

Background: XPC protein is ubiquitinated, but the ubiquitination does not lead to significant proteolysis of XPC.
Results: Ubiquitin-specific protease 7 is a deubiquitinating enzyme (DUB) for XPC.
Conclusion: USP7 deubiquitination prevents XPC from undergoing UV-induced and VCP/p97-regulated XPC proteolysis.
Significance: USP7 and VCP/p97 involvement in XPC regulation is crucial for understanding how nucleotide excision repair is executed and regulated in eukaryotic cells.
Keywords: Deubiquitylation (Deubiquitination), DNA Damage Response, DNA Repair, Nucleotide Excision Repair, Ubiquitylation (Ubiquitination), Ubiquitin-specific Protease 7, Valosin-containing Protein, Xeroderma Pigmentosum Complementation Group C Protein
Abstract
Ubiquitin specific protease 7 (USP7) is a known deubiquitinating enzyme for tumor suppressor p53 and its downstream regulator, E3 ubiquitin ligase Mdm2. Here we report that USP7 regulates nucleotide excision repair (NER) via deubiquitinating xeroderma pigmentosum complementation group C (XPC) protein, a critical damage recognition factor that binds to helix-distorting DNA lesions and initiates NER. XPC is ubiquitinated during the early stage of NER of UV light-induced DNA lesions. We demonstrate that transiently compromising cellular USP7 by siRNA and chemical inhibition leads to accumulation of ubiquitinated forms of XPC, whereas complete USP7 deficiency leads to rapid ubiquitin-mediated XPC degradation upon UV irradiation. We show that USP7 physically interacts with XPC in vitro and in vivo. Overexpression of wild-type USP7, but not its catalytically inactive or interaction-defective mutants, reduces the ubiquitinated forms of XPC. Importantly, USP7 efficiently deubiquitinates XPC-ubiquitin conjugates in deubiquitination assays in vitro. We further show that valosin-containing protein (VCP)/p97 is involved in UV light-induced XPC degradation in USP7-deficient cells. VCP/p97 is readily recruited to DNA damage sites and colocalizes with XPC. Chemical inhibition of the activity of VCP/p97 ATPase causes an increase in ubiquitinated XPC on DNA-damaged chromatin. Moreover, USP7 deficiency severely impairs the repair of cyclobutane pyrimidine dimers and, to a lesser extent, affects the repair of 6-4 photoproducts. Taken together, our findings uncovered an important role of USP7 in regulating NER via deubiquitinating XPC and by preventing its VCP/p97-regulated proteolysis.
Introduction
The genome of eukaryotic cells is constantly challenged by diverse and ubiquitous DNA-damaging agents. Repair of DNA damage enables cells to overcome genotoxicity, retain normal cellular functions, and maintain genomic integrity. Among the known repair pathways, nucleotide excision repair (NER)4 eliminates a broad variety of helix-distorting DNA lesions, including UV light-induced cyclobutane pyrimidine dimers (CPD) and 6-4 photoproducts (6-4PPs), as well as chemically induced bulky adducts. Mammalian NER consists of two distinct subpathways: global genomic repair (GGR), which operates throughout the genome, and transcription-coupled repair, which eliminates DNA damage from transcribed DNA strands of transcriptionally active genes (1, 2). Impaired NER activity is associated with several rare autosomal recessive genetic disorders, such as xeroderma pigmentosum (XP) and Cockayne syndrome (CS). Seven XP complementation groups, XP-A to XP-G, have been identified. The corresponding genes have been cloned, and their gene products have been characterized functionally and biochemically.
The XPC gene encodes a damage recognition factor specific to the GGR subpathway of NER (3). XPC protein complexes in vivo with one of the two homologs of yeast Rad23 protein, hRad23B/A (4–6). The XPC-hRad23B/A protein complex recognizes DNA damage and initiates the assembly of dual incision machinery. In DNA damage recognition, the XPC-hRad23B/A complex serves as a structure-specific DNA binding factor for various helix-distorting DNA lesions. Interestingly, the XPC complex appears to recognize lesion-containing secondary DNA structures rather than lesions themselves (7). The nature of the lesion has little effect on the binding affinity of the XPC complex (8). For instance, the XPC complex is equally capable of binding to DNA substrates that are not repaired in vivo by NER, suggesting that XPC may be a general sensor of DNA lesions.
Besides XPC, UV light-damaged DNA-binding protein (DDB) is another damage recognition factor specific to GGR. DDB is a heterodimeric complex consisting of DDB1 (p127) and DDB2 (p48). Loss of DDB activity because of mutation in the DDB2 gene is associated with the XP-E group (9–11). DDB is part of an E3 ubiquitin ligase complex containing cullin 4A (Cul4A) and Roc1 in association with the COP9 signalosome (12). The DDB-Cul4A E3 complex ubiquitinates XPC in response to UV light-induced DNA damage (13, 14). The ubiquitination appears to enhance the damage binding of XPC rather than alter its specificity. Interestingly, the ubiquitination of XPC does not lead to significant XPC degradation by proteolysis (13). The XPC ubiquitination is, presumably, reversed via deubiquitination.
The cellular deubiquitination processes are carried out by a class of enzymes called deubiquitinases or deubiquitinating enzymes (DUBs). The DUBs remove polyubiquitin chains from protein substrates and, thereby, prevent the substrates from undergoing ubiquitin-mediated proteasomal degradation. The human genome encodes ∼79 DUBs that are predicted to be active in opposing the function of E3 ubiquitin ligases (15). For example, ubiquitin-specific protease 7 (USP7 or HAUSP (herpesvirus-associated ubiquitin specific protease)) has been known as a DUB for tumor suppressor p53 and Mdm2 (16, 17), presumably processing lysine 48-linked ubiquitin conjugates, which mediate proteasomal degradation. USP7 deubiquitinates Mdm2 and prevents Mdm2 from undergoing proteasomal degradation, and Mdm2, in turn, ubiquitinates and degrades p53. Therefore, USP7 disruption leads to stabilization of p53 (18). Nevertheless, the specific DUB(s) involved in the regulation of XPC is/are currently unknown. In this study, we identified USP7 as a DUB for XPC. We provide evidence showing that USP7 physically interacts with and deubiquitinates XPC in vitro and in vivo. In the absence of the USP7 function, UV irradiation induces ubiquitin-mediated XPC proteolysis assisted by ubiquitin-selective chaperone valosin-containing protein (VCP)/p97, which is thought to function as a segregase to separate ubiquitinated proteins from tightly bound partners. Consequently, USP7 disruption severely impairs GGR of CPD and, to a lesser extent, affects the repair of 6-4PP.
EXPERIMENTAL PROCEDURES
DNA Constructs and Antibodies
USP7 constructs were obtained from Dr. Yanhui Xu (Department of Biochemistry, Fudan University Medical School). Rabbit anti-XPC and anti-CPD antibodies were as described previously (14, 19). Anti-γH2AX antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-FLAG M2-agarose beads and anti-FLAG M2 antibody were purchased from Sigma-Aldrich (St. Louis, MO). Anti-Myc SC-40, monoclonal anti-ubiquitin FK2, anti-VCP/p97, anti-USP7, and monoclonal anti-6-4PP (64 M-2) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), EMB Millipore (Billerica, MA), Abcam, Bethyl Laboratories (Montgomery, TX) and MBL International (Woburn, MA), respectively.
Cell Culture and Transfection
HCT116, HCT116-p53−/−, and HCT116-USP7−/− cells were obtained from the laboratory of Bert Vogelstein and grown in McCoy's 5A medium supplemented with 10% FCS and antibiotics at 37 °C in a humidified atmosphere of 5% CO2, whereas HeLa cells were maintained in DMEM. For transfection, exponentially growing cells were plated at a desired seeding density. Plasmid DNAs were transfected into the HeLa or HCT116 cells lines using FuGENE6 transfection reagents (Promega, Madison, WI). siRNA transfection was conducted using Lipofectamine 2000 reagents (Invitrogen).
RNA Interference
USP7 siRNA (5′-ACCCUUGGACAAUAUUCCUdTdT-3′), USP14 siRNA (5′-AGAAAUGCCUUGUAUAUCA-3′), Uch37 siRNA (5′-CUUAGAGCAACAUCC UAAU-3′), and control siRNA (5′-UUCUCCGA ACGUGUCACGUdT-3′) were synthesized by Thermo Scientific (Lafayette, CO).
GST Pulldown Assays
The GST, GST-S5a, and GST-USP7 fusion proteins were expressed in the Escherichia coli BL21 strain. Bacterial extracts were made in lysis buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, 1 mm DTT, and 1% Triton X-100) with or without 1% sarkosyl. Equal amounts of GST fusion proteins were immobilized on glutathione-Sepharose 4B beads in binding buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 0.1% (v/v) Triton X-100). The loaded beads were incubated with whole cell extracts containing ∼1.0 mg protein made from 20 J/m2 UV light-treated HCT116 cells in RIPA buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, and protease inhibitors). After incubation at 4 °C for 16 h, the beads were washed with RIPA buffer and boiled in SDS sample buffer. The bound proteins were analyzed by Western blotting.
Cellular Fractionation and Coimmunoprecipitation
Cellular fractionation was conducted as described by Anindya et al. (20), with modifications. Briefly, cells (∼107) were lysed with 1 ml (∼5× cell volume) of cytoplasmic lysis buffer (10 mm Tris-HCl (pH 7.9), 0.34 m sucrose, 3 mm CaCl2, 2 mm magnesium acetate, 0.1 mm EDTA, 1 mm DDT, 0.5% Nonidet P-40, and protease inhibitor mixture). Nuclei were pelleted by centrifugation at 3500 × g for 15 min and washed with cytoplasmic lysis buffer without Nonidet P-40 and then lysed in 1 ml of nuclear lysis buffer (20 mm HEPES (pH 7.9), 3 mm EDTA, 10% glycerol, 1.5 mm MgCl2, 150 mm KOAc, and protease inhibitors). The nucleoplasmic fractions were separated by centrifugation at 15,000 × g for 30 min, and the pellet was resuspended in 0.2 ml of nuclease incubation buffer (150 mm HEPES (pH 7.9), 1.5 mm MgCl2, 150 mm KOAc, and protease inhibitors) and incubated with 50 units of benzonase (25 units/μl) for 30 min at room temperature. The soluble chromatin fraction was collected by centrifugation at 20,000 × g for 30 min, whereas the insoluble chromatin fraction was dissolved in SDS sample buffer. Coimmunoprecipitation was done using soluble chromatin or whole cell lysates in RIPA buffer with appropriate antibodies at 4 °C overnight. The immunocomplexes were captured by protein A Plus G-agarose beads and analyzed by Western blotting.
Recombinant USP7 Enzymes
Recombinant His-USP7 and USP7 catalytic domains (USP7-CDs) were obtained from Boston Biochem (Cambridge, MA). The recombinant proteins were produced in Sf9 insect cells and purified to ≥95% purity by SDS-PAGE. The FLAG-tagged WT-USP7 and CS, M1, or M2 mutants were transiently expressed in HCT116 cells by 48-h transfection using FuGENE6 reagents. After a PBS wash, the transfected cells were lysed in E1A buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100) in the presence of protease inhibitors. The cell lysates were incubated with anti-FLAG-M2 beads at 4 °C overnight. The beads were washed three times with TBS buffer (50 mm Tris-HCl (pH 7.4) and 150 mm NaCl), and the bound proteins were eluted with FLAG peptide as described in the protocol of the manufacturer.
In Vitro Deubiquitination Assay
The His-USP7, USP7-CD, purified WT-USP7, or mutant USP7 were incubated in a 10- or 20-μl reaction with chromatin substrates (containing ∼6 μg of protein) in DUB buffer (50 mm Tris-Cl (pH 8.0), 50 mm NaCl, 1 mm EDTA, 5% glycerol, and 10 mm DTT) at 37 °C for the desired time periods. When HBX 41108 was used, the recombinant USP7 was incubated with HBX 41108 for 30 min at room temperature before the addition of substrates. The deubiquitination reactions were stopped by boiling in 1× SDS sample buffer and analyzed for XPC or USP7 by Western blotting.
Micropore UV Irradiation, Immunofluorescence, and Immunoslot-blot Analysis
The experiments were conducted according to a method established in our laboratory (14). Briefly, the cells were washed once with PBS, and a 5-μm isopore polycarbonate filter was placed on top of the cell monolayer, followed by UV irradiation at a desired dose, and cells were maintained in a suitable medium for the indicated time periods. The cells were washed twice with cold PBS, permeabilized with 0.5% Triton X-100/PBS for about 8 min on ice as needed, and/or fixed with 2% paraformaldehyde in 0.5% Triton X-100 at 4 °C for 30 min. The fixed cells were rinsed with twice with cold PBS, blocked with 20% normal goat serum in 0.1% Triton X-100/PBS, and stained with an appropriate primary antibody as well as with FITC-, Alexa Fluor 488-, or Texas Red-conjugated secondary antibodies. The coverslips were mounted in Vectashield mounting medium with DAPI. The fluorescence images were obtained with a Nikon fluorescence microscope (E80i, Tokyo, Japan) and processed with SPOT software. The CPD and 6-4PP in genomic DNA were examined by immunoslot-blot assay as described earlier, with some modifications (19, 21). The damage levels were on the basis of quantitative band intensities against reference standards using ImageJ software.
RESULTS
Identification of USP7 as a Candidate DUB for XPC Deubiquitination
XPC is a chromatin-associated nuclear protein that is ubiquitinated promptly within damaged chromatin in response to UV irradiation. Using RNAi to probe for DUB(s) specific to XPC deubiquitination, we mainly focused on DUBs residing in the nucleus, e.g. USP14, Uch37, and USP7. Knockdown of USP14 and Uch37 (Fig. 1, A and B) did not increase the ubiquitinated forms of XPC. Both USP14 and Uch37 are known proteasome-associated DUBs. XPC deubiquitination is apparently not directly associated with DUBs in proteasomal processing. Interestingly, knockdown of USP7 led to a moderate increase in the ubiquitinated forms of XPC, observed at 2, 4, and 8 h post-UV irradiation (Fig. 1C). We further checked the effect of the USP7-specific inhibitor HBX 41108 on ubiquitination of XPC. It has been shown that HBX 41108 stabilizes p53-ubiquitin conjugates in the presence of USP7 (22). Indeed, pretreatment of HCT116 with HBX 41108 at 2, 4, and 8 μm promoted the accumulation of ubiquitinated XPC, albeit at lower levels (Fig. 1D), whereas 16 μm HBX 41108 increased the polyubiquitination of XPC. Interestingly, HBX 41108 caused a slight decrease in USP7 but a slight increase in p53 levels. Because RNAi and HBX 41108 might not fully diminish the USP7 function, we focused our subsequent analysis on stable USP7-deficient HCT116 cells (18). As expected, USP7 disruption stabilized p53 and led to a subsequent increase in the p53 downstream target proteins p21 and DDB2 (Fig. 2A). However, p53 disruption diminished p21 and DDB2 expression and decreased XPC, in agreement with the observation that XPC is also a p53 downstream transcriptional regulatory target (23). In HCT116 cells, ubiquitinated forms of XPC were detected from 2–8 h post-UV irradiation and showed a typical UV light-induced accumulation at earlier times, followed by a steady decrease. On the other hand, in HCT116-USP7−/− cells, both the ubiquitinated and native forms of XPC exhibited a dramatic decrease at 2 h, followed by a slow recovery by 8 h. Blockage of XPC degradation by the proteasome inhibitor MG132 significantly restored native XPC (Fig. 2B). Although the XPC levels were slightly lower in HCT116-USP7−/− cells (Fig. 2B), UV irradiation caused a dramatic (>75%) decrease in total XPC at 2 h, with a gradual time-dependent recovery compared with the unirradiated control from HCT116-USP7−/− cells (Fig. 2C). However, UV irradiation did not significantly affect the total XPC level in parental HCT116 cells. MG132 treatment did not increase but decreased the accumulation of ubiquitinated XPC in both cells. This is due to the dependence of XPC ubiquitination on the proteasomal degradation of the preceding ubiquitinated DDB occupying the lesion site and impairing the damage handover to XPC and its ubiquitination (14, 24). This may also result from ubiquitin pool depletion or ubiquitin stress caused by proteasome inhibition. We probed this response further by using a selective irreversible proteasome inhibitor, lactacystin, in lieu of MG132. Lactacystin, like MG132, also demolished the ubiquitinated XPC (Fig. 2D). The veracity of the ubiquitinated forms of XPC was established by affinity tag analysis. The results showed that histidine-tagged ubiquitin was incorporated into modified XPC (Fig. 2E, lanes 1–3). Moreover, the proteasomal ubiquitin receptor S5a binds to both the ubiquitinated and native XPC forms (Fig. 2E, lanes 5–9). Taken together, these findings verified the UV-induced XPC ubiquitination and indicated that USP7 disruption exacerbated the degradation of XPC following UV irradiation. Therefore, we infer that USP7 is a specific DUB for XPC.
FIGURE 1.
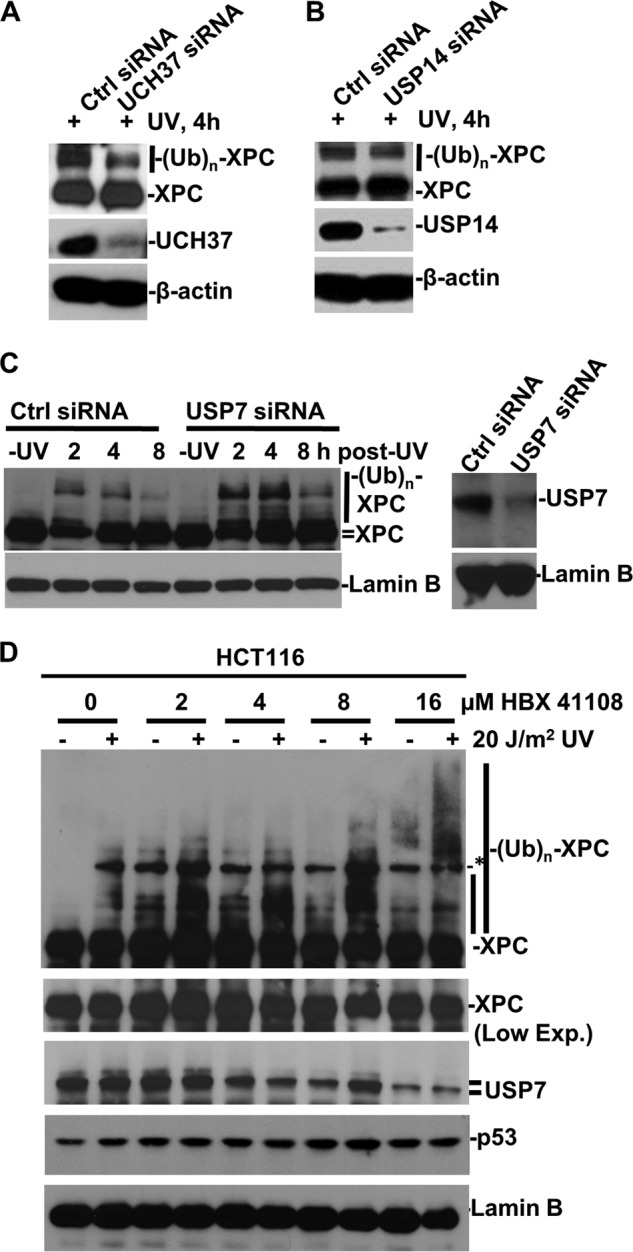
Effect of DUB knockdown and USP7 inhibition on XPC ubiquitination. A–C, HeLa cells were transfected with control (Ctrl), UCH37- (A), USP14- (B), or USP7-specific (C) siRNA for 48 h. The transfected cells were exposed to UV light at 20 J/m2 and harvested at 2 or 4 h post-UV irradiation. XPC, UCH37, USP14, or USP7 were examined in cellular protein extracts by Western blotting. The β-actin blots served as loading controls. D, inhibition of USP7 affects XPC ubiquitination. HCT116 cells were treated with the USP7 inhibitor HBX 41108 at the indicated concentration for 24 h, exposed to UV light at 20 J/m2, and harvested 2 h post-UV irradiation. XPC, USP7, p53, and lamin B were examined in cellular protein extracts by Western blotting. Low Exp., low exposure in protein detection. The asterisk indicates a nonspecific protein band that cross-reacts with anti-XPC antibody. (UB)n, multiple (n) Ub moieties.
FIGURE 2.
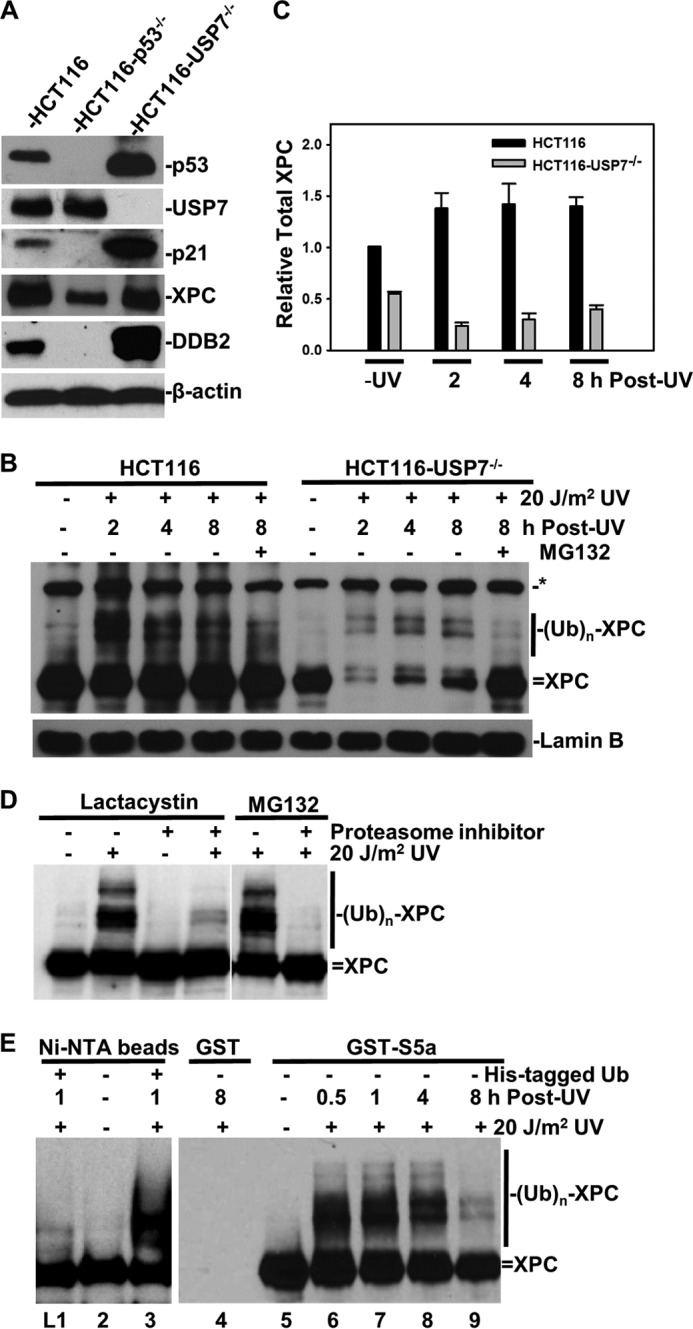
USP7 disruption affects XPC deubiquitination and degradation. A, detection of p53, USP7, p21, DDB2, and XPC in parental HCT116, HCT116-p53−/−, and USP7−/− cells. B, HCT116 and HCT116-USP7−/− cells were UV-irradiated at 20 J/m2, treated with the proteasome inhibitor MG132 at 10 μm or vehicle, and harvested at the indicated time points. The XPC and lamin B proteins were detected by Western blotting. The asterisk indicates a nonspecific protein band as in Fig. 1D. (UB)n, multiple (n) Ub moieties. C, the relative XPC amount, in comparison with non-UV controls from HCT116 cells, was calculated from density quantification of the autograph using ImageJ software. Mean ± S.E. were calculated from three independent experiments. D, HeLa cells were UV-irradiated at 20 J/m2, treated with MG132 or lactacystin at 10 μm or vehicle, and harvested 2 h Post-UV. The whole cell extracts were detected for XPC by Western blotting. E, HeLa cells were transiently transfected with expression constructs for histidine-tagged ubiquitin and whole cell extracts made in RIPA buffer from transfected or control cells. Pulldown assays were performed with nickel-nitrilotriacetic acid (Ni-NTA) beads (lanes 1 and 3). GST pulldown assays were performed with RIPA extracts from UV-irradiated HeLa cells using GST and GST-S5a immobilized glutathione beads (lanes 4–9).
Physical Interaction of USP7 with XPC Is Important for Targeted XPC Deubiquitination
The UV-induced dramatic XPC degradation in HCT116-USP7−/− cells suggests that USP7 is involved in the regulation of XPC stability. To investigate whether USP7 acts directly on XPC, we next examined the physical interaction between USP7 and XPC. In a GST pulldown assay, GST fusion proteins containing the USP7 UBL1 domain, spanning amino acids 560–644, were able to bind XPC in vitro (Fig. 3, A and B). Interestingly, the catalytic domain (CD) and UBL1 cooperatively enabled the binding of fusion protein to both native as well as ubiquitinated XPC, in agreement with the ability of the USP7 CD to bind to the ubiquitin moiety. In coimmunoprecipitation assays, XPC was seen to associate with both USP7 isoforms in soluble chromatin fractions irrespective of the UV-induced DNA damage (Fig. 3, C and D).
FIGURE 3.
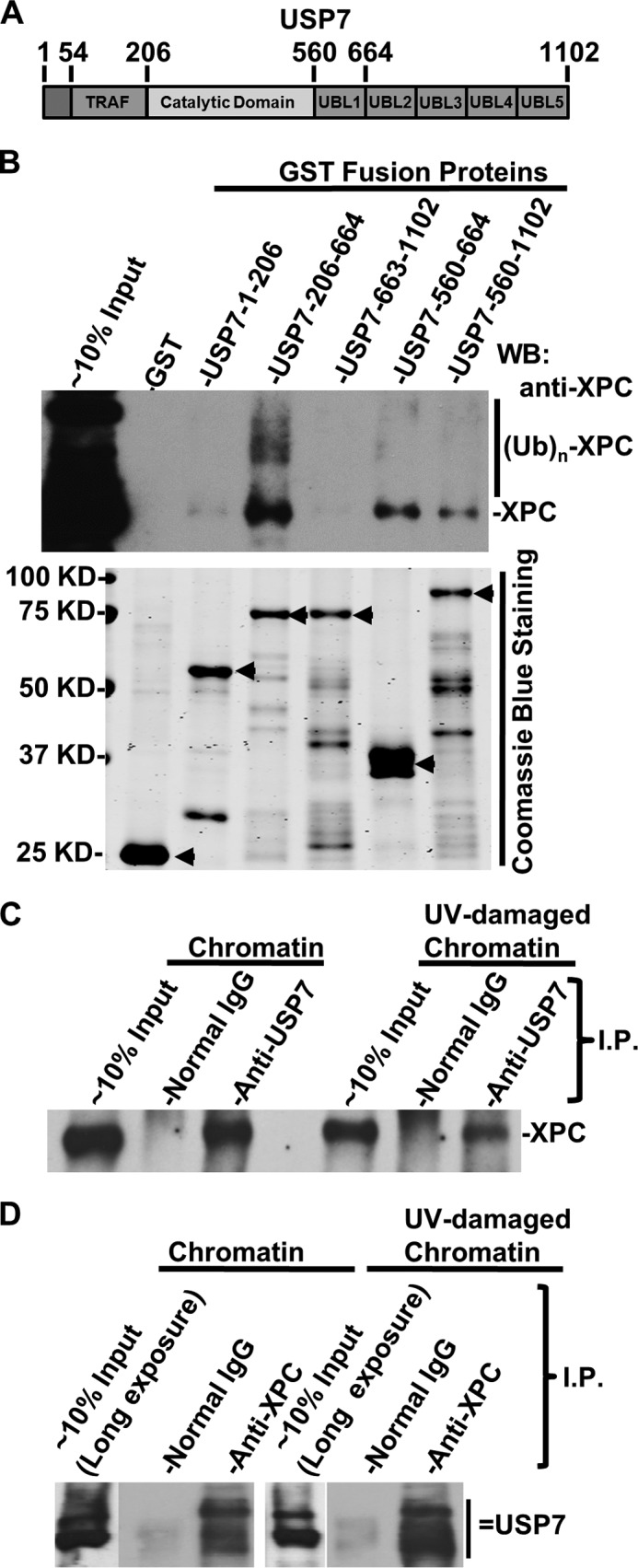
USP7 interacts with XPC. A, the structural domains of USP7. TRAF, tumor necrosis factor receptor-associated factor domain. B, USP7 interacts with XPC in vitro. GST pulldown assays were conducted using whole cell lysates of HCT 116 in RIPA buffer and GST fusion proteins containing the indicated segments of USP7. Bottom panel, GST fusion proteins containing various segments of USP7, which were expressed in E. coli., loaded onto GST beads, separated by SDS-PAGE, and detected by Coomassie Blue staining. WB, Western blot; (UB)n, multiple (n) Ub moieties. C and D, USP7 associates with XPC in vivo. Immunoprecipitation (I.P.) and Western blot analysis were performed using soluble chromatin factions made from HCT 116 cells with or without UV irradiation and suitable anti-USP7 and XPC antibodies. Normal rabbit IgG served as control. The long exposure of 10% input in Fig. 2D was to show a weaker upper USP7 species.
To evaluate the contribution of the catalytic and UBL1 domains of USP7 in USP7-XPC association in vivo, we used two UBL1 mutants and one CS mutant. As depicted in Fig. 4A, UBL1 M1 contains a tryptophan-to-serine substitution at amino acid 623 (W623S) and a phenylalanine-to-serine substitution at 661 (F661S). UBL1 M2 contains multiple amino acid substitutions (25), although CS contains a cysteine-to-serine substitution at 223 (C223S) that inactivates the deubiquitination activity of USP7. The coimmunoprecipitation experiments showed that WT-USP7 was able to bind to XPC in vivo. However, mutations of M1, M2, and CS abolished the in vivo interactions of USP7 with XPC (Fig. 4, B–E). Considering the ability of the USP7 CD to bind the ubiquitin moiety, it is possible that the C223S mutation affects this binding. In essence, these results suggest that the collaboration of the UBL1 and CD domains is important for the binding of USP7 to XPC in vivo.
FIGURE 4.

UBL1 and the catalytic domains of USP7 mediate the in vivo interaction between XPC and USP7. A, primary amino acid sequence of the USP7 UBL1 domain. The arrows indicate sequence alterations in UBL1 mutant M1 and M2. B–E, interaction between XPC and WT or mutant USP7 in vivo. FLAG-tagged WT-USP7 (B) and the M1, M2, and CS mutants (C–E, respectively) were transiently expressed in HCT116 cells. Immunoprecipitation was performed using anti-FLAG-agarose gels and cell lysates made in E1A buffer, followed by Western blot analysis for the presence of XPC. HCT116 cells without transfected cells were used as control.
We next examined the in vivo activity of USP7 and its mutants toward XPC by transient expression of the FLAG-tagged USP7 in HCT116 cells. The cells were chosen to avoid an XPC degradation situation, which could complicate data interpretation. The 2-h time point was chosen for these experiments because, in HCT116 cells, ubiquitinated forms of XPC peaked at this time point (Fig. 2B). As shown in Fig. 5, the exogenous FLAG-tagged USP7 proteins were detected by anti-FLAG antibody. In these experiments, film overexposures to show the FLAG-tagged USP7 in the 1-μg lane resulted in saturated bands at higher amounts (3 and 5 μg) of the expression construct. To ensure a dose response in USP7 transfection and expression, we further examined both exogenous and endogenous USP7 with anti-USP7 antibody. In the latter experiments, the USP7 proteins exhibited a clear dose-dependent increase upon transfection of different amounts of expression constructs. More importantly, WT-USP7 eliminated the UV light-induced ubiquitinated forms of XPC in a dose-dependent manner, albeit with a slight discernable increase in the unmodified forms of XPC, whereas none of the CS, M1, and M2 mutants were able to decrease but slightly increased the XPC-ubiquitin conjugates in a dominant negative manner. Because the UBL1 mutants M1 and M2 were unable to bind XPC in vivo, it is unclear why these mutants exhibit a dominant negative effect over the endogenous USP7. The combined results revealed the importance of both the catalytic activity of USP7 and the physical interaction between USP7 and XPC for deubiquitination of XPC in vivo.
FIGURE 5.
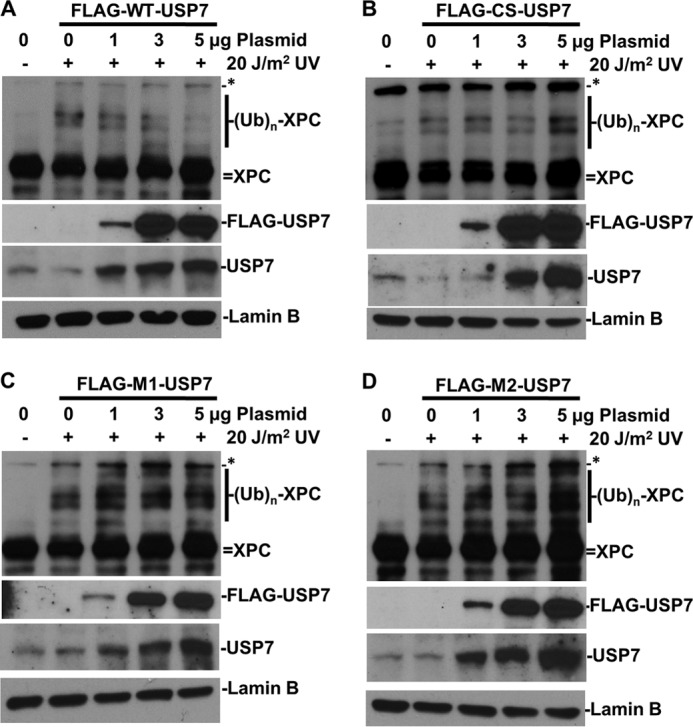
Overexpression of WT-USP7 accelerates XPC deubiquitination in vivo. A vector control or constructs for expressing FLAG-tagged WT or the catalytically inactive C223S mutant (CS) or the interaction-defective M1 and M2 mutant USP7 were transfected into HCT116 cells for 48 h. The transfected cells were UV-irradiated at 20 J/m2 and allowed to repair DNA for 2 h. The XPC, its ubiquitin conjugates, USP7, and FLAG-tagged USP7 were detected by Western blotting using XPC, USP7, and FLAG antibodies. The lamin B blots served as loading control. The asterisk indicates a nonspecific protein band as in Fig. 1D. A, FLAG-tagged WT-USP7. B, FLAG-tagged CS-USP7. C, FLAG-tagged M1-USP7. D, FLAG-tagged M2-USP7. (UB)n, multiple (n) Ub moieties.
USP7 Deubiquitinates XPC in Vitro
To directly examine the deubiquitination activity of USP7 toward XPC, we utilized a cell-free assay system for the in vitro deubiquitination. We used commercial recombinant USP7 against the freshly prepared soluble chromatin fraction from UV-irradiated HCT116-USP7−/− cells as substrates. The preparation contained the enriched XPC-ubiquitin conjugates. As shown in Fig. 6, A and B, compared with the reaction control where no USP7 was added, ubiquitinated XPC was reduced significantly in USP7 deubiquitinase-mediated reactions. His-USP7 efficiently deubiquitinated XPC at a concentration as low as 0.04 μm. We noticed that the amount of native XPC decreased slightly during the deubiquitination reaction. Because ubiquitin-mediated degradation by the 26 S proteasome is an ATP-dependent process and no ATP was supplied in the in vitro deubiquitination reactions, proteasomal degradation is probably not the cause of such a decrease in native XPC. We surmised that, despite an indistinguishable migration pattern, the band of “native” XPC in the soluble chromatin fraction may also harbor some degree of ubiquitination. In reactions with USP7-CD, which contains only the catalytic domain, USP7-CD was also able to deubiquitinate XPC. This is an interesting observation because USP7-CD is incapable of deubiquitinating p53 in vitro (26). The time course of the deubiquitination reaction indicated that the deubiquitination occurred at a faster rate. That is, the deubiquitination was obvious within 30 min, and the ubiquitin conjugates were undetectable by 120 min (Fig. 6C). Again, deubiquitination of the native XPC band was clearly observed in these time course reactions. When the native XPC bands collapsed, two discernable bands were distinguishable at this position. A similar response is also apparent in Fig. 2B. This phenomenon is consistent with the binding of native XPC to the proteasomal ubiquitin receptor S5a (Fig. 6E), supporting the possible presence of ubiquitinated species in the native XPC band. We also noticed that USP7 modestly reduced the protein bands smaller in size than native XPC in deubiquitination reactions. Although these bands were only detected at a longer exposure, it is quite possible that USP7 substrates other than XPC did exist in preparations of chromatin fractions. Therefore, we further examined the deubiquitinating specificity of USP7 toward XPC using the USP7-specific inhibitor HBX 41108 in deubiquitination reactions. The inhibition of USP7 could be seen at lower inhibitor concentrations. It became apparent at 10 μm HBX 41108 in the in vitro deubiquitination assay (Fig. 6D). Compared with the inhibitory effects of HBX 41108 on USP7-mediated p53 deubiquitination (22), HBX 41108 appears to inhibit USP7-mediated XPC deubiquitination with lesser efficiency. Therefore, USP7 may deubiquitinate XPC-ubiquitin conjugates with a greater proficiency than p53-ubiquitin conjugates.
FIGURE 6.

USP7 deubiquitinates XPC-ubiquitin conjugates in vitro. Deubiquitination assays were performed using soluble chromatin (Sol.Chr.) factions isolated from the UV-irradiated HCT116-USP7−/− cells and recombinant His-tagged USP7 (His-USP7) or the USP7 catalytic domain (USP7-CD) in DUB buffer. The reaction mixtures, containing ∼6 μg of soluble chromatin and the indicated amount of His-USP7 or USP7-CD, were incubated at 37 °C and stopped at 120 min. XPC and its ubiquitin conjugates were detected by Western blotting for XPC. The asterisk indicates a nonspecific protein band as in Fig. 1D. A, His-tagged USP7. B, the USP7 catalytic domain. C, time course of the deubiquitination reaction with 0.2 μm His-tagged USP7. D, inhibition of USP7-mediated XPC deubiquitination by HBX 41108. Lane 1L shows lane 1 at a lower exposure. E, XPC deubiquitination assays were performed using affinity-purified WT-USP7 or CS-, M1-, and M2-USP7 mutant proteins. (UB)n, multiple (n) Ub moieties.
We further reinforced our conclusion by testing the in vitro deubiquitination activity of USP7 mutants. The FLAG-tagged USP7 proteins were transiently expressed and purified with anti-FLAG affinity gel. The results showed that WT-USP7 of our preparation reduced XPC ubiquitination in a dose-dependent manner (Fig. 6E), despite that fact that the activity of our WT-USP7 preparation was not as robust as commercial recombinant USP7. Consistent with the lack of in vivo deubiquitination (Fig. 5), the catalytically inactive CS mutant did not show any deubiquitinating activity toward XPC, whereas mutations of M1 and M2 rendered USP7 inactive for XPC deubiquitination in vitro (Fig. 6E), confirming that the physical interaction is important for USP7-mediated XPC deubiquitination. Considering the role of the USP7 UBL1 domain in the USP7-XPC physical interaction, it is odd that USP7-CD displayed deubiquitinating activity in vitro (Fig. 6B), whereas mutants M1 and M2 did not. A possible explanation is that USP7 C-terminal UBL domains other than UBL1 may have a regulatory function in USP7 deubiquitination toward XPC. This caveat notwithstanding, the combined results demonstrate that USP7 displays a deubiquitinating activity toward XPC-ubiquitin conjugates in vitro.
VCP/p97 Participates in Processing Ubiquitinated XPC in Damaged Chromatin When USP7 Is Disrupted
UV-induced XPC degradation in HCT116-USP7−/− cells provides us an unique opportunity to understand how the ubiquitinated XPC is processed in vivo. We next examined XPC status in HCT116-USP7−/− cells under the influence of the proteasome inhibitor MG132 or the VCP/p97 inhibitor DBeQ. Small molecule DBeQ is identified as a potent, specific, and reversible inhibitor of VCP/p97 ATPase (27). As shown in Fig. 7A, DBeQ stabilized XPC and its ubiquitin conjugates at earlier time points and significantly restored the XPC level at 8 h, albeit less effectively than MG132. Like MG132, DBeQ was not able to restore XPC-ubiquitin conjugates at late time point (8 h), probably because of ubiquitin stress caused by these chemicals. These results suggest that VCP/p97 ATPase plays an accessory role in assisting ubiquitin-mediated proteolysis of XPC in HCT116-USP7−/− cells. Next we examined the XPC status within damaged chromatin under conditions of VCP/p97 inhibition by DBeQ pretreatment of the cells. The XPC-ubiquitin conjugates, detectable in soluble chromatin fraction, were increased significantly upon UV irradiation and enhanced further by 4-h DBeQ pretreatment (Fig. 7B). Moreover, the examination of VCP/p97 protein itself showed that, upon UV irradiation, a portion of VCP/p97 indeed translocated to soluble chromatin and that this VCP/p97 translocation was enhanced further by DBeQ treatment.
FIGURE 7.

A USP7 defect leads to UV light-induced XPC degradation that requires VCP/p97. A, effect of the VCP/p97 inhibitor DBeQ on XPC ubiquitination and degradation in HCT116-USP7−/− cells. HCT116-USP7−/− cells were UV-irradiated at 20 J/m2, treated with vehicle (dimethyl sulfoxide, DMSO), MG132, or DBeQ at 10 μm, and harvested at the indicated time points. XPC and histone H2A were detected by Western blotting. B, VCP/p97 inhibition leads to accumulation of ubiquitinated XPC and VCP/p97 on UV-damaged chromatin. HCT116-USP7−/− cells were pretreated with vehicle or DBeQ at 10 μm for 4 h, UV-irradiated at 20 J/m2, and kept under VCP/p97 inhibition for an additional 2 h. Nucleoplasmic (Nucl.), soluble chromatin (Sol.Chr.), and insoluble chromatin (Insol.Chr.) fractions were isolated by chromatin fractionation. The XPC, lamin B, and VCP/p97 proteins were detected by Western blotting in the indicated chromatin fractions containing the same amount of proteins. (UB)n, multiple (n) Ub moieties. C, VCP/p97 colocalizes with XPC at DNA damage spots. VCP/p97 and XPC were visualized in HeLa cells by immunofluorescence using specific antibodies. Scale bar = 10 μm. D, VCP/p97 colocalizes with γH2AX at DNA damage spots. VCP/p97 and γH2AX were visualized in HeLa cells. E, visualization of VCP/p97, γH2AX, and FK2 foci (the foci of ubiquitin conjugates detected by the anti-ubiquitin antibody FK2) in HCT116 and HCT116-USP7−/− cells. Scale bar = 10 μm.
To ascertain whether VCP/p97 is actually recruited to UV-induced damage sites, we next employed a micropore irradiation assay, which can deliver UV light locally and allow the assessment of UV damage responses within these localized areas of the cell nucleus. The results showed that VCP/p97 (red) formed discrete subnuclear spots at the UV damage sites where XPC (green) was specifically recruited (Fig. 7, C and D). Also, γH2AX foci were visualized at the same spots, indicating that VCP/p97 translocates to DNA repair sites. In both parental HCT116 and HCT116-USP7−/− cells, VCP/p97 translocated to damage sites and colocalized with γH2AX foci (Fig. 7E). Interestingly, the formation of FK2 foci (sites of ubiquitin conjugates detected by anti-ubiquitin FK2 antibody) was severely compromised in these cells. It is known that FK2 foci are formed through lysine 63-linked ubiquitination of H2A/γH2AX by the concerted action of RNF8 and RNF168 E3 ligases (28). However, it is currently unclear why loss of USP7 has a profound impact on the formation of FK2 foci. Nevertheless, translocation and intact VCP/p97 recruitment to the damaged chromatin in HCT116-USP7−/− cells suggests that H2A/γH2AX ubiquitination is at least not the trigger for VCP/p97 translocation.
USP7 Disruption Causes Accelerated Dispersal of XPC from DNA Damage Sites and Affects the Repair of UV Light-induced Lesions
We next compared the formation of XPC and γH2AX foci following micropore UV irradiation in parental HCT116 and HCT116-USP7−/− cells (Fig. 8, A and B). The γH2AX foci were detected in ∼30 and 40% of HCT116 cells at 0.5 and 2 h, respectively. In contrast, γH2AX foci were seen in ∼20 and 40% of HCT116-USP7−/− cells at the same time points, indicating a delay in the formation of γH2AX foci because of USP7 disruption. As expected, micropore UV irradiation induced the formation of XPC foci at 0.5 h in ∼60% of HCT116 cells (Fig. 8C). Formation of XPC foci at 0.5 h in HCT116-USP7−/− cells was ∼55%, indicating a similar formation of XPC foci at an early stage of DNA repair in both parental and HCT116-USP7−/−. At 2 h, 45% of HCT116 cells still remained positive for XPC foci. By contrast, the number of XPC foci dropped significantly to 16% 2 h post-irradiation in HCT116-USP7−/− cells. Therefore, we conclude that USP7 disruption causes an accelerated and premature dispersal of XPC from UV-induced DNA damage sites because of XPC degradation.
FIGURE 8.
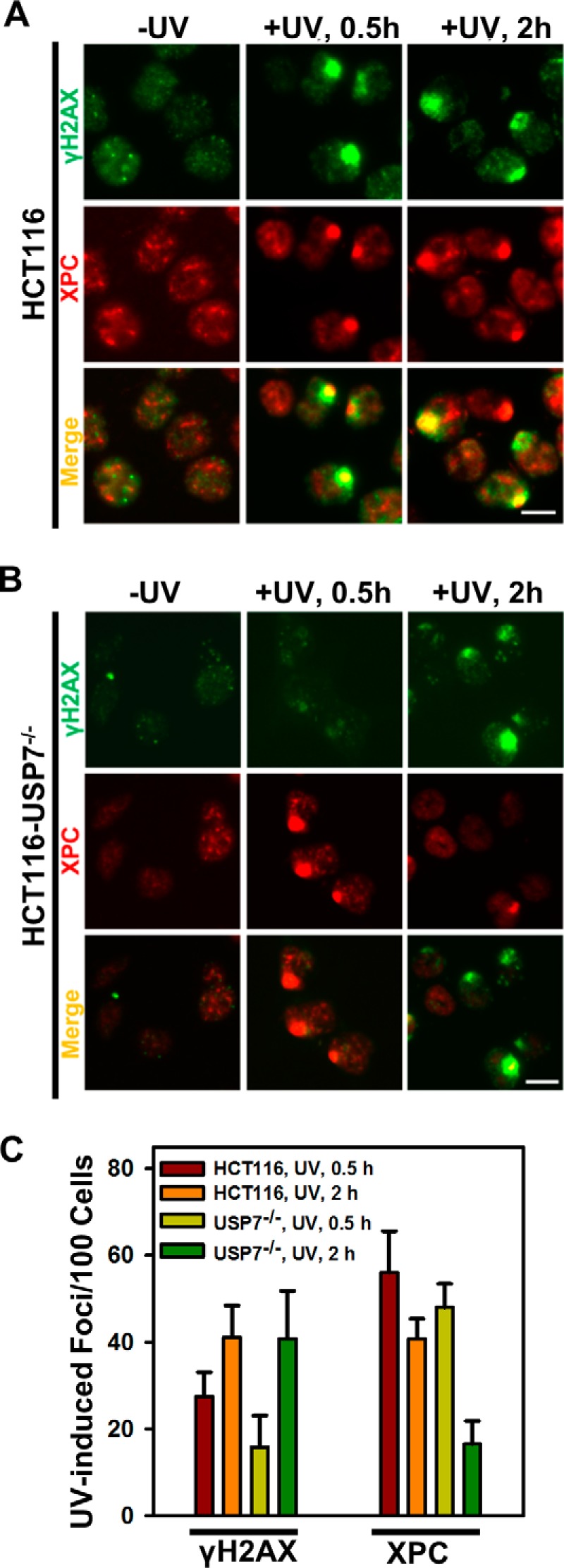
USP7 deficiency leads to accelerated dispersal of XPC from DNA damage spots. A, XPC and γH2AX foci were visualized at 0.5 and 2 h after 100 J/m2 micropore UV irradiation in HCT116 cells. Scale bar = 10 μm. B, XPC and γH2AX foci were visualized at 0.5 and 2 h after 100 J/m2 micropore UV irradiation in HCT116-USP7−/− cells. C, the quantitative data were from the analysis of XPC and γH2AX foci from HCT116 and HCT116-USP7−/− cells. Mean ± S.E. was calculated from four to six microscopic fields of three independent experiments.
Finally, we assessed the impact of USP7 disruption on the GGR pathway which requires XPC to initiate the assembly of the preincision complex on UV-induced photolesions. As shown in Fig. 9, A and B, the removal of CPD was impaired severely in HCT116-USP7−/− cells compared with that in parental HCT116 cells. For example, ∼43% CPD remained in parental HCT116 cells after a repair time of 24 h. In contrast, virtually no CPDs were repaired in the same period in HCT116-USP7−/− cells. Also, the 6-4PP remaining was ∼72, 34, and 6% in parental HCT116 cells at the 2, 4, and 8 h time points, respectively. In comparison, the 6-4PP remaining was ∼69, 56, and 17% in HCT116-USP7−/− cells, indicating a modest but critical decrease in processing of 6-4PP, which is known for its relatively faster rate of repair. In essence, the USP7 disruption severely compromises the slow repair of CPDs and also impacts the fast repair of 6-4PP. Because USP7 has other cellular functions, the relationship of USP7 deficiency impacting GGR through XPC deubiquitination remains correlative. The exact contribution of USP7-mediated XPC deubiquitination in GGR warrants further studies.
FIGURE 9.
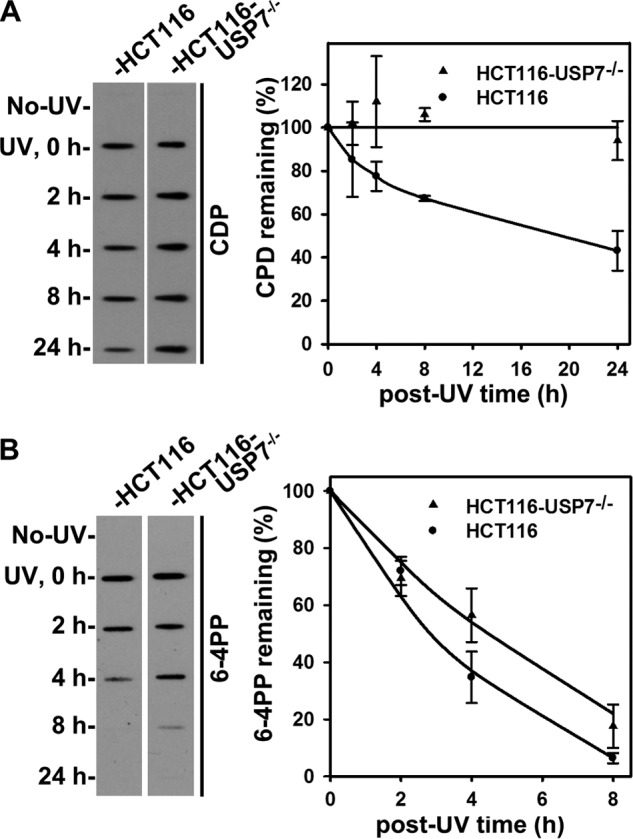
A USP7 defect impairs the repair of UV light-induced photolesions. Shown is an assessment of CPD repair and 6-4PP repair within genomic DNA of HCT116 and HCT116-USP7−/− cells. The cells were starved in serum-free medium for 24 h, UV-irradiated at 20 J/m2, and allowed to repair DNA for the indicated time periods. Identical amounts of genomic DNA from UV-irradiated cells were subjected to immunoslot-blot analysis of CPD or 6-4PP using the corresponding antibodies. The quantitative data indicate mean ± S.E. from four to six independent immunoslot-blot experiments. A, CPD repair. B, 6-4PP repair.
DISCUSSION
USP7 was originally identified as an enzyme that deubiquitinates both p53 and Mdm2 and modulates p53 stability in cells (16, 17). Subsequent studies have revealed more examples in which USP7 affects stability of other proteins, including UHRF1 (25). Recently, a newly identified UV light-sensitive syndrome protein, UVSSA (UV-stimulated scaffold protein A), has been reported to recruit USP7 to regulate transcription-coupled repair by modulating the stability of Cockayne syndrome group B protein and, possibly, RNA polymerase II (29–31). The results presented in this report support a new role of USP7 in the regulation of XPC and, therefore, the GGR pathway. Essentially, USP7 actuates deubiquitination of XPC to prevent its ubiquitin-mediated proteolysis to allow the timely assembly of downstream repair factors and efficient removal of UV light-induced CPDs. Therefore, USP7 plays a critical role in both the GGR and transcription-coupled repair subpathways of NER.
Mechanistically, the orchestration of the NER pathway is quite intricate, and its modulation by USP7 adds another regulatory layer to this process. In the past, our laboratory and others have documented the nuances of UV-induced XPC ubiquitination by the DDB-Cul4A E3 complex (13, 14). The ubiquitinated XPC was relatively stable and was detectable without proteasomal inhibition by MG132, which normally allows the accumulation and detection of unstable or fast-degrading ubiquitin conjugates. On the contrary, XPC ubiquitination after UV irradiation decreased upon proteasomal inhibition (14). This is probably due to the relative stability of ubiquitinated XPC and the cellular ubiquitin stress caused by proteasomal inhibition (Fig. 2D). Because the DDB-Cul4A E3 complex for XPC ubiquitination is also responsible for DDB2 ubiquitination and degradation, the non-lysine 48 linkages of ubiquitin conjugates on XPC are implausible. In addition to this dilemma, XPC ubiquitination did not lead to significant XPC degradation and appeared to be reversible. Therefore, cellular mechanisms must be operational that protect ubiquitinated XPC from being processed via the proteasome. Indeed, hRad23A/B, the molecular partner of XPC in cells, has been shown to help the stabilization of XPC (32). This may explain why ubiquitinated XPC is relatively stable, but it cannot explain how ubiquitinated XPC is converted back to its unmodified state. In this study, we identified USP7 as a DUB for XPC. Disruption of USP7 leads to UV light-induced XPC degradation, suggesting that USP7 plays an important role in protecting XPC from proteolysis and allowing XPC to recycle in the repair process. Nevertheless, we also found that, even in HCT116-USP7−/− cells, ubiquitinated XPC was eventually deubiquitinated when VCP/p97 was inhibited (Fig. 7A). Therefore, when USP7 is absent, the possible involvement of other DUB(s) in XPC deubiquitination cannot be ruled out. Additionally, in the DDB-Cul4A E3 complex, DDB2 is an adapter for XPC, and DDB2 itself is ubiquitinated and degraded by the proteasome. Therefore, even though the same E3 mediates ubiquitination of both XPC and DDB2 proteins, USP7 may only be involved in deubiquitinating XPC.
The DDB complex binds to 6-4PP with a relatively high affinity and specificity (33). Because DDB2 rapidly translocates to damage sites in UV-irradiated cells, even without a functional XPC (34), DDB is probably the first NER factor that recognizes 6-4PP in vivo. The 6-4PPs are also considered to be major triggers for the formation of XPC foci at DNA damage sites in vivo (35). In our study, USP7 disruption, despite DDB2 up-regulation, modestly decreased 6-4PP repair, whereas it severely impacted the repair of CPDs. The USP7 disruption also led to UV light-induced XPC degradation and earlier dispersal of XPC from DNA damage sites. Therefore, 6-4PP could furnish the trigger for UV light-induced XPC degradation in HCT116-USP7−/− cells. The failure of recycling XPC is apparently the cause for severely comprised CDP repair because of USP7 disruption.
UV-induced XPC degradation was observed in HCT116-USP7−/− but not in parental HCT116 cells. The involvement of VCP/p97 in such a process, however, provokes an unanswered question in the ubiquitin field. How are ubiquitinated proteins in chromatin processed or presented to the proteasome? Under VCP/p97 inhibition by DBeQ, more ubiquitinated XPC proteins were detected in the soluble chromatin fraction. It appears that, in this context, ubiquitinated XPC cannot be delivered properly to the proteasome without the assistance of VCP/p97. Perhaps ubiquitinated XPC, having a high affinity toward damaged DNA, is embedded tightly in chromatin in complexes with hHRad23, DDB-Cul4A E3, or other NER factors. Removal of the protein complex containing these DNA repair factors from chromatin requires VCP/p97 function. VCP/p97 recognizes ubiquitinated proteins through interaction with a plethora of cofactors and adapters, including UBXD7 (36–38). A recent study demonstrated that the VCP/p97 core adapters Npl4 and Ufd1 are essential for recruitment of VCP/p97 to UV lesion spots (39), whereas UBXD7 is required for processing Lys-48 ubiquitinated DDB2 in UV light-damaged chromatin. It would be interesting to know whether UBXD7 is involved in proteasomal processing ubiquitin conjugates of XPC in UV light-damaged chromatin in HCT116-USP7−/− cells.
Our detection of VCP/p97 at UV-irradiated damage sites confirmed the earlier observation that VCP/p97 is recruited to DNA damage (40, 41). In these studies, DNA damage was created in BrdU-sensitized cells by laser microirradiation, and VCP/p97 recruitment has been shown to be dependent on RNF8. Whether RNF168 is involved in VCP/p97 recruitment is a matter of debate. In this study, USP7 disruption did not affect VCP/p97 recruitment but, surprisingly, diminished the formation of FK2 foci. Because the lysine 63-linked ubiquitin conjugates of H2A and γH2AX, which form FK2 foci, were generated by concerted action of RNF8 and RNF168 E3 ligases, the involvement of RNF168 in VCP/p97 recruitment to UV lesion spots is very unlikely. Interestingly, a recent study by Puumalainen et al. (39) described that VCP/p97 recruitment to UV lesion spots depends on DDB2 and Cul4A/B, demonstrating that ubiquitination by the DDB-Cul4A E3 complex generates a signal for VCP/p97 recruitment. The same study indicated that VCP/p97 function is important for the timely extraction of DDB2 and XPC from damaged chromatin. Knockdown of VCP/p97, Npl4, or Ufd1 by siRNA leads to chromatin retention of DDB2 and XPC-ubiquitin conjugates. These discoveries are concurrent with our findings that VCP/p97 is involved in proteasomal processing of XPC-ubiquitin conjugates in HCT116-USP7−/− cells. Together, these findings suggest that proteasomal processing of XPC-ubiquitin only occurs when USP7 is disrupted, whereas the extracting XPC-ubiquitin conjugates from chromatin by VCP/p97 operate regardless of USP7 status.
In summary, this study identifies USP7 as a DUB for XPC. A previous finding that XPC was stabilized in primary mouse embryonic fibroblasts from Cul4A−/− mice corroborates our study and is consistent with the function of USP7-dependent XPC deubiquitination in normal cells (42). In a projected model of USP7-mediated XPC regulation (Fig. 10), the USP7 function is required for converting XPC to its unmodified states and preventing XPC from undergoing proteolysis because of UV damage-provoked, DDB-Cul4A-mediated XPC ubiquitination. We also demonstrated the involvement of VCP/p97 in extracting ubiquitinated XPC from damaged chromatin for proteasomal degradation. Because USP7 and VCP/p97 inhibitors are being developed for cancer therapy, it would be important to fully understand the regulatory underpinning of USP7 and VCP/p97 functions in NER and other DNA damage repair pathways. For example, it would be interesting to know whether USP7 and VCP/p97 are involved in the regulation of transcription-coupled repair. It has been shown that VCP/p97 is a regulator of damage-dependent destruction of CDT1, RNA polymerase II (38), and Set8 (43). We anticipate that VCP/p97 may play a broader role in processing ubiquitinated protein clients in damaged chromatin. It would be highly informative to identify new substrates and adapters of VCP/p97 in the regulation of DNA damage responses.
FIGURE 10.
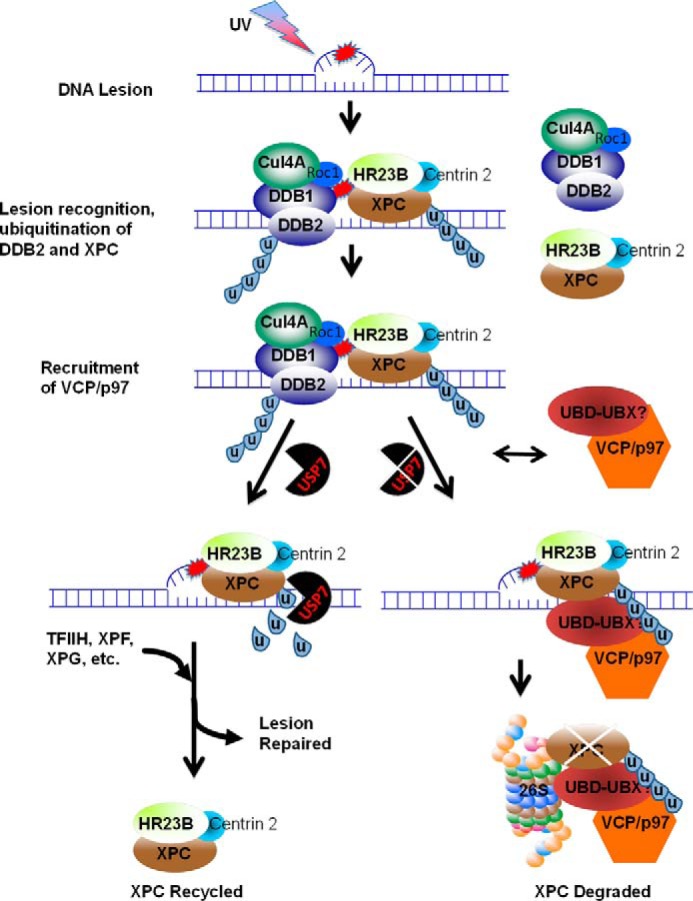
Hypothetical model for the regulation of XPC by USP7 and VCP/p97. In response to UV light-induced DNA damage, the DDB-Cul4A E3 and XPC-hRad23A/B-Centrin 2 complexes are recruited to sites of photolesions. Both DDB2 and XPC are ubiquitinated by DDB-Cul4A E3 ligase. In the presence of USP7, DDB2 is degraded by the proteasome, and the lesions are handed over to XPC. The ubiquitinated XPC binds tightly to DNA damage with a higher affinity than native XPC to allow the timely assembly of transcription factor II H (TFIIH) and other components into the preincision complex. The USP7 deubiquitinates XPC-ubiquitin conjugates, allowing the assembly of the final productive preincision complex, which does not contain XPC. XPC deubiquitination not only converts XPC to its unmodified state but also prevents XPC from proteasomal degradation, allowing XPC to recycle. In the absence of USP7, although photolesions are detected by the DDB and XPC complexes, the final preincision complex fails to assemble productively. The VCP/p97 complex can be recruited independently to damage sites via its adapter, UBD-UBX, interacting with ubiquitinated XPC and/or the E3 ligase complex. The XPC-ubiquitin conjugates are extracted by the VCP/p97 complex from damaged chromatin and presented to the proteasome for proteolysis.
Acknowledgments
We thank Dr. Bert Vogelstein for the HCT116, HCT116-p53−/−, and HCT116-USP7−/− cell lines. We also thank Dr. Yanhui Xu (Department of Biochemistry, Fudan University Medical School) and Dr. Yang Shi (Department of Cell Biology, Harvard Medical School) for the expression constructs of USP7.
This work was supported, in whole or in part, by National Institute of Health Public Health Service Grants ES2388 and ES12991.
- NER
- nucleotide excision repair
- CPD
- cyclobutane pyrimidine dimer
- 6-4PP
- 6-4 photoproduct
- GGR
- global genomic repair
- XP
- xeroderma pigmentosum
- DDB
- UV light-damaged DNA-binding protein
- DUB
- deubiquitinating enzyme
- RIPA
- radioimmune precipitation assay
- CD
- catalytic domain
- CS
- Cockayne syndrome
- UBL
- ubiquitin-like.
REFERENCES
- 1. de Laat W. L., Jaspers N. G., Hoeijmakers J. H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785 [DOI] [PubMed] [Google Scholar]
- 2. Ford J. M., Hanawalt P. C. (1997) Role of DNA excision repair gene defects in the etiology of cancer. Curr. Top. Microbiol. Immunol. 221, 47–70 [DOI] [PubMed] [Google Scholar]
- 3. Venema J., van Hoffen A., Karcagi V., Natarajan A. T., van Zeeland A. A., Mullenders L. H. (1991) Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol. Cell Biol. 11, 4128–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Araki M., Masutani C., Takemura M., Uchida A., Sugasawa K., Kondoh J., Ohkuma Y., Hanaoka F. (2001) Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J. Biol. Chem. 276, 18665–18672 [DOI] [PubMed] [Google Scholar]
- 5. Masutani C., Araki M., Sugasawa K., van der Spek P. J., Yamada A., Uchida A., Maekawa T., Bootsma D., Hoeijmakers J. H., Hanaoka F. (1997) Identification and characterization of XPC-binding domain of hHR23B. Mol. Cell Biol. 17, 6915–6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shivji M. K., Eker A. P., Wood R. D. (1994) DNA repair defect in xeroderma pigmentosum group C and complementing factor from HeLa cells. J. Biol. Chem. 269, 22749–22757 [PubMed] [Google Scholar]
- 7. Sugasawa K., Shimizu Y., Iwai S., Hanaoka F. (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair 1, 95–107 [DOI] [PubMed] [Google Scholar]
- 8. Shell S. M., Hawkins E. K., Tsai M. S., Hlaing A. S., Rizzo C. J., Chazin W. J. (2013) Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair 12, 947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chu G., Chang E. (1988) Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242, 564–567 [DOI] [PubMed] [Google Scholar]
- 10. Hwang B. J., Ford J. M., Hanawalt P. C., Chu G. (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. U.S.A. 96, 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nichols A. F., Ong P., Linn S. (1996) Mutations specific to the xeroderma pigmentosum group E Ddb- phenotype. J. Biol. Chem. 271, 24317–24320 [DOI] [PubMed] [Google Scholar]
- 12. Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A. F., Tanaka K., Nakatani Y. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 [DOI] [PubMed] [Google Scholar]
- 13. Sugasawa K., Okuda Y., Saijo M., Nishi R., Matsuda N., Chu G., Mori T., Iwai S., Tanaka K., Tanaka K., Hanaoka F. (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400 [DOI] [PubMed] [Google Scholar]
- 14. Wang Q. E., Zhu Q., Wani G., El-Mahdy M. A., Li J., Wani A. A. (2005) DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 33, 4023–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 16. Meulmeester E., Maurice M. M., Boutell C., Teunisse A. F., Ovaa H., Abraham T. E., Dirks R. W., Jochemsen A. G. (2005) Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 18, 565–576 [DOI] [PubMed] [Google Scholar]
- 17. Li M., Chen D., Shiloh A., Luo J., Nikolaev A. Y., Qin J., Gu W. (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653 [DOI] [PubMed] [Google Scholar]
- 18. Cummins J. M., Rago C., Kohli M., Kinzler K. W., Lengauer C., Vogelstein B. (2004) Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428, 1. [DOI] [PubMed] [Google Scholar]
- 19. Wani A. A., D'Ambrosio S. M., Alvi N. K. (1987) Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem. Photobiol. 46, 477–482 [DOI] [PubMed] [Google Scholar]
- 20. Anindya R., Aygün O., Svejstrup J. Q. (2007) Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell 28, 386–397 [DOI] [PubMed] [Google Scholar]
- 21. Arab H. H., Wani G., Ray A., Shah Z. I., Zhu Q., Wani A. A. (2010) Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS ONE 5, e11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colland F., Formstecher E., Jacq X., Reverdy C., Planquette C., Conrath S., Trouplin V., Bianchi J., Aushev V. N., Camonis J., Calabrese A., Borg-Capra C., Sippl W., Collura V., Boissy G., Rain J. C., Guedat P., Delansorne R., Daviet L. (2009) Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 8, 2286–2295 [DOI] [PubMed] [Google Scholar]
- 23. Hastak K., Adimoolam S., Trinklein N. D., Myers R. M., Ford J. M. (2012) Identification of a functional in vivo p53 response element in the coding sequence of the xeroderma pigmentosum group C gene. Genes Cancer 3, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El-Mahdy M. A., Zhu Q., Wang Q. E., Wani G., Praetorius-Ibba M., Wani A. A. (2006) Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J. Biol. Chem. 281, 13404–13411 [DOI] [PubMed] [Google Scholar]
- 25. Ma H., Chen H., Guo X., Wang Z., Sowa M. E., Zheng L., Hu S., Zeng P., Guo R., Diao J., Lan F., Harper J. W., Shi Y. G., Xu Y., Shi Y. (2012) M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc. Natl. Acad. Sci. U.S.A. 109, 4828–4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faesen A. C., Dirac A. M., Shanmugham A., Ovaa H., Perrakis A., Sixma T. K. (2011) Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 44, 147–159 [DOI] [PubMed] [Google Scholar]
- 27. Chou T. F., Brown S. J., Minond D., Nordin B. E., Li K., Jones A. C., Chase P., Porubsky P. R., Stoltz B. M., Schoenen F. J., Patricelli M. P., Hodder P., Rosen H., Deshaies R. J. (2011) Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartocci C., Denchi E. L. (2013) Put a RING on it: regulation and inhibition of RNF8 and RNF168 RING finger E3 ligases at DNA damage sites. Front Genet. 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schwertman P., Lagarou A., Dekkers D. H., Raams A., van der Hoek A. C., Laffeber C., Hoeijmakers J. H., Demmers J. A., Fousteri M., Vermeulen W., Marteijn J. A. (2012) UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 44, 598–602 [DOI] [PubMed] [Google Scholar]
- 30. Zhang X., Horibata K., Saijo M., Ishigami C., Ukai A., Kanno S., Tahara H., Neilan E. G., Honma M., Nohmi T., Yasui A., Tanaka K. (2012) Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 44, 593–597 [DOI] [PubMed] [Google Scholar]
- 31. Nakazawa Y., Sasaki K., Mitsutake N., Matsuse M., Shimada M., Nardo T., Takahashi Y., Ohyama K., Ito K., Mishima H., Nomura M., Kinoshita A., Ono S., Takenaka K., Masuyama R., Kudo T., Slor H., Utani A., Tateishi S., Yamashita S., Stefanini M., Lehmann A. R., Yoshiura K., Ogi T. (2012) Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 44, 586–592 [DOI] [PubMed] [Google Scholar]
- 32. Ng J. M., Vermeulen W., van der Horst G. T., Bergink S., Sugasawa K., Vrieling H., Hoeijmakers J. H. (2003) A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 17, 1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujiwara Y., Masutani C., Mizukoshi T., Kondo J., Hanaoka F., Iwai S. (1999) Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem. 274, 20027–20033 [DOI] [PubMed] [Google Scholar]
- 34. Wakasugi M., Kawashima A., Morioka H., Linn S., Sancar A., Mori T., Nikaido O., Matsunaga T. (2002) DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem. 277, 1637–1640 [DOI] [PubMed] [Google Scholar]
- 35. Volker M., Moné M. J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J. H., van Driel R., van Zeeland A. A., Mullenders L. H. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213–224 [DOI] [PubMed] [Google Scholar]
- 36. Meyer H. (2012) p97 complexes as signal integration hubs. BMC. Biol. 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. den Besten W., Verma R., Kleiger G., Oania R. S., Deshaies R. J. (2012) NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat. Struct. Mol. Biol. 19, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Verma R., Oania R., Fang R., Smith G. T., Deshaies R. J. (2011) Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell 41, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puumalainen M. R., Lessel D., Rüthemann P., Kaczmarek N., Bachmann K., Ramadan K., Naegeli H. (2014) Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 5, 3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Acs K., Luijsterburg M. S., Ackermann L., Salomons F. A., Hoppe T., Dantuma N. P. (2011) The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18, 1345–1350 [DOI] [PubMed] [Google Scholar]
- 41. Meerang M., Ritz D., Paliwal S., Garajova Z., Bosshard M., Mailand N., Janscak P., Hübscher U., Meyer H., Ramadan K. (2011) The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 13, 1376–1382 [DOI] [PubMed] [Google Scholar]
- 42. Liu L., Lee S., Zhang J., Peters S. B., Hannah J., Zhang Y., Yin Y., Koff A., Ma L., Zhou P. (2009) CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 34, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raman M., Havens C. G., Walter J. C., Harper J. W. (2011) A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell 44, 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]