biotechnology: vaccines, gene therapy and antibacterials. Trends Biotechnol. 24, 212–218 [DOI] [PubMed] [Google Scholar]
- 20. Fischlechner M., Donath E. (2007) Viruses as building blocks for materials and devices. Angew. Chem. Int. Ed. Engl. 46, 3184–3193 [DOI] [PubMed] [Google Scholar]
- 21. Tao P., Mahalingam M., Marasa B. S., Zhang Z., Chopra A. K., Rao V. B. (2013) In vitro and in vivo delivery of genes and proteins using the bacteriophage T4 DNA packaging machine. Proc. Natl. Acad. Sci. U.S.A. 110, 5846–5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinmetz N. F., Lin T., Lomonossoff G. P., Johnson J. E. (2009) Structure-based engineering of an icosahedral virus for nanomedicine and nanotechnology. Curr. Top. Microbiol. Immunol. 327, 23–58 [DOI] [PubMed] [Google Scholar]
- 23. Strable E., Finn M. G. (2009) Chemical modification of viruses and virus-like particles. Curr. Top. Microbiol. Immunol. 327, 1–21 [DOI] [PubMed] [Google Scholar]
- 24. Dröge A., Santos M. A., Stiege A. C., Alonso J. C., Lurz R., Trautner T. A., Tavares P. (2000) Shape and DNA packaging activity of bacteriophage SPP1 procapsid: protein components and interactions during assembly. J. Mol. Biol. 296, 117–132 [DOI] [PubMed] [Google Scholar]
- 25. Oliveira L., Tavares P., Alonso J. C. (2013) Headful DNA packaging: bacteriophage SPP1 as a model system. Virus Res. 173, 247–259 [DOI] [PubMed] [Google Scholar]
- 26. Isidro A., Henriques A. O., Tavares P. (2004) The portal protein plays essential roles at different steps of the SPP1 DNA packaging process. Virology 322, 253–263 [DOI] [PubMed] [Google Scholar]
- 27. Plisson C., White H. E., Auzat I., Zafarani A., São-José C., Lhuillier S., Tavares P., Orlova E. V. (2007) Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26, 3720–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Auzat I., Dröge A., Weise F., Lurz R., Tavares P. (2008) Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol. 70, 557–569 [DOI] [PubMed] [Google Scholar]
- 29. Lurz R., Orlova E. V., Günther D., Dube P., Dröge A., Weise F., van Heel M., Tavares P. (2001) Structural organisation of the head-to-tail interface of a bacterial virus. J. Mol. Biol. 310, 1027–1037 [DOI] [PubMed] [Google Scholar]
- 30. Alonso J. C., Lüder G., Stiege A. C., Chai S., Weise F., Trautner T. A. (1997) The complete nucleotide sequence and functional organization of Bacillus subtilis bacteriophage SPP1. Gene. 204, 201–212 [DOI] [PubMed] [Google Scholar]
- 31. Haima P., Bron S., Venema G. (1987) The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol. Gen. Genet. 209, 335–342 [DOI] [PubMed] [Google Scholar]
- 32. Dröge A., Tavares P. (2000) In vitro packaging of DNA of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 296, 103–115 [DOI] [PubMed] [Google Scholar]
- 33. Becker B., de la Fuente N., Gassel M., Günther D., Tavares P., Lurz R., Trautner T. A., Alonso J. C. (1997) Head morphogenesis genes of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 268, 822–839 [DOI] [PubMed] [Google Scholar]
- 34. Isidro A., Santos M. A., Henriques A. O., Tavares P. (2004) The high-resolution functional map of bacteriophage SPP1 portal protein. Mol. Microbiol. 51, 949–962 [DOI] [PubMed] [Google Scholar]
- 35. Poh S. L., el Khadali F., Berrier C., Lurz R., Melki R., Tavares P. (2008) Oligomerization of the SPP1 scaffolding protein. J. Mol. Biol. 378, 551–564 [DOI] [PubMed] [Google Scholar]
- 36. Philo J. S. (1997) An improved function for fitting sedimentation velocity data for low-molecular-weight solutes. Biophys. J. 72, 435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jakutytė L., Baptista C., São-José C., Daugelavičius R., Carballido-López R., Tavares P. (2011) Bacteriophage infection in rod-shaped gram-positive bacteria: evidence for a preferential polar route for phage SPP1 entry in Bacillus subtilis. J. Bacteriol. 193, 4893–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cole C., Barber J. D., Barton G. J. (2008) The Jpred3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dröge A. (1998) Capsidmorphogenese des Bakteriophagen SPP1. Doctoral thesis, Technische Universität Berlin, Berlin, Germany [Google Scholar]
- 41. Bella J., Eaton M., Brodsky B., Berman H. M. (1994) Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science 266, 75–81 [DOI] [PubMed] [Google Scholar]
- 42. Bella J., Brodsky B., Berman H. M. (1995) Hydration structure of a collagen peptide. Structure 3, 893–906 [DOI] [PubMed] [Google Scholar]
- 43. Cabré F., Canela E. I., Canela M. A. (1989) Accuracy and precision in the determination of Stokes radii and molecular masses of proteins by gel filtration chromatography. J. Chromatogr. 472, 347–356 [DOI] [PubMed] [Google Scholar]
- 44. Seifter S., Gallop P. M. (1962) Collagenase from Clostridium histolyticum: collagen + H2O → peptides gelatin + H2O → peptides. Methods Enzymol. 5, 659–665 [Google Scholar]
- 45. Orlova E. V., Gowen B., Dröge A., Stiege A., Weise F., Lurz R., van Heel M., Tavares P. (2003) Structure of a viral DNA gatekeeper at 10 Å resolution by cryo-electron microscopy. EMBO J. 22, 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wallace B. A., Janes R. W. (2001) Synchrotron radiation circular dichroism spectroscopy of proteins: secondary structure, fold recognition and structural genomics. Curr. Opin. Chem. Biol. 5, 567–571 [DOI] [PubMed] [Google Scholar]
- 47. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 48. Persikov A. V., Ramshaw J. A., Brodsky B. (2005) Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 280, 19343–19349 [DOI] [PubMed] [Google Scholar]
- 49. Xu Y., Keene D. R., Bujnicki J. M., Höök M., Lukomski S. (2002) Streptococcal Scl1 and Scl2 proteins form collagen-like triple helices. J. Biol. Chem. 277, 27312–27318 [DOI] [PubMed] [Google Scholar]
- 50. Mohs A., Silva T., Yoshida T., Amin R., Lukomski S., Inouye M., Brodsky B. (2007) Mechanism of stabilization of a bacterial collagen triple helix in the absence of hydroxyproline. J. Biol. Chem. 282, 29757–29765 [DOI] [PubMed] [Google Scholar]
- 51. Ghosh N., McKillop T. J., Jowitt T. A., Howard M., Davies H., Holmes D. F., Roberts I. S., Bella J. (June 6, 2012) Collagen-like proteins in pathogenic E. coli strains. PLoS ONE 10.1371/journal.pone.0037872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leikina E., Mertts M. V., Kuznetsova N., Leikin S. (2002) Type I collagen is thermally unstable at body temperature. Proc. Natl. Acad. Sci. U.S.A. 99, 1314–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cao H., Xu S.-H. (2008) Purification and characterization of type II collagen from chick sternal cartilage. Food Chem. 108, 439–445 [DOI] [PubMed] [Google Scholar]
- 54. Kranz J. K., Schalk-Hihi C. (2011) Protein thermal shifts to identify low molecular weight fragments. Methods Enzymol. 493, 277–298 [DOI] [PubMed] [Google Scholar]